

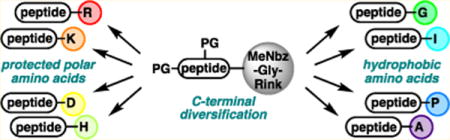
Abstract
Sequence diversification at the C terminus is traditionally limited by significant epimerization of the C-terminal residue during its activation toward nucleophilic attack, thus mandating repetition of the peptide synthesis for each targeted variation. Here, we accomplish divergent C-terminal elongation of a single peptide substrate with concomitant resin cleavage via displacement of an N-acyl urea moiety. Sterically hindered amino acids such as Ile and Pro are well-tolerated in this approach, which proceeds reasonable conversion and no detectable epimerization of the starting peptide’s C-terminal amino acid.
Graphical abstract
INTRODUCTION
Methods for the rapid diversification of peptide sequences are important for the optimization of peptide lead targets.1 Because of the tendency to epimerize the C-terminal residue2 during the carboxylic acid activation,3 sequences that vary at the C- terminal amino acid4 have not been readily accessible without repetition of the SPPS for each desired C-terminal amino acid. Recently, we reported a method for C-terminal derivatization, as outlined in Scheme 1.5 First, peptide 1 is generated on solid support using a methyl diaminobenzoyl (MeDbz) linker.6 After activation to form the N-acyl urea (MeNbz, 2), the peptide was either cleaved with a nucleophile7 to generate protected peptide 3, or it was cleaved at Gly using trifluoroacetic acid (TFA), and then the unprotected peptide (4) was functionalized.6,8 During this work, we found that the C-terminal residue is not epimerized during the modification reaction, even for sensitive residues such as His or Cys.9 Therefore, we viewed this approach as ideal for backbone sequence diversification at the C terminus.
Scheme 1.
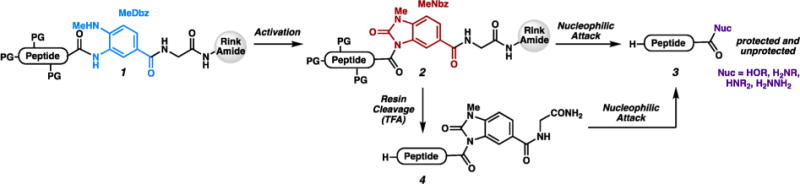
Our Previous C-terminal Functionalization of Peptides
In our previous work,5 we reported efficient addition of primary amine nucleophiles to MeNbz-appended peptides to generate secondary C-terminal amides. We observed reduced reactivity for secondary amines and α-branched alcohols during resin-cleaving reactions. However, solution-phase functionalization reactions proceeded more quickly. We expected that α-branched primary amines (e.g., amino acids) would be significantly less reactive than unbranched primary amines. This hypothesis was consistent with Pascal’s observation that Gly N-acyl benzimidazole could be substituted by leucine methyl ester in solution to form a dipeptide over 20 h.10
RESULTS AND DISCUSSION
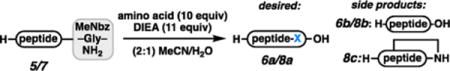
We began investigations toward developing a general method for C-terminal peptide elongation using unprotected amino acids as nucleophiles in solution-phase reactions. As shown in Table 1, a variety of amino acids can be employed as nucleophiles in the elongation of MeNbz-appended peptides in solution. For our initial evaluations, H-AWA-MeNbz-Gly-NH2 (5) was employed. Upon treatment with 10 equiv of amino acid and 11 equiv of Hünig’s base, complete conversion was observed in all cases to form either the corresponding elongated peptide (6a) or a mixture of 6a and hydrolysis product 6b.11 Water was employed as a cosolvent to improve the solubility of the unprotected amino acid nucleophiles relative to acetonitrile alone. In the absence of added water, reactivity was poor.12 The non-branched nucleophile Gly reacted to form the elongated peptide H-AWAG-OH exclusively (entry 1). However, when Ala was employed as a nucleophile, ∼1:1 elongation to hydrolysis was observed (entry 2). In our previous work, we found that the secondary Weinreb amine reacted much more slowly than primary amine nucleophiles. Therefore, we were not surprised to find that when Pro was the nucleophile, only 29% elongated peptide was observed. Surprisingly, the α- and β-branched Ile reacted analogously to Gly, giving only the C-terminal elongation product H-AWAI-OH (entry 4). Unprotected Trp required only 30 min and led exclusively to elongated peptide H-AWAW-OH, presumably because the unprotected nitrogen in the indole increases the amino acid’s solubility (entry 5).13 Additionally, it is conceivable that a transient π–π interaction with the MeNbz group assists in the addition.14 We also employed the test peptide H-AKTWA-MeNbz-Gly-NH2 (7) to determine whether the elongation was efficient enough to modify unprotected peptides bearing nucleophilic side chains. Unfortunately, only Gly reacted quickly enough to outcompete macrolactam formation, affording 8a as the major product (entry 6). All α-branched amino acids led primarily to macrocycle 8c (entries 7–9).
Table 1.
Solution Phase C-Terminal Peptide Elongation
| |||||||
|---|---|---|---|---|---|---|---|
| entrya | peptide | amino acid (X) | time (h) | conversion (%)b | a:b:c (%)c | ||
| 1 | AWA (5) | H-Gly (G) | 1 | >99 | >99 | <1 | n/a |
| 2 | AWA (5) | H-Ala (A) | 1 | >99 | 52 | 48 | n/a |
| 3 | AWA (5) | H-Pro (P) | 6 | >99 | 29 | 71 | n/a |
| 4 | AWA (5) | H-IIe (I) | 1 | >99 | >99 | <1 | n/a |
| 5 | AWA (5) | H-Trp (W) | 0.5 | >99 | >99 | <1 | n/a |
| 6 | AKTWA (7) | H-Gly (G) | 0.5 | >99 | >99 | <1 | <1 |
| 7 | AKTWA (7) | H-Ala (A) | 0.5 | >99 | <1 | <1 | >99 |
| 8 | AKTWA (7) | H-IIe (I) | 2 | >99 | 12 | 3 | 85 |
| 9 | AKTWA (7) | H-Pro (P) | 6 | >99 | <1 | <1 | >99 |
All reactions were performed on 20 mg of crude peptide in 300 μL of solvent, rt = 24 ± 1 °C.
Conversion indicates degree of consumption of peptide-MeNbz-Gly-NH2.
Based on relative ratio of HPLC/MS data.
On the basis of these results, it was clear that a useful strategy for C-terminal elongation would need to proceed directly from the resin-bound MeNbz peptide, where nucleophilic side chain groups would be protected. Thus, MeDbz-linked peptide 9 was activated and treated with 10 equiv of Gly and 11 equiv of DIEA in 500 μL DMF affording exclusively desired peptide 10a (Table 2). However, the conversion in this case was very low (5%, entry 1). To improve the reactivity of the amino acids, water was again employed as a cosolvent, improving the conversion to 39%, which was promising but certainly not synthetically viable (entry 2). We next increased the amount of Hünig’s base employed, leading to 76% conversion over 4 h and a 91:9 ratio of elongation (10a) to hydrolysis (10b, entry 3). The combination of NaSPh and ethyl-3-mercaptopropionate led to complete consumption of 9; however, the alkyl thioester was the sole observed product (entry 4).15 Finally, treatment with NaSPh in a 5:1 mixture of DMF:H2O allowed complete reactivity of the MeNbz group. A 94:6 ratio of elongated product to MeNbz hydrolysis (10a:10b) was observed (entry 5). A similar trend in reactivity was observed for Ile (entries 6–7) and Ala (entries 10–11) with the absence of NaSPh. However, addition of NaSPh did not fully displace the MeNbz group, which was previously observed with Gly. To improve the reactivity, a mixture of NaSPh and mercaptophenylacetic acid (MPAA) in a 5:1 mixture of DMF/Na2HPO4 buffer at pH 7.2 was used to help solubilize the amino acid (entries 9 and 13). Pro displaced MeNbz with complete conversion in the absence of any thiol additive (entry 15), presumably because it is more nucleophilic than the primary amines. As mentioned earlier, secondary amines were not ideal nucleophiles in this chemistry.5 The improved reactivity of Pro is likely caused by the more exposed nitrogen lone pair in the cyclic amine relative to the linear amine.16 For all amino acids tested under the optimized conditions, hydrolysis rates were maintained below 15%.
Table 2.
On-Resin Elongation To Access Protected Peptide Acids

| |||||
|---|---|---|---|---|---|
| entrya | amino acid (X) | DIEA (equiv) | additive | % conversionb (% isolated yield)c | 10a:10bd |
| 1e | Gly (G) | 11 | 5 | >99:<1 | |
| 2g | Gly (G) | 11 | 3 | 94:06 | |
| 3 | Gly (G) | 21 | 76 | 91:09 | |
| 4f | Gly (G) | 21 | NaSPh/HS(CH2)3CO2Et | >99 | n.o. |
| 5h | Gly (G) | 21 | NaSPh | >99 (39) | 94:06 |
| 6 | IIe (I) | 11 | 20 | 88:12 | |
| 7 | IIe (I) | 21 | 72 | 88:12 | |
| 8 | IIe (I) | 21 | NaSPh | 76 | 85:15 |
| 9g | IIe (I) | 21 | NaSPh/MPAA | 94 (22) | 90:10 |
| 10 | Ala (A) | 11 | 19 | 81:19 | |
| 11 | Ala (A) | 21 | 77 | 77:13 | |
| 12 | Ala (A) | 21 | NaSPh | 93 | 72:28 |
| 13g | Ala (A) | 21 | NaSPh/MPAA | 94 (29) | 86:14 |
| 14 | Pro (P) | 11 | 34 | 62:38 | |
| 15i | Pro (P) | 21 | >99 (44) | 88:12 | |
| 16 | Pro (P) | 21 | NaSPh | 94 | 83:17 |
| 17j | Pro (P) | 21 | NaSPh | >99 | 87:13 |
All reactions were conducted on 20 mg of resin in 600 μL of (5:1) DMF:H2O, rt = 24 ± 1 °C.
Conversion based on integration of remaining peptide on activated linker after nucleophile cleavage in MS data.
Yield based on final loading of (50–100 mg) WA-MeDbz-Gly-Rink resin.
Relative ratios at 254 nm.
Reaction was conducted in 500 μL of DMF.
100 μL of ethyl-3-mercaptopropionate was used, giving exclusively the alkyl thioester.
A solution of 600 μL of 5:1 DMF:modified NCL buffer (pH 7.2) was used.
Went to 73% conversion on a larger scale.
Went to 97% conversion on a larger scale.
Treated for 4 h twice.
We previously established that Hünig’s base is mild enough to avoid epimerization during conversion of MeNbz peptides to the corresponding acids, esters, and amides.5 However, we hypothesized that the phenyl thioester, which is generated in situ, might be more prone to epimerization than the MeNbz peptides. The intermediate thioester will be longest-lived in the presence of the slowest nucleophile. Thus, we tested the extent of epimerization observed during elongation with Ile and Pro (entries 9 and 17 in Table 2). Interestingly, 14% epimerization was observed in the presence of NaSPh in a modified native chemical ligation (NCL) buffer with Ile (see Table 3, entry 1).17 Repetition of this experiment without NaSPh lowered the epimerization to 1%, but the conversion also dropped to 38% (entry 2). However, treatment with NaSPh but no Hünig’s base afforded the elongated peptide with no epimerization and 50% conversion (entry 3), supporting our hypothesis that the aryl thioester is susceptible to epimerization in the presence of excess base. Under the conditions of entry 7, Table 2 (using 21 equiv Hünig’s base in 5:1 DMF:H2O), the conversion was improved to 72% while maintaining no epimerization (entry 4, Table 3). To access AWAI with complete conversion, the reaction was shaken for 18 h instead of 4 with minimal epimerization (entry 5). The epimerization was not reduced by exposing the resin to the conditions twice and combining the samples, but complete conversion could be achieved by this approach (entry 6). We were interested in the susceptibility to epimerization during Pro addition because of its increased basicity. Both entries 15 and 17 in Table 2 gave complete conversion to AWAP, so the epimerization was evaluated under those conditions. In the presence of NaSPh in 5:1 DMF:H2O, 3% epimerization was observed. Removing NaSPh did not significantly lower the epimerization in this case (entries 7 and 8, Table 3).
Table 3.
Epimerization of C-Terminal Alanine

| |||||||
|---|---|---|---|---|---|---|---|
| entrya,b | amino acid (X) | DIEA (equiv) | additive | cosolvent | t (h) | conversion (%)c | epimerization (%D-Ala)d |
| 1e | IIe (I) | 21 | NaSPh/MPAA | Na2HPO4(aq) | 4 | 94 | 14 |
| 2e | IIe (I) | 21 | MPAA | Na2HPO4(aq) | 4 | 38 | 1 |
| 3e | IIe (I) | NaSPh/MPAA | Na2HPO4(aq) | 4 | 50 | <1 | |
| 4 | IIe (I) | 21 | H2O | 4 | 72 | <1 | |
| 5 | IIe (I) | 21 | H2O | 18 | >99 | 2 | |
| 6 | IIe (I) | 21 | H2O | 4 (2×) | >99 | 2 | |
| 7 | Pro (P) | 21 | NaSPh | H2O | 4 | 94 | 3 |
| 8 | Pro (P) | 21 | H2O | 4 | >99 | 2 | |
All reactions were conducted on 10–20 mg of resin in 300–600 μL of (5:1) DMF:solvent, rt = 24 ± 1 °C.
For Ile, protecting groups were removed for better assay separation.
Conversion based on integration of remaining peptide on activated linker after nucleophile cleavage in MS data.
Relative ratios at 254 nm.
Na2HPO4 buffer at pH 7.2.
Having established conditions for maximizing conversion and minimizing epimerization, we next evaluated the elongation in the context of a longer peptide. We expected that the reactions would become slower as the peptide length was increased, leading to more non-elongated hydrolysis byproducts. We tested the optimized reaction conditions from Table 2 on H-LYRAGLRAY-MeNbz-Gly-Rink (11). Except for Gly, the ratio of elongation to hydrolysis products was worse for this substrate (Table 4, entries 1–4). Unfortunately, most of the carboxylic acid containing peptides (12b–15b) were challenging to separate from the desired peptides (12a–15a) by RP-HPLC. Originally, water was added to aid in solubilizing the amino acid nucleophiles. We envisioned that replacing water with an alcohol would lead to the ester byproduct (15c), which could be readily separated from the elongated acids. Therefore, a few representative alcoholic cosolvents were evaluated. We previously found that i-PrOH reacted more slowly than primary alcohols,9 so we reasoned that it would minimize ester byproduct formation. Gratifyingly, in the presence of NaSPh, these conditions reduced byproduct formation to 6% of the ester (15c), which was indeed readily separated from the elongated acid LYRAGLRAYP (15a, entry 5). No LYRAGLRAY acid byproduct (15b) was observed. Under these conditions, the peptide AWA was 4% epimerized (Table SI-01). When NaSPh was omitted, no epimerization was observed for AWA.11 However, for LYRAGLRAY, undesired hydrolysis by adventitious water to form 15b increased under these conditions (entry 6). Because hexafluoroisopropanol (HFIP) can be useful for solubilizing peptides, we evaluated it as a cosolvent as well. Treatment with NaSPh and Hünig’s base in the presence of DMF and HFIP led to substantial amounts of the HFIP ester, presumably because the pKa of HFIP is significantly lower than that of isopropanol (entry 7).18 Additionally, truncated acid byproducts were observed, likely because the HFIP ester is prone to hydrolysis upon quenching with MeCN/H2O. Minor epimerization (4%) was detected in the elongation of AWA under these conditions. Omission of NaSPh prevented epimerization in the AWA elongation and did not significantly alter the product ratios for LYRAGLRAY elongation (entry 8 and Figure 1).11
Table 4.
LYRAGLRAY Elongation to Access Protected Peptide Acids

| |||||
|---|---|---|---|---|---|
| entrya | amino acid (X) | % conversionb (% isolated yield)c | cosolvent | productd | a:b:ce |
| 1 | Gly (G) | >99 | H2O | Boc-LYRAGLRAYG (12a) | 91:09:n/a |
| 2f | IIe (I) | >99 | NCL buffer | Boc-LYRAGLRAYl (13a) | 74:26:n/a |
| 3f | Ala (A) | >99 | NCL buffer | Boc-LYRAGLRAYA (14a) | 79:21:n/a |
| 4g | Pro (P) | >99 (9) | H2O | Boc-LYRAGLRAYP (15a) | 70:30:n/a |
| 5 | Pro (P) | 97 | iPrOH | Boc-LYRAGLRAYP (15a) | 94:<1:06 |
| 6g,h | Pro (P) | 89 (11) | iPrOH | Boc-L YRAGLRAYP (15a) | 83:09:08 |
| 7 | Pro (P) | >99 | HFIP | Boc-LYRAGLRAYP (15a) | 81:07:12 |
| 8h | Pro (P) | >99 (10) | HFIP | Boc-LYRAGLRAYP (15a) | 83:05:12 |
All reactions were conducted on 20 mg of resin in 600 μL of (5:1) DMF:H2O, rt = 24 ± 1 °C.
Conversion based on integration of remaining peptide on activated linker after nucleophile cleavage in MS data.
Yield based on initial loading of 100 mg of MeDbz-Gly-Rink resin and represents 3 steps (SPPS, activation, and elongation).
Italicized letters indicate the amino acid residue is protected.
Relative ratios at 254 nm.
Used 500 μL of (5:1) DMF:modified NCL buffer containing MPAA, pH = 7.2.
Treated for 4 h twice.
NaSPh was not used.
Figure 1.
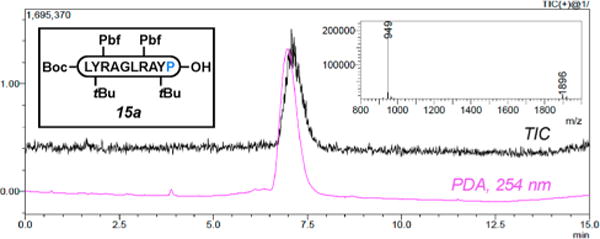
RP-HPLC trace at 254 nm and ESI-MS spectrum of pure Boc-LY(tBu)R(Pbf)AGLR(Pbf)AY(tBu)P-OH (15a) accessed via Table 4, entry 8.
Until this point, we focused on hydrophobic amino acids as nucleophiles. To further probe the scope of the chemistry, side-chain protected polar amino acids were evaluated (Table 5). We expected that the bulky protecting group on Arg(Pbf) would be most challenging to the system. Indeed, the HFIP ester was the major product, suggesting slow addition of the amino acid (entry 1). Similarly, Lys(Boc) led to 18% desired peptide 17a (entry 2). Happily, both Asp(OtBu) and His(Trt) gave complete conversion and reasonable ratios of product to ester (81 and 85% desired, entries 3 and 4). These results were consistent with those of Pro elongation in HFIP (i.e., Table 4, entry 8). In cases where low ratios of elongation product are observed, reasonable quantities of the elongated peptide could be cleanly isolated with no contamination from hydrolysis products (i.e., Table 5, entry 2), making the method viable for rapid access to C-terminal modifications for target validation purposes.11 A representative trace from the addition of aspartic acid t-butyl ester is provided in Figure 2. Taken together, a structurally representative set of amino acids permit the formation of the desired elongated products.
Table 5.
Elongation Employing Side-Chain Protected Amino Acids

| |||||
|---|---|---|---|---|---|
| entrya | amino acid (X) | PG | % conversionb (% isolated yield)c | productd | a:b:ce |
| 1 | Arg (R) | Pbf | 67 | Boc-LYRAGLRAYR (16a) | 12:11:77 |
| 2 | Lys (K) | Boc | >99 (5) | Boc-LYRAGLRA YK (17a) | 18:06:66 |
| 3 | Asp (D) | tBu | >99 (7) | Boc-LYRAGLRAYD (18a) | 81:04:15 |
| 4 | His (H) | Trt | >99 | Boc-LYRAGLRAYH (19a) | 85:03:12 |
All reactions were conducted on 50 mg of resin in 600 μL of (5:1) DMF:HFIP, rt = 24 ± 1 °C.
Conversion based on integration of remaining peptide on activated linker after nucleophile cleavage in MS data.
Yield based on initial loading of 50 mg of MeDbz-Gly-Rink resin and represents 3 steps (SPPS, activation, and elongation).
Italicized letters indicate the amino acid residue is protected.
Relative ratios at 190 nm.
Figure 2.
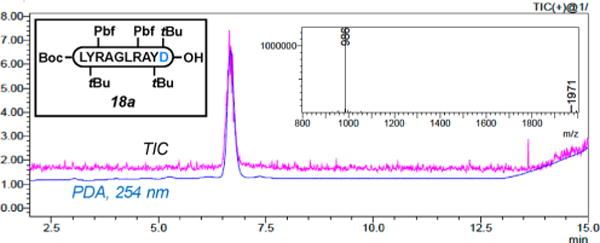
RP-HPLC trace at 254 nm and ESI-MS spectrum of pure Boc-LY(tBu)R(Pbf)AGLR(Pbf)AY(tBu)D(tBu)-OH (18a) accessed via Table 5, entry 3.
In summary, we developed conditions for the divergent elongation of peptides at the C terminus by displacement of the MeNbz group by various amino acids. This approach is surprisingly tolerant to steric effects, with good conversions observed for challenging amino acids such as Ile and Pro. Tolerable levels of epimerization (2–3%) and high conversions are observed upon elongation for extended times or in the presence of NaSPh. Alternatively, conversion can be sacrificed to preserve the stereointegrity of the C-terminal amino acid of the starting peptide by omitting NaSPh. Water serves as a helpful cosolvent, increasing conversion significantly, but leads to challenging separation of the elongated acid from the starting peptide acid. Use of isopropanol as the cosolvent minimizes this issue. Overall, this approach offers access to sequences with point mutations to the C terminus without the need to repeat the SPPS for every variation needed. These studies provide a framework of the scope and limitations of this approach to rapidly generating a library of C-terminally varied targets. Efforts to exploit the mild reactivity of N-acyl ureas for other synthetic challenges are ongoing in our laboratory.
EXPERIMENTAL SECTION
General Information
Solid-phase peptide synthesis was performed on a Biotage SP Wave Initiator+, and all peptides were synthesized on Rink Amide Resin (0.55 mmol/g from Chem-Impex Int’l. Inc.). 1H NMR spectra and 13C NMR spectra were recorded on a Varian MR-400, Agilent MR-400, Varian V-500, or an Agilent DD2-600 MHz instrument with a multinuclear broadband probe at ambient temperature unless otherwise stated. Chemical shifts are reported in parts per million relative to residual solvent peaks.19 All 13C spectra are recorded with complete proton decoupling. All HPLC analyses and purifications were performed on a custom reverse phase Shimadzu liquid chromatograph mass spectrometer (LCMS-2020), which can toggle between analytical and semipreparative columns. This instrument has a photodiode array (PDA) detector (D2 & W lamp) which collects a range of wavelengths in place of a traditional single channel UV detector. RP-HPLC-MS mobile phases (MeCN and H2O) contained 0.1% formic acid. Analytical HPLC was performed on a Phenomenex Kinetex C18 column (5 μm, 250 × 4.6 mm) and a Thermo Scientific Hypersil Gold C8 column (5 μm, 250 × 4.6 mm). Semi-preparative HPLC was conducted using a Thermo Scientific Hypersil Gold C8 column (5 μm, 150 × 10 mm). All yields refer to chromatographically and spectroscopically pure products. ESI-HRMS were recorded using a Waters LCT Premier Xe time-of-flight mass spectrometer.
Reagents and Materials
Unless otherwise specified, all commercially available reagents were used without further purification. Anhydrous Et2O, PhMe, n-hexane, MeCN, DMF, DMSO, and CH2Cl2 were purchased from Fisher; THF was purchased from EMD, and PhH was purchased from Sigma-Aldrich. These were passed through a commercial solvent purification system (two columns of alumina) and used without further drying. HPLC grade MeCN and H2O were purchased from Fisher. Triethylamine, diisopropylamine, pyridine, and Hünig’s base were distilled over CaH2 immediately prior to use. All L-amino acids and HATU were purchased from Chem-Impex Int’l. Inc. unless otherwise noted. 4-Mercaptophenylacetic acid and 4-nitrophenyl chloroformate were purchased from Alfa Aesar. 4-Fluoro-3-nitrobenzoic acid and Tris(2-carboxyethyl)phosphine·HCl were purchased from Oakwood Chemical. Methylamine (33 wt %) in absolute ethanol, guanidine·HCl, H-Pro-OH, and H-Ala-OH were purchased from Sigma-Aldrich. Sodium thiophenylate was purchased from Santa Cruz Biotechnology. Ethyl 3-mercaptopropionate was purchased from TCI America. H-Gly-OH was purchased from J. T. Baker (Fisher).
General Procedures
Semi-Automated Microwave Synthesis of Peptides
(1) Reactor vials [vial size (volume range allowed)]: 2 mL reactor vial (0.8–1.1 mL), 5 mL reactor vial (1.6–3.2 mL), and 10 mL reactor vial (3.2–6.4 mL). (2) Swell + Heat: DMF was added and vortexed at 1200 rpm for 20 min at 70 °C. The solvent was then removed over 1 min followed by two DMF washes (DMF was added and the suspension was vortexed at 600 rpm for 45 s, followed by the removal of solvent (over 2 min)). (3) Coupling: A solution of Fmoc-aa-OH (5 equiv), HATU (4.9 equiv), and DIEA (10 equiv) in DMF was made immediately prior to addition to the reaction vial containing the resin. Once the solution was added, the suspension was heated to 75 °C for 5 min with a vortex rate of 1200 rpm. After the reaction, the solution was removed (over 2 min), and the resin was rinsed with DMF 4 times (after addition of DMF, the suspension was agitated at a vortex rate of 1200 rpm for 1 min; solvent removal was at a rate of 2 min). (4) Fmoc removal (deprotection): The reactor vial was filled with 20% piperidine in DMF. The suspension was vortexed at 1200 rpm for 3 min at RT. The solvent was removed followed by addition of 20% piperidine in DMF. The suspension was vortexed again at 1200 rpm for 10 min at RT. The solvent was removed over 2 min, followed by 4 DMF washes (after addition of DMF, the suspension was agitated at a vortex rate of 1200 rpm for 1 min; solvent removal was at a rate of 2 min). (5) Wash: DMF was added to the reaction vial and agitated at a vortex rate of 1200 rpm for 1 min. The solvent was removed over 1 min, repeated for a total of 4 times. (6) Final wash: resin was rinsed with CH2Cl2 (3 × 1 mL) and MeOH (3 × 1 mL). (7) Drying for storage/weighing: After the final wash, the resin was placed on the lyophilizer overnight for drying.
MeDbz Activation
All activations were completed using a previously established protocol.6b Five equivalents of 4-nitrophenyl chloroformate was dissolved in 500 μL CH2Cl2, added to the preswelled resin, and agitated for 1 h. The solvent was removed, and the resin was rinsed with CH2Cl2 (3×). Then, 250 μL of 0.5–1 M DIEA/DMF was added and agitated for 15–30 min. The solvent was removed, and the resin was rinsed with DMF and CH2Cl2 (2× each).
Calculating Conversion of On Resin Reactions
After the nucleophilic cleavage was conducted, the resin was subjected to 95:2.5:2.5 TFA:TIPS:H2O to cleave the remaining MeNbz-Gly and unreacted peptide-MeNbz-Gly. The solvent was concentrated using a constant stream of air until a small amount of residue was left. Then, cold diethyl ether was added to the vial to crash out the crude peptide. After centrifugation, the ether was decanted off, and the solid was dissolved in 1:1 MeCN:H2O and lyophilized. The lyophilized solid was then dissolved in 20% MeCN/H2O and analyzed via RP-HPLC-MS using a gradient of 20–80% MeCN/H2O over 15 min. Conversion was calculated based on the ratio of H2N-peptide-MeNbz-Gly-NH2 to H2N-MeNbz-Gly-NH2.
Sodium Phosphate Buffer (with Guanidine).20
Na2HPO4 (56.6 mg, 0.4 mmol) was dissolved in 1 mL of water. Guanidine·HCl (1.146 g, 12 mmol) was added and dissolved and then diluted to 2 mL with water. The pH was brought to 7 with a solution of NaOH (5 M, 20 μL). MPAA (67 mg, 0.4 mmol) was then added along with 1 mL of water to fully dissolve it. The pH was then brought to 7.2 by adding NaOH (5 M, 135 μL).
Sodium Phosphate Buffer (Guanidine-Free).20
Na2HPO4 (56.6 mg, 0.4 mmol) was dissolved in 1 mL of water. MPAA (67 mg, 0.4 mmol) was added along with 1 mL of water to dissolve it. The pH was then brought to 7.2 by adding NaOH (5 M, 125 μL).
Boc-AW(Boc)AG-OH (10a-Gly)
Fluffy white solid (4.7 mg, 39% yield). 1H NMR (600 MHz, DMSO-d6) δ 8.12 (s, 1H), 7.98 (dd, J = 33.3, 7.4 Hz, 1H), 7.81 (d, J = 7.9 Hz, 1H), 7.67 (d, J = 8.2 Hz, 1H), 7.47 (s, 1H), 7.30 (q, J = 7.2, 6.7 Hz, 1H), 7.23 (q, J = 6.8 Hz, 1H), 6.96 (dd, J = 27.6, 7.2 Hz, 1H), 4.58 (dd, J = 14.0, 5.8 Hz, 1H), 4.36–4.25 (m, 1H), 3.91–3.84 (m, 1H), 3.79–3.68 (m, 3H), 3.11 (d, J = 11.0 Hz, 1H), 2.93 (dd, J = 14.9, 8.3 Hz, 1H), 1.60 (s, 9H), 1.34 (s, 8H), 1.21 (d, J = 7.0 Hz, 2H), 1.11 (dq, J = 16.0, 9.0, 8.1 Hz, 4H). 13C NMR (151 MHz, DMSO) δ 172.6, 172.3, 171.1, 170.5, 155.0, 149.1, 134.7, 130.4, 124.2, 122.4, 119.5, 116.1, 114.5, 83.4, 78.2, 52.4, 51.9, 50.1, 48.1, 40.8, 28.2, 27.7, 18.3, 18.1; HRMS (ESI+) m/z calcd for C29H41N5O9Na [M + Na]+ 626.2802, found 626.2786.
Boc-AW(Boc)AI-OH (10a-Ile)
Fluffy white solid (4.4 mg, 22% yield). 1H NMR (499 MHz, DMSO-d6) δ 8.21 (d, J = 7.1 Hz, 1H), 8.01 (d, J = 8.0 Hz, 1H), 7.88 (d, J = 8.2 Hz, 1H), 7.83 (d, J = 8.0 Hz, 1H), 7.65 (d, J = 7.6 Hz, 1H), 7.49 (s, 1H), 7.29 (t, J = 7.7 Hz, 1H), 7.21 (t, J = 7.5 Hz, 1H), 6.92 (d, J = 7.3 Hz, 1H), 4.66–4.53 (m, 1H), 4.37 (q, J = 6.9 Hz, 1H), 4.17 (dd, J = 8.2, 5.8 Hz, 1H), 3.92–3.83 (m, 1H), 3.13–3.02 (m, 1H), 2.97–2.87 (m, 1H), 1.76 (dq, J = 13.7, 7.7, 7.2 Hz, 1H), 1.60 (s, 9H), 1.33 (s, 8H), 1.20 (t, J = 6.9 Hz, 3H), 1.10 (d, J = 7.0 Hz, 4H), 0.84 (t, J = 7.8 Hz, 6H). 13C NMR (126 MHz, DMSO) δ 172.9, 172.6, 172.1, 170.6, 155.0, 149.1, 134.7, 130.4, 124.2, 122.4, 119.4, 116.2, 114.6, 83.4, 78.2, 56.3, 51.8, 50.2, 48.0, 36.5, 28.2, 27.7, 27.4, 24.7, 18.2, 15.6, 11.4; HRMS (ESI+) m/z calcd for C33H49N5O9Na [M + Na]+ 682.3428, found 682.3428.
Boc-AW(Boc)AA-OH (10a-Ala)
Fluffy white solid (5.5 mg, 29% yield). 1H NMR (600 MHz, DMSO-d6) δ 8.20 (s, 1H), 8.01 (d, J = 7.6 Hz, 1H), 7.80 (d, J = 7.8 Hz, 1H), 7.67 (s, 1H), 7.48 (s, 1H), 7.30 (t, J = 7.5 Hz, 1H), 7.26–7.18 (m, 1H), 6.94 (d, J = 6.4 Hz, 1H), 4.59 (s, 1H), 4.28 (d, J = 6.6 Hz, 1H), 4.16 (s, 1H), 3.90–3.85 (m, 1H), 3.08 (s, 1H), 2.92 (dd, J = 14.8, 8.3 Hz, 1H), 1.60 (s, 9H), 1.33 (s, 7H), 1.28–1.22 (m, 3H), 1.19 (d, J = 6.9 Hz, 3H), 1.10 (d, J = 7.3 Hz, 4H). 13C NMR (151 MHz, DMSO) δ 174.0, 172.6, 171.6, 170.4, 155.0, 149.1, 134.7, 130.4, 124.2, 122.4, 119.5, 116.2, 114.6, 83.4, 78.2, 51.9, 50.1, 47.9, 47.6, 28.2, 27.7, 18.2, 18.08, 17.3; HRMS (ESI+) m/z calcd for C30H43N5O9Na [M + Na]+ 640.2958, found 640.2940.
Boc-AW(Boc)AP-OH (10a-Pro)
Fluffy white solid (5.7 mg, 44% yield). 1H NMR (499 MHz, DMSO-d6) δ 12.43 (s, 1H), 8.23 (d, J = 7.1 Hz, 1H), 8.01 (d, J = 8.1 Hz, 1H), 7.81 (d, J = 8.0 Hz, 1H), 7.65 (d, J = 7.8 Hz, 1H), 7.48 (s, 1H), 7.30 (t, J = 7.2 Hz, 1H), 7.22 (t, J = 7.5 Hz, 1H), 6.93 (d, J = 7.4 Hz, 1H), 4.60 (d, J = 5.8 Hz, 1H), 4.53–4.43 (m, 1H), 4.23 (dd, J = 8.8, 4.1 Hz, 1H), 3.96–3.84 (m, 1H), 3.49 (d, J = 6.7 Hz, 2H), 3.11–2.98 (m, 1H), 2.91 (dd, J = 13.9, 7.6 Hz, 1H), 2.18–2.04 (m, 1H), 1.94–1.77 (m, 3H), 1.61 (s, 9H), 1.35 (s, 8H), 1.18 (d, J = 6.7 Hz, 3H), 1.11 (d, J = 7.0 Hz, 3H). 13C NMR (151 MHz, DMSO) δ 173.3, 172.5, 170.0, 155.0, 149.1, 134.6, 130.4, 124.2, 122.3, 119.4, 116.1, 114.6, 83.4, 78.2, 59.7, 58.6, 51.9, 50.0, 49.5, 46.3, 28.6, 28.2, 27.7, 24.5, 18.1, 16.9; HRMS (ESI+) m/z calcd for C32H45N5O9Na [M + Na]+ 666.3115, found 666.3115.
Supplementary Material
Acknowledgments
The authors thank the National Institutes of Health (Grant R00-GM097095), the National Science Foundation (CAREER Award supporting the training of undergraduate researcher R.E.S.: CHE-1554752), and Wayne State University for generous financial support (startup funds to J.L.S., Rumble-Schaap Fellowship to C.A.A.). The authors also acknowledge the NSF for an instrument grant funding the 600 MHz NMR used in this work (CHE-0840413).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.7b02655.
Detailed procedures for all reactions, RP-HPLC-MS spectra for all compounds, and 1H and 13C NMR spectra for reported compounds (PDF)
ORCID
Christine A. Arbour: 0000-0001-6056-296X
Jennifer L. Stockdill: 0000-0003-4238-6530
Notes
The authors declare no competing financial interest.
References
- 1.(a) Songster MF, Barany G. In: Methods in Enzymology, Solid-Phase Peptide Synthesis. Fields GB, editor. Vol. 289. Academic Press; Orlando: 1997. pp. 126–174. [DOI] [PubMed] [Google Scholar]; (b) Thieriet N, Guibé F, Albericio F. Org Lett. 2000;2:1815–1817. doi: 10.1021/ol0058341. [DOI] [PubMed] [Google Scholar]; (c) Kent SBH. J Pept Sci. 2015;21:136–138. doi: 10.1002/psc.2754. [DOI] [PubMed] [Google Scholar]; (d) Wu B, Wijma HJ, Song L, Rozeboom HJ, Poloni C, Tian Y, Arif MI, Nuijens T, Quaedflieg PJLM, Szymanski W, Feringa BL, Janssen DB. ACS Catal. 2016;6:5405–5414. [Google Scholar]
- 2.(a) Fujiwara Y, Akaji K, Kiso Y. Chem Pharm Bull. 1994;42:724–726. doi: 10.1248/cpb.42.724. [DOI] [PubMed] [Google Scholar]; (b) Henkel L, Zhang B, Bayer E. Liebigs Ann Recl. 1997;10:2161–2168. [Google Scholar]; (c) Angell YM, Alsina J, Albericio F, Barany G. J Pept Res. 2002;60:292–299. doi: 10.1034/j.1399-3011.2002.02838.x. [DOI] [PubMed] [Google Scholar]; (d) Hibino H, Miki Y, Nishiuchi Y. J Pept Sci. 2014;20:30–35. doi: 10.1002/psc.2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Carpino LA, Ionescu D, El-Faham A. J Org Chem. 1996;61:2460–2465. [Google Scholar]; (b) El-Faham A, Albericio F. Chem Rev. 2011;111:6557–6602. doi: 10.1021/cr100048w. [DOI] [PubMed] [Google Scholar]; (c) Zhang Y, Muthana SM, Farnsworth D, Ludek O, Adams K, Barchi JJ, Jr, Gildersleeve JC. J Am Chem Soc. 2012;134:6316–6325. doi: 10.1021/ja212188r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Popovic S, Bieräugel H, Detz RJ, Kluwer AM, Koole JAA, Streefkerk DE, Hiemstra H, van Maarseveen JH. Chem – Eur J. 2013;19:16934–16937. doi: 10.1002/chem.201303347. [DOI] [PubMed] [Google Scholar]
- 4.Camarero JA, Hackel BJ, De Yoreo JJ, Mitchell AR. J Org Chem. 2004;69:4145–4151. doi: 10.1021/jo040140h. [DOI] [PubMed] [Google Scholar]
- 5.Arbour CA, Saraha HY, McMillan TF, Stockdill JL. Chem Eur J. 2017;23:12484–12488. doi: 10.1002/chem.201703380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Blanco-Canosa JB, Dawson PE. Angew Chem, Int Ed. 2008;47:6851–6855. doi: 10.1002/anie.200705471. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Blanco-Canosa JB, Nardone B, Albericio F, Dawson PE. J Am Chem Soc. 2015;137:7197–7209. doi: 10.1021/jacs.5b03504. [DOI] [PubMed] [Google Scholar]; (b1) Mahto SK, Howard CJ, Shimko JC, Ottesen JJ. ChemBioChem. 2011;12:2488–2494. doi: 10.1002/cbic.201100472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Acosta GA, Royo M, de la Torre BG, Albericio F. Tetrahedron Lett. 2017;58:2788–2791. [Google Scholar]
- 8.(a) Siman P, Karthikean V, Nikolov M, Fischle W, Brik A. Angew Chem, Int Ed. 2013;52:8059–8063. doi: 10.1002/anie.201303844. [DOI] [PubMed] [Google Scholar]; (b) Zheng JS, Tang S, Qi YK, Wang ZP, Liu L. Nat Protoc. 2013;8:2483–2495. doi: 10.1038/nprot.2013.152. [DOI] [PubMed] [Google Scholar]; (c) Li H, Dong S. Sci China: Chem. 2017;60:201–213. [Google Scholar]
- 9.Arbour CA, Kondasinghe TD, Saraha HY, Vorlicek TL, Stockdill JL. Chem Sci. 2018;9:350–355. doi: 10.1039/c7sc03553e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pascal R, Chauvey D, Sola R. Tetrahedron Lett. 1994;35:6291–6294. [Google Scholar]
- 11.See the Supporting Information for details.
- 12.Dunn MS, Ross FJ, Read LS. J Biol Chem. 1933;103:579–595. [Google Scholar]
- 13.Gekko K, Ohmae E, Kameyama K, Takagi T. Biochim Biophys Acta, Protein Struct Mol Enzymol. 1998;1387:195–205. doi: 10.1016/s0167-4838(98)00121-6. [DOI] [PubMed] [Google Scholar]
- 14.Gallivan JP, Dougherty DA. Proc Natl Acad Sci U S A. 1999;96:9459–9464. doi: 10.1073/pnas.96.17.9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elashal HE, Sim YE, Raj M. Chem Sci. 2017;8:117–123. doi: 10.1039/c6sc02162j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kanzian T, Nigst TA, Maier A, Pichl S, Mayr H. Eur J Org Chem. 2009;2009:6379–6385. [Google Scholar]
- 17.The authors note that the pH was 8 in these reactions, which are run in a phosphine-free ligation buffer. This suggests that great care should be taken to maintain the pH near 7.2 during NCL reactions to avoid epimerization at the ligation site.
- 18.pKa of HFIP:; Apffel A, Chakel JA, Fischer S, Lichtenwalter K, Hancock WS. Anal Chem. 1997;69:1320–1325. doi: 10.1021/ac960916h. [DOI] [PubMed] [Google Scholar]; i-PrOH:; Reeve W, Erikson CM, Aluotto PF. Can J Chem. 1979;57:2747–2754. [Google Scholar]
- 19.Fulmer GR, Miller AJM, Sherden NH, Gottlieb HE, Nudelman A, Stoltz BM, Bercaw JE, Goldberg KI. Organometallics. 2010;29:2176–2179. [Google Scholar]
- 20.Modified from:; Aussedat B, Fasching B, Johnston E, Sane N, Nagorny P, Danishefsky SJ. J Am Chem Soc. 2012;134:3532–3541. doi: 10.1021/ja2111459. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.