

Abstract
The European Medicines Agency's (EMA) product‐specific bioequivalence guidelines outline harmonized regulatory requirements for studies to demonstrate bioequivalence for products that may have particular needs due to their pharmacokinetics, in addition to those outlined in general guidance. As such they are potentially very useful to the pharmaceutical industry in the development of generic medicinal products and to regulatory authorities for harmonized decision‐making. Since their introduction in 2013, EMA product‐specific bioequivalence guidelines continue to increase in number, and as of June 2017, encompass a number of different pharmacotherapeutic groups and pharmaceutical forms. This article further elucidates the processes involved for stakeholders and reviews the Agency's experience with the development of these guidelines, including the scientific issues witnessed with their advancement. A comparison with the United States Food and Drug Administration approach to similar guidelines is also provided.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ PSBGLs have been in existence in the EU since 2013. This is the first review into their development accompanied by an analysis on the issues seen with their advancement to date.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ This study is aimed at elucidating the reasons and processes behind the EMA's development of PSBGLs.
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE
☑ The latest European regulatory advances for generic drug development are highlighted. This study provides transparency into the development of PSBGLs in Europe, alongside a primary insight into their purpose in terms of harmonization of bioequivalence assessment.
HOW THIS MIGHT CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE
☑ This article outlines the process of PSBGL development which serves to consolidate requirements for the demonstration of bioequivalence for specific products with complex pharmacokinetics. Therefore, harmonized regulatory decision‐making and drug development for bioequivalence studies will be achieved through the continued advancement of these guidelines.
Facilitating access to medicines for the benefit of patients across Europe is a core activity of the European Medicines Agency (EMA) and its Scientific Committees. This is achieved in part through the publication of scientific guidelines that make explicit the regulator's views on requirements for marketing authorization in the European Union (EU), e.g., those for the investigation of bioequivalence.1
Two medicinal products (a generic and a reference) containing the same active substance are considered bioequivalent, if they are pharmaceutically equivalent or pharmaceutical alternatives, and if their bioavailabilities (rate and extent) after administration in the same molar dose lie within acceptable predefined limits. These limits are set to ensure comparable in vivo performance, i.e., similarity in terms of safety and efficacy.1
Any medicinal product containing a new active substance that falls within the scope of the legislation outlining the procedures for authorization of medicinal products in the EU2 has to undergo authorization via the centralized application procedure (e.g., treatment of cancer, diabetes, neurodegenerative disorder, acquired immune deficiency syndrome). For all medicinal products that undergo such a procedure, including generic medicinal products, a final scientific opinion on authorization is made by the EMA's Committee for Medicinal Products for Human Use (CHMP). Once granted by the European Commission, the centralized marketing authorization is valid in all EU Member States and Iceland, Norway, and Liechtenstein.
Generic medicinal products can, however, also be submitted for marketing authorization in the EU through a choice of three other application procedures in addition to the centralized route, i.e., via a decentralized or a national and/or mutual recognition route. For these procedures, the scientific assessment is concluded by the national competent authorities in the Member States, facilitated where appropriate by the Co‐ordination Group for Mutual Recognition and Decentralized Procedures – Human (CMDh). Where scientific issues arise in these procedures, the CMDh may refer it to the CHMP for their opinion. The authorization obtained through a national procedure is valid only in the Member State involved, whereas in a decentralized or mutual recognition procedure a number of Member States, as determined by the pharmaceutical company developing the generic medicinal product, may be involved. The pharmaceutical company may also consider factors such as the expected patient population and hence market‐size, procedure timelines, and costs in deciding which route to take.
Importantly, the same evidence requirements for bioequivalence underpin all these regulatory frameworks. The underlying legal basis for all specifies that for authorization as a generic a number of conditions need to be met, including the demonstration of bioequivalence with the reference medicinal product by appropriate studies.3 Such studies are always designed in a manner that takes account of a given generics clinical use, drug substance and product characteristics, formulation, site and mechanism of action, and the current scientific knowledge of the drug's solubility and permeability/absorption capacity so that its pharmacokinetics can best be characterized compared to the reference product.
The overarching EU requirements for the design, conduct, and evaluation of bioequivalence studies for immediate release dosage forms with systemic action are detailed in the CHMP Guideline on Investigation of Bioequivalence.1 Other related EU guidelines include those for modified release formulations and for the validation of bioanalytical methods, but this list is not exhaustive and individual guidelines will cross‐refer to others, as appropriate.4, 5 In addition, the CHMP Pharmacokinetics Working Party (PKWP) started in 2009 to publish positions addressing specific scientific issues for products in relation to the demonstration of bioequivalence that were not clearly addressed in existing guidance. This was done in a reactive, ad‐hoc manner, as issues arose in the context of the assessment of a regulatory submission.6 Delays may have resulted and, therefore, the EMA considered a more proactive and streamlined approach was needed to specify requirements for products that might encounter issues based on what is currently known of their pharmacokinetics. Consequently, the EMA developed individual guidance documents termed product‐specific bioequivalence guidelines (PSBGLs) that take account of the specific factors applicable to individual products.
Development process of PSBGL
There are several steps involved in the development process of a PSBGL, the first of which is product selection. For this, twice a year the CMDh select batches of five products based on high‐level criteria e.g., known complex pharmacokinetics, anticipated large number of generic applications, recurring requests for scientific advice (including at a national level). The second step is the drafting of individual guidelines. This is performed by the PKWP starting with a review of relevant information from the reference product. This information is used to determine the critical study design aspects for bioequivalence demonstration, which are outlined in Table 1 for oral immediate release products.
Table 1.
Requirements for oral immediate release bioequivalence demonstration
| Criterion | Detail |
|---|---|
| BCS classification | Outlining the documented class and any necessary background information on solubility or permeability, taking into consideration if a Biopharmaceutics Classification System (BCS) based biowaiver is possible. |
| Bioequivalence study design | Design features generally include the study design and population, number of studies required, conditions of administration (fasting/fed or both) and strength(s) to be investigated. |
| Analyte | Type of analyte to be measured and the medium to be investigated, also whether an enantioselective analytical method is required. |
| Bioequivalence assessment | The main pharmacokinetic variables required to demonstrate bioequivalence and the related acceptance criteria. |
The Biopharmaceutics Classification System (BCS) categorizes a drug substance in one of the four classes (I–IV), based on the intestinal permeability and solubility of the drug substance. In Europe, BCS Class I (highly permeable and highly soluble) and Class III (low permeable and highly soluble) substances are eligible for BCS‐class‐based biowaivers. Additional comparative in vitro dissolution data between the generic medicinal product and the reference medicinal product acts as a surrogate for in vivo bioequivalence.1 The relevant section of the PSBGLs on BCS states the known BCS class at the time of drafting the guideline. The remaining sections (bioequivalence study design, analyte, and bioequivalence assessment) subsequently detail the characteristics required to demonstrate bioequivalence if a BCS biowaiver is not feasible or not applied for. It is considered not feasible to address all noncritical study design aspects in the guidelines, e.g., the number of subjects and their characteristics, sampling timepoints, and duration of the studies, as that would result in a repetition of what is already mentioned in the guidelines.
The remaining steps (3–8) in the development of a PSBGL are presented in Figure 1. Of note, other working parties within the EMA can be consulted if a specific issue is identified, e.g., if a drug is considered to have a narrow therapeutic index (NTI), then the margins for the pivotal pharmacokinetic variables AUC (area under the concentration–time curve) and/or Cmax (the maximal observed plasma concentration) are tightened (e.g., published PSBGLs for everolimus, sirolimus, and tacrolimus with 90% confidence interval 90.00–111.11% for AUC). The EMA, therefore, considers it necessary to define whether a drug may be considered to have an NTI on a case‐by‐case basis requiring careful deliberation that includes the consultation of a CHMP Therapeutic Working Party. For drugs considered as highly variable drug products (e.g., entacapone), the Cmax can be widened to a maximum of 69.84–143.19%, to account for a large intrasubject variability.1
Figure 1.

Development lifecycle of PSBGL.
Before finalization, each guideline undergoes a 3‐month public consultation (step 5). In line with the general approach for the development of EU scientific guidelines, stakeholders, including marketing authorization holders for reference medicinal products, can comment on the proposed guidelines through public consultation.7 Each comment received during the consultation period is considered by the PKWP and is published in an overview document along with how it is addressed or otherwise in the final guideline (step 6). PSBGLs may therefore be considered as a scientific, transparent, and predictable support for industry and regulators alike for generic medicinal product development.
RESULTS
Since the introduction of the first batch of PSBGLs in 2013, 41 final guidelines have been published by the EMA, as of 30 June 2017. These are detailed in Figure 2.
Figure 2.
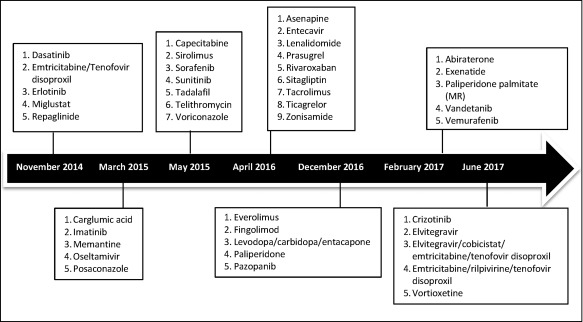
Dates of final adoption by CHMP for the PSBGL.
Pharmacotherapeutic groups
Seventeen different pharmacotherapeutic groups are represented in the PSBGLs to date, with antineoplastic agents being most frequent, followed by immunosuppressants and antivirals for systemic use (Figure 3).
Figure 3.
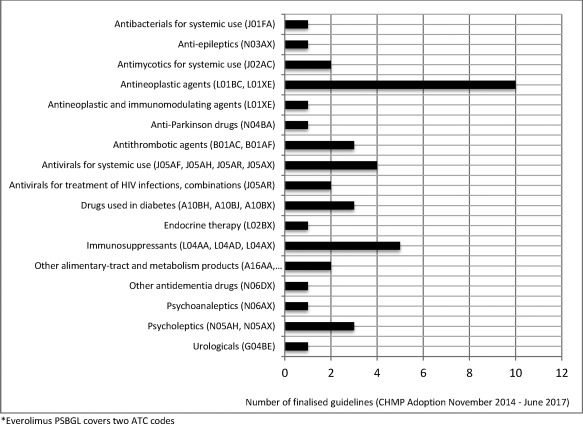
Categorization of all finalized PSBGL by pharmacotherapeutic group based on Anatomical Therapeutic Chemical (ATC) classification system.
Pharmaceutical forms
The initial focus of the PSBGLs was on immediate release formulations for oral use, as these were anticipated to be technically the most straightforward.8 Since their introduction in 2013, film‐coated tablets, coated tablets, immediate release tablets, and tablets make up 51% of the total number of finalized PSBGLs, with capsules making up an additional 18%. Subsequently, guidelines for suspensions for injections (6%), oral solutions, and oral suspensions (16%) have also been developed. The remainder of the finalized guidelines concern sublingual, dispersible tablets, oro‐dispersible, and prolonged‐release tablet formulations. Therefore, the selection of products is no longer focused on pharmaceutical form and future guidelines may also cover, e.g., liposomal formulations for intravenous administration.
Public consultation
Comments were received during public consultation for 24 of the 41 finalized guidelines. Most of the comments (88) were received from marketing authorization holders and pharmaceutical companies, with a limited number from contract research organizations (8), academics (2), and other national regulatory agencies (14). Comments have concerned all aspects of requirements including those relating to BCS classification, the number of studies to be conducted, study design (e.g., crossover, multiple dose), the strength to be used, whether studies should be conducted under fasting and/or fed conditions, and the main pharmacokinetic variables to be assessed. Stakeholders have also commented more generally on the structure, design, and use of the guideline and on the need for transparency in the prioritization of products for guideline development.9 All comments are taken into account, when deemed necessary by the PKWP, in finalizing the guidelines.
Overview with generic medicinal product development
As of May 2017, 14 of the 41 published guidelines have associated generic medicinal products on the EU market. These have been authorized via the four different routes (centralized or decentralized or national and/or mutual recognition) (Table 2).
Table 2.
PSBGL with associated generic medicinal products on the EU market
| Medicinal product | C | DCP | N | MR |
|---|---|---|---|---|
| Capecitabine film‐coated tablets 150 mg and 500 mg | Yes | Yes | Yes | Yes |
| Emtricitabine/tenofovir disoproxil film‐coated tablets 200 mg/245 mg | Yes | Yes | Yes | No |
| Entecavir film‐coated tablets 0.5 mg and 1 mg and oral solution 0.05 mg/ml | No | Yes | No | No |
| Erlotinib film‐coated tablets 25 mg, 100 mg and 150 mg | No | Yes | Yes | No |
| Imatinib hard capsules 50 mg and 100 mg and film‐coated tablets 100 mg and 400 mg | Yes | Yes | Yes | Yes |
| Levodopa/carbidopa/entacapone film‐coated tablets 200 mg/50 mg/200 mg, 175 mg/43.75 mg/200 mg, 150 mg/37.5 mg/200 mg, 125 mg/31.25 mg/200 mg, 100 mg/25 mg/200 mg, 75 mg/18.75 mg/200 mg and 50 mg/12.5 mg/200 mg | No | Yes | Yes | No |
| Memantine film‐coated tablets 5 mg, 10 mg, 15 mg and 20 mg and oral solution 5 mg | Yes | Yes | Yes | Yes |
| Miglustat hard capsules 100 mg | Yes | Yes | Yes | Yes |
| Oseltamivir hard capsules 30 mg, 45 mg and 75 mg and powder for oral suspension 6 mg/ml and 12 mg/ml | Yes | No | No | No |
| Repaglinide tablets 0.5 mg, 1 mg and 2 mg | Yes | Yes | Yes | Yes |
| Sitagliptin film‐coated tablets 25 mg, 50 mg and 100 mg | No | Yes | No | No |
| Tadalafil film‐coated tablets 2.5 mg, 5 mg, 10 mg and 20 mg | Yes | Yes | Yes | No |
| Voriconazole tablets 50 mg and 200 mg | Yes | Yes | Yes | Yes |
| Zonisamide hard capsules 25 mg, 50 mg and 100 mg and orodispersible tablets 25 mg, 50 mg, 100 mg and 300 mg | Yes | Yes | Yes | No |
C, Centralized; DCP, Decentralized; N, National; MR, Mutual Recognition.
DISCUSSION
Impact of product‐specific bioequivalence guidelines on generic authorization
The availability of PSBGLs serves to potentially facilitate the development of suitable generic medicinal products, which may be considered as beneficial for public health in terms of access to medicines. In addition, when the requirements of the PSBGLs are adhered to, appropriately designed bioequivalence studies can be conducted thereby reducing the number of inappropriate or “failed” bioequivalence studies conducted with unnecessary exposure of subjects to medicines.
The EMA anticipates that for pharmaceutical companies these guidelines will facilitate from the outset the design of study programs that meet the expectations of EU regulators across all submission routes, hence allowing for better predictability in terms of the assessment of the study design during the authorization process.8 How this might best be assessed in practice, however, currently poses a challenge, as the obvious option of just looking at the numbers of associated generic medicinal products in the marketplace, authorized by each of the four application procedures in the EU, is complicated by the fact that for some of the PSBGLs, the reference product is still under market exclusivity. It is therefore still too early to assess impact in this way. Of note, finalizing PSBGLs ahead of expiry of market exclusivity is intended to allow for the development of generics in a timely manner.
Also of note, the guidelines are not intended to just make generics available, but instead to harmonize standards and streamline the assessment of their development. Therefore, a more targeted approach would be to determine whether the PSBGL study design requisites were followed in the development of associated generics. This would require a crosscheck of how the conducted studies that are submitted in applications match the requirements detailed in the guidelines. Such an impact assessment is made more complex by the four options for licensing routes for generics in the EU. This is, however, a potential area for future research when more experience is gained with the use of these guidelines.
Harmonization across regulatory agencies
The different routes by which generics may be licensed in the EU mean that scientific consistency is paramount to create certainty for pharmaceutical companies seeking their development. By further harmonizing standards through the development of guidelines at the level of individual products, a significant proportion of this uncertainty is proactively addressed and the need for CMDh referrals to CHMP is expected to be reduced. Of note, the European harmonization of study designs for the demonstration of bioequivalence through the medium of the PSBGLs further extends internationally to Australia, where the Therapeutic Goods Administration (TGA) adopts and refers to the EMA PSBGLs for generic drug development in Australia.10 The EMA considers that such harmonization of accepted standards across multiple regulatory agencies at the individual product level is something that can potentially be taken forward by international regulatory agencies to supplement existing efforts at harmonization between agencies (e.g., at International Council for Harmonization (ICH) level involving EMA, US Food and Drug Administration (FDA), Pharmaceuticals and Medical Devices Agency (PMDA)).
Comparison to FDA guidance development
Similar to the EMA, the FDA product‐specific guidance includes recommendations on the type of studies to be conducted, including the study design, requirements for food intake, strength and subjects to be investigated, and the analytes to be measured. In contrast to the product‐specific EMA guidelines, FDA guidances also outline a waiver request of in vivo testing and the dissolution test method and sampling times required.11 Moreover, there can be differences between the FDA and EMA requirements for bioequivalence on aspects outlined in the individual guidelines, e.g., the EMA recommends for bioequivalence of fingolimod capsules, that one single dose study under fasting conditions should be carried out with a crossover or parallel study design. In contrast, the FDA recommends two single‐dose studies (one fasted and one fed) with both studies carried out in a two‐way crossover design. In addition, the FDA requires the parent and the active metabolite to be analytically measured, whereas the EMA only requires data on the parent compound.12, 13
As of December 2016, the FDA has more than 1,500 product‐specific guidances for generic drug development, while the EMA has 41 finalized.11, 14 Factors influencing this disparity in numbers may include: 1) the FDA having separate guidance for specific dosage forms (e.g., minocycline hydrochloride capsules v's tablet), whereas the EMA groups different dosage forms for similar active substances in the same PSBGL (e.g., vortioxetine hydrobromide immediate release tablets and vortioxetine lactate oral drops solution)11, 15; 2) the EMA beginning this initiative in late 2013 as compared to 2007 for the FDA; and 3) the FDA is mandated to develop product‐specific guidances in specific circumstances and in response to requests from sources including the pharmaceutical industry are also considered.11, 14, 16
The FDA revises their guidance based on postmarketing reports of adverse events with the reference medicinal product, literature reports, new study analysis, and reports of therapeutic in‐equivalence in approved Abbreviated New Drug Applications (ANDA).14 The EMA has had its first revision of a number of the PSBGLs in 2017 based on new scientific information or new product strengths being authorized. For products for which bioequivalence requirements have changed, public consultation is undertaken (e.g., prasugrel, tadalafil) before finalizing any revised guideline.17, 18 Similarly, the scientific information reflected in a PSBGL may supersede previously published information in the public domain, e.g., BCS classification at the time of marketing authorization of the reference product as stated in the European public assessment reports (EPAR) published on the EMA website.
Conclusion
Bioequivalence studies are the basis for the approval of generic medicinal products. The PSBGLs give specific advice on how bioequivalence should be demonstrated to support scientific consistency in their conduct and assessment. In essence, the pharmaceutical industry is provided with guidance on generating relevant data and potentially improving the number of successful and well‐conducted bioequivalence studies. Overall, the PSBGLs can allow for timely and specifically guided generic drug development with a standardized bioequivalence study design, which can thereby enable the availability of generic medicinal products.
METHODS
The EMA's electronic database of products (SIAMED and ARTICLE 57 Database) was searched to determine if generic medicinal products have been authorized for the respective product as named in the PSBGLs. The International Non‐proprietary Name (INN) used in the PSBGLs was used as the search term in the database. The searches were filtered to ensure only the dosage forms and corresponding strengths mentioned in the PSBGLs were accounted for.
All finalized guidelines and overview of comment documents available on the EMA website were reviewed to determine conclusions on comments received and to assist in the timeline determination. Internal EMA procedures were reviewed to illustrate the process development and to elucidate the issues accounted for during the guideline development.
CONFLICT OF INTEREST
The authors are employees of the European Medicines Agency or of a National Competent Authority. The authors declare no involvement, financial or otherwise, that might potentially bias their work.
FUNDING
This initiative received no specific grant from any funding agency in the public, commercial, or not‐for‐profit sectors.
AUTHOR CONTRIBUTIONS
J.O.S., K.B., M.B., T.S., and J.W. wrote the article; J.O.S and K.B. designed the research; J.O.S. performed the research; J.O.S., K.B., M.B., T.S., and J.W. analyzed the data.
DISCLAIMER
The views expressed in this article are the personal views of the authors and may not be understood or quoted as being made on behalf of or reflecting the position of the regulatory agencies or other organizations with which the authors are affiliated.
ACKNOWLEDGMENTS
The authors thank the observers of the PKWP who have supported the development of the PSBGL and also Efthymios Manolis, Quirine Fillekes, and Milton Bonelli for constructive comments. Pharmacokinetics Working Party: Ridha Belaiba (ANSM, France); Eva‐Gil Berglund (MPA, Sweden); Susan Cole (MHRA, UK); Alfredo García‐Arieta (AEMPS, Spain); Sotiris Michaleas (Ministry of Health Pharmaceutical Services, Cyprus); Janet Mifsud (Medicines Authority, Malta); Jan Neuhauser (AGES, Austria); Henrike Potthast (BfArM, Germany); Carolien Versantvoort (MEB, The Netherlands).
References
- 1. European Medicines Agency . Committee for Medicinal Products for Human Use (CHMP), Guideline on the Investigation of Bioequivalence CPMP/EWP/QWP/1401/98 Rev. 1/ Corr ** (2010). < http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/01/WC500070039.pdf > Accessed 12 June 2017.
- 2. European Parliament and the Council of the European Union . Annex to Regulation No 726/2004 of the European Parliament and of the Council of 31 March 2004. < http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2004:136:0001:0033:en:pdf >. Accessed 8 November 2017.
- 3. European Parliament and the Council of the European Union . Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001. < http://www.ema.europa.eu/docs/en_GB/document_library/Regulatory_and_procedural_guideline/2009/10/WC500004481.pdf >. Accessed 12 June 2017.
- 4. European Medicines Agency . Committee for Medicinal Products for Human Use (CHMP), Guideline on the pharmacokinetic and clinical evaluation of modified release dosage forms EMA/CPMP/EWP/280/96 Corr1 (2014). < http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2014/11/WC500177884.pdf >. Accessed 16 June 2017.
- 5. European Medicines Agency . Committee for Medicinal Products for Human Use (CHMP), Guideline on bioanalytical method validation EMEA/CHMP/EWP/192217/2009 Rev. 1 Corr. 2** (2011) < http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2011/08/WC500109686.pdf >. Accessed 16 June 2017.
- 6. European Medicines Agency . Pharmacokinetic Working Party (PKWP), Clinical pharmacology and pharmacokinetics: questions and answers. < http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/q_and_a/q_and_a_detail_000179.jsp&mid=WC0b01ac0580aff2ec >. Accessed 12 June 2017.
- 7. European Medicines Agency . Procedure for European Union Guidelines and related documents within the Pharmaceutical Legislative Framework. EMEA/P/24143/2004 Rev. 1 corr. < http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/10/WC500004011.pdf >. Accessed 16 June 2017.
- 8. European Medicines Agency . Committee for Medicinal Products for Human Use (CHMP). Concept paper on the development of product‐specific guidance on demonstration of bioequivalence EMA/CHMP/423137/2013. < http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/07/WC500147001.pdf >. Accessed 12 June 2017.
- 9. European Medicines Agency . Overview of comments received on “draft sorafenib product‐specific bioequivalence guidance” CHMP/PKWP/EMA/423707/2013. < http://www.ema.europa.eu/docs/en_GB/document_library/Overview_of_comments/2015/07/WC500189277.pdf >. Accessed 1 May 2017.
- 10. Australian Government Department of Health Therapeutic Goods Administation . < https://www.tga.gov.au/clinical-efficacy-and-safety-guidelines >. Accessed 16 June 2017.
- 11. U.S. Food and Drug Administration (FDA) . Product‐Specific Guidances for Generic Drug Development. < https://www.fda.gov/drugs/guidancecomplianceregulatoryinformation/guidances/ucm075207.htm >. Accessed 28 July 2017.
- 12. U.S. Food and Drug Administration (FDA) . Product‐Specific Guidances for Generic Drug Development. Draft Guidance on Fingolimod. < https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM270382.pdf >. Accessed 8 November 2017.
- 13. European Medicines Agency . Fingolimod capsules 0.5 mg product‐specific bioequivalence guidance EMA/CHMP/154812/2016. <http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2017/01/WC500219288.pdf >. Accessed 8 November 2017.
- 14. U.S. Food and Drug Administration (FDA) . Office of Generic Drugs. 2016 OGD Annual Report. < https://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/AbbreviatedNewDrugApplicationANDAGenerics/UCM542929.pdf >. Accessed 12 June 2017.
- 15. European Medicines Agency . Clinical pharmacology and pharmacokinetics. Product‐specific bioequivalence guidance. < http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000625.jsp&mid=WC0b01ac0580848f74 >. Accessed 28 July 2017.
- 16. U.S. Food and Drug Administration (FDA) . Lionberger R. GDUFA Regulatory Science Update. 9 February 2015. < https://www.fda.gov/downloads/forindustry/userfees/genericdruguserfees/ucm434325.pdf >. Accessed 12 June 2017.
- 17. European Medicines Agency . Prasugrel product‐specific bioequivalence guidance EMA/CHMP/158772/2016/Rev.1†. < http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_001654.jsp&mid=WC0b01ac0580848f74 >. Accessed 4 August 2017.
- 18. European Medicines Agency . Tadalafil product‐specific bioequivalence guidance EMA/CHMP/315234/2014/Rev.1†. < http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_001428.jsp&mid=WC0b01ac0580848f74 >. Accessed 4 August 2017.