

Abstract
Full control over multiple competing coupling sites would enable straightforward access to densely functionalized compound libraries. Historically, the site selection in Pd0‐catalyzed functionalizations of poly(pseudo)halogenated arenes has been unpredictable, being dependent on the employed catalyst, the reaction conditions, and the substrate itself. Building on our previous report of C−Br‐selective functionalization in the presence of C−OTf and C−Cl bonds, we herein complete the sequence and demonstrate the first general arylations and alkylations of C−OTf bonds (in <10 min), followed by functionalization of the C−Cl site (in <25 min), at room temperature using the same air‐ and moisture‐stable PdI dimer. This allowed the realization of the first general and triply selective sequential C−C coupling (in 2D and 3D space) of C−Br followed by C−OTf and then C−Cl bonds.
Keywords: arenes, C−C coupling, chemoselectivity, homogeneous catalysis, palladium(I) dimer
Polyfunctionalized arenes are ubiquitous building blocks in pharmaceuticals, materials, and natural products.1 To meet the ever‐increasing demands for diverse compound libraries and the exploration of chemical space in a two‐ and three‐dimensional fashion, highly modular, streamlined, and diversifiable approaches in the assembly of densely functionalized arenes are desired.2 Ideally, these features are paired with the potential for automation to realize high‐throughput operations. In this context, owing to the relative mildness and functional group tolerance, site‐selective and sequential Pd‐catalyzed cross‐couplings of polyhalogenated arenes are a highly desired technology.3 However, while the approximate reactivity order of C−I>C−Br≈C−OTf>C−Cl is frequently quoted,3 the actual site selectivity of Pd0‐catalyzed cross‐coupling reactions is the result of a complex interplay of the ligand/catalyst, the reaction conditions (additive, solvent), and, most importantly, the steric and electronic bias of the substrate itself.4 As such, coupling conditions identified for a given substrate frequently fail if slight structural or electronic modifications are introduced (see below). However, to access libraries of compounds in a programmable fashion, a general, a priori predictable, and fully selective technology will be required. Ideally, this is paired with practical features, such as rapid conversion, robustness (e.g., air tolerance), and broadness for maximal diversity (C(sp2) and C(sp3) coupling partners). In this context, Pd0‐based cross‐couplings5 frequently suffer from oxygen sensitivity, extended reaction times (hours), substrate specificity in site selectivity, and additional mechanistic challenges, such as β‐hydride elimination in C(sp2)−C(sp3) couplings.6 We therefore focused our attention on alternative oxidation states that bear potential for bimetallic synergy and alternative mechanisms.7 In this context, our group recently reported an a priori predictable and rapid C(sp2)−C(sp2) and C(sp2)−C(sp3) coupling of aromatic C−Br bonds in the presence of C−OTf and C−Cl bonds under open‐flask conditions using the air‐stable PdI dimer 1 (Figure 1).8 With selective C−Br functionalization realized, the next challenge towards modular functionalization lies in the selective manipulation of the C−OTf and C−Cl sites in a rapid and general manner. We herein present a solution to this challenge.
Figure 1.
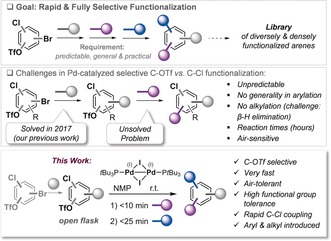
Challenges (top) in site‐selective couplings and this work (bottom).
While there are isolated reports of Pd0‐catalyzed C−OTf arylations in the presence of C−Cl bonds, the corresponding alkylation appears essentially unexplored, and there has been no demonstration of tolerance to diverse functional groups, nor generality in terms of steric/electronic bias on the substrate or coupling partner.9 As such, we initially set out to explore the potential generality of existing methods, and investigated a Pd(OAc)2/PCy3‐catalyzed Suzuki coupling protocol previously identified to enable selective arylation of 4‐chlorophenyl triflate at room temperature.4g We found that while the previously reported 4‐chlorophenyl triflate provided excellent C−OTf coupling with 2‐tolylboronic acid (90 %, as reported previously),4g an alternative substitution pattern was much less efficient, resulting in much lower conversion into the product (<10 %; see the Supporting Information, Table S1). Even when maintaining a 1,4‐substitution pattern of the Cl and OTf groups, much lower conversions were observed when varying the substrate structure, and the reaction also appeared to be intolerant to different boronic acid coupling partners, with a significant drop in conversion when moving from 2‐tolyl‐ (90 %) to 4‐methoxyphenyl‐ (26 %) or alternative boronic acids. Overall, these data suggest that reoptimization would be needed for alternative targets; there is no generality. As such, we set out to explore the possibility of PdI‐catalyzed selective functionalizations of C−OTf over C−Cl bonds.
Our previous work had indicated that solvent polarity can impact the nature of the active species, and hence the preference of C−OTf versus C−Cl functionalization.4d We reasoned that in the presence of coordinating additives, “Pd‐ate” complexes could be generated by dynamic ligand exchange from Pd0 in the polar media, which would preferentially react with C−OTf bonds. While our current data suggest that PdI cannot directly react with C−Cl and C−OTf bonds owing to high activation barriers, we envisioned that the dinuclear entity could act as a precursor to a defined bis‐ligated “ate” complex through rapid activation.7a, 10
Consequently, we investigated the effectiveness of a number of polar solvents in facilitating chemoselective Negishi cross‐coupling at C−OTf with the air‐stable PdI dimer 1. To our delight, the use of N‐methyl‐2‐pyrrolidinone (NMP) led to completely selective C−OTf over C−Cl functionalization in less than 10 min at room temperature. Notably, no efforts were made to avoid oxygen; the reaction was run in an open flask.11 These newly found data were very promising as the coupling is more rapid and robust than any previous method.
We next explored whether the procedure is general and applicable for a wide range of substrates (Scheme 1). To our delight, we were able to perform selective arylation, regardless of whether the C−Cl and C−OTf bonds were positioned ortho (4, 13), meta (3, 9) or para (2, 7, 8, 12) to each other, or whether additional steric hindrance or functional groups present (5, 6). Heterocyclic (7, 8, 13), nitrile (9), nitro (10), fluoro (11), ketone (5), and aldehyde (6) moieties were well tolerated. Moreover, not just arylation (using both heterocyclic and non‐heterocyclic arenes, 19–21), but also the more challenging alkylation could be achieved selectively at C−OTf (over C−Cl) in an equally rapid (10 min) and efficient manner in a reaction vessel open to air. Pleasingly, we were able to selectively couple both primary (14–16) and secondary (17, 18, 22, 23) alkyl groups in high yields; of particular note is the successful incorporation of a challenging branched secondary alkyl group (23), which is especially prone to isomerization. To the best of our knowledge, this is the first general site‐selective alkylation of C−OTf bonds in the presence of C−Cl bonds.
Scheme 1.
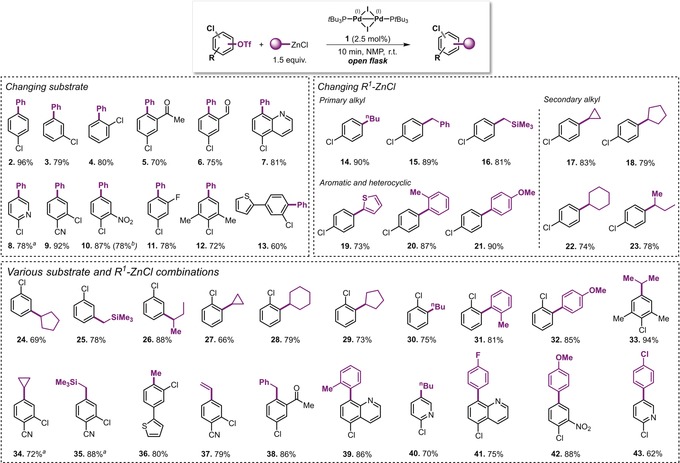
Scope of the chemoselective C−OTf (over C−Cl) Negishi coupling in NMP enabled by PdI dimer 1. Reaction conditions: Substrate (0.4 mmol), PdI dimer 1 (0.01 mmol), and RZnCl (0.6 mmol) in NMP, reaction mixture stirred for 10 min in a flask open to air. [a] RZnCl (0.46 mmol) was used. [b] Reaction performed on larger scale (1 g, 3.27 mmol).
Lastly, a number of different substrate and RZnCl combinations were examined, highlighting that this technique can be used to generate structurally diverse compound libraries (24–43). For example, ortho‐ and meta‐substituted chlorophenyl triflates underwent successful alkylation with both primary and secondary alkyl groups (24–30), and many of these compounds had not been reported previously (e.g., 33–42).
To explore the scalability of this method, we performed the C−OTf‐selective coupling with 1 g of starting material; pleasingly, product 10 was isolated in 78 % yield (Scheme 1). It should also be noted that while NMP was a very effective solvent, alternative polar solvents proved to be equally effective.12 As an example, for 4‐chlorophenyl triflate, exclusive C−OTf selectivity and high yields of the product 2 were obtained in DMI (90 %), DMPU (76 %), and DMAc (92 %).
Having achieved chemoselective C−OTf over C−Cl functionalization through a general, rapid, and air‐tolerant procedure, we next sought to develop a facile C−Cl functionalization using the PdI dimer 1 to eventually enable functionalization at all three sites using the same catalyst. The use of ArCl electrophiles in cross‐coupling reactions is generally more difficult, sometimes requiring high temperatures (up to 100 °C) and/or extended reaction times (up to 72 h).4g, 13 Encouraged by the high reactivities seen with 1, we slowly added PhZnCl (over 15 min) to a mixture of 1 and 2‐chloronitrobenzene in NMP at room temperature, and allowed the mixture to react for an additional 10 min. Pleasingly, we observed efficient C−Cl coupling and isolated 44 (Scheme 2) in 89 % yield.
Scheme 2.
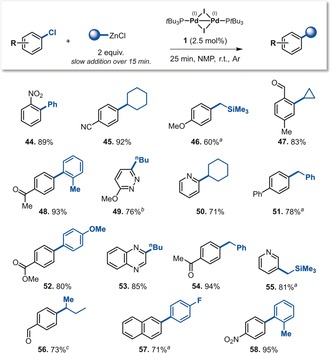
Scope of the PdI‐catalyzed C−Cl coupling. Reaction conditions: Chloroarene (0.4 mmol) and PdI dimer 1 (0.01 mmol) in NMP under Ar, RZnCl (0.8 mmol) added slowly over 15 min; then the reaction mixture was stirred for 10 min at room temperature, unless stated otherwise. [a] At 80 °C. [b] At 50 °C. [c] Contains 15 % of the linear isomer.
We next tested the C−Cl coupling more widely, assessing electron‐rich and ‐poor arenes, and examining sp2 and sp3 C−C bond formations; pleasingly, in each case, high yields of the desired products were obtained after only 25 min under argon (Scheme 2). While Negishi cross‐coupling reactions of Ar−Cl bonds have been reported,14 our method is a more rapid, general, and operationally simple alternative; for example, with Pd0 catalysis, products 48 and 58 were previously obtained in 50 % and 92 %, respectively, after 2 h at 100 °C.14d In contrast, these compounds were obtained in 93 % and 95 % yield after only 25 min at room temperature with our method.
With the ability to functionalize all three sites in a general fashion using a single catalyst and the defined reactivity sequence of Br>OTf>Cl established, we next explored the sequential functionalization of a) Br then OTf, b) OTf then Cl, and c) Br then Cl. These sequences were performed in a one‐pot process at room temperature, with a total reaction time of 15–35 min (Scheme 3). For (a), the cross‐coupling partner was added to a solution of a bromoaryl triflate and PdI dimer 1 in THF. After 5 min, NMP was added to the same pot (producing 1:1 THF/NMP) followed by R2ZnCl, yielding the bis‐functionalized arene in excellent yields in a total reaction time of 15 min at room temperature.
Scheme 3.
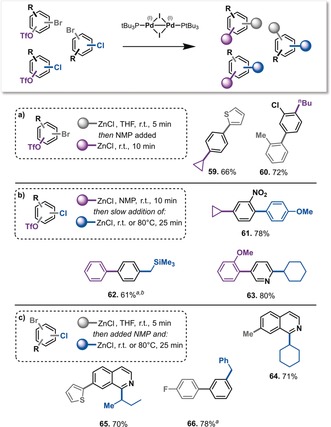
Sequential double functionalization of di(pseudo)halogenated arenes. Reaction conditions: Substrate (0.4 mmol), PdI dimer 1 (0.01 mmol), and R1ZnCl (0.4–0.8 mmol, see the Supporting Information for details). [a] C−Cl coupling done at 80 °C. [b] Reaction mixture filtered through silica prior to C−Cl functionalization.
For (b) (C−OTf then C−Cl functionalization), efficient coupling was also achieved in a one‐pot fashion, using NMP as the solvent throughout. Finally, for coupling sequence (c) (C−Br then C−Cl), a procedure analogous to (a) was applied (although under argon and over 25 min to facilitate C−Cl coupling). Overall, the desired bis‐functionalized products were obtained in good yields, with the introduction of both aryl and alkyl groups being demonstrated.
With all combinations allowing selective and sequential functionalizations, we next studied the functionalization of all three sites (Scheme 4). We achieved C−Br followed by C−OTf functionalization in a total of 15 min in a one‐pot process with the vessel open to air (as described above), and subsequently purified the resulting chloroarene intermediate. The C−Cl functionalization step was then performed in NMP under an argon atmosphere. This sequence enabled the synthesis of the trisubstituted product in high overall yields in a total reaction time of only 40 min, using the air‐stable PdI dimer 1 for all three functionalization steps.
Scheme 4.
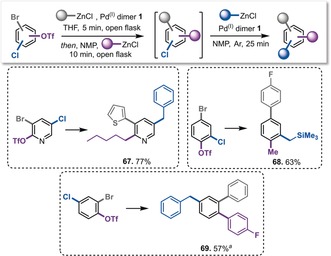
Sequential triple functionalization of tri(pseudo)halogenated arenes. Reaction conditions: Substrate (0.4 mmol), PdI dimer 1 (0.01 mmol), R1ZnCl (0.44–0.8 mmol, see the Supporting Information for details). [a] The chloroaryl triflate was isolated prior to C−OTf functionalization.
In conclusion, we have developed a predictable and operationally simple procedure to alkylate and arylate aromatic C−OTf sites in the presence of C−Cl bonds within 15 min at room temperature under air, which is also independent of the electronic or steric bias imposed by the substrate. Key to the success of this strategy was the use of an air‐ and moisture‐stable PdI dimer along with NMP (or DMPU, DMI, or DMAc) as the solvent. The same solvent/catalyst combination also allowed for subsequent C−Cl functionalization at room temperature in 25 min. Overall, the newly developed technology led to full control over all three coupling sites in the order C−Br>C−OTf>C−Cl, with triply selective and sequential functionalization occuring in only 40 min, using the same PdI dimer. Given its generality, robustness, speed, and full selectivity, we anticipate widespread applications of this method, including its implementation in automated approaches to rapidly access molecular complexity in a programmable fashion.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank RWTH Aachen University and the European Research Council (ERC‐637993) for funding.
S. T. Keaveney, G. Kundu, F. Schoenebeck, Angew. Chem. Int. Ed. 2018, 57, 12573.
Contributor Information
Dr. Sinead T. Keaveney, http://www.schoenebeck.oc.rwth-aachen.de/.
Prof. Dr. Franziska Schoenebeck, Email: franziska.schoenebeck@rwth-aachen.de.
References
- 1.
- 1a. Transition Metal-Catalyzed Couplings in Process Chemistry: Case Studies From the Pharmaceutical Industry (Eds.: J. Magano, J. R. Dunetz), Wiley, Hoboken, 2013; [Google Scholar]
- 1b. New Trends in Cross-Coupling: Theory and Applications (Ed.: T. Colacot), RSC Catalysis Series, Cambridge, 2015. [Google Scholar]
- 2.For examples of iterative cross-couplings through selective transmetalation, see:
- 2a. Crudden C. M., Ziebenhaus C., Rygus J. P. G., Ghozati K., Unsworth P. J., Nambo M., Voth S., Hutchinson M., Laberge V. S., Maekawa Y., Imao D., Nat. Commun. 2016, 7, 11065; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b. Li J., Ballmer S. G., Gillis E. P., Fujii S., Schmidt M. J., Palazzolo A. M. E., Lehmann J. W., Morehouse G. F., Burke M. D., Science 2015, 347, 1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.For reviews, see:
- 3a. Almond-Thynne J., Blakemore D. C., Pryde D. C., Spivey A. C., Chem. Sci. 2017, 8, 40; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Fairlamb I., Chem. Soc. Rev. 2007, 36, 1036; [DOI] [PubMed] [Google Scholar]
- 3c. Schröter S., Stock C., Bach T., Tetrahedron 2005, 61, 2245. [Google Scholar]
- 4.
- 4a. Proutiere F., Lyngvi E., Aufiero M., Sanhueza I. A., Schoenebeck F., Organometallics 2014, 33, 6879; [Google Scholar]
- 4b. Tran T. D. et al., ChemMedChem 2014, 9, 1378; [DOI] [PubMed] [Google Scholar]
- 4c. Hassan Z., Hussain M., Villinger A., Langer P., Tetrahedron 2012, 68, 6305; [Google Scholar]
- 4d. Proutiere F., Schoenebeck F., Angew. Chem. Int. Ed. 2011, 50, 8192; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 8342; [Google Scholar]
- 4e. Milbank J. B. J., Pryde D. C., Tran T. D., WO2011004276, 2011;
- 4f. Espino G., Kurbangalieva A., Brown J. M., Chem. Commun. 2007, 1742; [DOI] [PubMed] [Google Scholar]
- 4g. Littke A. F., Dai C. Y., Fu G. C., J. Am. Chem. Soc. 2000, 122, 4020; [Google Scholar]
- 4h. Kamikawa T., Hayashi T., Tetrahedron Lett. 1997, 38, 7087. [Google Scholar]
- 5.For recent reviews on Pd-catalyzed cross-coupling reactions, see:
- 5a. Ruiz-Castillo P., Buchwald S. L., Chem. Rev. 2016, 116, 12564; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Johansson Seechurn C. C. C., Kitching M. O., Colacot T. J., Snieckus V., Angew. Chem. Int. Ed. 2012, 51, 5062; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 5150. [Google Scholar]
- 6.For examples of recent developments that avoid β-H elimination (without site selectivity), see:
- 6a. Atwater B., Chandrasoma N., Mitchell D., Rodriguez M. J., Pompeo M., Froese R. D. J., Organ M. G., Angew. Chem. Int. Ed. 2015, 54, 9502; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 9638; [Google Scholar]
- 6b. Yang Y., Niedermann K., Han C., Buchwald S. L., Org. Lett. 2014, 16, 4638; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6c. Giannerini M., Fañanás-Mastral M., Feringa B. L., Nat. Chem. 2013, 5, 667. [DOI] [PubMed] [Google Scholar]
- 7.For our mechanistic work on the feasibility of dinuclear metal catalysis, see:
- 7a. Aufiero M., Scattolin T., Proutiere F., Schoenebeck F., Organometallics 2015, 34, 5191; [Google Scholar]
- 7b. Yin G., Kalvet I., Schoenebeck F., Angew. Chem. Int. Ed. 2015, 54, 6809; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 6913; [Google Scholar]
- 7c. Kalvet I., Bonney K. J., Schoenebeck F., J. Org. Chem. 2014, 79, 12041; [DOI] [PubMed] [Google Scholar]
- 7d. Bonney K. J., Proutiere F., Schoenebeck F., Chem. Sci. 2013, 4, 4434. [Google Scholar]
- 8.
- 8a. Kalvet I., Magnin G., Schoenebeck F., Angew. Chem. Int. Ed. 2017, 56, 1581; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 1603; [Google Scholar]
- 8b. Kalvet I., Sperger T., Scattolin T., Magnin G., Schoenebeck F., Angew. Chem. Int. Ed. 2017, 56, 7078; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 7184. [Google Scholar]
- 9.Alkylation of C−OTf in the presence of C−Cl bonds has been achieved with three substrates under Pd catalysis; see:
- 9a. Dupuy S., Zhang K.-F., Goutierre A.-S., Baudoin O., Angew. Chem. Int. Ed. 2016, 55, 14793; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 15013 (with β-H elimination/isomerization occurring); [Google Scholar]
- 9b. Corley E. G., Conrad K., Murry J. A., Savarin C., Holko J., Boice G., J. Org. Chem. 2004, 69, 5120 (using a Cu co-catalyst); for examples of selective arylation, see: [DOI] [PubMed] [Google Scholar]
- 9c. Hassan Z., Al-Shidhani S., Al-Ghafri A., Al-Harrasi A., Hussain J., Csuk R., Tetrahedron Lett. 2015, 56, 7141; [Google Scholar]
- 9d. Zhou C., Liu Q., Li Y., Zhang R., Fu X., Duan C., J. Org. Chem. 2012, 77, 10468; [DOI] [PubMed] [Google Scholar]
- 9e. Ishikawa S., Manabe K., Tetrahedron 2010, 66, 297; [Google Scholar]
- 9f. Tomohiro M., Yoshiaki K., Satoko S., Takahiro U., Ai S., Asami T., Yusuke K., Koichi E., Utpal B., Takanori K., Atsushi K., Yasunari M., Hironao S., Chem. Eur. J. 2007, 13, 5937.17444548 [Google Scholar]
- 10.
- 10a. Proutiere F., Aufiero M., Schoenebeck F., J. Am. Chem. Soc. 2012, 134, 606; for a perspective, see: [DOI] [PubMed] [Google Scholar]
- 10b. Paton R. S., Brown J. M., Angew. Chem. Int. Ed. 2012, 51, 10448; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 10598; for selected examples of labile PdI dimers as precatalysts, see: [Google Scholar]
- 10c. Melvin P. R., Nova A., Balcells D., Dai W., Hazari N., Hruszkewycz D. P., Shah H. P., Tudge M. T., ACS Catal. 2015, 5, 3680; [Google Scholar]
- 10d. Colacot T. J., Platinum Met. Rev. 2009, 53, 183; [Google Scholar]
- 10e. Christmann U., Pantazis D. A., Benet-Buchholz J., McGrady J. E., Maseras F., Vilar R., J. Am. Chem. Soc. 2006, 128, 6376; [DOI] [PubMed] [Google Scholar]
- 10f. Stambuli J., Kuwano R., Hartwig J. F., Angew. Chem. Int. Ed. 2002, 41, 4746; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 4940. [Google Scholar]
- 11.Owing to the moisture sensitivity of organometallic reagents, safety precautions should be made when using an open flask setup.
- 12. Bisz E., Szostak M., Green Chem. 2017, 19, 5361. [Google Scholar]
- 13.
- 13a. Froese R. D. J., Lombardi C., Pompeo M., Rucker R. P., Organ M. G., Acc. Chem. Res. 2017, 50, 2244; [DOI] [PubMed] [Google Scholar]
- 13b. Fu G. C., Acc. Chem. Res. 2008, 41, 1555; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13c. Martin R., Buchwald S. L., Acc. Chem. Res. 2008, 41, 1461; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13d. Marion N., Navarro O., Mei J., Stevens E. D., Scott N. M., Nolan S. P., J. Am. Chem. Soc. 2006, 128, 4101. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Liu Z., Dong N., Xu M., Sun Z., Tu T., J. Org. Chem. 2013, 78, 7436; [DOI] [PubMed] [Google Scholar]
- 14b. Sase S., Jaric M., Metzger A., Malakhov V., Knochel P., J. Org. Chem. 2008, 73, 7380; [DOI] [PubMed] [Google Scholar]
- 14c. Milne J. E., Buchwald S. L., J. Am. Chem. Soc. 2004, 126, 13028; [DOI] [PubMed] [Google Scholar]
- 14d. Dai C., Fu G. C., J. Am. Chem. Soc. 2001, 123, 2719. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary