

Abstract
Ewing sarcoma (EWS) is a soft tissue and bone tumor that occurs primarily in adolescents and young adults. In most cases of EWS, the chimeric transcription factor, EWS‐FLI1 is the primary oncogenic driver. The epigenome of EWS cells reflects EWS‐FLI1 binding and activation or repression of transcription. Here, we demonstrate that EWS‐FLI1 positively regulates the expression of proteins required for serine‐glycine biosynthesis and uptake of the alternative nutrient source glutamine. Specifically, we show that EWS‐FLI1 activates expression of PHGDH, PSAT1, PSPH, and SHMT2. Using cell‐based studies, we also establish that EWS cells are dependent on glutamine for cell survival and that EWS‐FLI1 positively regulates expression of the glutamine transporter, SLC1A5 and two enzymes involved in the one‐carbon cycle, MTHFD2 and MTHFD1L. Inhibition of serine‐glycine biosynthesis in EWS cells impacts their redox state leading to an accumulation of reactive oxygen species, DNA damage, and apoptosis. Importantly, analysis of EWS primary tumor transcriptome data confirmed that the aforementioned genes we identified as regulated by EWS‐FLI1 exhibit increased expression compared with normal tissues. Furthermore, retrospective analysis of an independent data set generated a significant stratification of the overall survival of EWS patients into low‐ and high‐risk groups based on the expression of PHGDH, PSAT1, PSPH, SHMT2, SLC1A5, MTHFD2, and MTHFD1L. In summary, our study demonstrates that EWS‐FLI1 reprograms the metabolism of EWS cells and that serine‐glycine metabolism or glutamine uptake are potential targetable vulnerabilities in this tumor type.
Keywords: de novo serine‐glycine biosynthesis, Ewing sarcoma, glutamine, metabolism, ROS
Abbreviations
- AICAR
N1‐(β‐D‐Ribofuranosyl)‐5‐aminoimidazole‐4‐carboxamide
- AMPK
AMP‐activated Ser/Thr protein kinase
- ANOVA
analysis of variance
- ATF4
Activating transcription factor 4
- CBR5884
Ethyl 5‐[(2‐furanyl carbonyl)amino]‐3‐methyl‐4‐thiocyanato‐2‐thiophenecarboxylate
- ChIP
chromatin immunoprecipitation
- CMV
Cytomegalovirus
- EWS
Ewing sarcoma
- ETS
E26‐transformation specific
- EWSR1
EWS RNA binding protein 1
- EWS‐FLI1
EWSR1‐FLI1 fusion
- EZH2
enhancer of zeste 2 polycomb repressive complex 2
- FBS
fetal bovine serum
- FDR
false discovery rate
- FLI1
Fli‐1 proto‐oncogene, ETS transcription factor
- GEO
Gene Expression Omnibus
- GLI1
GLI family zinc finger 1
- GPNA
L‐Glutamic acid γ‐(p‐nitroanilide) hydrochloride
- GSH
L‐Glutathione, reduced
- GSSG
Glutathione disulfide
- IGV
Integrative Genomics Viewer
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- L‐DON
6‐diazo‐5‐oxo‐L‐nor‐Leucine
- MTHFD1
methylenetetrahydrofolate dehydrogenase 1
- MTHFD2
methylenetetrahydrofolate dehydrogenase 2
- MTHFD1L
methylenetetrahydrofolate dehydrogenase 1 like
- NADH
Nicotinamide adenine dinucleotide
- NADPH
Nicotinamide adenine dinucleotide phosphate
- NCBI
National Center for Biotechnology Information
- NRF2
nuclear factor, erythroid 2 like 2
- NR0B1
nuclear receptor subfamily 0 group B member 1
- NSCLC
non‐small cell lung cancer
- NCT503
N‐(4,6‐Dimethylpyridin‐2‐yl)‐4‐(4‐(trifluoromethyl)‐benzyl)‐piperazine‐1‐carbothioamide
- PARP1
poly(ADP‐ribose) polymerase 1
- PBS
phosphate buffered saline
- PHGDH
phosphoglycerate dehydrogenase
- PSAT1
phosphoserine aminotransferase 1
- PSPH
phosphoserine phosphatase
- RPM
Reads per million
- ROS
Reactive oxygen species
- SHMT1
serine hydroxymethyltransferase 1
- SHMT2
serine hydroxymethyltransferase 2
- SLC1A4
solute carrier family 1 member 4
- SLC1A5
solute carrier family 1 member 5
- STR
short tandem repeat
- TCA
Tricarboxylic acid
- TDO2
tryptophan 2,3‐dioxygenase
- TPM
Transcripts per kilobase million
- ULA
ultra‐low attachment
1. INTRODUCTION
Cancer cells must activate or enhance metabolism to satisfy the energy and biosynthetic needs of rapid proliferation and growth in nutrient and oxygen‐poor environments. Alterations in metabolic processes used by cancer cells include high rates of glycolysis and cytosolic lactic acid fermentation (the Warburg effect), or the utilization of non‐essential amino acids as alternate nutrient sources or as metabolites. Examples of amino acids some tumor types use to support their increased metabolic requirements include glutamine, serine, or glycine (reviewed in ref.1, 2, 3, 4, 5).
The serine‐glycine biosynthesis pathway (Figure 1A) involves the conversion of 3P‐glycerate to 3P‐hydroxy‐pyruvate by phosphoglycerate dehydrogenase (PHGDH), the conversion of 3P‐hydroxy‐pyruvate to 3P‐serine by phosphoserine aminotransferase 1 (PSAT1), and phosphoserine phosphatase (PSPH) converts 3P‐serine to serine. Serine hydroxymethyltransferase 1 (SHMT1) and serine hydroxymethyltransferase 2 (SHMT2) catalyze the conversion of serine to glycine in the cytosol and mitochondria, respectively. Serine‐glycine biosynthesis is not only linked to the generation of macromolecules like nucleotides, lipids, and proteins, but it also regulates cellular redox homeostasis and methylation reactions via methionine and one‐carbon folate cycles.1, 6 Recent studies of melanoma, and breast and lung cancers revealed genetic changes that result in the utilization of the serine‐glycine biosynthesis pathway to support cell growth.7, 8, 9
Figure 1.
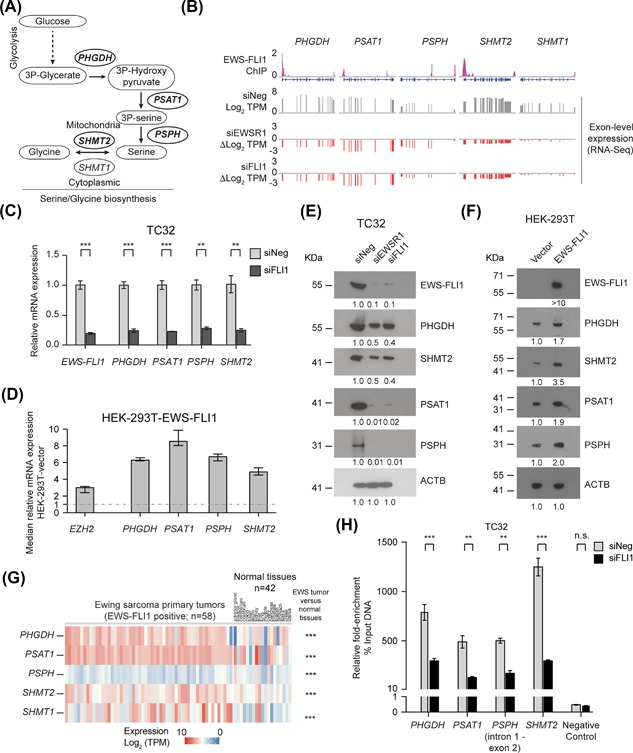
EWS‐FLI1 regulates the expression of multiple serine‐glycine biosynthesis genes. (A) A schematic of the de novo serine‐glycine biosynthesis pathway. (B) Exon level expression (Log2 TPM) and fold‐change (ΔLog2 TPM) of the expression of PHGDH, PSAT1, PSPH, SHMT2, and SHMT1 in control (siNeg) and EWS‐FLI1‐silenced TC32 cells (siEWSR1 or siFLI1) matched with EWS‐FLI1 ChIP‐seq data from Bilke et al.40 (C) qPCR analysis of the relative expression (compared to siNeg‐transfected cells) of the indicated genes in EWS‐FLI1‐depleted TC32 cells (siFLI1, 48 h; mean ± s.e.m., n = 3). (D) qPCR analysis of the relative expression (normalized to vector control cells) of the indicated genes in HEK‐293T cells stably expressing EWS‐FLI1; median and range of three technical replicas). (E) Immunoblots of the indicated proteins in lysates prepared from TC32 cells, siRNA‐transfected as indicated. (F) Immunoblots of the indicated proteins in lysates prepared from vector or EWS‐FL11‐HEK‐293T cells. (G) The expression of PHGDH, PSAT1, PSPH, SHMT2, and SHMT1 (Log2, TPM) in a panel of EWS primary tumors (EWS‐FLI positive; (n = 58) and normal tissues (n = 42). (H) Fold‐enrichment of EWS‐FLI1‐co‐precipitating DNA determined by qPCR of genomic DNA spanning putative EWS‐FLI1 binding sites. Data shown were calculated as a percent of input DNA and the fold‐enrichment of the anti‐FLI1 antibody compared to IgG; triplicate samples of each treatment in each experiment. (C, G, and H) Unpaired t‐test, adjusted for multiple comparisons **q < 0.01, ***q < 0.001
The genetic basis for activation of serine‐glycine biosynthesis pathway in cancer includes amplification of the PHGDH locus or its transcriptional deregulation. Overall, 16% of all cancers exhibit a gain of the chromosome 1p12 region that contains the PHGDH locus,7, 10 including a sizeable proportion of melanomas and breast cancers.7, 8 Furthermore, approximately 70% of estrogen receptor‐negative breast cancers overexpress PHGDH protein. In non‐small cell lung cancer (NSCLC), the transcription factor NRF2 alters the expression of ATF4 that in turn upregulates PHGDH.9 Importantly, the inhibition of PHGDH or de novo serine‐glycine biosynthesis in cell lines with elevated PHGDH expression results in decreased cell viability, indicating that these cells are dependent on serine‐glycine biosynthesis for cell survival.7, 8, 9, 11
The genetic reprogramming of some cancer types to make use of glutamine as an alternative nutrient source includes increased expression of proteins that act as transporters of amino acids, such as SLC1A5 (ASTC2),12, 13, 14 or the upregulation of enzymes that catalyze the metabolism of glutamine, for example, glutaminase.15 Proliferating cancer cells use glutamine as a nitrogen donor for the synthesis of nucleotide precursors, and following the conversion to glutamate, the generation of the amino acids alanine and aspartate.4, 16, 17 The conversion to glutamate also enables cells to use glutamine as a carbon source for the production of α‐ketoglutarate through the activity of glutamine dehydrogenase or an aminotransferase, including PSAT1.4, 16, 17 Strategies to exploit the dependence of some tumor types on glutamine that are under development include the use of glutamine transport or enzyme inhibitors.18, 19, 20
Ewing sarcoma (EWS), a soft tissue and bone tumor, primarily occurs in adolescents and young adults. In most cases of EWS, the initiating genetic event involves a chromosomal translocation that fuses the 5′ end of the EWSR1 gene to the 3′ end of a member of the ETS (E26‐transformation specific) family of genes, FLI1. The EWS‐FLI1 fusion gene expresses an oncogenic chimeric transcription factor that deregulates the expression of many hundreds of genes. The epigenome of EWS cells reflects the changes in the regulatory state of genes associated with EWS‐FLI1 binding and activation or repression of transcription.21, 22, 23 Examples of genes linked to the oncogenic activity of EWS‐FLI1 include other regulators of transcription such as NR0B1,24, 25, 26 GLI1,27, 28, 29 and EZH2,30, 31 and enzymes such as PARP1.32 Few studies have assessed the regulation of metabolism by EWS‐FLI1, though one study showed that EWS‐FLI1 deregulates tryptophan metabolism by suppressing the expression of TDO2,33 and a second recent study demonstrated EWS‐FLI1 regulates proteins involved in serine‐glycine biosynthesis.34
In our study, we confirm EWS‐FLI1 controls the expression of the major enzymes involved in de novo serine‐glycine biosynthesis but extend those earlier findings by establishing that EWS‐FLI1 regulates the expression of these genes directly. Furthermore, we show that EWS‐FLI1 also regulates the glutamine transporter, SLC1A5/ASCT2, and enzymes involved in the one‐carbon cycle. Moreover, we demonstrate that EWS cells are sensitive to inhibition of PHGDH function and preferentially depend on exogenous glutamine rather than serine and glycine for cell proliferation and are thus susceptible to glutamine antagonism. Importantly, EWS primary tumors exhibit deregulation of these critically interrelated metabolic genes. Our study suggests that it may be possible to exploit the EWS‐FLI1 reprogramming of EWS cell metabolism as a therapeutic vulnerability.
2. MATERIALS AND METHODS
2.1. Cell culture and reagents
We obtained SKNMC (ATCC: HTB‐10™) cells from ATCC, Manassas, VA. Tim Triche (The Saban Research Hospital, Children's Hospital of Los Angeles, Los Angeles, CA) supplied the TC32 cell line. Dr. Ashish Lal (Genetics Branch, CCR, NCI) provided HCT116 and HEK‐293T cells. Unless otherwise stated, we cultured TC32 and SKNMC cells in RPMI medium and HCT116, and HEK‐293T cells in DMEM medium, (Thermo Fisher Scientific, Waltham, MA), supplemented with 10% fetal bovine serum (FBS), at 37°C, 5% CO2. We confirmed the identity of cell lines not purchased directly from ATCC by short tandem repeat (STR) analysis performed by ATCC and determined cells to be mycoplasma free using the MycoAlert Plus system (Lonza, Walkersville, MD). For transgene expression studies, we cloned the EWS‐FLI1 (type 1 (7/6) fusion) cDNA into a C‐terminal 3xFLAG‐tag vector (pDest‐312, Protein Expression Laboratory, Leidos Biomedical Research, Inc. Frederick National Laboratory for Cancer Research), transfected cells using Lipofectamine 2000 (Thermo Fisher Scientific) and selected for stably expressing cells using puromycin (2 μg/mL) (Thermo Fisher Scientific).
We purchased CBR5884 (Ethyl 5‐[(2‐furanyl carbonyl)amino]‐3‐methyl‐4‐thiocyanato‐2‐thiophenecarboxylate) and AICAR (N1‐(β‐D‐Ribofuranosyl)‐5‐aminoimidazole‐4‐carboxamide) from Tocris Bioscience (Ellisville, MO). Cayman Chemical (Ann Arbor, MI) supplied L‐DON (6‐diazo‐5‐oxo‐L‐nor‐leucine) and GSH (L‐glutathione, reduced). We obtained L‐glutamic acid γ‐(p‐nitroanilide) hydrochloride (GPNA) from Santa Cruz Biotechnology, (Santa Cruz, CA). NCT503 (SML1659), tiron, and the metabolites, glucose, glutamine, serine, and glycine were from Sigma‐Aldrich (St. Louis, MO). We dissolved the metabolites, L‐DON, GSH, and GPNA in phosphate buffered saline (PBS) and all other compounds in DMSO at room temperature. For RNAi studies, we purchased siRNAs from Thermo Fisher Scientific (Ambion) or Qiagen (Germantown, MD) and transfected cells using 20 nM siRNA complexed with RNAi‐Max (Thermo Fisher Scientific). To deplete EWS‐FLI1 expression, we used siRNAs we have validated previously that target either the EWSR1 (siEWSR1.1 5′‐GCCUCCCACUGGUUAUACUtt‐3′, Ambion, S4888) or the FLI1 (siFLI1.1 5′‐CAAACGAUCAGUAAGAAUAtt‐3′, Ambion, S5266) derived portions of the fusion transcript.35 To silence the expression of ATF, PHGDH, or SLC1A5 we used the following siRNAs: siATF4 5′‐CAGCGTTGCTGTAACCGACAA‐3′ (Qiagen, SI03019345); siPHGDH.1 5′‐CACGACAGGCTTGCTGAATGA‐3′ (Qiagen, SI00090384); siPHGDH.2 5′ ‐TGGGATGAAGACTATAGGGTA‐3′ (Qiagen, SI00090405); siSLC1A5.1 5′‐UAGGUGGUAGAGUAUGAGCga‐3′ (Ambion, S12916) siSLC1A5.2 5′‐AAAGAGUAAACCCACAUCCtc‐3′ (Ambion, S12918).
2.2. Gene expression and chromatin immunoprecipitation (ChIP) analysis
For real‐time PCR analysis, we extracted total RNA from cells using Maxwell simply RNA system (Promega, Madison, WI), and synthesized cDNA using the iScript cDNA synthesis kit (Bio‐Rad, Hercules, CA) following the manufacturer's protocol using the levels of 18S ribosomal RNA or ACTB as a reference. We used the following primer sets for SYBR green based qPCR: EWS‐FLI1: Forward primer 5′‐ACCCCAAACTGGATCCTACAG‐3′, Reverse Primer 5′‐GGCCGTTGCTCTGTATTCTTAC‐3′; EWSR1 Forward primer 5′‐TACTCTCAGCAGAACACCTATGG‐3′, Reverse Primer 5′‐GTGGTCCTGTCGGAATGAACT‐3′; 18S ribosomal RNA: Forward primer 5′‐GTAACCCGTTGAACCCCATT‐3′, Reverse Primer 5′‐CCATCCAATCGGTAGTAGCG‐3′; and ACTB Forward primer 5′‐GGCACCCAGCACAATGAA‐3′, Reverse Primer 5′‐CCGATCCACACGGAGTACTTG‐3′ were custom synthesized (Thermo Fisher Scientific) and from Qiagen: PHGDH (QT00083279), PSAT1 (QT00074424), PSPH (QT00081067), SHMT2 (QT00012754), SHMT1 (QT00072499), SLC1A4 (QT00042595), SLC1A5 (QT00083909), ATF4 (QT00074466), MTHFD1 (QT00056966), MTHFD2 (QT00081592), and MTHFD1L (QT00043421).
For analysis of whole transcriptome profiles, we transfected cells (4 × 105 cells per well of a 6‐well plate) grown overnight with 20 nM siRNA complexed with 5 μL RNAi‐Max and harvested cells 24 or 48 h post‐transfection. We purified total RNA using the Maxwell 16 LEV SimplyRNA cells kit (Promega), confirmed decreased expression of EWS‐FLI1 and selected target genes by qPCR analysis (Figure S1A) and prepared poly‐A selected RNA libraries that we sequenced on an Illumina HiSeq2000 (Illumina, San Diego, CA). We aligned RNA‐sequencing (RNA‐seq) reads to the hg19 reference genome using Linux TopHat. We calculated gene expression values as fragments per kilobase (kb) of transcript per million mapped reads (TPM) using Cufflinks and gene level coordinates derived using the University of California, Santa Cruz genome browser. We determined differential gene expression using Cuffdiff36 and confirmed reversal of the expression of multiple EWS‐FLI1 gene targets (Figures S1B and S1C). The RNA‐seq data is available at the Gene Expression Omnibus (GEO) portal (NCBI, NIH) using accession number GSE103843. Tang et al37 used the same methods and analysis pipeline to generate gene expression data for control shRNA and doxycycline‐inducible EWS‐FLI1 shRNA expressing A673 cells.38
The acquisition and molecular profiling of EWS primary tumor samples and normal tissues were described previously.39 All specimens for sequencing were obtained from patients with appropriate consent from the local institutional review board in accordance with the Children's Oncology Group and the National Cancer Institute; see the previous study for additional details.39 The complete dataset is available at dbGaP Study Accession Number: phs000768.v2.p1.
We collected ChIP‐seq data from a published study,40 and generated coverage density maps (tiled data files) by counting the number of reads that mapped to each 25 base pair (bp) window using Integrative Genomics Viewer (IGV) tools (http://software.broadinstitute.org/software/igv/). We performed ChIP‐PCR studies using the Magna‐ChIP Kit (Millipore, Billerica, MA) as per manufacturer's instructions and either an anti‐FLI1 antibody (ab15289, Abcam, Cambridge, MA) or normal rabbit IgG antibody (02‐6102, Thermo Fisher Scientific) overnight as described previously.35 Two independent experiments were performed, one in which we normalized samples to IgG antibody and one in which normalized samples to the percentage input DNA. The latter experiment included analysis of a gene desert region of DNA (Chr12) as a negative control. The primers used for ChIP‐PCR (synthesized by Thermo Fisher Scientific) were as follows: PHGDH (chr1:120,254,509‐120,254,623; exon 1) Forward 5′‐GGAGGAGGAGGAGGAGATGA‐3′, Reverse 5′‐GGCCGCTGTGAGTAGAAGTA‐3′; PSAT1 (chr9:80,912,035‐80,912,278; exon 1), Forward 5′‐CCGGCTGCAGACTCTCAC‐3′, Reverse 5′‐GGAATCCGACTGCCACAC‐3′; PSPH (chr7:56,119,389‐56,119,569; exon 1) Forward 5′‐GACGACCTGTGGCCCAAT‐3′, Reverse 5′‐TGGGGTTCAGGGTCTTCAC‐3′; PSPH (chr7:56,101,810‐56,102,021; intron 1 and exon 2) Forward 5′‐GGGTGGCTTCTGATGAGTTC‐3′, Reverse 5′‐GGAGCTCTTTTCCTCTGCAA‐3′; SHMT2 (chr12:57,623,491‐57,623,688; exon 1 and intron 1) Forward 5′‐GGTCTTCTTCCGACAGCTTG‐3′, Reverse 5′‐AGCCAAGAGAACCCCAGAGT‐3′; SLC1A5 (chr19:47,291,658‐47,291,755; Exon 1) Forward 5′‐CCTGGGTCTTGGACACTGAG‐3′, Reverse 5′‐ACCCTCCCGGACCTAAGAG‐3′; MTHFD2 (chr2:74,425,700‐74,425,783, Exon 1) Forward 5′‐ACGAGGCCGCAGTATAACC‐3′, Reverse 5′‐AGAAGTCGCAGCCATAGACC‐3′; MTHFD1L (chr6:151,187,679‐151,187,750, Exon 1) Forward 5′‐AGCTCCCTGGTGTTGTGC‐3′, Reverse 5′‐GGACGTCAGCCACAGACC‐3′; negative control (chr12: 61,273,954‐61,274,043) Forward 5′‐AGGGATTTTTATGAGCATTCCA‐3, Reverse 5′AGCAGGTAAAGGTCCATATTTCA‐3′.
2.3. Immunoblotting
We prepared total lysates from cells post‐treatment using cell extraction buffer (FNN0011, Thermo Fisher Scientific) and performed immunoblotting using the following antibodies: anti‐ACTB (sc‐47778) from Santa Cruz Biotechnology, Santa Cruz, CA; anti‐ATF4 (ab85049), anti‐FLI1 (ab15289), anti‐PHGDH (ab13428), anti‐PSAT1 (ab96136), anti‐PSPH (ab96414), anti‐SLC1A5 (ab187692), anti‐H2AX (ab10475), and anti‐γH2AX (pS139) (ab2893) from Abcam; anti‐SHMT2 (12762), anti‐MTHFD1L (14999), and anti‐MTHFD2 (41377) from Cell Signaling Technology, Danvers, MA.
2.4. Cell starvation experiments
We grew cells in their respective complete medium containing 10% dialyzed FBS for 96 h before initiation of starvation experiments. For serine, glycine, and glucose deprivation studies, we cultured cells in RPMI lacking serine, glycine, and glucose (Teknova, Hollister, CA) and supplemented with 11.11 mM glucose, 0.134 mM glycine, or 0.28 mM serine as required. For glutamine deprivation experiments, we cultured cells in RPMI lacking glutamine (Thermo Fisher Scientific) and supplemented with 2.0 mM glutamine from outside for replenishment. For cells growing in DMEM, we used glucose and glutamine‐free DMEM (Thermo Fisher Scientific) and supplemented with either 25 mM glucose or 4 mM glutamine as required. Similarly, for serine and glycine deprivation studies, we cultured cells in serine‐glycine free DMEM (US Biological Life Sciences, Salem, MA), and we supplemented with 0.4 mM of serine and glycine as needed. We added 10% dialyzed FBS (Thermo Fisher Scientific) to all starvation media.
2.5. Metabolism assays and ROS analysis
We assayed α‐ketoglutarate, glycine, formate, GSH/GSSG and NADP/NADPH ratios, pyruvate, and hexokinase activity using the respective colorimetric or fluorometric assays (BioVision, Milpitas, CA) as per the manufacturer's instructions generating standard curves for each experiment as specified in the supplied protocols. We estimated protein concentrations and pre‐normalized metabolic levels or enzyme activity by using equal amounts of protein for each measurement and assayed at least three biological replicas in each experiment using an Ensight plate reader (Perkin Elmer, Waltham, MA) for all assays. We measured the generation of ROS was using fluorescent dye carboxy‐H2DCFDA (Thermo Fisher Scientific) as per the manufacturer's instructions and analyzed the flow cytometry data using FlowJo software (Version 10.0.7) (FlowJo, LLC, Ashland, OR).
2.6. Functional and 3D spheroid assays
We measured caspase activation and cell viability using the CaspaseGlo 3/7 and the CellTiter‐Glo® Luminescent assay systems respectively (Promega) per manufacturer's instruction using at least three biological replicas of each treatment.
We performed spheroid studies using a magnetic 3D‐spheroid system from Nano3D Biosciences, Inc (Houston, TX). Briefly, we magnetized cells before spheroid assembly using magnetic nanoparticles as per manufacturer's instructions (NanoShuttle‐PL, Nano3D Biosciences). Following overnight treatment with magnetic nanoparticles, cells were trypsinized, counted, and plated in replenished media at a concentration of 250 cells/well in 384‐well ultra‐low attachment (ULA) flat bottom plates (Greiner Bio‐one #781976). For spheroid formation, we placed the plates on top of a magnetic spheroid drive (Nano3D Biosciences). To facilitate complete media changes, after 48 h in culture, we placed the plates on a magnetic holding drive (Nano3D Biosciences) that retains the spheroids within each well and replaced the growth media. We cultured spheroids for an additional 24 to 48 h in complete or nutrient depleted media. We analyzed cell viability (48 h) using the CellTiter‐Glo® reagent (Promega) supplemented with 20% trypsin to help disrupt the spheroids. We measured caspase activation (24 h) using CaspaseGlo 3/7 (Promega). During incubation with the CaspaseGlo 3/7 reagent, we disrupted the spheroids structures by pipetting. We obtained images of spheroids using a Celigo microwell image cytometer and software (Nexcelom Bioscience, MA).
We performed spheroid‐based clonogenic assays as described by Senkowski et al.41 Briefly, we plated TC32 cells in 384‐well ULA round bottom plates, and cultured spheroids for 96 h followed by 72 h of drug treatment. After drug treatment, we collected each spheroid, dispersed the cells that form the spheroid, and plated the resulting single cell suspension into the well of a six‐well plate. We incubated cells for seven days in the drug‐free medium, after which we fixed cell colonies with 100% ethanol and stained with crystal violet (0.04% crystal violet, 2% methanol in PBS). We performed each experiment in biological triplicate. We determined the number of colonies using a Celigo microwell image cytometer and software.
2.7. Statistical analysis
We used Prism (GraphPad, La Jolla, CA) to perform unpaired t‐test analysis (adjusted using the Benjamini and Hochberg procedure or Holm‐Sidak method) or one‐ or two‐way analysis of variance (ANOVA) with a Bonferroni's test to correct for multiple comparisons to the same control group. We considered a False Discovery Rate (FDR) q‐ or P‐value <0.05 significant. For the analysis of tumor or cell line versus normal tissue expression data, we performed unpaired t‐test analysis correcting for multiple comparisons (Holm‐Sidak method). We also used Prism to calculate IC50 values. We employed GSEA (http://www.broadinstitute.org/gsea/index.jsp) to investigate the enrichment of genes associated with KEGG pathways using the molecular signature database (MSigDB v5.1; http://software.broadinstitute.org/gsea/msigdb). We performed the analysis using a weighted enrichment statistic and ranked genes using the Log2 ratio of expression for transfected or treated cells to control cells. We analyzed the resulting landscape plots for peaks in the tails of the ranked gene lists, considering an FDR q‐value <0.05 as significant.
3. RESULTS
3.1. The EWS‐FLI1 oncoprotein regulates the expression of multiple serine‐glycine biosynthesis genes
To identify metabolic pathways regulated by EWS‐FLI1, we interrogated the expression profiles of EWS‐FLI1‐silenced TC32 EWS cells (Figure S1). Gene set enrichment analysis (GSEA) revealed decreased expression of multiple genes involved in metabolic pathways involving fructose and mannose; cysteine and methionine; and glycine, serine, and threonine; and the citrate and TCA cycles (Figure S2A) following depletion of EWS‐FLI1. GSEA showed no significant enrichment for changes in the expression of genes that EWS‐FLI1 represses, though consistent with a recent study,33 we did observe increased expression of TDO2 following silencing of EWS‐FLI1 (Figure S2B).
Consideration of the function of individual genes within each of the metabolic pathways identified by gene set enrichment analysis (GSEA) focused our attention on genes involved in serine‐glycine metabolism (Figure 1A). Genes encoding four members of the serine‐glycine biosynthesis pathway exhibited decreased expression following depletion of EWS‐FLI1 (Figure 1B). Specifically, three genes encoding enzymes required for serine biosynthesis: phosphoglycerate dehydrogenase (PHGDH), phosphoserine aminotransferase 1 (PSAT1), phosphoserine phosphatase (PSPH); and one of the two genes that encode enzymes that catalyze the conversion of serine to glycine, serine hydroxymethyltransferase 2 (SHMT2). SHMT2 catalyzes the conversion of serine to glycine in the mitochondria, while SHMT1 catalyzes the same reaction in the cytoplasm. We observed minimal changes in the expression of SHMT1 following silencing of EWS‐FLI1.
To validate our RNA‐sequencing data, we depleted EWS‐FLI1 in TC32 cells and a second EWS cell line, SKNMC, or silenced EWSR1 or ectopically expressed EWS‐FLI1 in HEK‐293T cells and analyzed the expression of the serine‐glycine genes by qPCR. EWS‐FLI1‐depleted EWS cells (siFLI1) exhibited significantly reduced levels of PHGDH, PSAT1, PSPH, and SHMT2 mRNA (Figures 1C and S3A). The application of a siRNA targeting the EWSR1 portion of the EWS‐FLI1 transcript (siEWSR1) generated comparable results in both TC32 and SKMNC cells (Figure S3B). EWS cells express little or no full‐length FLI1 but do express wild‐type EWSR1 and so to exclude any involvement of EWSR1 in the expression of the serine‐glycine synthesis genes we examined EWSR1‐depleted HEK‐293T cells (Figure S3C). Depletion of EWSR1 did not affect the expression of PHGDH, PSAT1, PSPH, or SHMT2 (Figure S3C). In contrast, HEK‐293T cells expressing EWS‐FLI1 exhibited increased mRNA levels of the same genes compared to control cells (Figure 1D). We observed comparable changes in the expression of PHGDH, PSAT1, PSPH, and SHMT2 proteins using the same perturbations of EWS‐FLI1 expression (Figures 1E, 1F, and S2D). Consistent with our RNA‐seq data, our qPCR analysis indicated silencing of EWS‐FLI1 did not affect the expression of SHMT1 (Figure S2E). Interrogation of expression profiles of control and EWS‐FLI1 (shRNA) depleted‐A673 EWS cells developed as part of an independent study 37 validated our results (Figure S2F), but interestingly, this system indicated a decrease in the expression of SHTM1 not observed following silencing of EWS‐FLI1 in TC32 cells.
Next, we assessed the clinical relevance of our findings by interrogating RNA‐seq data from 58 primary EWS tumors (EWS‐FLI1 positive) and 42 normal tissues (Figure 1G) and 33 EWS cell lines (EWS‐FLI1 positive) (Figure S4A). Compared to muscle and most other tissue types, transcript levels of PHGDH, PSAT1, PSPH, and SHMT2 were much higher in EWS tumor samples (Figure 1G). We observed a similar pattern of expression in EWS cell lines (Figure S4A). EWS tumors exhibited higher levels of SHMT1 compared with normal tissues (Figure 1G), but in EWS cell lines we observed more variation in the expression of SHMT1 (Figure S4A). Overall, our findings confirm observations by Tanner and co‐workers that also demonstrated altered expression of serine‐glycine synthesis genes following depletion of EWS‐FLI1, but as that previous study did not determine whether EWS‐FLI1 regulates the expression of these genes directly or indirectly, we next investigated this question, focusing on the regulation of PHGDH, PSPH, PSAT1, and SHMT2.
ATF4 is a key regulator of amino‐acid biosynthesis, and in non‐small cell lung carcinoma it is the effector transcription factor that activates expression of the serine‐glycine pathway.9 To determine whether ATF4 functions as the regulator of serine‐glycine biosynthesis genes in EWS cells, we silenced ATF4 (siATF4) in TC32 and SKMNC cells. Depletion of ATF4 in EWS cells did not affect PHGDH, PSPH, PSAT1, or SHMT2 mRNA levels (Figure S4B). Furthermore, silencing of EWS‐FLI1 (siFLI1) had no significant effect on ATF4 expression (Figure S4C). We thus hypothesized that EWS‐FLI1 regulates the expression of multiple serine‐glycine biosynthesis genes directly. To test this hypothesis, we first interrogated reported EWS‐FLI1 ChIP‐seq data generated using A673 EWS cells.40 Published data indicated substantial binding of EWS‐FLI1 at sites within the regulatory regions of PHGDH, PSAT1, PSPH, and SHMT2 (Figure 1B). To validate the binding of EWS‐FLI1 to specific regulatory regions within these four serine‐glycine biosynthesis genes, we performed ChIP‐PCR analysis of control and siFLI1‐transfected TC32 cells. Our results confirmed EWS‐FLI1 binding of the same gene regulatory regions in the PHGDH, PSAT1, PSPH, and SHMT2 genes in TC32 cells as reported for A673 EWS cells (Figures 1H and S4D). Together, our data suggest that EWS‐FLI1 is a direct driver of the transcriptional regulation of multiple enzymes involved in serine‐glycine biosynthesis.
3.2. EWS cells use glutamine as a nutrient source
We next hypothesized that the activation of de novo serine‐glycine synthesis should enable EWS cells to proliferate in the absence of exogenous sources of serine or glycine. To test that hypothesis, we measured the viability of TC32 and SKNMC cells after 24 or 48 h of growth in control medium or medium depleted of serine and glycine. For comparison, we grew HCT116 cells (a colorectal cancer cell line) under the same conditions. HCT116 cells depend on exogenous serine or glycine for cell proliferation.42 EWS cells showed no reduction in viability when grown in the absence of serine and glycine, whereas we observed reduced viability of HCT116 cells grown in serine‐glycine depleted medium (Figure 2A).
Figure 2.
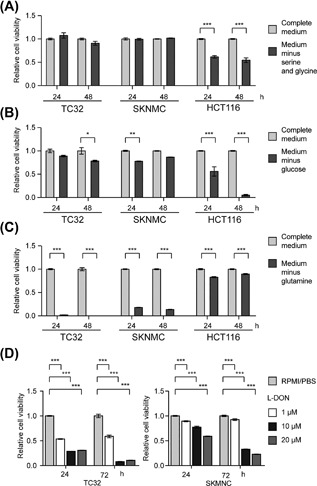
EWS cells use glutamine as a nutrient source. (A) Relative cell viability of TC32, SKNMC, and HCT116 cells measured after 24 or 48 h of serine‐glycine deprivation as indicated (mean ± SEM of three independent experiments). (B) Relative cell viability of TC32, SKNMC, and HCT116 cells measured after 24 or 48 h of glucose deprivation as indicated (mean ± SEM of three independent experiments). (C) Relative cell viability (mean ± SEM of three independent experiments) of TC32, SKNMC, and HCT116 cells measured after 24 or 48 h of glutamine deprivation as indicated. (D) Relative cell viability of TC32 and SKNMC cells measured after the indicated treatment with L‐DON (mean ± SEM of three independent experiments). (A‐D) Two‐way ANOVA (adjusted P‐value) *P < 0.05, **P < 0.01, ***P < 0.001
In comparison with HCT116 cells, we saw minimal effects of the depletion of glucose on the growth of EWS cells, suggesting that EWS cells are less dependent on exogenous glucose than the control cells (Figure 2B). In contrast, depletion of glutamine from growth medium reduced the viability of EWS cells significantly (Figure 2C). Depletion of glutamine minimally affected the viability of HCT116 cells (Figure 2C). To confirm the relative dependency of EWS cells on glutamine compared with glucose, we assessed the effects of L‐DON, a glutamine antagonist, and AICAR, an activator of AMP‐activated Ser/Thr protein kinase (AMPK) signaling that induces glucose starvation, on the viability of EWS cells (Figures 2D and S5). L‐DON mediated significant decreases in the viability of TC32 cells at all concentrations tested, and we observed time‐dependent decreases in the viability of TC32 and SKNMC cells confirming the importance of glutamine for EWS cell survival (Figure 2D). AICAR induced minimal changes in cell viability consistent with the resistance of EWS cells to glucose deprivation (Figures S5A and S5B) but did mediate decreased viability of HCT‐116 control cells (Figure S5C).
3.3. EWS cells uptake extracellular glutamine via the glutamine transporter SLC1A5
To determine whether EWS‐FLI1 regulates the expression of genes involved in glutamine metabolism or glutamine uptake directly, we interrogated the expression profiles of EWS‐FLI1‐depleted EWS cells. We observed no evidence that EWS‐FLI1 regulates the expression of genes involved in glutamine metabolism or most glutamine transporters. However, we did detect a significant downregulation in the levels of the genes encoding the glutamine transporters SLC1A4 (ASCT1) and SLC1A5 (ASCT2) (Figures 3A and S6A). Analysis of control and EWS‐FLI1 (shRNA) depleted‐A673 EWS cells derived data 37 also indicated decreased expression of SLC1A4 and SLC1A5 (Figure S6B). EWS‐FLI1 positive EWS primary tumors and cell lines expressed an elevated level of SLC1A4 compared with normal tissues (Figure 3B). EWS‐FLI1 positive EWS cell lines exhibited increased expression of SLC1A5 versus normal control samples, but SLC1A5 expression was within the range observed in normal tissues.
Figure 3.
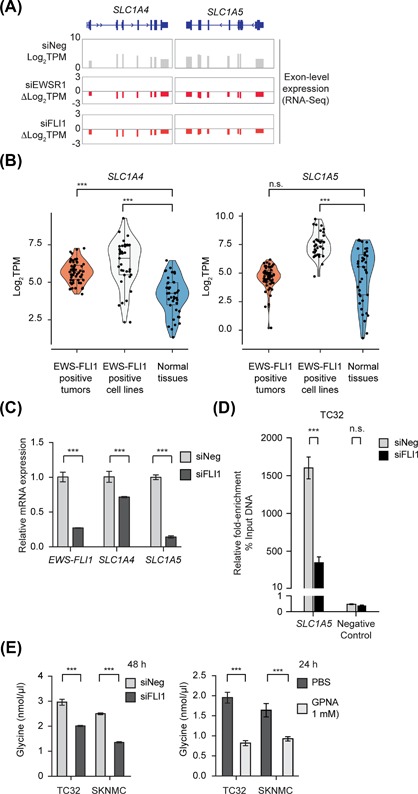
EWS cells uptake extracellular glutamine via the glutamine transporter SLC1A5. (A) Exon level expression and fold‐change from RNA‐seq analysis (Log2 TPM) of the expression of SLC1A4 and SLC1A5 in control (siNeg) and EWS‐FLI1‐silenced TC32 cells (siEWSR1 or siFLI1). (B) Violin scatter plots (including median and quartiles) of the expression of SLC1A4 and SLC1A5 in EWS‐FLI1 positive EWS primary tumors (n = 58), EWS‐FLI1 positive EWS cell lines (n = 33) and normal tissues (n = 42). (C) qPCR analysis of the expression of the indicated genes (relative to siNeg‐transfected cells, mean ± s.e.m., n = 3) in EWS‐FLI1‐silenced TC32 cells (48 h). (D) Fold‐enrichment of EWS‐FLI1‐co‐precipitating DNA determined by qPCR of genomic DNA spanning putative EWS‐FLI1 binding sites. Data shown were calculated as a percent of input DNA and the fold‐enrichment of the anti‐FLI1 antibody compared to IgG; triplicate samples of each treatment in each experiment. (E) Glycine levels in siRNA or compound‐treated TC32 and SKNMC cells (mean ± SEM of three independent experiments). (B) Unpaired adjusted t‐test ***P < 0.001 n.s. = non‐significant.; (C and E) Two‐way ANOVA (adjusted P‐value) ***P < 0.001; (D) Unpaired t‐test, corrected for multiple comparisons ***P < 0.001
To obtain evidence as to whether EWS‐FLI1 regulates one or both SLC1A4 and SLC1A5 directly, we first confirmed the effects of silencing EWS‐FLI1 on their expression using qPCR analysis. qPCR analysis detected a small decrease in SLC1A4 expression following depletion of EWS‐FLI1, but a more substantial decrease in the expression of SLC1A5 (Figure 3B) that we also observed in SKMNC cells (Figure S6C) and at a protein level (Figure S6D) and so we focused our next studies on this glutamine transporter. Using published A673‐ChIP‐seq data,40 we identified a potential regulatory sequence within the SLC1A5 gene bound by EWS‐FLI1. ChIP‐PCR‐based analysis of this DNA region in TC32 cells showed decreased binding of EWS‐FLI1 following the silencing of EWS‐FLI1 (Figures 3D and S6E). We were unable to identify any regulatory sequence within the SLC1A4 gene bound by EWS‐FLI1.
To assess whether EWS cells depend on SLC1A5 function for serine‐glycine biosynthesis, we assayed intracellular glycine levels in EWS cells depleted of EWS‐FLI1 (Figure 3E) or exposed to the SLC1A5 inhibitor, L‐γ‐Glutamyl‐p‐nitroanilide (GPNA, 1 mM).12, 43, 44 Both the decreased expression of EWS‐FLI1 and the addition of GPNA lowered the intracellular levels of glycine in TC32 and SKNMC cells, suggesting that SLC1A5 positively modulates intracellular levels of glycine.
3.4. De novo biosynthesis of serine and glycine feeds into essential metabolic pathways
De novo serine‐glycine biosynthesis supports the growth and proliferation of cancer cells by providing metabolites for several metabolic pathways. For example, α‐ketoglutarate formed as an anaplerotic intermediate of PSAT1 transamination of glutamate to 3‐phosphohydroxypyruvate, refuels about 50% of the TCA cycle (Figure 4A).8, 45 Depletion of EWS‐FLI1 expression in TC32 or SKNMC cells resulted in a significant decrease in α‐ketoglutarate (Figure 4B). Furthermore, HEK‐293T cells expressing EWS‐FLI1 exhibited increased levels of α‐ketoglutarate relative to control cells (Figure 4C). In contrast, reduced expression of EWS‐FLI1 did not alter pyruvate levels or hexokinase enzyme activity, suggesting that when grown in standard cell culture conditions the activity of the fusion oncoprotein has little or no direct effect on glycolysis or glucose metabolism (Figures S7A and S7B). To validate that the generation of α‐ketoglutarate occurs as a byproduct of serine‐glycine biosynthesis, we silenced PHGDH in TC32 and SKNMC cells and measured α‐ketoglutarate levels (Figure 4D). EWS cells depleted of PHGDH exhibited a significant reduction in total α‐ketoglutarate levels (Figure 4D), and we observed similar effects when we treated TC32 cells with increasing concentrations of the PHGDH inhibitors CBR5884 or NCT503 (Figure 4E). Our results suggest that EWS‐FLI1‐mediated serine‐glycine biosynthesis generates the anaplerotic pool of α‐ketoglutarate needed for EWS cell proliferation.
Figure 4.
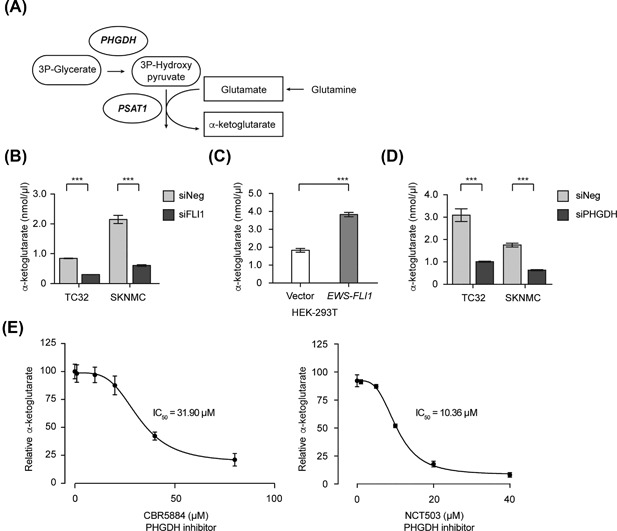
De novo biosynthesis of serine and glycine feeds into essential metabolic pathways. (A) A schematic illustrating the generation of α‐ketoglutarate from glutamine. (B) α‐ketoglutarate levels in TC32 and SKNMC cells 48 h post‐transfection of either siNeg or siFLI1 (mean ± SEM of three independent experiments). (C) α‐ketoglutarate levels in stably transfected HEK‐293T cells (mean ± SEM of three independent experiments). (D) α‐ketoglutarate levels in TC32 and SKNMC cells 48 h post‐transfection of either siNeg or siPHGDH (mean ± SEM of three independent experiments). (E) Relative α‐ketoglutarate levels in TC32 cells 24 h post‐treatment with either CBR‐5884 or NCT‐503 (relative to DMSO‐treated cells, mean ± SEM of three independent experiments). (B) and (D) Unpaired t‐test, adjusted for multiple comparisons ***q < 0.001 (). (C) Unpaired t‐test ***P < 0.001
3.5. The EWS‐FLI1 oncoprotein regulates the expression of genes required for the mitochondrial one‐carbon cycle
Serine acts as the primary carbon donor for the mitochondrial folate cycle, donating its side‐chain carbon to folate, consequently generating glycine and methylene‐tetrahydrofolate (mTHF) as a part of the one‐carbon cycle (Figure 5A).3, 6 Since folate intermediates cannot cross the mitochondrial membrane without cleavage to formate, we measured formate levels as a readout of one‐carbon cycle activity in EWS cells. The silencing of EWS‐FLI1 in EWS cells reduced formate levels, as did the application of the PHGDH inhibitors CBR5884 or NCT503 (Figure 5B).
Figure 5.
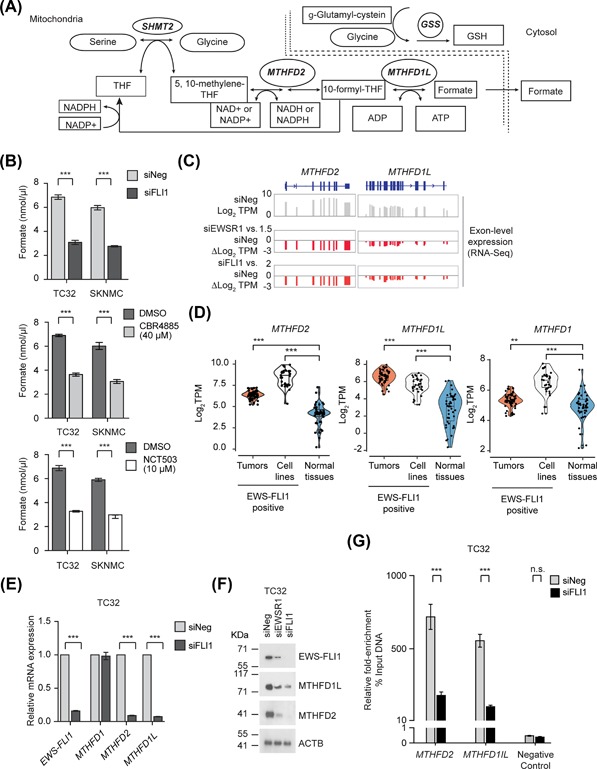
The EWS‐FLI1 oncoprotein regulates the expression of genes required for the mitochondrial one‐carbon cycle. (A) A schematic illustrating the incorporation of serine and glycine into the one‐carbon cycle and production of reducing equivalents for cells. (B) The levels of formate in TC32 and SKNMC cells 48 h post‐transfection of either siNeg or siFLI1; treatment with CBR5884 40 μM for 24 h; or treatment with NCT503 10 μM for 24 h (mean ± SEM of three independent experiments). (C) Exon level expression and fold‐change (Log2 TPM) from RNA‐seq of the expression of MTHFD2 and MTHFD1L in control (siNeg) and EWS‐FLI1‐silenced (siFLI1) TC32 cells. (D) Violin plots (including median and quartiles) of the expression of MTHFD2 and MTHFD1L in EWS‐FLI1 positive EWS primary tumors (n = 58), EWS‐FLI1 positive EWS cell lines (n = 33) and normal tissues (n = 42). (E) qPCR analysis of the expression of the indicated genes (relative to siNeg‐transfected cells, mean ± s.e.m., n = 3) in EWS‐FLI1‐silenced TC32 cells (48 h). (F) Immunoblots of the indicated proteins in lysates prepared from TC32 cells, siRNA‐transfected as indicated. (G) Fold‐enrichment of EWS‐FLI1‐co‐precipitating DNA determined by qPCR of genomic DNA spanning putative EWS‐FLI1 binding sites. Data shown were calculated as a percent of input DNA and the fold‐enrichment of the anti‐FLI1 antibody compared to IgG; triplicate samples of each treatment in each experiment. (B, E, and G) Two‐way ANOVA (adjusted P‐value) ***P < 0.001. (D) Unpaired t‐test, adjusted for multiple comparisons; **q < 0.01, ***q < 0.001
We next assessed whether EWS‐FLI1 also regulates enzymes that catalyze steps in the one‐carbon cycle directly, focusing on the cytoplasmic enzyme MTHFD1, and the mitochondrial enzymes MTHFD2 and MTHFD1L. Our RNA‐seq analysis of EWS‐FLI1‐depleted TC32 (Figure 5C) and results from the A673‐silenced EWS‐FLI1 cells generated by Tang et al (Figure S7C) both indicated a substantial decrease in the expression of MTHFD2. However, the data for MTHFD1 and MTHFD1L were less consistent. Following silencing of EWS‐FLI1 in TC32 cells, we observed a robust decrease in MTHFD1L expression (Figure 5C), but not MTHFD1, while the silencing of EWS‐FLI1 in A673 cells showed changes in MTHFD1 expression, but not MTHFD1L (Figure S7C). Analysis of the expression of the methylenetetrahydrofolate dehydrogenase genes in EWS tumors and cell lines (Figures 5D) showed significant overexpression of MTHFD1, but overall the increased expression of MTHFD2 and MTHFD1L were much higher than the range of expression observed in normal tissues, and so we selected to focus on further study of these two genes. PCR‐based and immunoblot analysis confirmed that depletion of EWS‐FLI1 reduces the expression of MTHFD2 and MTHFD1L (Figures 5E, 5F, and S7D) and we also observed reduced binding of EWS‐FLI1 to sites within the promoters of MTHFD2 and MTHFD1L (Figures 5G and S7E).
3.6. The inhibition of serine‐glycine synthesis in EWS cells results in ROS production and DNA damage
Serine‐glycine biosynthesis feeds into the one‐carbon cycle to generate the NADPH and GSH reducing equivalents (Figure 5A). To assess the redox state of EWS cells following depletion of EWS‐FLI1 or inhibition of PHGDH, we measured the ratios of NADPH/NADP and reduced versus oxidized glutathione (GSH to GSSG) following depletion of EWS‐FLI1 or inhibition of PHGDH (Figures 6A and 6B). The silencing of EWS‐FLI1 (siFLI1) or the application of the PHGDH inhibitor CBR5884 reduced the relative ratios of NADPH/NADP and GSH/GSSG in TC32 and SKNMC cells. Our data suggest the regulation of serine‐glycine biosynthesis by EWS‐FLI1 affects the generation of reducing equivalents by EWS cells.
Figure 6.
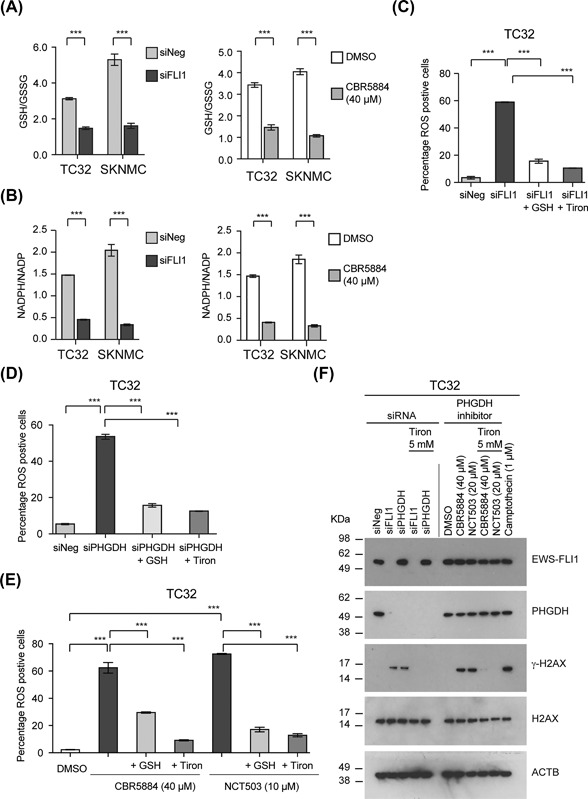
The inhibition of serine‐glycine synthesis in EWS cells results in ROS production and DNA damage. (A) The GSH/GSSG ratio of EWS cells 48 h post‐transfection with either siNeg or siFLI1 or after treatment with CBR5884, 40 μM for 24 h (mean ± SEM of three independent experiments for all data). (B) The NADPH/NADP ratio of EWS cells 48 h post‐transfection with either siNeg or siFLI1 or after treatment with CBR5884, 40 μM for 24 h (mean ± SEM of three independent experiments). (C) The percentage of ROS positive TC32 cells following treatment with siNeg or siFLI1 for 48 h (mean ± SEM of three independent experiments). (D) The percentage of ROS positive TC32 cells following treatment with siNeg or siPHGDH for 48 h (mean ± SEM of three independent experiments). (E) The percentage of ROS positive TC32 cells following treatment with CBR‐5884 or NCT‐503 for 24 h (mean ± SEM of three independent experiments). (F) Immunoblots of the indicated proteins in TC32 cells either transfected with siNeg, siFLI1, or siPHGDH for 48 h or treated with DMSO, CBR5884, or NCT‐503 for 24 h. Where indicated, we added tiron (5 mM) to cells for the last 10 h. As a control, we used the induction of γH2AX following treatment of TC32 cells with Camptothecin (1 μM, 24 h). (A and B) Two‐way ANOVA (adjusted P‐value) ***P < 0.001. (C, D, and E) One‐way ANOVA (adjusted P‐value) ***P < 0.001
Since NADPH and GSH are mediators of redox balance, we next assayed for the presence of reactive oxidant species (ROS) in EWS cells (percentage positive) following depletion of EWS‐FLI1 or PHGDH (Figures 6C and 6D) or treatment of cells with either CBR5884 (40 μM) or NCT‐503 (10 μM) (Figure 6E). All four perturbations significantly increased the percentage of ROS positive cells. The addition of exogenous reduced GSH (5 mM) or the superoxide scavenger and antioxidant tiron (5 mM) decreased the number of ROS positive cells we detected.
The induction of ROS can result in DNA damage. Perturbation of EWS‐FLI1 or PHGDH expression or treatment with CBR5884 or NCT503 induced DNA damage as measured by the phosphorylation of γ‐H2AX (Figure 6F). Consistent with our previous findings, tiron (5 mM) inhibited the phosphorylation of γ‐H2AX observed following depletion or inhibition of PHGDH or silencing of EWS‐FLI1.
3.7. The inhibition of glutamine uptake or PHGDH induces apoptosis in EWS cells and reduces EWS cell growth
The induction of ROS and DNA damage can result in apoptosis, and so we next assessed whether EWS cells cultured in the absence of glutamine exhibit activation of caspase 3/7, a marker of apoptotic cell death. EWS cells grown in media depleted of glutamine exhibited significant activation of caspase 3/7 whereas cells grown in the absence of serine and glycine or glucose exhibited no evidence of apoptosis (Figure 7A). Exposure of EWS cells to the glutamine metabolism inhibitor L‐DON also triggered the activation of caspase 3/7 (Figure 7B). Importantly, the addition of tiron blocked the activation of caspase 3/7 by L‐DON suggesting that the induction of ROS is responsible for the apoptosis of EWS cells. We observed a similar activation of caspase 3/7 when we treated EWS cells with the PHGDH inhibitors CBR5884 and NCT‐503 (Figure 7C). Tiron blocked the activation of caspase 3/7 by CBR5884 and NCT‐503.
Figure 7.
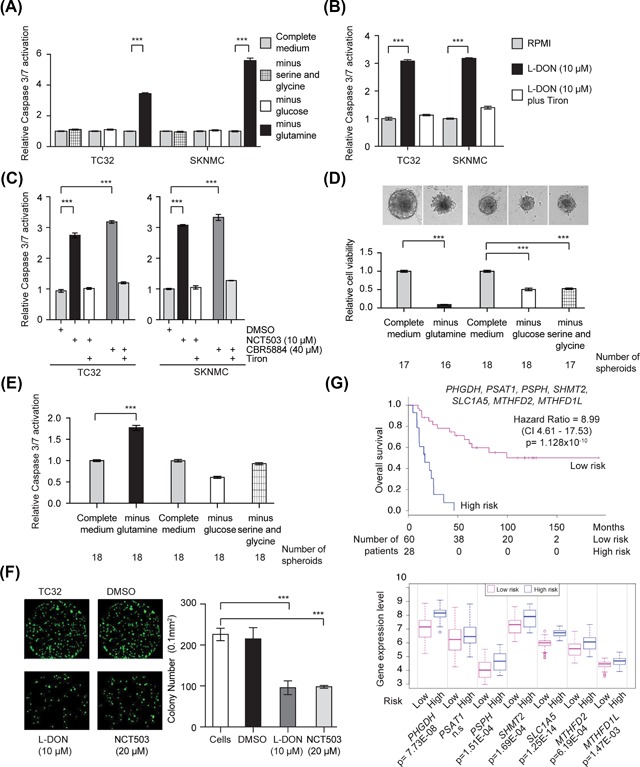
The inhibition of glutamine uptake or PHGDH induces apoptosis in EWS cells and reduces EWS cell growth. (A) Caspase3/7 activation in TC32 and SKNMC cells 48 h after growth in the indicated conditions (mean ± SEM of three independent experiments). (B) Relative caspase 3/7 activation measured in TC32 and SKNMC cells 48 h post treated with either PBS or L‐DON (10 μM) (mean ± SEM of three independent experiments). (C) Caspase 3/7 activity measured in TC32 and SKNMC cells treated for 48h hours with either DMSO, NCT503 or CBR5884 (mean ± SEM of three independent experiments). (B and C) We added tiron (5 mM) to cells for the last 10 h. (D) The viability of TC32 spheroids grown under the indicated conditions. Indicated are the number of spheroids per treatment condition (mean ± SEM). (E) Caspase 3/7 activation in TC32 spheroids 24 h after growth in the indicated conditions. Indicated are the number of spheroids per treatment condition (mean ± SEM). F. Colony formation assay using 7‐day‐old TC32 spheroids pre‐treated with the indicated concentrations of L‐DON or NCT‐503 for 72 h and then grown for seven days in drug‐free medium. We imaged and counted colonies using a Celigo microwell image cytometer (mean ± SEM of three independent experiments). (G) Kaplan‐Meier curves for the overall survival of patients with EWS separated into high‐ and low‐risk groups, and the expression of the genes indicated in these two groups (SurvExpress, Cox Survival Analysis, maximized risk groups).48, 49 (A and B) Two‐way ANOVA (adjusted P‐value) ***P < 0.001. (C) One‐way ANOVA (adjusted P‐value) *P < 0.0; **P < 0.001; ***P < 0.001; (D) One‐way ANOVA (adjusted for multiple comparisons) ***P < 0.001
Next, we assessed whether EWS cells grown as spheroids similarly require exogenous glutamine for cell survival. We selected to use cells grown as spheroids as this culture system allows for the establishment of nutrient gradients that have the potential to recapitulate those seen in tumors in vivo.46, 47 First, we compared the viability of 4‐day‐old TC32 spheroids grown for 48 h in media depleted of glutamine, glucose, and serine/glycine to spheroids cultured in complete media (Figure 7D). Consistent with our findings in monolayer culture, glutamine starvation had a significant effect on the cell viability of TC32 cells grown as spheroids. In spheroid culture, glucose starvation and serine/glycine starvation also affected the overall viability of TC32 cells, but these effects were not as great as those observed following glutamine starvation. Furthermore, we only observed Caspase 3/7 activation in TC32 spheroids following glutamine starvation (Figure 7E). To confirm the dependency of EWS cells on glutamine, as well as de‐novo serine/glycine biosynthesis, in spheroid culture, we assessed the colony formation ability of TC32 cells dispersed from 7‐day‐old spheroids treated for 72 h with L‐DON or NCT503 (Figure 7F). Treatment with L‐DON decreased the number of colony‐forming cells versus controls. Treatment with NCT503 also reduced the number of colony‐forming cells by about 50% versus controls.
Finally, we expanded a previous assessment that examined the overall survival of patients with EWS correlated with expression of several metabolic genes (PHGDH, PSPH, SHMT2, and MTHFD2)[34], to include the additional metabolic genes identified by our study as regulated by EWS‐FLI1. Analysis of a deposited EWS dataset generated by Savola and co‐workers 48 using the SurvExpress tool 49 showed highly significant separation (hazard ratio 8.99; P = 1.128 × 10−10) of the overall survival of EWS patients into low‐ and high‐risk groups based on the expression of PHGDH, PSPH, PSAT1, SHMT2, SLC1A5, MTHFD2, and MTHFD1L (Figure 7G). Analysis of the Savola dataset indicates increased expression of SLC1A5 is associated with a higher risk of poor survival. Together, these data suggest that the deregulation of multiple metabolism genes supports the growth of EWS tumors.
4. DISCUSSION
Here, we report that the fusion transcription factor EWS‐FLI1 regulates the expression of multiple enzymes required for de novo serine‐glycine biosynthesis, specifically PHGDH, PSAT1, PSPH, and SHMT2. We also demonstrate that EWS‐FLI1 regulates expression of the amino acid transporter SLC1A5, and two enzymes that function in the mitochondrial one‐carbon cycle, MTHFD2 and MTHFD1L. Depletion of EWS‐FLI1, or inhibition of serine‐glycine biosynthesis through the application of PHGDH inhibitors, results in ROS accumulation, DNA damage, and apoptotic cell death (Figure 8).
Figure 8.
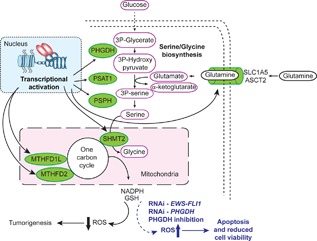
The regulation of multiple metabolic genes by EWS‐FLI1 supports EWS tumorigenesis
Our study complements and extends a recent report by Tanner and colleagues 34 that also described the regulation of de novo serine‐glycine biosynthesis by EWS‐FLI1. Tanner and colleagues presented data using shRNA‐based silencing of EWS‐FLI1 that indicated EWS‐FLI1 regulates PHGDH, PSAT1, and PSPH expression, and that depletion of EWS‐FLI1 decreases the expression of SHMT1/2. Importantly, Tanner and co‐workers show PHGDH expression is high in EWS primary tissue samples and that overall survival also correlates with PHGDH expression. Our study confirms these findings (Figure 1B‐F), and further shows using ChIP‐PCR analysis that EWS‐FLI1 regulates the expression of PHGDH, PSAT1, and PSPH directly (Figure 1G). Interestingly, our data suggest SHMT2, but not SHMT1, is a direct target of EWS‐FLI1. Our analysis of RNA‐seq data from primary EWS tumors is broadly consistent with that presented by Tanner and colleagues, particularly the elevated levels of PHGDH expression (Figure 1H). Together, these studies establish de novo serine‐glycine biosynthesis as an important contributor to EWS tumorigenesis.
The metabolic reprogramming of cancer cells can also involve the utilization of alternative nutrient sources. Consistent with the ability of EWS cells to synthesize serine and glycine de novo, EWS cells grown in medium depleted of these amino acids exhibit no change in viability (Figure 2A). However, EWS cells were sensitive to the depletion of glutamine (Figures 2C and 2D) and treatment with the glutamine antagonist L‐DON (10 to 20 μM; Figure 2E). Previously, Bachmaier and colleagues reported L‐DON (100 μM) decreases EWS‐FLI1 protein levels due to altered O‐GlcNAcylation of the fusion protein.50 While another study, conducted by Olsen and co‐workers, linked the sensitive of EWS and neuroblastoma cells to L‐DON to MYC activity.51 Our study suggests a further reason for the sensitivity of EWS cells to glutamine antagonism, by demonstrating that EWS‐FLI1 transcriptionally regulates glutamine uptake and that EWS cells require exogenous glutamine for glycine synthesis. Collectively, our data suggests EWS cells preferentially utilize exogenous glutamine as a nutrient source and that this may be one of the mechanisms EWS cells use to survive in a nutrient‐poor environment. Inhibitors of SLC1A5/ASCT2 are under development for potential clinical application.52, 53 Our data suggest that as the next generation of these inhibitors become available, they should be assessed in EWS model systems. In the future, it will also be interesting to determine the response of EWS cells to inhibitors of glutaminase, for example, the glutaminase inhibitor CB‐389 is the focus of several ongoing clinical trials in adults with solid tumors (eg, NCT02071862).
Interestingly, we demonstrated through nutrient deprivation and inhibitor studies that EWS cells exhibit little dependence on glucose in comparison with control cells (Figures 2B and S3B). We also observed no changes in total pyruvate or hexokinase levels (Figure S5A and B) following depletion of EWS‐FLI1. Our results differ from previously published findings. Tanner and colleagues reported an increased utilization of glucose by glycolysis following depletion of EWS‐FLI1 that they linked to the derepression of the expression of the hexokinase enzyme.34 A separate study showed glucose deprivation in combination with hypoxia affected the viability of EWS cells.54 We speculate that these divergent results reflect the use of different growth conditions and assays, but future studies will be required to clarify the reasons for these differences.
The additional catalytic functions of PSAT1 and SHMT2 link the serine‐glycine biosynthesis pathway to other metabolic pathways. PSAT1, the second enzyme in the serine‐glycine biosynthesis pathway also catalyzes the generation of α‐ketoglutarate, a key metabolite in TCA cycle, from glutamate. Using gain‐ or loss‐of‐function studies we show EWS‐FLI1 regulates α‐ketoglutarate levels and importantly, inhibition of PHGDH activity recapitulates the decrease in α‐ketoglutarate observed following depletion of EWS‐FLI1 (Figure 4). Also, in addition to catalyzing the conversion of serine to glycine, SHMT1 and SHMT2 catalyze the reversible conversion of tetrahydrofolate (THF) to methylene tetrahydrofolate (meTHF) as part of the one‐carbon cycles in the cytoplasm and mitochondria, respectively. Our analyses show regulation of SHMT2 expression by EWS‐FLI1 (Figure 1), but not SHMT1. Furthermore, we show that other enzymes involved in one‐carbon metabolism, specifically, MTHFD2 and MTHD1L are transcriptional targets of EWS‐FLI1, but not their cytosolic counterpart MTHFD1 (Figure 5). These data suggest EWS‐FLI1 favors activation of enzymes associated with the mitochondrial one‐carbon cycle, which provides the reducing equivalents GSH and NADPH. Depletion of EWS‐FLI1 or the silencing or inhibition of PHGDH reduced the GSH/GSSG and NADPH/NADP ratios in EWS cells resulting in increased production of reactive oxygen species in EWS cells (Figure 6). Our findings suggest a mechanistic basis for previous observations that have described mitochondrial stress34 or DNA damage55 following depletion of EWS‐FLI1 in EWS cells.
In conclusion, our study extends a previous study that reported EWS cells utilize the serine‐glycine biosynthesis pathway by showing EWS‐FLI1 directly regulates the enzymes that catalyze this pathway and that EWS cells also depend on the import of glutamine. Targeting the dependence of EWS cells on the serine‐glycine pathway and exogenous glutamine could form the basis of an innovative approach for the treatment of this aggressive tumor type.
Supporting information
Additional supporting information may be found online in the Supporting Information section at the end of the article.
Figure S1. Characterization of EWS‐FLI1 depleted TC32 cells.
Figure S2. Depletion of EWS‐FLI1 alters the expression of genes associated with multiple metabolic pathways.
Figure S3. EWS‐FLI1 regulates expression of multiple serine‐glycine biosynthesis genes.
Figure S4. The regulation of the expression of serine‐glycine synthesis genes in Ewing sarcoma cells does not require ATF4.
Figure S5. AICAR, an activator of AMPK, has minimal effect on the viability of EWS cells.
Figure S6. The expression of glutamine metabolism and transporter genes in EWS‐FLI1‐depleted TC32 cells.
Figure S7. Glycolysis and cytosolic one‐carbon metabolism are unaffected by the activity of EWS‐FLI1.
ACKNOWLEDGMENTS
Federal funds from the Center for Cancer Research, NCI, NIH, DHHS: ZIA BC 011704 supported this work: Discovery of genes required for the expression or activity of fusion oncogenes, Caplen, N.J. Cancer Prevention Research Institute of Texas (CPRIT) grant RP130397 also funded part of this work. We thank Young Song, Sivasish Sindiri, Vineela Gangalapudi, Hsien‐Chao Chou, and Manoj Tyagi (Oncogenomics Section, Genetics Branch, CCR), and Tamara Jones (Functional Genetics Section, Genetics Branch, CCR) for computational or technical assistance. We also thank Yves Pommier and colleagues, (Developmental Therapeutics Branch, CCR), for making A673/shEWS‐FLI1 RNA‐seq data available to us and the Laboratory of Cancer Biology and Genetics, CCR for access to a Celigo microwell image cytometer. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the US Government.
Sen N, Cross AM, Lorenzi PL, et al. EWS‐FLI1 reprograms the metabolism of Ewing sarcoma cells via positive regulation of glutamine import and serine‐glycine biosynthesis. Molecular Carcinogenesis. 2018;57:1342–1357. 10.1002/mc.22849
Nirmalya Sen current address: Ramalingaswami Fellow, Rajiv Gandhi Centre for Biotechnology (Government of India, Ministry for Science and Technology), Jagathy, Thiruvananthapuram, India.
REFERENCES
- 1. Amelio I, Cutruzzola F, Antonov A, Agostini M, Melino G. Serine and glycine metabolism in cancer. Trends Biochem Sci. 2014;39:191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. 2016;23:27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yang M, Vousden KH. Serine and one‐carbon metabolism in cancer. Nat Rev Cancer. 2016;16:650–662. [DOI] [PubMed] [Google Scholar]
- 4. Zhang J, Pavlova NN, Thompson CB. Cancer cell metabolism: the essential role of the nonessential amino acid, glutamine. EMBO J. 2017;36:1302–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Newman AC, Maddocks ODK. Serine and functional metabolites in cancer. Trends Cell Biol. 2017;27:645–657. [DOI] [PubMed] [Google Scholar]
- 6. Locasale JW. Serine, glycine and one‐carbon units: cancer metabolism in full circle. Nat Rev Cancer. 2013;13:572–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Locasale JW, Grassian AR, Melman T, et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet. 2011;43:869–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Possemato R, Marks KM, Shaul YD, et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature. 2011;476:346–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. DeNicola GM, Chen PH, Mullarky E, et al. NRF2 regulates serine biosynthesis in non‐small cell lung cancer. Nat Genet. 2015;47:1475–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Beroukhim R, Mermel CH, Porter D, et al. The landscape of somatic copy‐number alteration across human cancers. Nature. 2010;463:899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jain M, Nilsson R, Sharma S, et al. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science. 2012;336:1040–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hassanein M, Hoeksema MD, Shiota M, et al. SLC1A5 mediates glutamine transport required for lung cancer cell growth and survival. Clin Cancer Res. 2013;19:560–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jeon YJ, Khelifa S, Ratnikov B, et al. Regulation of glutamine carrier proteins by RNF5 determines breast cancer response to ER stress‐inducing chemotherapies. Cancer Cell. 2015;27:354–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bolzoni M, Chiu M, Accardi F, et al. Dependence on glutamine uptake and glutamine addiction characterize myeloma cells: a new attractive target. Blood. 2016;128:667–679. [DOI] [PubMed] [Google Scholar]
- 15. Gao P, Tchernyshyov I, Chang TC, et al. C‐Myc suppression of miR‐23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458:762–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. De Vitto H, Perez‐Valencia J, Radosevich JA. Glutamine at focus: versatile roles in cancer. Tumour Biol. 2016;37:1541–1558. [DOI] [PubMed] [Google Scholar]
- 17. Still ER, Yuneva MO. Hopefully devoted to Q: targeting glutamine addiction in cancer. Br J Cancer. 2017;116:1375–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Le A, Lane AN, Hamaker M, et al. Glucose‐independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 2012;15:110–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gross MI, Demo SD, Dennison JB, et al. Antitumor activity of the glutaminase inhibitor CB‐839 in triple‐negative breast cancer. Mol Cancer Ther. 2014;13:890–901. [DOI] [PubMed] [Google Scholar]
- 20. Wang Q, Hardie RA, Hoy AJ, et al. Targeting ASCT2‐mediated glutamine uptake blocks prostate cancer growth and tumour development. J Pathol. 2015;236:278–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Riggi N, Knoechel B, Gillespie SM, et al. EWS‐FLI1 utilizes divergent chromatin remodeling mechanisms to directly activate or repress enhancer elements in Ewing sarcoma. Cancer Cell. 2014;26:668–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tomazou EM, Sheffield NC, Schmidl C, et al. Epigenome mapping reveals distinct modes of gene regulation and widespread enhancer reprogramming by the oncogenic fusion protein EWS‐FLI1. Cell Rep. 2015;10:1082–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sheffield NC, Pierron G, Klughammer J, et al. DNA methylation heterogeneity defines a disease spectrum in Ewing sarcoma. Nat Med. 2017;23:386–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kinsey M, Smith R, Iyer AK, McCabe ER, Lessnick SL. EWS/FLI and its downstream target NR0B1 interact directly to modulate transcription and oncogenesis in Ewing's sarcoma. Cancer Res. 2009;69:9047–9055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Garcia‐Aragoncillo E, Carrillo J, Lalli E, et al. DAX1, a direct target of EWS/FLI1 oncoprotein, is a principal regulator of cell‐cycle progression in Ewing's tumor cells. Oncogene. 2008;27:6034–6043. [DOI] [PubMed] [Google Scholar]
- 26. Mendiola M, Carrillo J, Garcia E, et al. The orphan nuclear receptor DAX1 is up‐regulated by the EWS/FLI1 oncoprotein and is highly expressed in Ewing tumors. Int J Cancer. 2006;118:1381–1389. [DOI] [PubMed] [Google Scholar]
- 27. Beauchamp E, Bulut G, Abaan O, et al. GLI1 is a direct transcriptional target of EWS‐FLI1 oncoprotein. J Biol Chem. 2009;284:9074–9082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zwerner JP, Joo J, Warner KL, et al. The EWS/FLI1 oncogenic transcription factor deregulates GLI1. Oncogene. 2008;27:3282–3291. [DOI] [PubMed] [Google Scholar]
- 29. Joo J, Christensen L, Warner K, et al. GLI1 is a central mediator of EWS/FLI1 signaling in Ewing tumors. PLoS ONE. 2009;4:e7608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Riggi N, Suva ML, Suva D, et al. EWS‐FLI‐1 expression triggers a Ewing's sarcoma initiation program in primary human mesenchymal stem cells. Cancer Res. 2008;68:2176–2185. [DOI] [PubMed] [Google Scholar]
- 31. Richter GH, Plehm S, Fasan A, et al. EZH2 is a mediator of EWS/FLI1 driven tumor growth and metastasis blocking endothelial and neuro‐ectodermal differentiation. Proc Natl Acad Sci U S A. 2009;106:5324–5329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brenner JC, Feng FY, Han S, et al. PARP‐1 inhibition as a targeted strategy to treat Ewing's sarcoma. Cancer Res. 2012;72:1608–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mutz CN, Schwentner R, Kauer MO, et al. EWS‐FLI1 impairs aryl hydrocarbon receptor activation by blocking tryptophan breakdown via the kynurenine pathway. FEBS Lett. 2016;590:2063–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tanner JM, Bensard C, Wei P, et al. EWS/FLI is a master regulator of metabolic reprogramming in ewing sarcoma. Mol Cancer Res. 2017;15:1517–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Grohar PJ, Kim S, Rangel Rivera GO, et al. Functional genomic screening reveals splicing of the EWS‐FLI1 fusion transcript as a vulnerability in ewing sarcoma. Cell Rep. 2016;14:598–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA‐s eq. Nat Biotechnol. 2013;31:46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tang SW, Bilke S, Cao L, et al. SLFN11 is a transcriptional target of EWS‐FLI1 and a determinant of drug response in ewing sarcoma. Clin Cancer Res. 2015;21:4184–4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Carrillo J, Garcia‐Aragoncillo E, Azorin D, et al. Cholecystokinin down‐regulation by RNA interference impairs Ewing tumor growth. Clin Cancer Res. 2007;13:2429–2440. [DOI] [PubMed] [Google Scholar]
- 39. Brohl AS, Solomon DA, Chang W, et al. The genomic landscape of the Ewing Sarcoma family of tumors reveals recurrent STAG2 mutation. PLoS Genet. 2014;10:e1004475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bilke S, Schwentner R, Yang F, et al. Oncogenic ETS fusions deregulate E2F3 target genes in Ewing sarcoma and prostate cancer. Genome Res. 2013;23:1797–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Senkowski W, Jarvius M, Rubin J, et al. Large‐Scale gene expression profiling platform for identification of context‐dependent drug responses in multicellular tumor spheroids. Cell Chem Biol. 2016;23:1428–1438. [DOI] [PubMed] [Google Scholar]
- 42. Labuschagne CF, van den Broek NJ, Mackay GM, Vousden KH, Maddocks OD. Serine, but not glycine, supports one‐carbon metabolism and proliferation of cancer cells. Cell Rep. 2014;7:1248–1258. [DOI] [PubMed] [Google Scholar]
- 43. Esslinger CS, Cybulski KA, Rhoderick JF. Ngamma‐aryl glutamine analogues as probes of the ASCT2 neutral amino acid transporter binding site. Bioorg Med Chem. 2005;13:1111–1118. [DOI] [PubMed] [Google Scholar]
- 44. van Geldermalsen M, Wang Q, Nagarajah R, et al. ASCT2/SLC1A5 controls glutamine uptake and tumour growth in triple‐negative basal‐like breast cancer. Oncogene. 2016;35:3201–3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Klysz D, Tai X, Robert PA, et al. Glutamine‐dependent alpha‐ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Sci Signal. 2015;8:ra97. [DOI] [PubMed] [Google Scholar]
- 46. Kunz‐Schughart LA, Doetsch J, Mueller‐Klieser W, Groebe K. Proliferative activity and tumorigenic conversion: impact on cellular metabolism in 3‐D culture. Am J Physiol Cell Physiol. 2000;278:C765–C780. [DOI] [PubMed] [Google Scholar]
- 47. Jamieson LE, Harrison DJ, Campbell CJ. Chemical analysis of multicellular tumour spheroids. Analyst. 2015;140:3910–3920. [DOI] [PubMed] [Google Scholar]
- 48. Savola S, Klami A, Myllykangas S, et al. High expression of complement component 5 (C5) at tumor site associates with superior survival in ewing's sarcoma family of tumour patients. ISRN Oncol. 2011;2011:168712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Aguirre‐Gamboa R, Gomez‐Rueda H, Martinez‐Ledesma E, et al. SurvExpress: an online biomarker validation tool and database for cancer gene expression data using survival analysis. PLoS ONE. 2013;8:e74250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bachmaier R, Aryee DN, Jug G, et al. O‐GlcNAcylation is involved in the transcriptional activity of EWS‐FLI1 in Ewing's sarcoma. Oncogene. 2009;28:1280–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Olsen RR, Mary‐Sinclair MN, Yin Z, Freeman KW. Antagonizing Bcl‐2 family members sensitizes neuroblastoma and Ewing's sarcoma to an inhibitor of glutamine metabolism. PLoS ONE. 2015;10:e0116998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Colas C, Grewer C, Otte NJ, et al. Ligand discovery for the alanine‐Serine‐Cysteine transporter (ASCT2, SLC1A5) from homology modeling and virtual screening. PLoS Comput Biol. 2015;11:e1004477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schulte ML, Khodadadi AB, Cuthbertson ML, Smith JA, Manning HC. 2‐Amino‐4‐bis(aryloxybenzyl)aminobutanoic acids: a novel scaffold for inhibition of ASCT2‐mediated glutamine transport. Bioorg Med Chem Lett. 2016;26:1044–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Knowles HJ, Schaefer KL, Dirksen U, Athanasou NA. Hypoxia and hypoglycaemia in Ewing's sarcoma and osteosarcoma: regulation and phenotypic effects of Hypoxia‐Inducible Factor. BMC Cancer. 2010;10:372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Grohar PJ, Segars LE, Yeung C, et al. Dual targeting of EWS‐FLI1 activity and the associated DNA damage response with trabectedin and SN38 synergistically inhibits Ewing sarcoma cell growth. Clin Cancer Res. 2014;20:1190–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found online in the Supporting Information section at the end of the article.
Figure S1. Characterization of EWS‐FLI1 depleted TC32 cells.
Figure S2. Depletion of EWS‐FLI1 alters the expression of genes associated with multiple metabolic pathways.
Figure S3. EWS‐FLI1 regulates expression of multiple serine‐glycine biosynthesis genes.
Figure S4. The regulation of the expression of serine‐glycine synthesis genes in Ewing sarcoma cells does not require ATF4.
Figure S5. AICAR, an activator of AMPK, has minimal effect on the viability of EWS cells.
Figure S6. The expression of glutamine metabolism and transporter genes in EWS‐FLI1‐depleted TC32 cells.
Figure S7. Glycolysis and cytosolic one‐carbon metabolism are unaffected by the activity of EWS‐FLI1.