

Summary
Regulation of RNA polymerase II (Pol II) elongation is a critical step in gene regulation. Here, we report that U1 snRNP recognition and transcription pausing at stable nucleosomes are linked through premature polyadenylation signal (PAS) termination. By generating RNA exosome conditional deletion mouse embryonic stem cells, we identified a large class of polyadenylated short transcripts in the sense direction destabilized by the RNA exosome. These PAS termination events are enriched at the first few stable nucleosomes flanking CpG islands and suppressed by U1 snRNP. Thus, promoter-proximal Pol II pausing consists of two processes: TSS-proximal and +1 stable nucleosome pausing, with PAS termination coinciding with the latter. While pausing factors NELF/DSIF only function in the former step, flavopiridol-sensitive mechanism(s) and Myc modulate both steps. We propose that premature PAS termination near the nucleosome-associated pause site represents a common transcriptional elongation checkpoint regulated by U1 snRNP recognition, nucleosome stability, and Myc activity.
INTRODUCTION
Divergent transcription is a hallmark of gene regulation across many species, generating protein coding transcripts and upstream antisense RNAs (uaRNAs) from active promoters (Core et al., 2008; Preker et al., 2008; Seila et al., 2008) and bidirectional enhancer RNAs (eRNAs) from enhancers. These classes of non-coding RNAs (ncRNAs) are low abundant and typically far less than protein-coding transcripts, partly due to targeting by the RNA exosome, a complex with 3´-to-5´ exonuclease activity (Kilchert et al., 2016). In addition to degrading these ncRNAs, the RNA exosome regulates rRNA/snoRNA maturation, degradation of improperly spliced transcripts, and nonsense mediated decay. Furthermore, recent studies have implicated the RNA exosome broadly in transcription regulation, resolution of RNA-DNA hybrids, R-loop, and maintenance of genomic integrity (Kilchert et al., 2016). The RNA exosome complex consists of 10 or 11 subunits and requires the core subunit Exosc3 (also known as Rrp40) for its major activities.
High levels of polyadenylation signal (PAS) motifs throughout the genome can signal transcription termination wherever there is an initiation event, leading to subsequent rapid destabilization of transcripts (Andersen et al., 2012). To ensure production of full-length coding transcripts, mRNA genes have evolved low levels of PAS motifs across the transcription unit and an enrichment for 5´ splice sites proximal to the transcription start site (TSS). Recognition of the 5´ splice site by U1 snRNP suppresses the use of nearby PAS motifs by the 3´ end processing machinery, promoting elongation and synthesis of mature RNA (Berg et al., 2012; Kaida et al., 2010). Thus, the relative frequency of U1 splicing signals and PAS motifs (U1-PAS axis) is thought to have important roles in modulating promoter directionality of divergent transcription (Almada et al., 2013; Ntini et al., 2013). Although the RNA exosome degrades uaRNAs, it is unclear how the RNA exosome contributes to transcription regulation in the sense mRNA direction and more specifically whether, under the influence of U1-PAS axis, the RNA exosome also degrades prematurely terminated RNAs in the sense direction.
Chromatin remodeling and histone modifications have been linked with regulation of divergent transcription (Bagchi and Iyer, 2016). In S. cerevisiae, histone chaperone CAF-I and other proteins in the H3K56ac chromatin-assembly pathway reduce divergent transcription of ncRNAs (Marquardt et al., 2014). Incorporation of the histone variant H2A.Z influences exosome-mediated destabilization of uaRNAs in mouse embryonic stem cells (mESC) (Rege et al., 2015). The promoter-proximal nucleosome is known to be a barrier for RNA polymerase II (Pol II) and Pol II frequently pauses at the first few nucleosomes (Mayer et al., 2015; Weber et al., 2014). Mammalian chromatin remodeler Chd1 and Drosophila H2A.Z have been reported to facilitate Pol II promoter escape at the promoter-proximal nucleosomes (Skene et al., 2014; Weber et al., 2014). Pol II pausing at TSS-proximal regions is a well-studied phenomenon of metazoan transcription and is regulated by several pause factors including DSIF and NELF and the pause release factor, positive transcription elongation factor b (P-TEFb) (Adelman and Lis, 2012).
The relationship between promoter-proximal premature termination in the U1-PAS axis, chromatin remodeling, and Pol II pausing has not been investigated in depth (Spies et al., 2009). To stabilize RNAs produced by these processes, we created an Exosc3 CRISPR conditional knockout mESC line. We found that the RNA exosome destabilizes a large class of polyadenylated short transcripts terminating in the first intron and U1 snRNP suppresses this premature PAS termination. Exosome-targeted PAS termination is dramatically enriched at the edges of promoter proximal regions devoid of stable nucleosomes, demarcated by CpG islands, and is associated with active regulation of chromatin remodeling and Pol II pausing. Our analysis further showed that these genomic domains mechanistically delineate two types of Pol II pausing: TSS proximal pausing and +1 stable nucleosome pausing. Overall, this study proposes an elongation checkpoint involving the convergence of the U1-PAS axis, exosome activity, Myc regulation, and Pol II pausing.
RESULTS
Conditional Depletion of Exosc3 Identifies Many Exosc3-Targeted Non-coding RNAs
To further identify exosome-targeted transient RNA species, we generated a doxycycline (dox)-inducible Exosc3 conditional knockout (CKO) mESC line. The CRISPR-Cas9 system was used to delete the entire endogenous Exosc3 gene in a mESC line expressing dox-inducible C-terminus FLAG-HA-tagged Exosc3 (Exosc3-FH) (Figures S1A–C and Table S1). After 2–3 days of dox withdrawal, Exosc3 mRNA levels decrease to barely detectable levels, confirming high efficiency of conditional Exosc3 depletion (Figure S1D). An increase in cell death was observed at this time point, suggesting Exosc3 is an essential gene.
We performed RNA-seq on rRNA-depleted RNA from Exosc3 CKO mESCs after 3 days of dox removal (Figure S1E). Metaplots of RNA-seq reads around the TSS of UCSC canonical transcripts revealed a 6-fold stabilization of uaRNAs upon Exosc3 depletion, but little change in overall RNA reads in the sense direction (Figure 1A). This is consistent with a previous report (Preker et al., 2008). Similarly, eRNAs from intergenic enhancers bound by Oct4, Sox2, and Nanog (OSN) were stabilized by 8-fold upon loss of Exosc3 (Figure 1B). Based on de novo transcript assembly (Figures S1F–G and S2A–D), we found that most uaRNAs, super-enhancer-associated enhancer RNAs (seRNA), and typical enhancer-associated enhancer RNAs (teRNA) were significantly upregulated upon loss of the RNA exosome (Fold change (FC) ≥ 2, FDR < 0.1) (Figure 1C).
Figure 1. Loss of Exosc3 upregulates many non-coding RNAs.
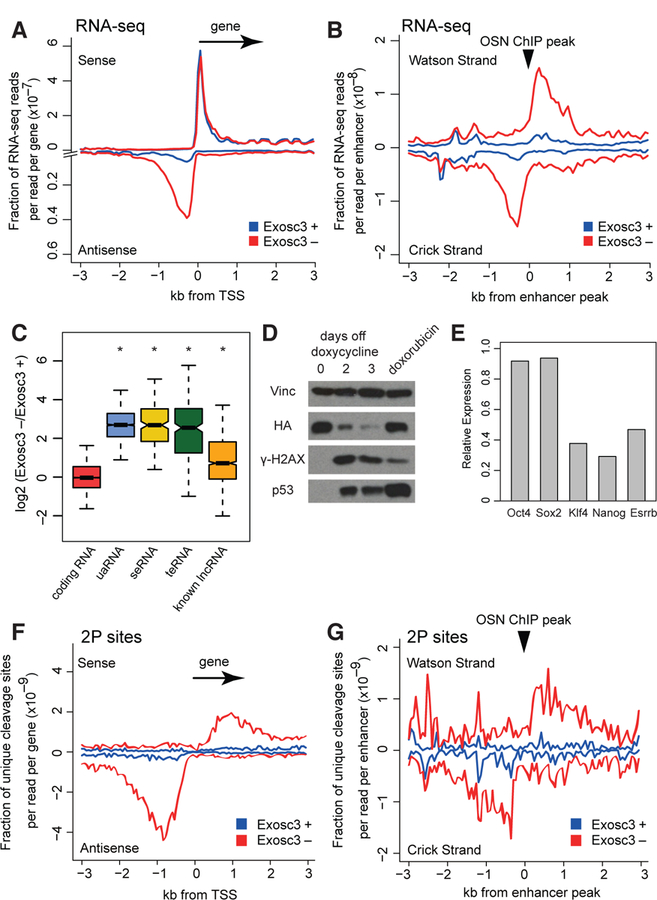
(A, B) Metaplots of RNA-seq reads around a 3 kb window flanking TSS of non-overlapping UCSC canonical genes (A) and centers of Oct4, Sox2, and Nanog (OSN) ChIP-seq peaks (B). Blue: Exosc3 +, on dox, Red: Exosc3 –, 3 days off dox.
(C) Boxplot showing expression changes of various RNA species upon Exosc3 depletion. * P < 0.001 with Wilcoxon signed ranked sum test.
(D) Western Blot for vinculin, HA-tagged Exosc3, γ-H2AX, and total p53. doxorubicin: 7 hour treatment with 1 μM doxorubicin.
(E) Relative expression of pluripotency genes upon Exosc3 depletion, determined by RNA-seq.
(F, G) Metaplots of mean unique cleavage sites (2P-seq) with PAS motifs around non-overlapping TSS of genes (F) and centers of OSN ChIP-seq peaks (G), normalized by library depth.
See also Figures S1–S3.
Gene set enrichment analysis (GSEA) revealed that p53-target genes were significantly changing upon Exosc3 depletion (Figures S2E–F), consistent with an increase in p53 protein level and cleaved caspase 3 signal (Figures 1D and S2G). An increase in γ-H2AX level was also observed (Figure 1D). Consistently, inhibition of RNA exosome is known to cause genomic instability (Kilchert et al., 2016). In addition, changes in genes linked with differentiation of mESCs were also detected by GSEA (Figures S2E–F). While Oct4 and Sox2 expression levels did not change, Klf4, Nanog, and Esrrb levels decreased (Figure 1E), suggesting a possible conversion from a naïve stem cell state to a primed stem cell state (Hackett and Surani, 2014).
Using poly(A)-primed sequencing (2P-seq) (Spies et al., 2013), we generated a genome-wide dataset of cleavage sites from polyadenylated transcripts. We focused on cleavage sites with nearby PAS motifs because they comprise the majority of cleavage sites (Figures S3A–B). The distribution of PAS variants of intronic 2P sites is similar to that of gene terminal 2P sites, suggesting that two reactions are similar if not identical. Cleavage sites without an upstream detectable PAS motif were detected less frequently (Figure S3C), suggesting they could be degradation intermediates.
Analysis of unique cleavage sites with the PAS motifs revealed global derepressive effects of Exosc3 loss on polyadenylated uaRNAs and eRNAs (Figures 1F–G), consistent with previous results (Almada et al., 2013). The half-lives of individual uaRNAs increased by 2–3 fold upon depletion of exosome activity following transcription arrest with flavopiridol (Figures S3D–E). 40% of annotated uaRNAs and 31% of defined eRNAs generated detectable cleavage sites with PAS motifs. These observations are consistent with additional PAS-independent pathways that degrade uaRNAs (Meola et al., 2016) and an integrator-dependent mechanism that controls the biogenesis of non-polyadenylated eRNAs (Lai et al., 2015).
PAS Termination in the First Intron
Unexpectedly, upon Exosc3 depletion, there was a dramatic increase in unique detectable 2P cleavage sites within the gene body peaking at 800 nts downstream of the TSS for approximately 3500 of all genes (Figure 1F). These sites overlap the position where PAS motif frequency reaches the intragenic background levels (Figure S3F), suggesting that prematurely-terminated sense transcripts with these PAS sites are additional substrates of the RNA exosome. Consistent with this conclusion, after filtering out exonic reads, we found that Exosc3 depletion causes an increase in intronic RNA-seq reads proximal to the first 5´ splice site, and this gradually diminishes over the first 2 kb of the first intron (Figure 2A).
Figure 2. Premature PAS termination in the first intron of sense transcripts.
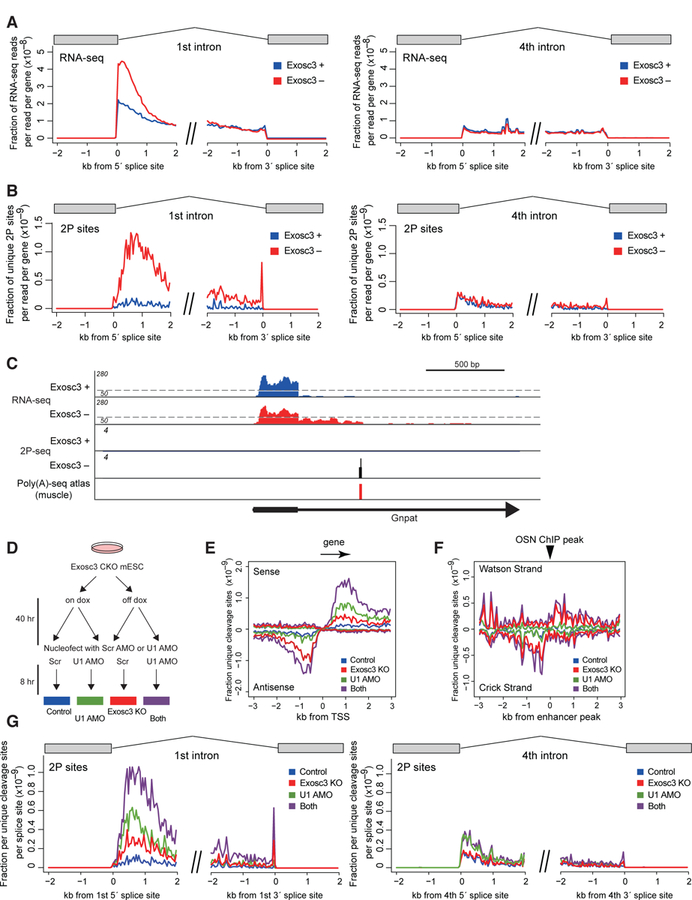
(A, B) Mean exon-removed RNA-seq signal (A) and unique cleavage sites (2P-seq) with canonical PAS motifs (B) flanking 5´ or 3´ splice sites of the first or fourth intron, normalized by library depth.
(C) Genome browser shot of Gnpat. For RNA-seq, scale changes at a dotted line. Previously reported PAS site in mouse tissues (Derti et al., 2012) is also shown.
(D) Experimental design for double treatment with Exosc3 depletion and U1 inhibition.
(E-G) Mean unique cleavage site signal (2P-seq) with PAS motifs around TSS (E), around OSN ChIP-seq peaks (F), and around the first or fourth 5´ and 3´ splice sites (G) after Exosc3 depletion and/or U1 inhibition.
See also Figure S3.
One potential explanation for the increase in the first intron signal is stabilization of lariat intermediates. However, this possibility is unlikely because there is no increase in intronic RNA-seq reads at the fourth intron (Figure 2A). This also suggests that the increase in reads in the first intron is not due to a general stimulation of transcription upon exosome depletion. The fourth intron is shown as an example but this is also the case for other introns in gene expression-normalized data (Figure S3G). Moreover, 2P cleavage sites stabilized by exosome depletion are found almost exclusively in the first intron and not in the fourth intron (Figure 2B). For instance, a profile of the representative gene Gnpat shows this stabilization in the first intron upon Exosc3 removal (Figure 2C). The increase in termination remains specific to the first intron after normalizing for gene expression (Figure S3H). Intriguingly, a few premature events overlapped with previously reported PAS sites in mouse tissues (Derti et al., 2012) and known cDNA annotations (premature Rad23b is AK163379, premature Pcf11 is BC048838, and premature Psmd14 is AK014293), consistent with these being contiguous transcripts from the TSS.
Suppression of Sense Direction PAS Termination by U1 snRNP
Inhibition of U1 snRNP activity is known to promote the use of early PAS motifs in mammalian cells. We next combined Exosc3 depletion and U1 snRNP inhibition by antisense morpholino oligonucleotide (AMO) (Figure 2D). Consistent with previous reports, in 2P-seq analysis, the effects of U1 inhibition were minor for uaRNAs and eRNAs (Figures 2E–F). In contrast, an increase in PAS cleavage sites was induced in the sense direction from the TSS by both Exosc3 depletion and inhibition of U1 recognition, and further augmented by their combination (Figure 2E). This suggests that U1 recognition suppresses production of exosome-sensitive PAS-terminated transcripts in the first intron. Similarly, the combinatorial effects of U1 inhibition and Exosc3 depletion were observed in the first intron but not in the fourth intron (Figure 2G). Unlike Exosc3 depletion, U1 inhibition led to about 2 fold increase in PAS-linked unique cleavage sites in the 4th intron, consistent with the idea that U1 suppresses the use of nearby PAS sites throughout the gene (Berg et al., 2012; Kaida et al., 2010; Oh et al., 2017).
Because sites of cleavage and polyadenylation can vary locally downstream of a PAS site, we combined neighboring cleavage sites within 25 nucleotides into reproducible cleavage clusters (hereafter “2P cluster” or “premature cluster”), and hereafter focused on 2P clusters with PAS motifs. Hierarchical clustering of 2P clusters showed that about half of the clusters showed significantly higher 2P-seq signals when both Exosc3 and U1 activity are reduced (Figures S4A–B). In contrast, almost all 2P clusters in uaRNAs were primarily Exosc3-responsive (Figure S4C).
Roles of cleavage and polyadenylation factor and Pabpn1 in PAS termination
3´ RACE and sequencing analysis using gene-specific primers for several genes confirmed that RNA terminated at the predicted site in the 1st intron (Figures 3A–B and S4D-E). We investigated the roles of cleavage and polyadenylation (CPA) factor Cpsf73 and Xrn2 nuclease in the processing of these transcripts with 2P clusters, since they are suggested to be involved in promoter-proximal premature termination (Brannan et al., 2012; Nojima et al., 2015; Wagschal et al., 2012). A recent mammalian native elongating transcript sequencing (mNET-seq) study reported that Xrn2 knockdown affects specifically Pol II termination in TSS-proximal region but not in transcription end site (TES) region, suggesting its unique contribution to premature termination (Nojima et al., 2015). Knockdown of Cpsf73 but not Xrn2 attenuated induction of premature transcripts with 2P clusters by Exosc3 depletion and U1 inhibition (Figures 3C and S5A), suggesting that usage of these promoter-proximal PAS motifs is actually coupled to termination mediated by CPA factors (“PAS termination”). In contrast, it is unlikely that the 5´-to-3´ exonuclease activity of Xrn2 is rate limiting in degrading premature PAS-terminated transcripts.
Figure 3. Roles of CPA factor and Papbn1 in premature PAS termination.
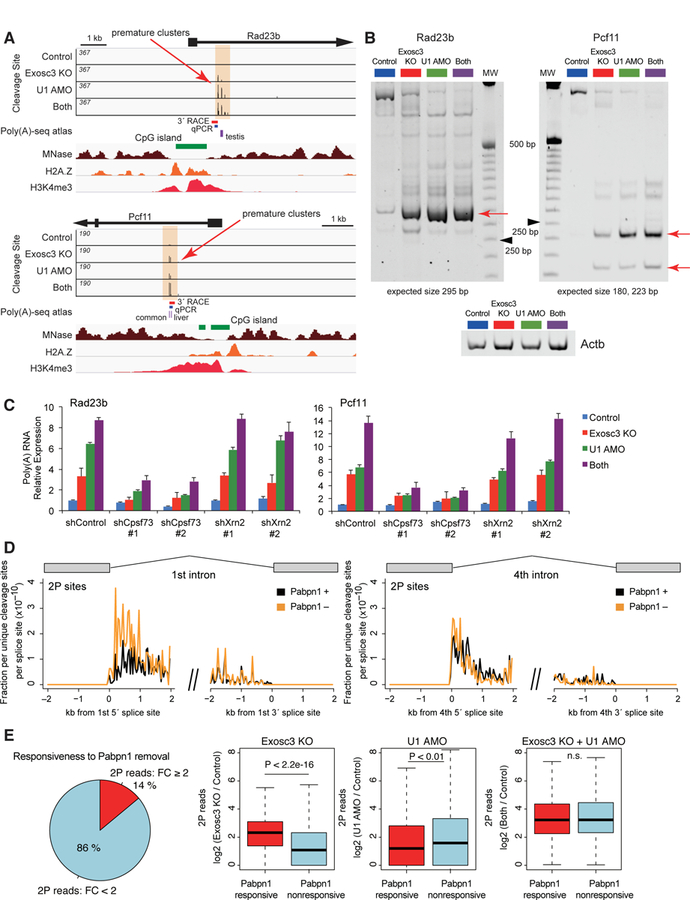
(A) Genome browser shot of Rad23b and Pcf11 showing 2P sites (top, orange shade), CpG island (green), MNase-seq (brown), H2A.Z ChIP-seq (orange), and H3K4me3 ChIP-seq (red). Previously reported PAS sites in mouse tissues (purple) and PCR products are also shown.
(B) Nested 3´ RACE analysis of premature termination events, Rad23b and Pcf11, on a nondenaturing polyacrylamide gel. Red arrows indicate the most frequent termination sites, which have been sequence validated. Ladder is 25 bp ladder, and black arrowheads indicate 250 bp. Actb is the loading control.
(C) Effects of knockdown of Cpsf73 or Xrn2 on induction of premature polyadenylated transcripts, determined by qRT-PCR using the fusion primer covering gene-specific sequence and poly(A) tail.
(D) Mean unique cleavage site signal (2P-seq) with PAS motifs around 5´ or 3´ splice sites of the first or fourth intron after Pabpn1 depletion, normalized by library depth.
(E) A relationship between the responses of 2P clusters upon Pabpn1 depletion, Exosc3 depletion, and U1 inhibition. The left pie chart shows percentage of Exosc3- or U1-sensitive 2P clusters sensitive (FC ≥ 2) or non-sensitive (FC < 2) to Pabpn1 removal. Right box plots show read changes in Pabpn1-responsive and Pabpn1-nonresponsive 2P clusters upon Exosc3 depletion, U1 inhibition, or both treatment.
See also Figures S4 and S5.
In addition, we explored the roles of nuclear poly(A)-binding protein Pabpn1 by generating a Pabpn1 conditional knockout mESC line (Figure S5B) as Pabpn1 mediates degradation of various nuclear ncRNAs (Bresson et al., 2015; Li et al., 2015). 2P-seq revealed that depletion of Pabpn1 also caused an increase in detectable cleavage sites in the first intron although to a lesser extent than Exosc3 deletion, and no increase in the fourth intron (Figure 3D). We divided Exosc3- or U1-sensitive 2P clusters into a Pabpn1-responsive group (FC ≥ 2) and a Pabpn1-nonresponsive group (FC < 2). We found that 2P signals of both groups increase upon inhibition of Exosc3 and U1, and that the Pabpn1-responsive group increases more upon Exosc3 depletion while the Pabpn1-nonresponsive group increases more upon U1 inhibition (Figure 3E). This suggests that Pabpn1, which recognizes poly(A) tracts, partly participates in destabilization of PAS termination transcripts probably by recruitment of the RNA exosome (Meola et al., 2016).
Enrichment of PAS Termination at the Edges of Stable Nucleosome Free Regions
Manual inspection of several genes suggested that the PAS-linked cleavage sites in the first intron are often found at the periphery of a CpG island, a region rich in H2A.Z and H3K4me3, and close to the edge of a region of low nucleosome occupancy in MNase-seq (Figure 3A). A genome-wide analysis revealed almost all expressed genes (FPKM > 0.5) with 2P clusters have promoters overlapping with CpG islands (P < 0.0001, hypergeometric test) (Figure 4A). Although genes with CpG-islands are typically expressed at higher levels than other genes (Ramirez-Carrozzi et al., 2009), there was no clear relationship between expression levels and the fraction of genes with 2P clusters above FPKM values of 1 (Figure 4B), thus suggesting that these observations were not due to an expression bias.
Figure 4. Premature PAS termination around +1/–1 stable nucleosomes demarcated by CpG islands.
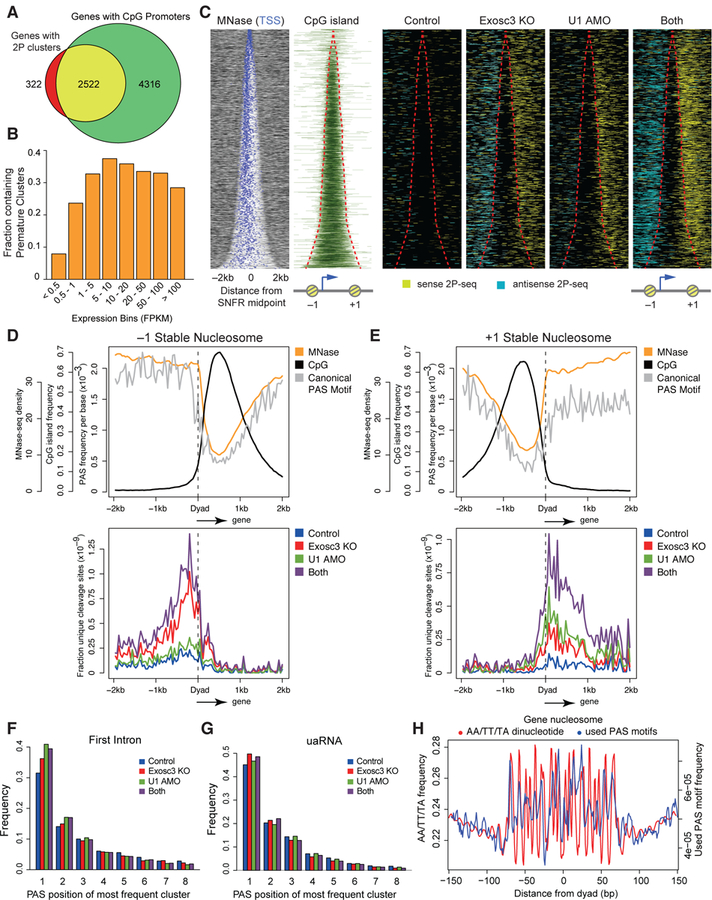
(A) Venn diagram demonstrating significant overlap of expressed genes with 2P clusters (FPKM > 0.5) and genes with promoters overlapping with annotated CpG islands.
(B) Fraction of genes with detectable premature cleavage events in different expression bins.
(C) Heatmap of MNase-seq, CpG islands, and PAS-linked cleavage sites (yellow: sense 2P-seq reads, light blue: antisense 2P-seq reads) around the SNFR midpoint for non-overlapping expressed genes with 2P clusters, ranked by increasing SNFR width. Red lines indicate SNFR edges.
(D, E) Metaplots of MNase-seq, CpG islands, and predicted canonical PAS motifs (top) and PAS-linked cleavage sites (bottom) around the dyad axis of the –1 (D) and +1 (E) stable nucleosome.
(F, G) Frequency of PAS position of the most frequently used cluster with AATAAA and ATTAAA motif at the first intron (F) and at defined uaRNAs (G).
(H) AA/TT/TA dinucleotide frequency (red) and frequency of unique used PAS motifs from cleavage clusters (blue) per gene body nucleosome in a 150 bp window from chemical mapping-defined dyad axis. Gene body nucleosomes are between TSS and 2kb upstream from the transcription end site (TES) of genes.
See also Figure S5.
In mammals, CpG islands are regions with unstable nucleosomes and frequently flanked by more stable nucleosomes. By analyzing the relative error of nucleosome occupancy in multiple MNase-seq datasets (Vainshtein et al., 2017) and incorporating the information of precise nucleosome dyad centers defined by chemical mapping (Voong et al., 2016), we generated a catalogue of invariant nucleosomes in mESCs. The resulting +1 and –1 stable nucleosome positions correlated strongly with a dramatic increase in resistance to MNase digestion at the boundary of CpG islands in MNase-seq (Figure 4C). We compared the distribution of cleavage sites, CpG islands, PAS motifs, and nucleosomes by aligning them around the center of these stable nucleosome free regions (SNFR) (Figures 4C–E and S5C).
Surprisingly, premature PAS termination events peaked immediately after the dyads of first stable nucleosomes, and extended through a downstream 1 kb window spanning approximately 4 nucleosomes in both sense and antisense directions (Figures 4D–E). We term this region where enhanced termination occurs the Stable Nucleosome Termination Area (SNTA). The PAS motif frequency strongly mirrors nucleosome positioning in both directions (Figures 4D–E), primarily due to the high GC content in the SNFR. While the frequency of PAS motifs remains constant across the gene body in the sense direction, premature PAS termination is restricted to the first few stable nucleosomes, i.e. SNTA (Figure 4E). In addition, the fraction of clusters at predicted canonical PAS motifs in the first intron were approximately 30%, 15% and 10% at the first, second, and third motif, irrespective of U1 inhibition or Exosc3 depletion (Figure 4F), suggesting that Pol II terminates most frequently at the first PAS motif in this region. Similar trends were observed for uaRNAs (Figure 4G). In comparisons of wide and narrow SNFRs, we observed a similar trend, but the effects of U1 inhibition were more apparent for wide SNFR genes (Figure S5D).
Nucleosome positioning is strongly influenced by AA/TT/TA dinucleotide sequences phased at 10 base pairs intervals (Voong et al., 2016). We found that both canonical PAS motifs used in premature termination events within the gene body and predicted PAS motifs closely mimic the periodic AA/TT/TA dinucleotide patterns (Figures 4H and S5E). These findings suggest that sequence contexts has a strong impact on nucleosome organization and PAS termination.
Association between PAS Termination and Chromatin Remodeling at +1 Stable Nucleosome
Nucleosome organization is influenced by various chromatin remodelers such as Chd1, Chd4, and Ep400. Among them, Chd1 have been linked with regulating the stalling of Pol II at promoter proximal nucleosomes (Skene et al., 2014). Using recently reported MNase digestion-coupled ChIP-seq datasets for various chromatin remodelers (de Dieuleveult et al., 2016), we investigated the relationship between PAS termination and chromatin remodeling. As previously reported (de Dieuleveult et al., 2016), most chromatin remodeling factors were enriched around the SNFR edges of genes with 2P clusters, aside from Chd2 being distributed across the gene body (Figures 5A and S6A). Despite no major difference in MNase-seq signal between genes with or without 2P clusters, genes with 2P clusters were more strongly bound by several chromatin remodelers including Chd1, Chd2, and Chd9 (Figures 5A and S6B), suggesting that +1 stable nucleosomes associated with PAS termination are actively marked by several chromatin remodelers. To test if the Chd1 remodeler is involved in influencing the frequency of PAS termination in this region, we examined the effects of knockdown of this factor. Knockdown of Chd1 augmented induction of premature transcripts with 2P clusters (Figure S6C), suggesting that suppression of Pol II elongation is linked to the stability of the nucleosome.
Figure 5. Active chromatin remodeling at +1 stable nucleosome of genes with PAS termination.
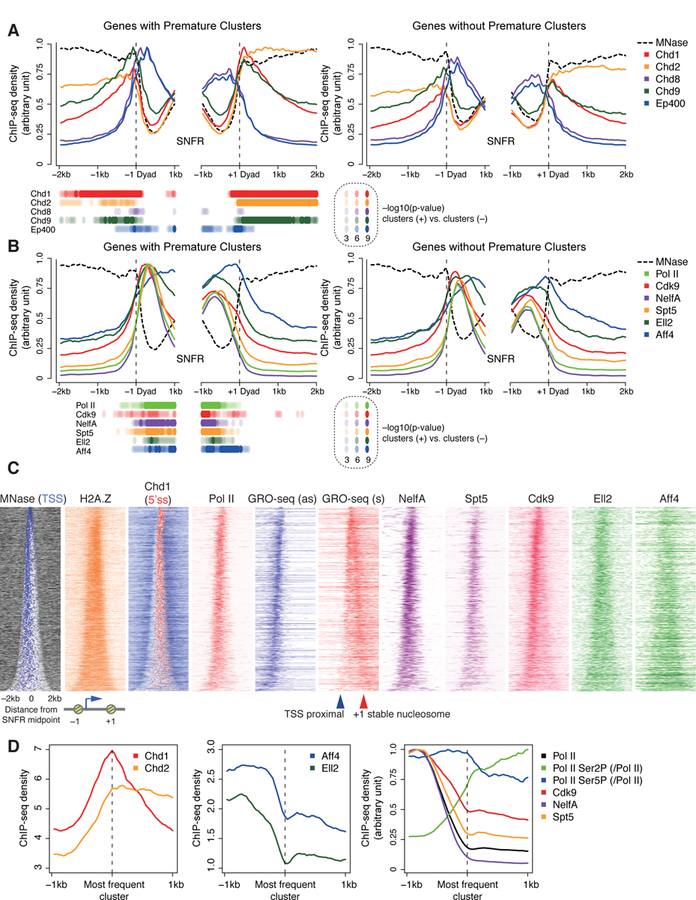
(A, B) Read coverage of MNase-seq and MNase digestion-coupled ChIP-seq of various chromatin remodelers (A) and ChIP-seq for Pol II and various pausing and elongation factors (B) around the –1 and + 1 stable nucleosome dyad axis, separated for genes with premature intron clusters (left) and expression-matched genes without premature intron clusters (right). P values with K-S test at each bin are displayed.
(C) Heatmap of MNase-seq, GRO-seq, and ChIP-seq, as in Figure 4C.
(D) Metaplots of Chd1, Chd2, SEC components, and other factors around the most frequent PAS-linked 2P clusters.
ChIP-seq and GRO-seq datasets are from (de Dieuleveult et al., 2016; Jonkers et al., 2014; Lin et al., 2011; Rahl et al., 2010; Seila et al., 2008; Whyte et al., 2013).
See also Figure S6.
We also investigated the distribution of various histone marks (Ji et al., 2015; Subramanian et al., 2013) (Figure S6D). While H2A.Z is reported to reduce the nucleosome barrier for Pol II (Weber et al., 2014), we observed no differences in H2A.Z signals between genes with or without premature termination (Figure S6E).
Premature PAS Termination Correlates with Active Pol II Pause Regulation
We further analyzed ChIP-seq datasets of various Pol II regulators and global run-on (GRO)-seq (Table S2) (Jonkers et al., 2014; Lin et al., 2011; Rahl et al., 2010; Seila et al., 2008; Whyte et al., 2013). The ChIP signal for Pol II was primarily distributed close to the TSS for genes with 2P clusters with some signals within SNFR (Figures 5B–C). In contrast, GRO-seq signals, which detect transcribing Pol II, were abundant at both TSS-proximal regions and the edges of the SNFR in the sense direction. Consistent with a previous report (Kellner et al., 2015), this GRO-seq pattern suggests that two types of Pol II pausing occur in the sense direction, especially for genes with wide SNFRs where the two pauses can be resolved: TSS-proximal pause and stable nucleosome pause. Two pausing factors, NelfA subunit of NELF and Spt5 subunit of DSIF, were enriched at the site of TSS-proximal paused Pol II, consistent with their roles in promoting the promoter-proximal pause (Figures 5B–C) (Adelman and Lis, 2012). Cdk9, a subunit of the P-TEFb complex that stimulates promoter-proximal pause release, accumulated at the TSS-proximal region in parallel with its substrates Pol II and DSIF, but was further distributed within the SNFR. Aff4 and Ell2, subunits of the Super Elongation Complex (SEC) associated with P-TEFb (Lin et al., 2011), were widely distributed from TSS to the SNFR edge. Interestingly, genes with premature PAS clusters had increased binding of Pol II, SEC components (Aff4 and Ell2), and NELF/DSIF, when compared to expression-matched controls (Figure 5B). These findings suggest that premature termination is associated with more active Pol II pause regulation at the edge of SNFRs.
Modifications of the C-terminal repeat domain (CTD) of Pol II at Ser5 and Ser2 reflect the Pol II status during elongation. We selected the most frequently used 2P cluster in the sense direction for each gene and constructed metaplots in order to better compare these modifications with respect to the site of PAS termination (Figure 5D). Similar to the SNFR view (Figure 5A), Chd1 accumulated at the most frequent 2P cluster, whereas the SEC, Aff4 and Ell2, diminished across the most frequently used 2P site (Figure 5D). Though the density of Pol II reached a nadir at this point, the density of Ser2 phosphorylation increased while that of Ser5 phosphorylation remained relatively constant. This suggests that a Ser2 kinase, such as Cdk9, is likely active at these 2P cluster sites.
PAS Termination is associated with a Flavopiridol-sensitive +1 Stable Nucleosome Pausing
Our results suggest that +1 stable nucleosomes associated with premature polyadenylation are marked by active chromatin remodeling and active Pol II pause regulation. To further investigate the relationship between premature termination and Pol II pausing, we focused on genes with wide SNFRs (distance between TSS and +1 dyad axis > 600 bps) since it is difficult to distinguish between a TSS proximal pause and a +1 stable nucleosome pause at genes with narrow SNFRs. Alignments of Pol II ChIP-seq around the TSS revealed a major pause immediately downstream of the TSS (Figure 6A, top, blue bar), followed by a less steep ramp around 300 to 900 bp from the TSS (orange bar) representing the +1 stable nucleosome pause, followed by gene body signal (green bar). Consistent with the results above, genes with premature clusters had increased Pol II ChIP signal near the promoter relative to expression-matched genes without 2P clusters. Alignments with the dyad of the first stable nucleosome showed that genes with premature clusters have a ramp of Pol II occupancy in front of the dyad and a peak of GRO-seq signal flanking the +1 stable nucleosome, and this phenomenon was less pronounced at genes without premature clusters (Figure 6B). This suggests genes with premature clusters are more likely to be targets of active pausing at the +1 stable nucleosome.
Figure 6. PAS termination and +1 stable nucleosome-associated Pol II pause regulation.
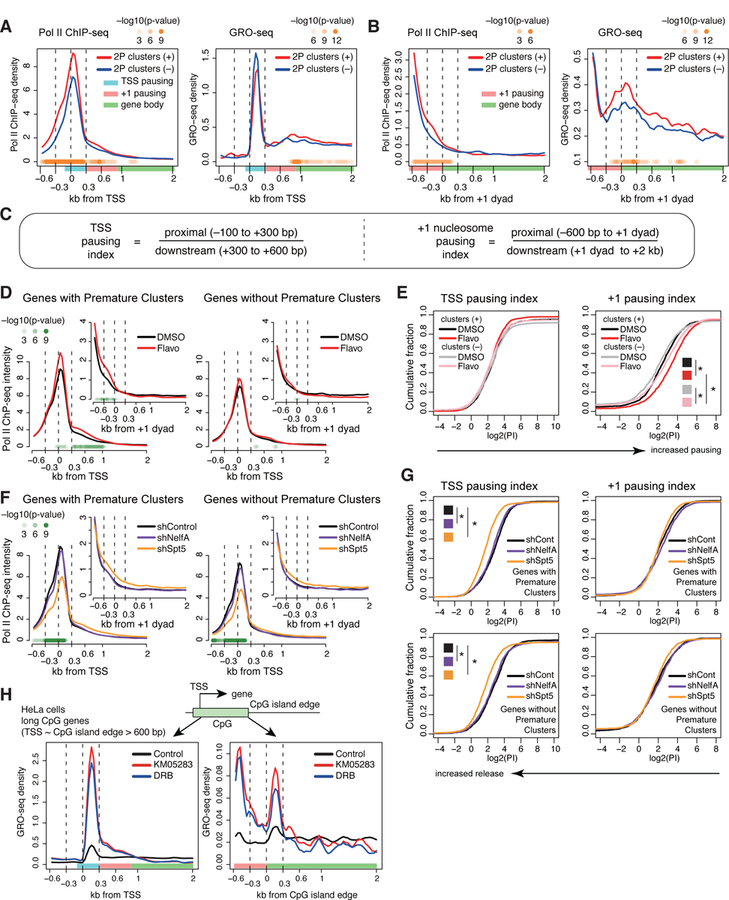
(A, B) Metaplots of mean Pol II ChIP-seq (left) or GRO-seq (right) read density around the TSS (A) or the +1 dyad (B) for wide SNFR genes with 2P clusters (red) and expression-matched wide SNFR genes without 2P clusters (blue). Wide SNFR: distance between TSS and +1 stable nucleosome dyad > 600 bps. P values with K-S test at each bin are displayed in panels (A), (B), (D), and (F, shSpt5 vs. shControl).
(C) Formulas for the two pausing indices.
(D) Metaplots of Pol II ChIP-seq density around the TSS or +1 dyad (inset) of wide SNFR genes with DMSO or flavopiridol treatment.
(E) Cumulative distribution plot of log2(pausing index) of the TSS proximal (left) and +1 stable nucleosome pause (right) for wide SNFR genes with 2P clusters and expression-matched wide SNFR genes without 2P clusters under DMSO or flavopiridol treatment. * P < 0.01 with K-S test.
(F) Metaplots of Pol II ChIP-seq density around the TSS or +1 dyad (inset) in shControl, shSpt5, and shNelfA mESCs.
(G) Cumulative distribution plot of log2(pausing index) of the TSS or +1 stable nucleosome pause for genes with 2P clusters (top) and genes without 2P clusters (bottom) in shControl, shSpt5, and shNelfA mESCs. * P < 0.01 with K-S test.
(H) Metaplots of GRO-seq density around the TSS and edges of CpG islands with KM05283 and DRB treatment in human HeLa cells. Long CpG island genes (distance between TSS and the edge of CpG island > 600 bps) were analyzed.
ChIP-seq and GRO-seq datasets are from (Jonkers et al., 2014; Laitem et al., 2015; Rahl et al., 2010; Seila et al., 2008). See STAR Methods for statistical tests. See also Figure S7.
We next closely compared differential sensitivity of Pol II pausing at genes with or without premature clusters to experimental modulation of Pol II pause regulators: treatment with flavopiridol (an inhibitor of Cdk9/Cdk12) or knockdown of DSIF and NELF, using previously published datasets (Rahl et al., 2010). Furthermore, to distinguish the effects on the TSS-proximal pause and +1 stable nucleosome-associated pause, we introduced two pausing indices based on Pol II ChIP-seq: a TSS Pausing index and a +1 Nucleosome Pausing index (Figure 6C). In this analysis, a higher pausing index suggests increased pausing.
The Cdk9 kinase of P-TEFb has a central role in promoter-proximal Pol II pausing kinetics by phosphorylating DSIF and NELF, and is blocked by flavopiridol. Treatment with flavopiridol resulted in statistically-significant increases in mean Pol II signals at both the TSS-proximal region and the immediate upstream region from the dyads of +1 nucleosomes at genes with premature clusters (Figure 6D). Comparisons of the TSS pausing index and the +1 nucleosome pausing index showed that flavopiridol-induced pausing is greater at the +1 nucleosome, and was even stronger at genes with premature clusters than genes without premature clusters (Figures 6E and S7A). On the other hand, knockdown of DSIF component Spt5 caused a substantial pause release effect only at TSS-proximal regions (Figures 6F–G and S7B), and there was no apparent difference between genes with and without premature clusters (Figures 6G and S7B). The effects of NelfA knockdown were very modest possibly due to incomplete knockdown. Results of re-analysis of Start-RNA-seq in NelfB knockout mESCs (Williams et al., 2015) were largely consistent with our observations (Figure S7C). These analyses unexpectedly highlight differential contributions of DSIF/NELF and flavopiridol-sensitive mechanism(s) to two Pol II pausing steps. DSIF seems to only influence pausing at the TSS-proximal site, whereas flavopiridol-sensitive mechanism(s) are active at both sites.
Re-analysis of GRO-seq datasets (Jonkers et al., 2014) revealed that flavopiridol treatment resulted in a substantial increase in promoter proximal pausing (Figure S7D) and induces a substantial drop in GRO-seq signal near the +1 stable nucleosome for both genes with premature clusters and those without (Figure S7E). Furthermore, re-analysis of previously reported GRO-seq datasets in human (Laitem et al., 2015) using other Cdk9 inhibitors, KM05283 and DRB, confirmed dual effects at the TSS-proximal regions and edges of CpG islands (Figure 6H), suggesting conservation of this mechanism in mouse and human.
R-loops regulated by the RNA exosome can affect Pol II elongation, subsequently influencing premature PAS termination (Kilchert et al., 2016). We assessed this possibility using precision nuclear run-on (PRO)-seq with modification (Mahat et al., 2016). Our preliminary PRO-seq analysis suggests that the increase in PAS termination transcripts upon Exosc3 depletion is mainly attributable to RNA stabilization, and not from increased pausing (Figures S7F–H, see STAR Methods).
Myc Regulates +1 Stable Nucleosome Pausing
Myc has been reported to regulate the release of Pol II from the promoter region in mESC (Rahl et al., 2010). According to classification of mESC genes based on association with transcription factor binding (Chen et al., 2008), we found that over 60 % of genes with 2P clusters fall into gene classes with Myc binding (Figure 7A, Class II and III). Myc-binding sites are preferentially found in CpG islands (Perna et al., 2012), consistent with a large overlap with genes sets with 2P clusters and CpG promoters (Figure 4A).
Figure 7. Myc regulates genes with PAS termination and +1 stable nucleosome Pol II pause.
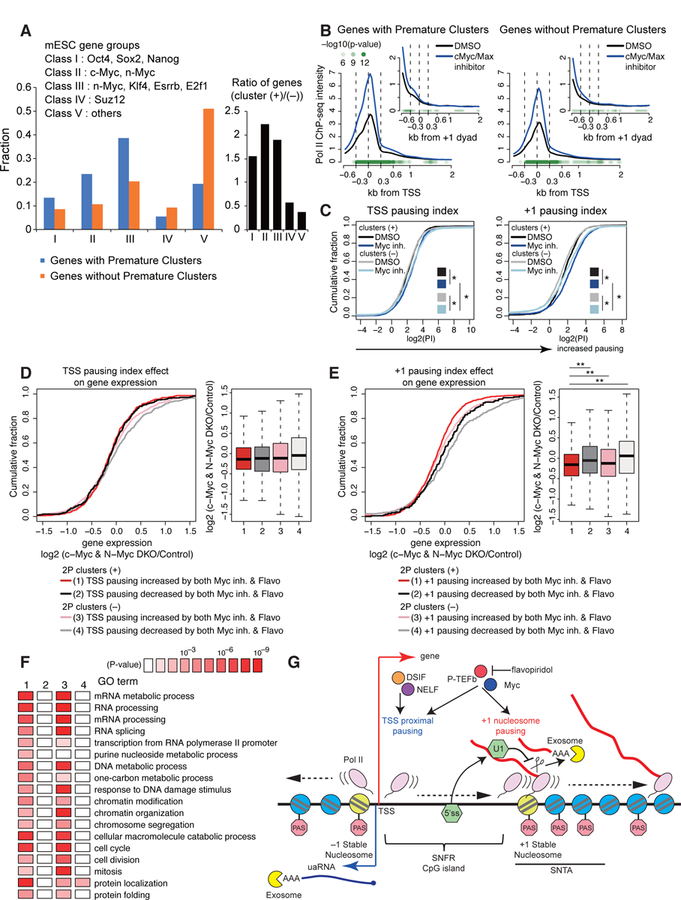
(A) Fraction of mESC gene groups for genes with premature intronic clusters (blue) or without premature intronic clusters (orange). Bar graph in black represents fold change.
(B, C) Metaplots of Pol II ChIP-seq density around the TSS or +1 dyad (B) and cumulative distribution plot of log2(pausing index) of the TSS proximal or +1 stable nucleosome pause (C) for wide SNFR genes with or without 2P clusters upon treatment with DMSO or Myc inhibitor, as shown in Figures 6D and 6E. ChIP-seq datasets are from (Rahl et al., 2010). See STAR Methods for statistical tests. * P < 0.01 with K-S test.
(D) Effects of TSS pause on Myc-dependent gene regulation. Cumulative distribution of log2 fold change of RNA expression in c-Myc and N-Myc double knockout (DKO) mESC is shown for wide SNFR genes with/without PAS termination and flavopiridol/Myc-sensitive TSS pausing.
(E) Cumulative distribution plot is shown as in panel (D) using +1 stable nucleosome pausing indices. ** P < 0.001 with K-S test.
(F) Gene ontology terms enriched in each gene sets as defined in panel (E). All expressed genes were analyzed.
(G) Model of +1 stable nucleosome-associated premature PAS termination.
See also Figure S7.
An examination of Pol II ChIP data upon treatment with a low-molecular-weight inhibitor of c-Myc/Max (Rahl et al., 2010) revealed that both genes with and without premature clusters showed roughly a 2-fold increase in Pol II occupancy at the TSS following Myc inhibition (Figures 7B–C and S7I). Strikingly, genes with premature clusters had an increase in +1 nucleosome pausing upon treatment with a Myc inhibitor, whereas there were much smaller changes at genes without premature clusters (Figures 7B–C and S7I), suggesting that Myc preferentially regulates the +1 stable nucleosome pause at genes with premature clusters.
Finally, we analyzed the relationship between Myc-regulated Pol II pausing and Myc-dependent gene regulation. Myc regulates diverse synthetic and metabolic processes and double knockout of c-Myc and N-Myc in mESCs induces a pluripotent dormant state (Scognamiglio et al., 2016). There is no statistical correlation between changes in genome wide gene expression upon Myc knockout and changes of the TSS pausing index upon Myc inhibition and flavopiridol treatment (Figure 7D). In contrast, genes with 2P clusters and increased +1 nucleosome pausing upon treatment with Myc inhibitor and flavopiridol have a greater decrease in mRNA expression following Myc knockout relative to other genes (Figure 7E). Consistent with this, genes with increased +1 nucleosome pausing following Myc inhibition and flavopiridol treatment are strongly linked to biological processes characteristic to Myc target genes, including RNA processing, DNA metabolism, chromatin modification, and cell cycle (Figure 7F). Interestingly, we also observed that loss of Exosc3 results in reduced expression of Myc-regulated target genes (Figure S2F). These data collectively suggest that Myc-dependent gene regulation is associated with regulation of the +1 stable nucleosome-associated pause.
Taken together, these findings strongly suggest that promoter proximal pausing consists of at least two distinct processes differentially regulated by multiple pausing regulators: TSS-proximal pausing and +1 stable nucleosome-mediated pausing (Figure 7G). NELF and DSIF primarily function in the former step, and flavopiridol-sensitive mechanism(s) and Myc have broader roles in the two types of pausing and are involved in the latter step. Furthermore, PAS termination is preferentially associated with active regulation of the latter step.
DISCUSSION
We have identified novel sense-direction short transcripts marked by PAS termination as targets of the RNA exosome for a large class of promoters. Similar short transcripts, uaRNAs, observed in the upstream antisense direction are also targets of the exosome. Thus, the RNA exosome rapidly degrades promoter-proximal terminated poly(A) RNA in both directions: suppressing uaRNAs and sense PAS-termination products.
We conjecture that premature PAS termination in the region of the first few stable nucleosome represents an important checkpoint of Pol II elongation in the sense direction (Figure 7G), which integrates several previously reported promoter-proximal events such as CpG island-associated pause (Kellner et al., 2015) and CPA factor-dependent promoter-proximal termination (Nojima et al., 2015). This study extends these reports by connecting the CPA factor-dependent termination to the first stable nucleosome and identifying its dependence on U1 snRNP recognition, and proposes the role as a general checkpoint for elongation. The frequency of this premature termination is suppressed by U1 snRNP presumably through recognition of 5´ splice site sequences near the TSS (Almada et al., 2013; Kaida et al., 2010). Importantly, in both directions, termination predominantly occurs at the edges of the stable nucleosome free regions, SNFR, as defined by micrococcal nuclease digestion. While previous reports described relationships between nucleosome organization (+1 nucleosome) and Pol II pausing, our findings indicate that each of the +1 and –1 stable nucleosomes demarcated by CpG islands are also regions of PAS termination. Genes with prominent sense PAS termination at the edge of SNFRs have enhanced pausing following treatment with flavopiridol, an inhibitor of Cdk9 kinase, or inhibition of Myc activity, suggesting this is an important regulatory step.
CpG islands overlap about 60–70 % of mammalian promoters. These segments are sites of less stable nucleosomes marked by H2A.Z and are bracketed by the –1 and +1 stable nucleosomes. PAS termination in the sense direction increases in frequency at the dyad of the first stable nucleosome and is prominent through the next few nucleosomes. These sense poly(A) RNAs commonly map to sequences immediately downstream of the first or second PAS (A[A/U]UAAA) related sequence in this region. Many of these RNAs are observed even in the presence of U1 snRNP and their abundance increases dramatically in the absence of exosome activity, indicating rapid degradation under normal conditions. Inhibition of U1 snRNP increases these RNAs in the presence of exosome activity, but the highest level of these RNAs is observed when both U1 snRNP and exosome activities are reduced. We picture these nucleosomes at the edge of the SNFRs forming a barrier to the elongating polymerase, pausing it, which enhances the rate of cleavage. U1 snRNP is important for bypassing this checkpoint potentially by suppressing the rate of cleavage, by recruiting chromatin rearranging factors such as Chd1, or by generating a processive polymerase complex through the recruitment of Pol II elongation factors such as SEC and FACT. Consistent with this, ChIP signals for the SEC subunit Aff4 accumulated at the edge of the SNFR. Ser2 phosphorylation of Pol II CTD, a substrate of Cdk9, increases near these PAS termination sites and inhibition of P-TEFb by flavopiridol promotes increased pausing at the edge of the SNFR, suggesting that Cdk9 also controls this pause step. After passage through this region, the Pol II elongation complex must be highly processive in order to transcribe genes mega base pairs in length. This transition could be pictured as a checkpoint where transcription is coupled to the necessary elongation factors that probably include the RNA splicing machinery. A recent report demonstrated that suppression of premature cleavage and polyadenylation by U1 snRNP, called U1 telescripting, is selectively required for long-distance transcription elongation in introns of large genes (Oh et al., 2017). Thus, U1 snRNP telescripting may have pleiotropic roles in transcriptional elongation: regulation of the +1 stable nucleosome-associated elongation checkpoint and subsequent prevention of Pol II termination in downstream large introns.
In this study, we are able to resolve features of two distinct pauses: TSS-proximal Pol II pausing and +1 nucleosome Pol II pausing, especially for wide SNFR genes. At promoters with short SNFRs, it is difficult to convincingly resolve these two pauses. However, since both U1-sensitive PAS termination and NELF/DSIF accumulation is observed at both long and short SNFRs, both pause processes probably occur at short SNFR promoters.
The finding of PAS termination in the sense direction harmonizes the concept of divergent transcription between upstream antisense and sense directions. We here demonstrated global stabilization of PAS terminated RNA by exosome depletion for uaRNAs. U1 snRNP inhibition does not significantly increase PAS termination of uaRNAs as 5´ splice sites are not commonly found near the initiation site in this direction. Furthermore, in both directions, PAS termination occurs when elongating Pol II encounters the first stable nucleosome. The –1 stable nucleosome is very close to the divergent TSS for CpG promoters and the uaRNAs are typically shorter than their sense counterparts. All of this strengthens the argument that a key feature which distinguishes sense from antisense transcription is the presence of a 5´ splice site in the sense direction, which engages U1 snRNP for further elongation and coupling to RNA splicing.
Myc promotes promoter-proximal pause release at many promoters in mESC by recruiting P-TEFb (Rahl et al., 2010) and we show that Myc also promotes passage through the pause at the first stable nucleosome. Promoters with premature PAS termination have increased nucleosome pausing that is further enhanced by flavopiridol treatment and Myc inhibition. Furthermore, deletion of c-Myc and N-Myc in mESCs preferentially reduces the level of mRNA expression from promoters with premature PAS termination. The biochemistry and biology of genes regulated by Myc may be associated with PAS termination. Both PAS termination and Myc binding are common at CpG promoters. Myc controls various synthetic and metabolic processes (Perna et al., 2012; Scognamiglio et al., 2016), and genes with CpG islands are enriched for housekeeping genes critical for the bio-synthetic capacity of cells (Ramirez-Carrozzi et al., 2009). Thus, Myc’s regulation of PAS termination at CpG promoters could be important for cell growth and other processes critical for tumorigenesis.
STAR METHODS
• CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Phillip A. Sharp (sharppa@mit.edu).
• EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Culture
V6.5 mESCs were grown under standard conditions without feeders (Almada et al., 2013). Cells were passaged every two days to avoid confluency. Exosc3 CKO clones were maintained in 0.1 μg/ml of dox. Pabpn1 CKO clones were maintained in 1 μg/ml of dox.
Generation of Exosc3 CKO and Pabpn1 CKO mESC Cell Lines
The Exosc3 CKO mESCs were generated through two steps (Figure S1A). To prepare lentivirus for conditional expression of Exosc3, HEK293T cells were transfected with packaging vectors VSV-G and dr8.91 and a lentivirus plasmid (pSLIK-Hygro) containing a hygromycin resistance cassette and doxycycline-inducible C-terminally tagged FLAG-HA mouse Exosc3 cDNA. Virus was collected days 2 and 3 post-transfection. V6.5 mESCs were seeded to be about 20% confluent and infected with virus and polybrene (1:2000 of 8 mg/ml polybrene stock). Cells were selected with 150 μg/ml of Hygromycin B, and single cell clones were isolated. Expression of FH-Exosc3 was validated using anti-HA antibodies (Roche 3F10).
Next deletion of endogenous Exosc3 gene was attempted by cotransfection of two CRISPR-Cas9 vectors (pX330) with sgRNAs (sgExosc3–2 and sgExosc3–5) flanking the Exosc3 gene. Heterozygotes were isolated and validated by PCR amplification across the deletion and subsequent sequencing. Heterozygotes were further transfected with two CRISPR-Cas9 vectors containing sgExosc3–3 and sgExosc3–6 to target the other allele under treatment with 0.1 μg/mL dox (Figure S1B). Subsequent clones were screened for shortened PCR product across the entire gene. Deletion of the two alleles was validated by DNA sequencing (Figure S1C). Finally, deletion of Exosc3 was further validated using qRT-PCR for the Exosc3 gene after 3 days of dox removal. The Exosc3 mRNA level in 0.1 μg/ml of dox was comparable to that in parental cells (Figure S1D). The Pabpn1 CKO mESCs were generated similarly. The sgRNA and primer sequences are described in Table S1.
• METHOD DETAILS
Western Blotting
Protein lysate was run on 1.5% NuPAGE Bis-Tris Gels using the NuPAGE Western Blotting System (ThermoFisher Scientific). The gels were transferred at 4 °C in 10% Methanol and 1x NuPAGE Transfer Buffer onto PVDF membranes. Membranes were blocked with 5% skim milk and then incubated with primary antibody in 5 % skim milk overnight. Blots were washed in PBS-T and incubated with ECL HRP secondary antibody in milk for an hour at 1:10000 dilution. Blots were further washed in PBS-T before imaged using Western Lightning Plus-ECL substrate.
qRT-PCR
Total RNA was extracted using TRIzol Reagent (ThermoFisher Scientific) and genomic DNA was removed using DNase Turbo (Ambion AM2238). For conventional qRT-PCR, RNA was reverse-transcribed using random hexamers and SuperScript III First-Strand Synthesis System (ThermoFisher Scientific) according to the manufacturer’s instructions. Quantitative PCR was performed with PowerUp SYBR Green Master Mix (Thermo Scientific) and the 7500 Fast Real-Time PCR System (Applied Biosystems).
For detection of polyadenylated PAS termination transcripts, total RNA was subjected to reverse transcription using the fusion primer including gene-specific sequence, oligo(dT)12, and adapter sequence and the SuperScript III First-Strand Synthesis System (Invitrogen; 1 hr at 55 °C). Subsequent PCR was performed using the gene-specific primer and the adapter primer. Results were normalized to Actb in standard qRT-PCR analysis. Sequences of primers are described in Table S1.
RNAi
shRNA oligonucleotides were cloned into PB-EF1α-GreenPuro-H1-MCS shRNA vector (System Biosciences PBSI506A-1). mESCs were cotransfected with the shRNA expression vector and CAGG-PBase vector using Lipofectamine 2000 (Invitrogen), selected by puromycin (Invitrogen, 2 μg/mL), and used for subsequent analysis. Sequences of oligonucleotides are described in Table S1.
RNA-seq
Total RNA was isolated with TRIzol Reagent and treated with DNase Turbo (Ambion AM2238) to remove genomic DNA contamination. RNAs that passed a Bioanalyzer RIN score of 8.5 were subsequently used to prepare libraries. RNAs were depleted of ribosomal RNAs using the RiboZero rRNA removal kit (Epicentre MRZH116), converted into stranded RNA-seq libraries with the Illumina Tru-Seq kit (Illumina RS-122–2101), and sequenced in paired end read mode using the Illumina NEXT-Seq 500.
Active Caspase 3 Assay
Exosc3 CKO cells were removed from dox for 0, 1, 2, or 3 days. Subsequently, we labelled cells using the FITC Active Caspase-3 Apoptosis kit (BD Pharmingen) as per manufacturer’s instructions, before FACS analysis for FITC positive cells.
Determination of uaRNA Half-Lives
Cells were maintained with dox or removed from dox for 2 days. Subsequently, cells were placed into mESC media containing 1 μg/ml flavopiridol dissolved in DMSO for 0 min, 5 min, 10 min, 20 min, 30 min, or 1 hr before harvesting in TRIzol. cDNA was generated using oligo-dT20 and SuperScript III reverse transcriptase. qRT-PCR was performed using primers in Table S1. Results was normalized to values at when time is 0. Averages across three experiments were used to determine fraction remaining. Half-lives were determined by fitting an exponential decay curve using R, starting with the formula: y= e–bx, and then finding the point such that y=0.5.
U1 Inhibition Experiment
Exosc3 CKO cells were either kept in dox or removed from dox for 40 hours. Cells were subsequently trypsinized, washed twice in PBS, and 5 million cells were nucleofected with 15 μM concentration of scrambled (Scr) control antisense morpholino oligonucleotide (AMO) or U1 AMO, antisense to U1 sequences recognizing the 5´ splice site (sequence in Table S1). Cells were seeded onto 10 cm dishes, and total RNA was harvested 8 hours later in TRIzol Reagent.
3´ End sequencing (2P-seq)
2P-seq was performed as described in (Spies et al., 2013). Briefly, total RNA is poly(A) selected using oligo-dT dynabeads. Subsequently, RNA was cleaved with trace levels of RNase T1 for 20 minutes at 22°C, inactivated, and cleaned up with an ethanol precipitation. The resulting RNA was reverse transcribed using IW-RT1p and the size selected for 200–400 nts on a polyacrylamide gel. Next the cDNA was circularized using CircLigase II (Epicentre), PCR amplified with primers IW-PCR-F.1 and IW-PCR-RPI, and further size selected to remove adapters, before sequencing from the poly(A) tail using IW-Seq-PE1.1 in single end read mode on the Illumina NEXT-Seq 500.
3´ RACE
DNase-treated RNA was reverse transcribed with Superscript III using the 3´ RACE adapter oligo. Subsequently, nested PCRs were performed using Phusion DNA Polymerase. In the first round, PCR buffer conditions included GC Buffer, 3% DMSO, 1 mM dNTP, and the 3´ RACE outer primer and gene specific outer primers. In the next round, PCR buffer conditions were similar but the 3´ RACE inner primer and gene specific inner primers were used instead. Products were run on a 5% nondenaturing polyacrylamide gel with 25 bp ladder (Life Technologies). 3´ RACE adapter oligo and primers were from FirstChoice RLM-RACE Kit (ThermoFisher Scientific). Sequences of PCR primers are described in Table S1.
PRO-seq
PRO-seq was modified from the published PRO-seq protocol described in (Mahat et al., 2016). Briefly, nuclei were isolated from mESCs as described using cell permeabilization, followed by run on and biotin enrichment. Individual libraries were ligated with 3´ barcoded adaptors and pooled into one tube, before completing addition enrichment for biotin and reverse transcription. Unlike regular PRO-seq which performs PCR amplification and size selection, we treated RNAs with a cocktail of RNase A and RNase H and phenol-chloroform extracted the ensuing single-stranded cDNA library. The library was sequenced on the Illumina NEXT-Seq 500.
• QUANTIFICATION AND STATISTICAL ANALYSIS
RNA-seq Analysis
All analyses were carried out using UCSC (NCBI37/mm9) mouse gene annotations. Paired end reads were first mapped to ribosomal RNA and various repetitive sequences such as U1 snRNA using Bowtie2 (Langmead and Salzberg, 2012), and then subsequently mapped to the mouse UCSC transcriptome and genome using STAR aligner (Dobin et al., 2013). The ensuing reads were filtered for uniquely mapping, properly paired reads, and subsequently potential PCR duplicates were removed using the Picard Suite MARKDUP (http://broadinstitute.github.io/picard). In genome browser shots, the reads are displayed. For metaplot alignments, we further processed the reads by selecting read 2 of the paired-end read (same direction as the RNA), and filtered away any overlapping miRNAs, tRNAs, repeats from repeatMasker, or snoRNA.
de novo Transcriptome Assembly
To obtain profiles of normally suppressed transcripts in the presence of RNA exosomes with accurate transcript architectures, the RNA transcriptome was assembled de novo using the Stringtie algorithm (Pertea et al., 2015) after pooling RNA-seq libraries, followed by various filtering steps to categorize transcript classes (Figures S1F–G). For instance, uaRNAs were defined as divergent transcripts with a 5´ end within 1 kb upstream and antisense of the closest gene TSS (Figure S2A), whereas convergent transcripts were antisense RNAs that overlapped the gene TSS (Figure S2B) (Mayer et al., 2015). Enhancer RNAs (eRNAs) were defined as transcripts overlapping a 1 kb window of an OSN enhancer peak (Figure S2C).
To identify non-coding RNAs genomewide, the two doxycycline replicates were collapsed into one file. Subsequently, Stringtie was run on this using the parameters –f 0.1 –c 5 –g 10 (Pertea et al., 2015). The resulting candidate transcripts were first removed for any transcript that overlapped UCSC canonical genes, snoRNAs, and known miRNA genes. Any candidate transcripts were then aligned against the antisense version of UCSC canonical genes, and divided into two categories: 1) convergent RNAs: those that started within the gene and was transcribed across the TSS of the canonical gene or 2) antisense RNAs: antisense transcripts that did not overlap the TSS. The remaining candidate transcripts were further analyzed for uaRNAs: transcripts that were antisense to the coding gene and started within 1 kb of the TSS. The remaining candidate transcripts were further subsegmented into eRNAs: transcripts that overlapped a flanking 1 kb window of called Oct4, Sox2, and Nanog binding sites described in a previous report (Whyte et al., 2013). Finally, the remaining candidate RNAs were filtered for de novo lncRNAs by removing previously annotated lncRNAs followed by running the Slncky algorithm (Chen et al., 2016).
We identified 3,336 high-confidence uaRNAs, with a median distance of 135 bp to the closest gene TSS (Figures S1G and S2D). While this number (29% of surveyed expressed genes) is less than the number of divergent promoters (68%) (Seila et al., 2008), our uaRNA definition excludes bidirectional genes and convergent transcripts and used more stringent thresholds.
In Figure 1C, previously identified long non-coding RNAs (lncRNAs) changed more modestly, 1.5 fold, upon Exosc3 depletion. Novel lncRNAs identified in this study were more significantly upregulated than previously identified lncRNAs (data not shown), but this class may be contaminated with eRNAs originating from enhancers other than Oct4/Sox2/Nanog enhancers. Nevertheless, this suggests genome-wide studies identifying lncRNAs may have missed lncRNAs that are normally degraded by the RNA exosome.
Differential Expression Analysis and Gene Set Enrichment Analysis
The number of reads per transcript was counted by using intersectBed of the Bedtools suite (Quinlan and Hall, 2010), only allowing for exonic or spliced reads. After filtering out for intervals with low numbers, differential transcripts were called using edgeR, where we normalized libraries using UQ normalization. Statistically significant transcripts were those with at least a two-fold change and a false-discovery rate less than 0.10. A substantial fraction (28%) of mRNA encoding protein changed upon Exosc3 depletion (log2 fold change ≥1, FDR < 0.1). For GSEA, genes were pre-ranked by log2(fold change) and the preranked algorithm was run against all gene sets (Subramanian et al., 2005).
Metaplots
We filtered the intervals for metaplot as follows. For metaplots around TSS, UCSC canonical genes were filtered to remove any genes that overlapped within 5 kb of the TSS. For metaplots at enhancers, we aligned against centers of all Oct4/Sox2/Nanog defined enhancer peaks (typical enhancers and super-enhancers) according to a previous report (Suzuki et al., 2017; Whyte et al., 2013). Subsequently, we filtered out any overlapping enhancers peaks within a 3 kb window and also any that overlapped a UCSC canonical gene. For metaplots at splice sites, UCSC canonical genes with at least 4 introns were identified to ensure a sufficiently large number of genes. We also removed any introns that had known snoRNAs and required introns be at least 2 kb long. Metaplots in Figures 5 and Figure S6 required that the gene must be expressed (FPKM>0.5). Metaplots in Figures 6 and 7 required two filter where a) the dyad axis must be at least 600 bps from the TSS and b) the gene must be expressed (FPKM>0.5). In addition, expression-matched gene sets without 2P clusters were used as control gene sets in Figures 5, 6, and 7. In Figures 5, 6, and 7, we confirmed that the changes in metaplots were not due to extreme outliers, as removing the 5% extremes resulted in similar results.
To create metaplots for RNA-seq, MNase-seq, or ChIP-seq, we counted the number of overlapping reads across non-overlapping bins that span the aligned region. The one exception is for splice sites, we did an additional filter where we removed any reads that overlapped annotated exons. Bins were normalized by:
For 2P-seq, we focused on unique PAS-linked cleavage sites rather than potential cleavage sites, because the low number of cleavage site positions created extremely spiky reads if we align uncollapsed reads. We counted the number of unique PAS-linked cleavage sites across non-overlapping bins that span the aligned region. Similar to RNA-seq, any cleavage sites that overlapped exons were removed if we were doing splice site alignments. Normalization for 2P-seq was challenging as we did not have spike ins. Normalization by number of detected unique sites is a challenge because a significant fraction of unique sites is located within genes, so any major shift (as expected with U1 inhibition) will misrepresent the number of unique cleavage sites. We chose to normalize by number of mapped 2P-sites which also factors in sequencing depth. In other words, bins were normalized by:
2P-seq Analysis
Read Processing
The cleavage site is defined as the last nucleotide before the addition of a poly(A) tail. The putative cleavage sites were further filtered to remove sequencing artifacts from internal A-stretches, and subdivided into those containing one of 36 PAS motif variants within an upstream 80 bp window (Almada et al., 2013) and those without such PAS motifs. The following steps were performed for read processing.
Reads were first quality filtered by trimmed of adapters with Trimmomatic (Bolger et al., 2014) and A stretches (>5 As) were removed if they were immediately downstream of first sequenced nucleotide. We interpreted these events as poly(A) tails that due to reverse transcription errors or biological reasons had a non-As added to the cDNA. Next, we mapped either filtered reads (set A) or filtered reads with the first 15 nts trimmed (set B) to the mm9 genome using STAR aligner, end-to-end mode. The trimming of first 15 nt was done to ensure that reads were not going to be lost due to mismatches at the 5´ end, which may involve non-templated nucleotides (such as uridines), which are added to some termination events. For both sets, the first mapped nucleotide was considered the cleavage site.
The two mapped libraries were combined as follows. If the read only aligned in set A or set B, the cleavage site was used as is. If the read aligned in both set A and set B, we subjected the mapped site to one further test. If the mapped cleavage site in set A overlaps the mapped cleavage site minus 15 nucleotides in set B, the position in set A was used. However, if the mapped cleavage site in set A differed substantially from the read in set B, we chose the site in set A as the mapped site. We attributed changes for this subset to the shorter read being harder to find exact matches, so preferred the mapped position of the longer read.
With the combined mapped cleavage sites, we then applied an internal priming filter, in which we removed reads with at least 7 adenosines in the 10 nucleotides 3´ of the cleavage site, or 13 adenosines in the downstream 20 nucleotides. The remaining cleavage sites were filtered so that it must have at least 2 different reads mapping to it and also to not overlap B2 SINE elements. Finally, we scored reads as PAS containing or not PAS containing by surveying the 80 nucleotides upstream of the cleavage site for the presence of the top 36 PAS motifs, as described in (Almada et al., 2013). Specifically, the top 2 canonical PAS motifs are AATAAA or ATTAAA. Next, we also look for known variants, AGTAAA or TATAAA. We subsequently look for the next 8 most frequent sites or PAS8 (AATATA, AATACA, CATAAA, GATAAA, AATGAA, ACTAAA, AAGAAA, AATAGA). Finally we look for the remaining 24 PAS variants.
Cleavage Cluster Pipeline
Cleavage sites from biological replicates of 2P-seq datasets were collapsed. Sites within 25 nucleotides of each other on the same strand were merged using Bedtools mergeBed (Quinlan and Hall, 2010). The tentative clusters were further merged across all 2P-seq datasets to create a combined cluster set with mergeBed, but this time only if they overlapped, creating a combined list of cleavage clusters.
Next we assigned whether the cleavage cluster was a PAS-linked or PAS independent cluster. To do this, the most abundant cleavage site within a cluster was called the max site. We looked up to 100 nucleotides upstream of the max site and looked for one of PAS36 motifs. Those with PAS36 motifs were called PAS-linked clusters whereas those without were PAS-independent clusters. Since most clusters were only present in one library, we assumed those were noise and focused on defining robust clusters. Robust clusters were those where at least three independent 2P-seq libraries had non-zero reads among the 12 libraries; genes with premature clusters were defined as genes with robust clusters overlapped intron 1 of the gene.
Hierachical Clustering
The number of counts across robust clusters in the first intron or uaRNAs were counted and normalized by library size. Subsequently, the robust clusters were subjected to hierarchical clustering using the Pearson correlation metric in Multiple Experiment Viewer. Hierarchical clustering of 2P clusters that overlapped the first intron and had at least 10 reads confirmed reproducibility among replicates (Figures S4A–C).
Identifying the Position of Most Frequently Used Cluster
To identify the most frequent cleavage cluster at both the first intron and at uaRNAs, we overlapped robust clusters found in each replicate to either the first intron of non-overlapping UCSC canonical genes or to a 3 kb window upstream of the TSS for non-overlapping UCSC canonical genes. The cluster with the most reads in each interval was called the most frequently used cleavage cluster.
All predicted A[A/T]TAAA sites were identified across non-overlapping UCSC canonical genes or uaRNAx, and ranked whereby position 1 is closest to the TSS. The most frequently used cleavage clusters were filtered for those with canonical PAS motifs (A[A/T]TAAA) and then assigned a position based on the ranked PAS motifs.
CpG Island Genes
Annotation of CpG island was downloaded from UCSC genome browser, and genes with CpG island promoters were defined as genes where CpG islands overlap TSS to +100 bp of UCSC canonical genes.
MNase-seq Analysis
To generate a catalogue of the invariant nucleosomes in mESC, we first utilized the recently developed NucTools algorithm, which integrates previously reported multiple MNase-seq datasets to define stable versus unstable nucleosomes using the relative error of nucleosome occupancy (Vainshtein et al., 2017). We further incorporated the information of precise nucleosome dyad centers recently defined by chemical mapping in mESC (Voong et al., 2016). The information of nucleosome dyads in mESCs was download from (Voong et al., 2016). To identify regions with stable nucleosomes, we analyzed five different mESC MNase-seq datasets (Teif et al., 2012; West et al., 2014; Zhang et al., 2014) using NucTools (Vainshtein et al., 2017). We determined stable nucleosome regions using stable_nucs_replicates.pl. A sliding window of 50 bp was used and stable regions were selected based on the relative error based on five replicates < 0.5. The dyads within NucTools-defined stable regions, which were most proximal to TSS, were regarded as dyads of +1/–1 stable nucleosomes and used for subsequent analysis. For heatmap analysis, reads from various datasets were assigned to nonoverlapping bins in a 2 kb flanking window around the SNFR for each gene containing a robust premature cleavage cluster. The intervals were sorted by increasing SNFR width. The datasets used in this study are summarized in Table S2.
ChIP-seq/GRO-seq Analysis and Pausing Indices
Sources of the datasets used in this study are described in Table S2. Reads were aligned to the mouse genome build mm9 or human genome build hg19 using bowtie as described previously (Suzuki et al., 2017). Pausing indices were calculated as shown in Figure 6C. The widths of intervals used to calculate pausing indices were determined from analysis of the Pol II ChIP-seq alignments in Figures 6B and 6D, taking into account the widths of a Pol II ramp and a flavopiridol-affected region upstream of the +1 dyad. In mouse GRO-seq analysis, normalization between datasets was done with uniquely aligned spike-in RNA reads (Jonkers et al., 2014). In Figure 6H, normalization was done using the reads in the 5´ external transcribed spacer of the 45S rRNA gene, as previously described (Laitem et al., 2015), and long CpG island genes (distance between TSS and the edge of CpG island > 600 bps) were analyzed.
Dinucleotide Frequency Analysis
Gene body nucleosomes were defined as nucleosome that was between TSS and (TES – 2 kb). The number of AA/TT/TA dinucleotides was counted in a 2 base pair sliding window along a 150 bp window flanking the dyad axis and divided by the total number of gene body nucleosomes. The predicted PAS frequency was identified by searching for A[A/T]TAAA on the same strand of the gene, using a sliding 6 bp window along the 150 bp window flanking the dyad axis, divided by total number of gene body nucleosomes. The used PAS frequency was identified by counting the number of PAS motif assigned to robust clusters in a sliding 6 bp window along the 150 bp window flanking the dyad axis, divided by total number of gene body nucleosomes.
PRO-seq Analysis
Sequenced reads were aligned to mm9 using bowtie2 (Langmead and Salzberg, 2012) and options -D 15 -R 2 -N 0 -L 20 -i S,1,0.75. Processed reads were resized to the 3´ most sequenced read, which represents the precise position of the RNA in the catalytic site. In PRO-seq analysis, Exosc3 depletion caused a modest increase of PRO-seq signals in the first half of first intron (Figure S7F), potentially reflecting a mixture of slowly transcribing or paused Pol II. Depletion of Exosc3 also elicited slight pausing effects at +1 stable nucleosomes, but this effect did not differ between genes with and without 2P clusters (Figures S7G–H), thus suggesting that the increase in PAS termination transcripts upon Exosc3 depletion is mainly attributable to RNA stabilization, and not from increased pausing.
Myc DKO mESC RNA-seq Analysis
RNA-seq in c-Myc and N-Myc double knockout mESCs was previously reported (Scognamiglio et al., 2016). Sample 2 (Control, c-MycΔ/Δ and N-MycΔ/fl) and sample 6 (DKO, 96 hours) were compared. Gene ontology (GO) analyses were performed using Database for Annotation, Visualization, and Integrated Discovery (DAVID; https://david.ncifcrf.gov) and GO BP (Biological Process) terms. Similar results were obtained for both all expressed genes and wide SNFR genes (Figure 7F).
Statistical Analysis
In Figures 1C and 3E, statistical significance for boxplots was evaluated with Wilcoxon signed ranked sum test.
In Figure 4A, statistical significance for Venn diagram overlaps was evaluated using the hypergeometric test (P < 0.0001).
In Figures 5A, 5B, 6A, 6B, 6D, 6F, 7B, and S6B, to show positional information of differences between groups, P values with Kolmogorov–Smirnov test at each bin are displayed (5A, 5B, and S6B: one-sided test for increases in genes with 2P clusters; 6A and 6B: two-sided test between genes with 2P clusters and genes without 2P clusters; 6D: one-sided test for increases upon flavopiridol treatment; 6F: one-sided test for decreases in shSpt5 relative to shControl; and 7B: one-sided test for increases upon Myc inhibitor treatment).
In Figures 5A (genes with 2P clusters vs. genes without 2P clusters) and 5B (genes with 2P clusters vs. genes without 2P clusters), Kolmogorov–Smirnov (K-S) tests were also performed across all bins. In Figure 5A, Chd1, Chd2, Chd8, Chd9, and Ep400 show increased binding for genes with 2P clusters (P < 0.05). In Figure 5B, Pol II, Spt5, Ell2, and Aff4 show increased binding for genes with 2P clusters (P < 0.05).
In Figures 6E and S7A, statistical significance for flavopiridol-mediated pause effects in +1 nucleosome pausing index was evaluated with Kolmogorov–Smirnov test, showing P < 0.01 in both gene sets with/without 2P clusters. In addition, +1 nucleosome pausing index of genes with 2P clusters upon flavopiridol treatment were significantly higher than those of genes without 2P clusters upon flavopiridol treatment (P < 0.01).
In Figures 6G and S7B, statistical significance for pause release effects in TSS pausing index was evaluated with Kolmogorov–Smirnov test, showing P < 0.01 in shNelfA and shSpt5 samples in both gene sets.
In Figures 7C and S7I, statistical significance for Myc-inhibition-mediated pause effects in TSS pausing index or +1 nucleosome pausing index was evaluated with Kolmogorov–Smirnov test, showing P < 0.01 in both gene sets with/without 2P clusters. In addition, in Figure 7C, TSS pausing index and +1 nucleosome pausing index of genes with 2P clusters upon Myc inhibition were significantly higher than those of genes without 2P clusters upon Myc inhibition (P < 0.01 with Kolmogorov–Smirnov test), supporting that Myc preferentially regulates the +1 stable nucleosome pause at genes with premature clusters.
In Figure 7E, statistical significance was evaluated with Kolmogorov–Smirnov test and displayed as P < 0.001 with asterisks.
In Figure S6C, statistical significance was evaluated with Student’s t-test and displayed as P < 0.05 with asterisks.
• DATA AND SOFTWARE AVAILABILITY
Data Resources
The accession number for the data for RNA-seq and 2P-seq reported in this paper is GEO: GSE100537.
Unprocessed Blot and Gel images in this manuscript have been deposited to Mendeley Data and are available at http://dx.doi.org/10.17632/vzv6n64kd8.1.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to members of the Sharp laboratory for discussion and critical review of the manuscript. A.C.C. is supported by NSERC PGS-D. H.I.S. is supported by the Uehara Memorial Foundation Research Fellowship and the Osamu Hayaishi Memorial Scholarship for Study Abroad. X.W. is a Helen Hay Whitney Foundation Fellow. We thank the Robert A. Swanson (1969) Biotechnology Center at the Koch Institute for Integrative Cancer Research at the Massachusetts Institute of Technology for technical support, specifically Stuart Levine and the staff of the MIT BioMicro Center/KI Genomic Core Facility. This work was supported by Program Project Grant P01CA042063 from the National Cancer Institute, by United States Public Health Service Grants R01-GM34277 and R01-CA133404 from the National Institutes of Health awarded to P.A.S., and, in part, by the Koch Institute Support (Core) Grant P30-CA14051 from the National Cancer Institute.
Footnotes
Declaration of Interests
The authors declare no competing financial interests.
References
- Adelman K, and Lis JT (2012). Promoter-proximal pausing of RNA polymerase II: emerging roles in metazoans. Nat Rev Genet 13, 720–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almada AE, Wu X, Kriz AJ, Burge CB, and Sharp PA (2013). Promoter directionality is controlled by U1 snRNP and polyadenylation signals. Nature 499, 360–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen PK, Lykke-Andersen S, and Jensen TH (2012). Promoter-proximal polyadenylation sites reduce transcription activity. Genes Dev 26, 2169–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagchi DN, and Iyer VR (2016). The Determinants of Directionality in Transcriptional Initiation. Trends Genet 32, 322–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg MG, Singh LN, Younis I, Liu Q, Pinto AM, Kaida D, Zhang Z, Cho S, Sherrill-Mix S, Wan L, et al. (2012). U1 snRNP determines mRNA length and regulates isoform expression. Cell 150, 53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brannan K, Kim H, Erickson B, Glover-Cutter K, Kim S, Fong N, Kiemele L, Hansen K, Davis R, Lykke-Andersen J, et al. (2012). mRNA decapping factors and the exonuclease Xrn2 function in widespread premature termination of RNA polymerase II transcription. Mol Cell 46, 311–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bresson SM, Hunter OV, Hunter AC, and Conrad NK (2015). Canonical Poly(A) Polymerase Activity Promotes the Decay of a Wide Variety of Mammalian Nuclear RNAs. PLoS Genet 11, e1005610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Shishkin AA, Zhu X, Kadri S, Maza I, Guttman M, Hanna JH, Regev A, and Garber M (2016). Evolutionary analysis across mammals reveals distinct classes of long non-coding RNAs. Genome Biol 17, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, Wong E, Orlov YL, Zhang W, Jiang J, et al. (2008). Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 133, 1106–1117. [DOI] [PubMed] [Google Scholar]
- Core LJ, Waterfall JJ, and Lis JT (2008). Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science 322, 1845–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Dieuleveult M, Yen K, Hmitou I, Depaux A, Boussouar F, Bou Dargham D, Jounier S, Humbertclaude H, Ribierre F, Baulard C, et al. (2016). Genome-wide nucleosome specificity and function of chromatin remodellers in ES cells. Nature 530, 113–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derti A, Garrett-Engele P, Macisaac KD, Stevens RC, Sriram S, Chen R, Rohl CA, Johnson JM, and Babak T (2012). A quantitative atlas of polyadenylation in five mammals. Genome Res 22, 1173–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett JA, and Surani MA (2014). Regulatory principles of pluripotency: from the ground state up. Cell Stem Cell 15, 416–430. [DOI] [PubMed] [Google Scholar]
- Ji X, Dadon DB, Abraham BJ, Lee TI, Jaenisch R, Bradner JE, and Young RA (2015). Chromatin proteomic profiling reveals novel proteins associated with histone-marked genomic regions. Proc Natl Acad Sci U S A 112, 3841–3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonkers I, Kwak H, and Lis JT (2014). Genome-wide dynamics of Pol II elongation and its interplay with promoter proximal pausing, chromatin, and exons. Elife 3, e02407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaida D, Berg MG, Younis I, Kasim M, Singh LN, Wan L, and Dreyfuss G (2010). U1 snRNP protects pre-mRNAs from premature cleavage and polyadenylation. Nature 468, 664–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellner WA, Bell JS, and Vertino PM (2015). GC skew defines distinct RNA polymerase pause sites in CpG island promoters. Genome Res 25, 1600–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilchert C, Wittmann S, and Vasiljeva L (2016). The regulation and functions of the nuclear RNA exosome complex. Nat Rev Mol Cell Biol 17, 227–239. [DOI] [PubMed] [Google Scholar]
- Lai F, Gardini A, Zhang A, and Shiekhattar R (2015). Integrator mediates the biogenesis of enhancer RNAs. Nature 525, 399–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laitem C, Zaborowska J, Isa NF, Kufs J, Dienstbier M, and Murphy S (2015). CDK9 inhibitors define elongation checkpoints at both ends of RNA polymerase II-transcribed genes. Nat Struct Mol Biol 22, 396–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, You B, Hoque M, Zheng D, Luo W, Ji Z, Park JY, Gunderson SI, Kalsotra A, Manley JL, et al. (2015). Systematic profiling of poly(A)+ transcripts modulated by core 3’ end processing and splicing factors reveals regulatory rules of alternative cleavage and polyadenylation. PLoS Genet 11, e1005166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C, Garrett AS, De Kumar B, Smith ER, Gogol M, Seidel C, Krumlauf R, and Shilatifard A (2011). Dynamic transcriptional events in embryonic stem cells mediated by the super elongation complex (SEC). Genes Dev 25, 1486–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahat DB, Kwak H, Booth GT, Jonkers IH, Danko CG, Patel RK, Waters CT, Munson K, Core LJ, and Lis JT (2016). Base-pair-resolution genome-wide mapping of active RNA polymerases using precision nuclear run-on (PRO-seq). Nat Protoc 11, 1455–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquardt S, Escalante-Chong R, Pho N, Wang J, Churchman LS, Springer M, and Buratowski S (2014). A chromatin-based mechanism for limiting divergent noncoding transcription. Cell 157, 1712–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer A, di Iulio J, Maleri S, Eser U, Vierstra J, Reynolds A, Sandstrom R, Stamatoyannopoulos JA, and Churchman LS (2015). Native elongating transcript sequencing reveals human transcriptional activity at nucleotide resolution. Cell 161, 541–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meola N, Domanski M, Karadoulama E, Chen Y, Gentil C, Pultz D, Vitting-Seerup K, Lykke-Andersen S, Andersen JS, Sandelin A, et al. (2016). Identification of a Nuclear Exosome Decay Pathway for Processed Transcripts. Mol Cell 64, 520–533. [DOI] [PubMed] [Google Scholar]
- Nojima T, Gomes T, Grosso ARF, Kimura H, Dye MJ, Dhir S, Carmo-Fonseca M, and Proudfoot NJ (2015). Mammalian NET-Seq Reveals Genome-wide Nascent Transcription Coupled to RNA Processing. Cell 161, 526–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ntini E, Jarvelin AI, Bornholdt J, Chen Y, Boyd M, Jorgensen M, Andersson R, Hoof I, Schein A, Andersen PR, et al. (2013). Polyadenylation site-induced decay of upstream transcripts enforces promoter directionality. Nat Struct Mol Biol 20, 923–928. [DOI] [PubMed] [Google Scholar]
- Oh JM, Di C, Venters CC, Guo J, Arai C, So BR, Pinto AM, Zhang Z, Wan L, Younis I, et al. (2017). U1 snRNP telescripting regulates a size-function-stratified human genome. Nat Struct Mol Biol 24, 993–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perna D, Faga G, Verrecchia A, Gorski MM, Barozzi I, Narang V, Khng J, Lim KC, Sung WK, Sanges R, et al. (2012). Genome-wide mapping of Myc binding and gene regulation in serum-stimulated fibroblasts. Oncogene 31, 1695–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, and Salzberg SL (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 33, 290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preker P, Nielsen J, Kammler S, Lykke-Andersen S, Christensen MS, Mapendano CK, Schierup MH, and Jensen TH (2008). RNA exosome depletion reveals transcription upstream of active human promoters. Science 322, 1851–1854. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahl PB, Lin CY, Seila AC, Flynn RA, McCuine S, Burge CB, Sharp PA, and Young RA (2010). c-Myc regulates transcriptional pause release. Cell 141, 432–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez-Carrozzi VR, Braas D, Bhatt DM, Cheng CS, Hong C, Doty KR, Black JC, Hoffmann A, Carey M, and Smale ST (2009). A unifying model for the selective regulation of inducible transcription by CpG islands and nucleosome remodeling. Cell 138, 114–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rege M, Subramanian V, Zhu C, Hsieh TH, Weiner A, Friedman N, Clauder-Munster S, Steinmetz LM, Rando OJ, Boyer LA, et al. (2015). Chromatin Dynamics and the RNA Exosome Function in Concert to Regulate Transcriptional Homeostasis. Cell Rep 13, 1610–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scognamiglio R, Cabezas-Wallscheid N, Thier MC, Altamura S, Reyes A, Prendergast AM, Baumgartner D, Carnevalli LS, Atzberger A, Haas S, et al. (2016). Myc Depletion Induces a Pluripotent Dormant State Mimicking Diapause. Cell 164, 668–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seila AC, Calabrese JM, Levine SS, Yeo GW, Rahl PB, Flynn RA, Young RA, and Sharp PA (2008). Divergent transcription from active promoters. Science 322, 1849–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene PJ, Hernandez AE, Groudine M, and Henikoff S (2014). The nucleosomal barrier to promoter escape by RNA polymerase II is overcome by the chromatin remodeler Chd1. Elife 3, e02042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spies N, Burge CB, and Bartel DP (2013). 3’ UTR-isoform choice has limited influence on the stability and translational efficiency of most mRNAs in mouse fibroblasts. Genome Res 23, 2078–2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spies N, Nielsen CB, Padgett RA, and Burge CB (2009). Biased chromatin signatures around polyadenylation sites and exons. Mol Cell 36, 245–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian V, Mazumder A, Surface LE, Butty VL, Fields PA, Alwan A, Torrey L, Thai KK, Levine SS, Bathe M, et al. (2013). H2A.Z acidic patch couples chromatin dynamics to regulation of gene expression programs during ESC differentiation. PLoS Genet 9, e1003725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki HI, Young RA, and Sharp PA (2017). Super-Enhancer-Mediated RNA Processing Revealed by Integrative MicroRNA Network Analysis. Cell 168, 1000–1014 e1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teif VB, Vainshtein Y, Caudron-Herger M, Mallm JP, Marth C, Hofer T, and Rippe K (2012). Genome-wide nucleosome positioning during embryonic stem cell development. Nat Struct Mol Biol 19, 1185–1192. [DOI] [PubMed] [Google Scholar]
- Vainshtein Y, Rippe K, and Teif VB (2017). NucTools: analysis of chromatin feature occupancy profiles from high-throughput sequencing data. BMC Genomics 18, 158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voong LN, Xi L, Sebeson AC, Xiong B, Wang JP, and Wang X (2016). Insights into Nucleosome Organization in Mouse Embryonic Stem Cells through Chemical Mapping. Cell 167, 1555–1570 e1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagschal A, Rousset E, Basavarajaiah P, Contreras X, Harwig A, Laurent-Chabalier S, Nakamura M, Chen X, Zhang K, Meziane O, et al. (2012). Microprocessor, Setx, Xrn2, and Rrp6 co-operate to induce premature termination of transcription by RNAPII. Cell 150, 1147–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber CM, Ramachandran S, and Henikoff S (2014). Nucleosomes are context-specific, H2A.Z-modulated barriers to RNA polymerase. Mol Cell 53, 819–830. [DOI] [PubMed] [Google Scholar]
- West JA, Cook A, Alver BH, Stadtfeld M, Deaton AM, Hochedlinger K, Park PJ, Tolstorukov MY, and Kingston RE (2014). Nucleosomal occupancy changes locally over key regulatory regions during cell differentiation and reprogramming. Nat Commun 5, 4719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, and Young RA (2013). Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams LH, Fromm G, Gokey NG, Henriques T, Muse GW, Burkholder A, Fargo DC, Hu G, and Adelman K (2015). Pausing of RNA polymerase II regulates mammalian developmental potential through control of signaling networks. Mol Cell 58, 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Vastenhouw NL, Feng J, Fu K, Wang C, Ge Y, Pauli A, van Hummelen P, Schier AF, and Liu XS (2014). Canonical nucleosome organization at promoters forms during genome activation. Genome Res 24, 260–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.