

Abstract
Pomalidomide is an immunomodulatory drug, and the dosage of 4 mg per day taken orally on days 1‐21 of repeated 28‐day cycles has been approved in the European Union and the United States to treat patients with relapsed/refractory multiple myeloma. In vitro data showed that pomalidomide is a substrate of multiple cytochrome P450 (CYP) isozymes and that its oxidative metabolism is mediated primarily by CYP1A2 and CYP3A4, with minor contributions from CYP2C19 and CYP2D6. The effect of CYP1A2 inhibition by fluvoxamine (a strong CYP1A2 inhibitor) and CYP1A2 induction by smoking on pomalidomide pharmacokinetics in healthy subjects has been assessed in 2 separate phase 1 open‐label, single‐dose studies. Following administration of a single oral dose of 4 mg pomalidomide, the plasma exposure when coadministered with fluvoxamine was 225.1% and 123.7% of that when administered alone for the total plasma exposure (AUC0‐inf) and the plasma peak exposure (Cmax), respectively. In smokers with elevated CYP1A2 activity demonstrated by high caffeine clearance (a marker of CYP1A2 induction), the AUC0‐inf was 32.3% lower, whereas the Cmax was 14.4% higher than that in nonsmokers. In addition, pomalidomide was safe and well tolerated as a single oral dose of 4 mg in healthy male smokers and nonsmokers ≥ 40 to ≤ 80 years old, and a single oral dose of 4 mg pomalidomide coadministered with multiple oral 50‐mg doses of the CYP1A2 inhibitor fluvoxamine compared with pomalidomide alone was safe and well tolerated by the healthy male subjects.
Keywords: pomalidomide, CYP1A2, inhibition, induction, drug‐drug interaction
The immunomodulatory drug agent pomalidomide has pleiotropic cytotoxic effects against multiple myeloma (MM) cells1, 2 in addition to antiproliferative,3, 4 antiangiogenic,5, 6, 7 and immunomodulatory activity.8, 9 Furthermore, pomalidomide has potent effects on key cytokines, including interleukin‐10, interferon‐γ, and tumor necrosis factor‐α.10 Pomalidomide has been studied for the treatment of various hematologic and nonneoplastic hematologic disorders.11, 12, 13 The dosage of 4 mg per day taken orally on days 1‐21 of repeated 28‐day cycles is approved (in combination with dexamethasone) in the European Union and the United States for the treatment of patients with multiple myeloma who have received ≥2 prior therapies, including lenalidomide and bortezomib (in the European Union; a proteasome inhibitor in the United States), and who have progressed on or within 60 days of completion of the last therapy or have disease progression in the last therapy.14, 15 This combination (pomalidomide plus low‐dose dexamethasone) significantly increased progression‐free survival and overall survival compared with high‐dose dexamethasone.11 Thrombocytopenia, neutropenia, and anemia were the most common grade 3/4 adverse events.11
Pomalidomide pharmacokinetics have been well characterized both in healthy subjects and in subjects with relapsed and refractory MM.16, 17, 18, 19, 20, 21, 22 Pomalidomide was absorbed with a maximum plasma concentration (Cmax) at a median time (tmax) between 2.0 and 3.0 hours. Systemic exposure to a single dose of pomalidomide as determined from the area under the concentration‐time curve (AUC) increased in an approximately dose‐proportional manner up to 50 mg. Pomalidomide has a mean apparent volume of distribution (Vz/F) between 62 and 138 L at steady state. Multiple‐dose exposure over the 0.5‐ to 2‐mg dose range was approximately dose proportional, with pomalidomide reaching steady state by day 3, and accumulation appeared minimal (less than 14.0% for both AUC and Cmax). The extent of plasma protein binding in pooled human male and female plasma ranged from 12.0% to 44.0%.14, 15 A [14C]‐pomalidomide study in healthy adult men23 showed that pomalidomide was extensively metabolized in vivo via multiple metabolic pathways, including oxidation, hydroxylation, and hydrolysis. The mean apparent terminal half‐life (t1/2) of pomalidomide was approximately 7.5 hours, and apparent total plasma clearance (CL/F) generally ranged from 6.5 to 10.8 L/h. Pomalidomide‐related material was eliminated predominantly through renal excretion (∼73.0% of the administered dose), with a low fraction of the dose excreted in urine as unchanged drug (overall < 5.0% of the administered dose).
In vitro, pomalidomide was metabolized via multiple pathways (the metabolites observed were formed primarily via oxidative hydroxylation with subsequent glucuronidation or hydrolysis of the parent compound), and several cytochrome P450 (CYP) enzymes were capable of metabolizing pomalidomide, namely, CYP1A2, CYP3A4, and, to a minor extent, CYP2C19 and CYP2D6. Previously, a phase 1 open‐label study was conducted to assess pomalidomide pharmacokinetics (PK) when coadministered with the CYP3A inhibitor alone and with the CYP3A inhibitor plus CYP1A2 inhibitor.21 The total drug exposure of a single 4‐mg dose of pomalidomide increased by 18.8% and 145.7% in the presence of the CYP3A inhibitor (ketoconazole) and in the presence of both CYP3A and CYP1A2 inhibitors (ketoconazole + fluvoxamine), respectively, compared with pomalidomide alone.21 Because of the possible synergistic effect between CYP450 enzymes, it is challenging to delineate quantitatively the pronounced increases of pomalidomide exposure to CYP1A2 inhibition and CYP3A inhibition.
Based on the US Food and Drug Administration (FDA) guidance on clinical drug interaction studies,24 if an investigational drug is a CYP1A2 substrate, the sponsor should consider conducting a study in smokers based on the intended patient population and the effect of CYP1A2 induction on the drug's exposure. Study A was conducted to evaluate the effect of CYP1A2 induction by smoking on pomalidomide PK in healthy subjects. In addition, following the prior CYP3A inhibition study, an understanding of the contribution of CYP1A2 inhibition alone to changes in exposure to pomalidomide was sought to complement the quantification of the contributions of CYP3A and CYP3A plus CYP1A2 established in the prior CYP3A inhibition study. study B was conducted to assess the effect of CYP1A2 inhibition by fluvoxamine alone on pomalidomide PK in healthy subjects.
Methods
All subjects provided written informed consent prior to screening. These studies were conducted and monitored in accordance with Celgene procedures and the study protocols. These procedures complied with the ethical principles of the International Conference on Harmonisation harmonized tripartite guideline E6 (R1): Good Clinical Practice, as required by the major regulatory authorities. The conduct also complied with the Declaration of Helsinki, Title 21 of the US Code of Federal Regulations, Parts 50 and 56, concerning informed consent and institutional review board regulations and applicable national, state, and local laws or regulations.
Study Design
CC‐4047‐CP‐011 (Study A)
Study A was an open‐label, nonrandomized study with 2 parallel cohorts. During study participation, healthy male smokers were required to smoke approximately 25 cigarettes per day for a total of 10 days (days 1 to 10) approximately evenly distributed throughout typical waking hours of each day. Nonsmokers did not smoke and were not in the presence of smokers while on site. One of the exploratory objectives of this study was to evaluate the relative CYP1A2 metabolism by caffeine (a commonly used probe substrate for CYP1A2 activity) clearance in healthy smokers and nonsmokers. To assess caffeine clearance, all subjects orally received a 200‐mg caffeine capsule on day 6 of each cohort, and on day 8, subjects received a single oral 4‐mg dose of pomalidomide. All subjects were on a methylxanthine‐free diet (other than caffeine administered for test purposes) for at least 1 week before and during the study (no chocolate, cocoa, soft drinks, coffee, tea, or foods containing these ingredients). Within no more than 21 days (day ‐21) and no less than 2 days (day ‐2) prior to the start of the first period, healthy male subjects underwent routine screening procedures including physical examination, 12‐lead electrocardiogram (ECG), vital signs, clinical laboratory safety tests (serum/plasma chemistry, hematology, and urinalysis), serology screen, and drug/alcohol/cotinine screen. Eligible subjects returned to the study center on day ‐1 for baseline assessments and to start the in‐clinic stay. On the morning of day 1 of each cohort, subjects who continued to be qualified for participation in the study were enrolled in cohort A (smokers) or cohort B (nonsmokers), and each subject in each cohort received the following oral dosing regimens: a single oral dose of 200 mg caffeine on day 6 and a single oral dose of 4 mg pomalidomide on day 8. Subjects fasted overnight (at least 10 hours) prior to the caffeine clearance test on day 6. Similarly, subjects fasted overnight prior to the 4‐mg pomalidomide dose on day 8 and continued to fast for at least 4 hours after dosing. During each cohort, subjects were housed at the study center from day ‐1 through the morning of day 10. Subjects were discharged from the study center on day 10 on completion of study procedures.
CC‐4047‐CP‐012 (Study B)
This was a single‐center, open‐label, nonrandomized study. The entire study consisted of a screening phase, a treatment period, and a follow‐up telephone call for safety. Within no more than 21 days (day ‐21) and no less than 2 days (day ‐2) prior to the start of the treatment period, subjects underwent routine screening procedures. These included the following: a 12‐lead ECG, vital signs, clinical laboratory safety tests (serum/plasma chemistry, hematology, and urinalysis), serology screen, and drug and alcohol screen. Eligible subjects returned to the study center on day ‐1 for baseline assessments and confirmation of enrollment criteria. Subjects who continued to be qualified for participation enrolled on the morning of day 1.
Each subject received the following oral dosing regimens: a single oral dose of 4 mg pomalidomide in the morning on day 1, dosing withheld from days 2 to 3, twice‐daily oral doses of 50 mg fluvoxamine (a known strong inhibitor of CYP1A2) from days 4 to 7, a single oral dose of 4 mg pomalidomide in the morning plus a twice‐day oral dose of 50 mg fluvoxamine on day 8, and twice‐daily oral doses of 50 mg fluvoxamine on days 9 and 10.
Blood Collection for Pharmacokinetic Analysis
In study A, serial blood samples (approximately 5 mL per blood draw) were collected at the following times: for pomalidomide (on day 8), predose (zero hour), 0.5, 1, 1.5, 2, 2.5, 3, 6, 8, 12, 24, 36, and 48 hours post–pomalidomide dose; and for caffeine and paraxanthine (on day 6), predose (zero hour), 0.5, 1, 2, 4, 6, 8, 10, and 12 hours post–caffeine dose.
In study B, serial blood samples (approximately 5 mL per blood draw) were collected at the following times: for pomalidomide, on both day 1 and day 8, predose (zero hour), 1, 2.5, 3, 6, 12, 24, 36, 48, 60, and 72 hours postdose; and for fluvoxamine, predose on day 1 and 4 hours postdose on days 4 to 10.
Safety Assessment
Safety was monitored throughout both studies. Safety evaluations included adverse event (AE) reporting, physical examinations, vital sign measurements, 12‐lead ECGs, and clinical laboratory safety tests. All concomitant medications were assessed and recorded throughout the study from the time the informed consent document (ICD) was signed until study completion (follow‐up safety telephone call). Adverse events and severe adverse events (SAEs) were assessed and recorded from the time the subject signed the ICD until study completion (follow‐up safety telephone call), and when made known to the investigator within 28 days after the last dose of investigational product (and those SAEs made known to the investigator at any time thereafter that were suspected of being related to the investigational product).
Bioanalytical Methodology
To determine human plasma samples for pomalidomide and CYP1A2 inhibitor (caffeine, paraxanthine, and fluvoxamine on pomalidomide) concentrations, the validated liquid chromatography‐tandem mass spectrometry (MS/MS) assays were used. The lower limit of quantification was 0.25 ng/mL for pomalidomide, 0.5 ng/mL for fluvoxamine, and 20 ng/mL for both caffeine and paraxanthine. These were subsequently processed by liquid‐liquid extraction and then analyzed using reverse‐phase high‐performance liquid chromatography (HPLC) with electrospray MS/MS detection.23
Pharmacokinetic Analyses
Noncompartmental PK parameters such as Cmax, tmax, AUC0‐t, AUC0‐inf, t1/2, CL/F, and Vz/F were calculated from the plasma concentration‐time data with PhoenixWinNonlin Professional version 6.3 (Pharsight, a Certara company, St. Louis, Missouri). Actual sampling times were used in the calculations. Descriptive statistics (n, mean, standard deviation [SD], coefficient of variation [CV%], geometric mean, geometric CV%, median, minimum, and maximum) were provided for concentrations at each time and for all PK parameters.
Statistical Analyses
In study A, sample size was based on empirical considerations, and no formal sample‐size calculation was performed. Twenty‐eight male subjects (14 smokers, 14 nonsmokers) were enrolled in the study. To assess the effect of smoking on the PK of pomalidomide, an analysis of variance (ANOVA) was performed on natural log‐transformed data of Cmax, AUC0‐t, and AUC0‐inf using MIXED procedures in SAS. The MIXED model contained the term cohort as a fixed effect. The geometric means, percent ratios of the geometric means (smoker/nonsmoker), and 90% confidence interval (CI) for the geometric mean ratio were calculated. The tmax was analyzed by nonparametric method to generate the difference in medians between treatment (smoker and nonsmoker) and 90%CIs of the median difference. To assess the effect of smoking on the PK of caffeine and paraxanthine/caffeine ratio, an ANOVA was performed on natural log‐transformed data of Cmax, AUC0‐t, and AUC0‐inf using MIXED procedures in SAS. The MIXED model contained the term cohort as a fixed effect. The geometric means, percent ratios of the geometric means (smoker/nonsmoker), and 90%CIs for the geometric mean ratios were provided. The tmax was analyzed by nonparametric method to generate the difference in medians between cohorts (smoker and nonsmoker) and 90%CI of the median difference.
In study B, no formal sample size calculation was performed. Fifteen healthy, adult male subjects were enrolled in the study. To assess the effect of fluvoxamine on the PK of pomalidomide, an ANOVA was performed on the natural log‐transformed AUC0‐t, AUC0‐inf, and Cmax to estimate the ratio of geometric means between the treatments (pomalidomide plus fluvoxamine versus pomalidomide) and its 90%CI. The ANOVA model included treatment (pomalidomide and pomalidomide plus fluvoxamine) as a fixed effect and subject as a random effect.
All safety assessments, including AEs, vital sign measurements, clinical laboratory information, concomitant medications, physical examinations, and ECG interpretations, were tabulated and summarized as appropriate.
Results
Demographic and Other Baseline Characteristics
A total of 28 subjects were enrolled in study A. Demographic data are presented in Table 1. Mean and minimum body mass index were slightly lower in smokers than in nonsmokers, whereas mean age and range were similar in smokers and nonsmokers. All enrolled subjects satisfied the inclusion and exclusion criteria, with no clinically significant abnormalities prior to administration of the first dose, and the investigator approved all the subjects for study participation.
Table 1.
Demographic and Other Baseline Characteristics
| Study A | Study B | |||
|---|---|---|---|---|
| Variable | Smokers (n = 14) | Nonsmokers (n = 14) | Total (n = 28) | Total (n = 15) |
| Age (y), mean (range) | 51.9 (40 to 67) | 50.4 (40 to 66) | 51.1 (40 to 67) | 34.2 (23.0 to 50.0) |
| Height (cm), mean (range) | 173. 9 (168.2 to 184.0) | 177.1 (162.0 to 191.7) | 175.5 (162.0 to 191.7) | 176.6 (164.0 to 188.1) |
| Weight (kg), mean (range) | 80.4 (50.0 to 116.0) | 91.2 (73.0 to 110.4) | 85.8 (50.0 to 116.0) | 84.3 (64.8 to 98.6) |
| BMI (kg/m2), mean (range) | 26.6 (17.7 to 36.1) | 29.1 (24.6 to 36.9) | 27.8 (17.7 to 36.9) | 27.0 (20.8 to 31.3) |
| Race, n (%) | ||||
| White | 13 (92.9) | 8 (57.1) | 21 (75.0) | 8 (53.3) |
| Black or African American | 1 (7.1) | 6 (42.9) | 7 (25.0) | 7 (46.7) |
| Ethnicity, n (%) | ||||
| Hispanic or Latino | 1 (7.1) | 1 (7.1) | 2 (7.1) | 4 (26.7) |
| Not Hispanic or Latino | 13 (92.9) | 13 (92.9) | 26 (92.9) | 11 (73.3) |
BMI, body mass index; n, number of subjects.
A total of 15 subjects were enrolled in study B, and 14 subjects (93.3%) completed the study. Demographic data are presented in Table 1. All subjects were male, with a mean age of 34.2 years. The majority were not Hispanic or Latino (73.3%). There were a similar number of black or African American and white subjects (7 and 8 subjects, respectively).
Effect of the CYP1A2 Inhibitor Fluvoxamine on Pomalidomide PK
Mean ± SD pomalidomide plasma concentration profiles from a single oral dose of 4 mg pomalidomide when administered alone and when administered with the CYP1A2 inhibitor fluvoxamine are presented in Figure 1. Mean pomalidomide plasma concentration‐time profiles were well characterized over the 72‐hour postdose sampling interval. A summary of the PK parameters of pomalidomide when administered alone and when administered with the CYP1A2 inhibitor fluvoxamine is presented in Table 2. By ANOVA analysis, the PK parameters were not equivalent when pomalidomide was administered with fluvoxamine and when pomalidomide was administered alone, demonstrated by total pomalidomide plasma exposure (AUC0‐inf), which increased by 125.1% when pomalidomide was administered with fluvoxamine compared with when administered alone (geometric mean, 1179.4 and 526.5 ng·h/mL, respectively). Peak exposure to pomalidomide (Cmax) increased by 23.7% when pomalidomide was administered with fluvoxamine compared with when administered alone (geometric mean, 59.5 and 49.2 ng/mL, respectively; see Tables 2 and 3). Mean t1/2 of pomalidomide when administered alone was less than that when administered with fluvoxamine (6.0 and 13.1 hours, respectively). Mean CL/F of pomalidomide when administered alone was greater than that when administered with fluvoxamine (7.6 and 3.4 L/h, respectively). The mean Vz/F of pomalidomide when administered alone was similar to that when administered with fluvoxamine (65.4 and 64.0 L, respectively). In general, the PK parameters from this study were similar to those from the previous drug interaction study (7.6‐8.3 L/h, 3.1‐3.3 hours, and 6.1 hours for CL/F, tmax, and t1/2 from a prior drug interaction study, respectively).21
Figure 1.
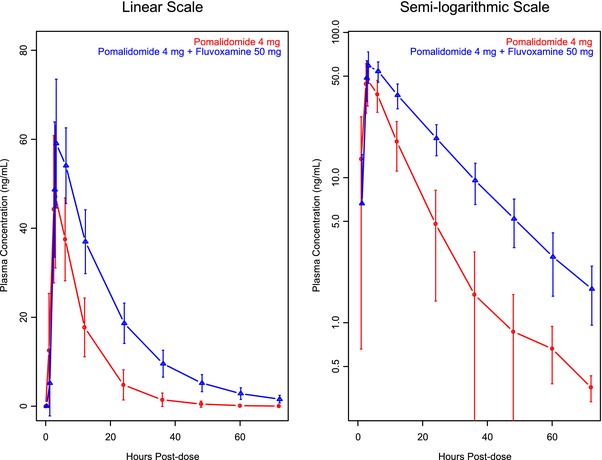
Mean ± SD plasma concentration of pomalidomide‐time profiles by treatment (study B).
Table 2.
Summary of Pomalidomide Plasma Pharmacokinetic Parameters (Study B)
| PK Parameters of Pomalidomide | Pomalidomide 4 mg Administered Alone, n = 15 | Pomalidomide 4 mg Administered With Fluvoxamine, n = 14 |
|---|---|---|
| AUC0‐inf (ng·h/mL) | 526.5 (33.2) | 1179.4 (23.4) |
| Cmax (ng/mL) | 49.2 (26.2) | 59.5 (21.9) |
| tmax (h)a | 3.0 (2.5, 6.0) | 4.5 (3.0, 6.0) |
| t1/2 (h) | 6.0 (28.9) | 13.1 (15.8) |
| CL/F (L/h) | 7.6 (33.2) | 3.4 (23.4) |
| Vz/F (L) | 65.4 (16.9) | 64.0 (16.8) |
Geometric mean (geometric CV%) data from descriptive statistics are presented.
AUC0‐inf, area under the plasma concentration‐time curve from time 0 to infinity; CL/F, apparent total plasma clearance; Cmax, maximum plasma drug concentration; n, number of subjects; t1/2, terminal half‐life; tmax, time to Cmax; Vz/F, apparent volume of distribution during the terminal phase when dosed orally.
Median (minimum, maximum).
Table 3.
Statistical Analysis of Pomalidomide Plasma Pharmacokinetic Parameters With and Without Fluvoxamine
| Pharmacokinetic Parameter (Unit) | Treatment | n | Geometric Meana | Ratio (%) of Geometric Means | 90%CI of Ratio of Geometric Means |
|---|---|---|---|---|---|
| AUC0‐inf (ng·h/mL) | Administered with fluvoxamine | 14 | 1185.2 | 225.1 | (198.0‐257.0) |
| Administered alone | 15 | 526.5 | |||
| Cmax (ng/mL) | Administered with fluvoxamine | 14 | 60.8 | 123.7 | (116.5‐131.3) |
| Administered alone | 15 | 49.2 |
AUC0‐inf, area under the plasma concentration‐time curve from time 0 to infinity; CI, confidence interval; Cmax, maximum plasma drug concentration; n, number of subjects.
Geometric means from analysis of variance analysis.
Pomalidomide Pharmacokinetics in Smokers Versus Nonsmokers
Mean plasma concentration‐time profiles of pomalidomide from smokers and nonsmokers are presented in Figure 2. Mean plasma concentration‐time profiles of pomalidomide in both populations were well characterized over the 48‐hour postdose sampling interval. A summary of the pomalidomide plasma PK parameters from smokers and nonsmokers is presented by smoking status in Table 4. By ANOVA analysis, the PK parameters were not equivalent in smokers and nonsmokers, as demonstrated by the AUC0‐inf, which decreased by 32.3% in smokers compared with in nonsmokers (geometric mean, 463.0 and 684.0 ng·h/L, respectively), whereas the Cmax increased by 14.4% in smokers compared with that in nonsmokers (geometric mean, 64.4 and 56.3 ng/mL, respectively); see Tables 4 and 5. The t1/2 of pomalidomide in smokers was less than that in nonsmokers (4.8 and 7.8 hours, respectively). The mean CL/F of pomalidomide in smokers was greater than that in nonsmokers (8.6 and 5.8 L/h, respectively). In general, the PK parameters from the nonsmokers were similar to those from the previous drug interaction study (7.6‐8.3 L/h, 3.1‐3.3 hours, and 6.1 hours for CL/F, tmax, and t1/2, respectively, from a prior drug interaction study).21
Figure 2.
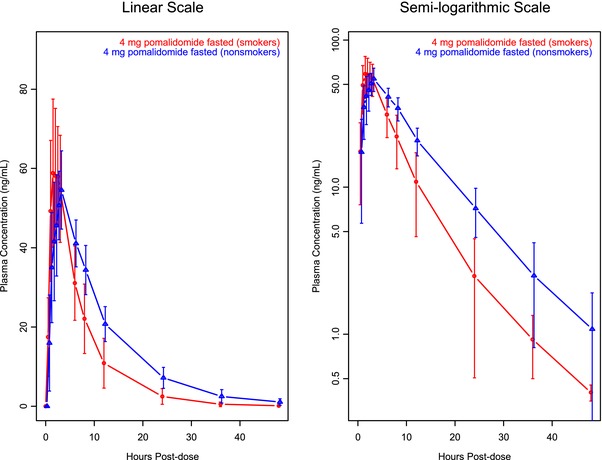
Mean ± SD pomalidomide plasma concentration‐time profiles: CYP1A2 induction effect on pomalidomide (study A).
Table 4.
Summary of Pomalidomide Plasma Pharmacokinetic Parameters by Smoking Status (Study A)
| Pomalidomide PK Parameters | Nonsmokers, n = 13 | Smokers, n = 14 |
|---|---|---|
| AUC0‐inf (ng·h/mL) | 684.4 (19.6) | 463.1 (32.5) |
| Cmax (ng/mL) | 56.3 (16.6) | 64.4 (18.7) |
| tmax (h)a | 3.0 (1.5, 6.0) | 1.8 (1.0, 3.0) |
| t1/2 (h) | 7.8 (18.3) | 4.8 (29.9) |
| CL/F (L/h) | 5.8 (19.6) | 8.6 (32.5) |
| Vz/F (L) | 65.8 (10.0) | 59.7 (24.9) |
Geometric mean (geometric CV%) data from descriptive statistics are presented.
AUC0‐inf, area under the plasma concentration‐time curve from time 0 to infinity; CL/F, apparent total plasma clearance; Cmax, maximum plasma drug concentration; n, number of subjects; t1/2, terminal half‐life; tmax, time to Cmax; Vz/F, apparent volume of distribution during the terminal phase when dosed orally.
Median (minimum, maximum).
Table 5.
Statistical Comparison of Pomalidomide Plasma Pharmacokinetic Parameters (AUC0‐inf and Cmax): Effect of CYP1A2 Induction
| Pharmacokinetic Parameter (Uunit) | Treatment | n | Geometric Mean | Comparison | Ratio (%) of Geometric Means | 90%CI of Ratio of Geometric Means |
|---|---|---|---|---|---|---|
| AUC0‐inf (ng·h/mL) | Smokers | 14 | 463.1 | Smokers/nonsmokers | 67.7 | (56.8‐80.6) |
| Nonsmokers | 13 | 684.4 | — | — | — | |
| Cmax (ng/mL) | Smokers | 14 | 64.4 | Smokers/nonsmokers | 114.4 | (101.9‐128.4) |
| Nonsmokers | 13 | 56.3 | — | — | — |
AUC0‐inf, area under the plasma concentration‐time curve from time 0 to infinity; Cmax, maximum plasma drug concentration; CI, confidence interval; n, number of subjects.
Relationship Between Smoking Status and Caffeine Clearance and the Correlation Between Caffeine Clearance and Pomalidomide Clearance
The relationships between smoking status and caffeine clearance and between caffeine clearance and pomalidomide clearance have been explored, and the results are presented in Figures 3 and 4.
Figure 3.
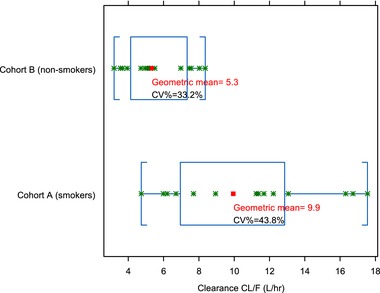
Relationship between smoking status and caffeine clearance. CL/F, apparent total plasma clearance.
Figure 4.
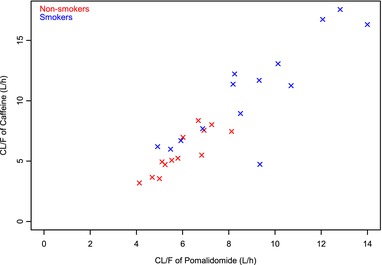
Scatterplot of caffeine clearance versus pomalidomide clearance. CL/F, apparent total plasma clearance.
Caffeine plasma clearance was higher in smokers than in nonsmokers (9.9 and 5.3 L/h, respectively). In addition, there was good correlation between caffeine clearance and pomalidomide clearance in vivo.
Safety
In study A, overall, 21 of 55 subjects (38.2%) reported 37 treatment‐emergent adverse events (TEAEs). There were 12 subjects (21.8%) who reported 22 TEAEs that were suspected by the investigator of being related to the study drug. There were 3 TEAEs of moderate severity (all instances of presyncope occurring near the time of blood sample collection), none of which the investigator considered related to the study drug. No subject experienced an SAE or severe TEAE, and no subjects withdrew from the study because of a TEAE. The incidence of TEAEs in smokers appeared higher than in nonsmokers. However, because TEAEs were recorded after caffeine administration on day 6 but prior to pomalidomide administration on day 8, these numbers overstated the number of TEAEs attributable to pomalidomide. When only TEAEs that occurred after pomalidomide dosing were considered, there was no difference in the incidence of TEAEs between smokers and nonsmokers. There were no deaths.
In study B, overall, only 1 of 15 subjects (6.7%) reported 1 TEAE. One subject (7.1%) reported 1 TEAE in the 50‐mg fluvoxamine treatment. No subject reported at least 1 TEAE related to the study drug. No deaths, SAEs, or TEAEs leading to discontinuation were reported. The TEAE of dry lip was reported by 1 subject (7.1%) in the 50‐mg fluvoxamine treatment and was the only TEAE reported during the study. The TEAE of dry lip was resolved by the end of the study. One subject (7.1%) in the 50‐mg fluvoxamine treatment reported a TEAE that was mild in severity and was not suspected of being related to the study drug. No moderate or severe TEAEs were reported during the study. One subject was discontinued as per management discretion because of inappropriate behavior while in the clinical unit.
Discussion
Evaluation of drug‐drug interactions (DDIs) is an important part of drug development because patients frequently use more than 1 medication at a time. A clinically relevant change in exposure of a coadministered drug can lead to loss of efficacy or an adverse drug reaction (ADR). In the general population, 20.0%‐30.0% of all ADRs have been attributed to DDIs.25, 26 Some severe DDIs have resulted in market withdrawals and major revisions to product labels. It becomes especially critical among oncology drugs as they are typically administered at or close to the maximum tolerated dose.27 Extensive metabolism of pomalidomide prior to elimination has been demonstrated in both in vitro and in vivo studies, suggesting that pomalidomide may have DDI potential as a victim drug.23 Prior in vivo DDI assessment showed that coadministration of ketoconazole, a strong inhibitor of both CYP3A isozymes and P‐glycoprotein, with pomalidomide induced a mild increase in pomalidomide exposure that was not considered clinically relevant. However, concomitant dosing of pomalidomide with both the strong CYP1A2 inhibitor fluvoxamine and the strong CYP3A inhibitor ketoconazole approximately doubled the mean exposure to pomalidomide in healthy men.21 A limitation of this study design was that it was not possible to determine whether the significant increase in pomalidomide exposure was because of CYP1A2 inhibition alone or of a synergistic interaction requiring strong concomitant CYP3A isozyme and CYP1A2 inhibition. Therefore, an understanding of the contribution of CYP1A2 inhibition alone to changes in exposure to pomalidomide is sought to complement the quantification of the contributions of CYP3A and CYP3A plus CYP1A2 established in the prior in vivo inhibition study.
Based on the FDA guidance on clinical drug interaction studies, if an investigational drug is a CYP1A2 substrate, the sponsor should consider conducting a study in smokers based on the intended patient population and the effect of CYP1A2 induction on the drug's exposure.24 Consistently, published literature has shown that polycyclic aromatic hydrocarbons are some of the major lung carcinogens found in tobacco smoke that are potent inducers of the hepatic cytochrome P450 (CYP) isoenzyme 1A2.28, 29, 30, 31, 32, 33, 34, 35, 36 However, there is considerable interindividual variability in both basal and induced CYP1A2 activity states, so creating a reproducible enzyme‐induced state can be challenging. The variability is considered multifactorial, with the potential to be affected by both intrinsic and extrinsic factors. Differences in the individual ability to metabolize CYP1A2 substrates has been linked to extrinsic and some intrinsic factors such as diet (eg, cruciferous vegetables, grilled/broiled meats), smoking (induction), contraceptive use (inhibition), and certain cancers.37, 38 Caffeine clearance has been described as the gold standard marker of CYP1A2 induction, and among the published methods, a saliva‐ or plasma‐based determination of the paraxanthine‐to‐caffeine ratio approximately 6 hours after a defined amount (usually 100‐200 mg) of caffeine intake is both convenient and fully validated.39 Phenotypic assessment of baseline and induced CYP1A2 activity using caffeine clearance was included in study A to allow evaluation of potential variability.
The primary objective of study A was to evaluate the effect of smoking on the PK of pomalidomide. One of the exploratory objectives was to evaluate relative CYP1A2 metabolism by caffeine (a commonly used probe substrate for CYP1A2 activity) clearance in healthy smokers and nonsmokers. The smokers selected to participate were heavy smokers and, per protocol, were required to smoke approximately 25 cigarettes per day prior to pomalidomide dosing. Caffeine clearance was used as a marker of CYP1A2 activity. Consistent with the published literature, it showed that the caffeine clearance was approximately 2‐fold higher in smokers than in nonsmokers (9.9 and 5.3 L/h, respectively), and as expected, smokers showed slightly higher intersubject variability on caffeine clearance (43.8% and 33.2%, respectively); see Figure 3. In addition, there was good correlation between caffeine clearance and pomalidomide clearance, supporting that CYP1A2‐mediated metabolism of pomalidomide is predominant in vivo (Figure 4). The Cmax of pomalidomide was 14.4% higher in smokers than that in nonsmokers, whereas the AUC0‐inf of pomalidomide was 32.3% lower in smokers than in nonsmokers (Table 5). The induction of CYP1A2 caused by smoking, however, still yielded exposure within the range of efficacious exposure seen in the general population.
Study B was a single‐center, open‐label, nonrandomized study. The primary objective of this study was to evaluate the PK of pomalidomide administered with the CYP1A2 inhibitor fluvoxamine compared with pomalidomide alone in healthy male subjects. For fluvoxamine coadministration versus pomalidomide alone, the ratio of AUC was greater than that of Cmax (225.1% and 123.7%, respectively). This suggests that the predominant effect of coadministration of fluvoxamine was on pomalidomide clearance by the inhibition of hepatic CYP1A2 and, to a lesser extent, on pomalidomide absorption by the inhibition of intestinal CYP1A2, consistent with the more pronounced increases in t1/2 and decreases in CL/F and less pronounced increases in tmax and Cmax. Pharmacokinetic results showed that fluvoxamine coadministration had both a statistically and clinically remarkable increase in pomalidomide exposure compared with administration of pomalidomide alone. The magnitude of the mean increase (approximate doubling of exposure [AUC]) caused by fluvoxamine alone was similar to that observed when the second inhibitor, ketoconazole (which inhibits CYP3A), was coadministered with fluvoxamine (the prior CYP3A inhibition). This result indicated that CYP1A2 inhibition or the interaction of CYP1A2 and CYP3A, but not CYP3A, primarily drives observed changes in pomalidomide exposure and clearance. When both inhibitors are coadministered with pomalidomide, the initial dose should be half the normal starting dose. When the CYP1A2 inhibitor is administered alone with pomalidomide, the initial dose should also be halved.
Overall, pomalidomide was safe and well tolerated as a single oral dose of 4 mg in healthy male smokers and nonsmokers ≥ 40 to ≤ 80 years old from study A, and a single oral dose of 4 mg pomalidomide coadministered with multiple oral 50‐mg doses of the CYP1A2 inhibitor fluvoxamine compared with pomalidomide alone was safe and well tolerated by the healthy male subjects in study B. No clinical laboratory parameter or abnormal 12‐lead ECG result was considered clinically significant or reported as a TEAE by the investigator, and no individual vital sign result was reported as a TEAE by the investigator from study B.
In conclusion, the current studies showed that the mean pomalidomide exposure when administered with fluvoxamine was 225.1% and 123.7% of that when administered alone for AUC0‐inf and Cmax, respectively. The AUC0‐inf was 32.3% lower in smokers than that in nonsmokers, whereas the Cmax was 14.4% higher in smokers than that in nonsmokers. Based on the PK results from these studies, pomalidomide prescribing information approved by the FDA recommends to “avoid concomitant use of strong CYP1A2 inhibitors. If a strong CYP1A2 inhibitor must be used, reduce POMALYST dose by 50%,” and “Cigarette smoking reduces pomalidomide AUC by 32% due to CYP1A2 induction. Advise patients that smoking may reduce the efficacy of pomalidomide.”
Declaration of Conflicting Interests
Yan Li, Liangang Liu, Xiaomin Wang, Chengyue Zhang, Josephine Reyes, Matthew Hoffmann, Maria Palmisano, and Simon Zhou are employees of and hold equity ownership in Celgene Corporation.
Funding
The two clinical pharmacology studies were sponsored and funded by Celgene Corporation.
References
- 1. Zhu D, Corral LG, Fleming YW, Stein B. Immunomodulatory drugs Revlimid (lenalidomide) and CC‐4047 induce apoptosis of both hematological and solid tumor cells through NK cell activation. Cancer Immunol Immunother. 2008;57(12):1849–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mitsiades N, Mitsiades CS, Poulaki V, et al. Apoptotic signaling induced by immunomodulatory thalidomide analogs in human multiple myeloma cells: therapeutic implications. Blood. 2002;99(12):4525–4530. [DOI] [PubMed] [Google Scholar]
- 3. Hideshima T, Chauhan D, Shima Y, et al. Thalidomide and its analogs overcome drug resistance of human multiple myeloma cells to conventional therapy. Blood. 2000;96(9):2943–2950. [PubMed] [Google Scholar]
- 4. Verhelle D, Corral LG, Wong K, et al. Lenalidomide and CC‐4047 inhibit the proliferation of malignant B cells while expanding normal CD34+ progenitor cells. Cancer Res. 2007;67(2):746–755. [DOI] [PubMed] [Google Scholar]
- 5. Gupta D, Treon SP, Shima Y, et al. Adherence of multiple myeloma cells to bone marrow stromal cells upregulates vascular endothelial growth factor secretion: therapeutic applications. Leukemia. 2001;15(12):1950–1961. [DOI] [PubMed] [Google Scholar]
- 6. Reddy N, Hernandez‐Ilizaliturri FJ, Deeb G, et al. Immunomodulatory drugs stimulate natural killer‐cell function, alter cytokine production by dendritic cells, and inhibit angiogenesis enhancing the anti‐tumour activity of rituximab in vivo. Br J Haematol. 2008;140(1):36–45. [DOI] [PubMed] [Google Scholar]
- 7. Lu L, Payvandi F, Wu L, et al. The anti‐cancer drug lenalidomide inhibits angiogenesis and metastasis via multiple inhibitory effects on endothelial cell function in normoxic and hypoxic conditions. Microvasc Res. 2009;77(2):78–86. [DOI] [PubMed] [Google Scholar]
- 8. Corral LG, Haslett PA, Muller GW, et al. Differential cytokine modulation and T cell activation by two distinct classes of thalidomide analogues that are potent inhibitors of TNF‐alpha. J Immunol. 1999;163(1):380–386. [PubMed] [Google Scholar]
- 9. Hayashi T, Hideshima T, Akiyama M, et al. Molecular mechanisms whereby immunomodulatory drugs activate natural killer cells: clinical application. Br J Haematol. 2005;128(2):192–203. [DOI] [PubMed] [Google Scholar]
- 10. Teo SK, Chen Y, Muller GW, et al. Chiral inversion of the second generation IMiD CC‐4047 (ACTIMID) in human plasma and phosphate‐buffered saline. Chirality. 2003;15(4):348–351. [DOI] [PubMed] [Google Scholar]
- 11. San Miguel J, Weisel K, Moreau P, et al. Pomalidomide plus low‐dose dexamethasone versus high‐dose dexamethasone alone for patients with relapsed and refractory multiple myeloma (MM‐003): a randomised, open‐label, phase 3 trial. Lancet Oncol. 2013;14(11):1055–1066. [DOI] [PubMed] [Google Scholar]
- 12. Daver N, Shastri A, Kadia T, et al. Phase II study of pomalidomide in combination with prednisone in patients with myelofibrosis and significant anemia. Leuk Res. 2014;38(9):1126–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Richardson PG, Siegel DS, Vij R, et al. Pomalidomide alone or in combination with low‐dose dexamethasone in relapsed and refractory multiple myeloma: a randomized phase 2 study. Blood. 2014;123(12):1826–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pomalidomide I. Summary of product characteristics. Uxbridge, UK: Celgene Europe; 2016. [Google Scholar]
- 15. Pomalidomide P. package insert. Summit, NJ: Celgene Corporation; 2016. [Google Scholar]
- 16. Dao K, Chtioui H, Lu Y, et al. Pharmacokinetics of pomalidomide in a patient receiving hemodialysis using a high‐cutoff filter. Am J Kidney Dis. 2017;69(4):553–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shimizu M, Suemizu H, Mitsui M, Shibata N, Guengerich FP, Yamazaki H. Metabolic profiles of pomalidomide in human plasma simulated with pharmacokinetic data in control and humanized‐liver mice. Xenobiotica. 2017;47(10):844–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Torigoe K, Nakayama N, Achiwa H. Pomalidomide (Pomalyst((R)) capsule 1 mg/2 mg/3 mg/4 mg): pharmacokinetics, pharmacodynamics and clinical study outcome. Nihon Yakurigaku Zasshi. 2016;148(3):154–161. [DOI] [PubMed] [Google Scholar]
- 19. Li Y, Zhou S, Hoffmann M, Kumar G, Palmisano M. Modeling and simulation to probe the pharmacokinetic disposition of pomalidomide R‐ and S‐enantiomers. J Pharmacol Exp Ther. 2014;350(2):265–272. [DOI] [PubMed] [Google Scholar]
- 20. Gay F, Mina R, Troia R, Bringhen S. Pharmacokinetic evaluation of pomalidomide for the treatment of myeloma. Expert Opin Drug Metab Toxicol. 2013;9(11):1517–1527. [DOI] [PubMed] [Google Scholar]
- 21. Kasserra C, Assaf M, Hoffmann M, et al. Pomalidomide: evaluation of cytochrome P450 and transporter‐mediated drug‐drug interaction potential in vitro and in healthy subjects. J Clin Pharmacol. 2014;55(2):168–178. [DOI] [PubMed] [Google Scholar]
- 22. Li Y, Wang X, O'Mara E, et al. Population pharmacokinetics of pomalidomide in patients with relapsed or refractory multiple myeloma with various degrees of impaired renal function. Clin Pharmacol. 2017;9:133–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hoffmann M, Kasserra C, Reyes J, et al. Absorption, metabolism and excretion of [14C]pomalidomide in humans following oral administration. Cancer Chemother Pharmacol. 2013;71(2):489–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. FDA. Clinical Drug Interaction Studies ‐ Study Design , Data Analysis, and Clinical Implications. FDA Guidance. 2017. [DOI] [PubMed] [Google Scholar]
- 25. Bertoli R, Bissig M, Caronzolo D, Odorico M, Pons M, Bernasconi E. Assessment of potential drug‐drug interactions at hospital discharge. Swiss Med Wkly. 2010;140:w13043. [DOI] [PubMed] [Google Scholar]
- 26. Murtaza G, Khan MY, Azhar S, Khan SA, Khan TM. Assessment of potential drug‐drug interactions and its associated factors in the hospitalized cardiac patients. Saudi Pharm J. 2016;24(2):220–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jansman FG, Reyners AK, van Roon EN, et al. Consensus‐based evaluation of clinical significance and management of anticancer drug interactions. Clin Ther. 2011;33(3):305–314. [DOI] [PubMed] [Google Scholar]
- 28. Anderson GD, Chan LN. Pharmacokinetic drug interactions with tobacco, cannabinoids and smoking cessation products. Clin Pharmacokinet. 2016;55(11):1353–1368. [DOI] [PubMed] [Google Scholar]
- 29. Laki S, Kalapos‐Kovacs B, Antal I, Klebovich I. [Importance of drug interactions with smoking in modern drug research]. Acta Pharm Hung. 2013;83(4):107–120. [PubMed] [Google Scholar]
- 30. Rouhos A, Raaska K. [Smoking and drug interactions]. Duodecim. 2012;128(10):1073–1080. [PubMed] [Google Scholar]
- 31. Molden E, Spigset O. [Tobacco smoking and drug interactions]. Tidsskr Nor Laegeforen. 2009;129(7):632–633. [DOI] [PubMed] [Google Scholar]
- 32. Kroon LA. Drug interactions with smoking. Am J Health Syst Pharm. 2007;64(18):1917–1921. [DOI] [PubMed] [Google Scholar]
- 33. Kroon LA. Drug interactions and smoking: raising awareness for acute and critical care providers. Crit Care Nurs Clin North Am. 2006;18(1):53–62, xii. [DOI] [PubMed] [Google Scholar]
- 34. Zevin S, Benowitz NL. Drug interactions with tobacco smoking. An update. Clin Pharmacokinet. 1999;36(6):425–438. [DOI] [PubMed] [Google Scholar]
- 35. Schein JR. Cigarette smoking and clinically significant drug interactions. Ann Pharmacother. 1995;29(11):1139–1148. [DOI] [PubMed] [Google Scholar]
- 36. Schein J. Smoking and drug interactions. Am J Public Health. 1994;84(6):1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dobrinas M, Cornuz J, Oneda B, Kohler Serra M, Puhl M, Eap CB. Impact of smoking, smoking cessation, and genetic polymorphisms on CYP1A2 activity and inducibility. Clin Pharmacol Ther. 2011;90(1):117–125. [DOI] [PubMed] [Google Scholar]
- 38. Dobrinas M, Cornuz J, Pedrido L, Eap CB. Influence of cytochrome P450 oxidoreductase genetic polymorphisms on CYP1A2 activity and inducibility by smoking. Pharmacogenet Genomics. 2012;22(2):143–151. [DOI] [PubMed] [Google Scholar]
- 39. Faber M, Jetter A, Fuhr U. Assessment of CYPA2 activity in clinical practice: why, how, and when? Pharmacol Toxicol. 2005;97:125–134. [DOI] [PubMed] [Google Scholar]