

Abstract
Anabasum is a synthetic analog of Δ8‐tetrahydrocannabinol (THC)‐11‐oic acid that in preclinical models of experimental inflammation exerts potent anti‐inflammatory actions with minimal central nervous system (CNS) cannabimimetic activity. Here we used a novel model of acute inflammation driven by i.d. UV‐killed E. coli in healthy humans and found that anabasum (5 mg) exerted a potent anti‐inflammatory effect equivalent to that of prednisolone in terms of inhibiting neutrophil infiltration, the hallmark of acute inflammation. These effects arose from the inhibition of the neutrophil chemoattractant LTB4, while the inhibition of antiphagocytic prostanoids (PGE2, TxB2, and PGF2α) resulted in enhanced clearance of inflammatory stimulus from the injected site. Anabasum at the higher dose of 20 mg possessed the additional properties of triggering the biosynthesis of specialized pro‐resolving lipid mediators including LXA4, LXB4, RvD1, and RvD3. Collectively, we demonstrate for the first time a striking anti‐inflammatory and pro‐resolution effects of a synthetic analog of THC in healthy humans.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ While nonsteroidal anti‐inflammatory drugs, steroids, and biologics dampen the cardinal signs of inflammation, they can derail pro‐resolution pathways, thereby propagating the underlying disease. Moreover, they exert organ‐based sides effects, with some compromising antimicrobial host defense. Thus, more tolerable pharmaceutics with novel mechanisms of actions and possessing equal or greater efficacy to the current regimes is required.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ Is anabasum, a novel cannabinoid receptor 2 agonist, anti‐inflammatory and pro‐resolution in a novel proof‐of‐concept model of UV‐killed E. coli‐triggered dermal inflammation in humans?
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE
☑ Anabasum is strikingly immune‐suppressive in humans. It inhibited neutrophil infiltration, caused enhanced clearance of the injected antigen, and also triggered the biosynthesis of special pro‐resolution lipid mediators leading to pronounced abatement of the inflammatory response.
HOW THIS MIGHT CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE
☑ Despite being potently immune‐suppressive, anabasum paradoxically triggered bacterial clearance while also activating pro‐resolution pathways, a first in humans. This, alongside its favorable safety profile, suggests that anabasum is a promising new drug for the treatment of inflammation‐driven diseases.
The current therapeutic strategy for treating chronic inflammatory diseases is based largely on inhibiting the factors that drive acute inflammation and includes nonsteroidal anti‐inflammatory drugs,1 steroids,2 and anti‐TNFα therapies.3 Although these medicines ameliorate some disease symptoms, they do not bring about a “cure” and are ineffective in a significant subset of patients. Furthermore, they can hamper endogenous homeostatic systems resulting in serious adverse effects including predisposing to infection, gastrointestinal toxicity, as well as cardiac and metabolic imbalances. Thus, there is a significant need to identify more effective and safer therapeutics to treat chronic inflammatory diseases. One emerging approach is to harness the body's own inflammatory resolution process for therapeutic gain.4
Consequently, attention has turned to the other end of the inflammatory spectrum, resolution, to understand the endogenous processes involved in switching off inflammation. Our approach was to identify novel internal counter‐regulatory systems that naturally terminate inflammation in order to provide new pharmacological targets that can be activated to treat excessive inflammation through a pro‐resolution pathway.5 Over the past decade, inflammatory resolution has been extensively studied and conclusively demonstrated to be an active process with quantifiable indices and specific physiological requirements.6 For instance, it has become clear that for effective resolution to occur the inflammatory stimulus must first be eliminated.7, 8 Thereafter, prior to resolution being initiated, proinflammatory cytokines/chemokines are catabolized,9, 10 while infiltrated effector neutrophils die through apoptosis,11 which allows for their nonphlogistic efferocytosis by cells of the monocyte/macrophage lineage.12
Having recently characterized a human model of resolution of inflammation triggered by intradermal injection of UV‐killed E. coli (UVkEc) into the forearms of healthy volunteers,13 we sought to test a drug, from the published literature, that possessed anti‐inflammatory as well as potential pro‐resolution properties. Anabasum is a synthetic analog of Δ8‐tetrahydrocannabinol (THC)‐11‐oic acid, the terminal metabolite of Δ8‐THC. Unlike Δ9‐THC, the psychoactive principle of cannabis, it shows potent anti‐inflammatory action with minimal central nervous system (CNS) cannabimimetic activity as a result of its CB2 receptor selectivity and limited penetration of the blood–brain barrier.14 The CB2 receptor is preferentially expressed on activated immune cells where it is believed to mediate the immune modulatory properties of cannabis. For instance, the anti‐inflammatory effects of anabasum have been demonstrated in a number of animal models including an arachidonic acid‐induced rodent paw model, where it blocked swelling,15 and in a rat adjuvant‐induced arthritis, where it suppressed joint swelling and prevented the development of cartilage and bone erosion that is typical of this aggressive model of chronic inflammation.16 Evidence of pro‐resolution properties of anabasum have also been demonstrated using synovial fibroblasts, where it triggers the biosynthesis of 15‐deoxy‐Δ12, 14‐PGJ2,17 originally shown by us to be expressed during the resolution phase of acute rat pleuritis18 and in a mouse peritonitis model, where it reduced the infiltration of neutrophils and stimulated the biosynthesis of the specialized pro‐resolution lipid mediator (SPM) lipoxin A4.19 In addition, anabasum has been shown to trigger the biosynthesis of lipoxin A4 from lipopolysaccharide (LPS)‐stimulated human whole blood and synovial cells.19 Collectively, in addition to being anti‐inflammatory, there is emerging evidence that anabasum may also activate pro‐resolution pathways.
Anabasum is currently being evaluated in double‐blinded phase II clinical trials for the treatment of cystic fibrosis, diffuse cutaneous systemic sclerosis (“systemic sclerosis”), dermatomyositis, and systemic lupus erythematosus, four indications in which inflammation contributes to disease progression (http://www.clinicaltrial.gov identifier: NCT02465450, NCT02465437, NCT02466243). We investigated whether anabasum ameliorates inflammation in a human model of acute inflammation and sought to determine the mechanism by which it works. Prednisolone was used as a positive control because of its clinical potency in treating diseases driven by overexuberant inflammation, and because it increases efferocytosis of apoptotic bodies, a key determinant of inflammatory resolution.20, 21 Anabasum exhibited potent anti‐inflammatory activity, being equivalent to prednisolone as defined by the inhibition of neutrophil numbers at the site of bacterial injection. Anabasum also triggered the biosynthesis of SPMs and brought about the clearance of the injected bacteria as measured by endotoxin levels, thereby paving the way for effective resolution to occur. This is the first study of its kind to demonstrate in humans that anabasum is a unique anti‐inflammatory drug that acts on both the onset and resolution cascades of inflammation.
RESULTS
Volunteer recruitment and compliance
Twenty‐eight volunteers were screened, and five volunteers were excluded. The reasons for exclusion were following: out of reference range blood counts (n = 2), high body mass index (BMI) (n = 1), needle phobia (n = 1), and loss to follow‐up (n = 1). The remaining 23 volunteers were randomized into the four study groups. In the placebo group, two volunteers were excluded. The first volunteer was excluded on day 3 because his serum creatinine levels went above the reference range. Follow‐up investigation by a study clinician revealed that it was due to dehydration. The other volunteer was excluded during analysis, having missed the scheduled 4‐h blister timepoint. In the 5 mg b.i.d. anabasum group, one volunteer was excluded on day 3 as his platelet count was found to be below the reference range. Follow‐up investigation by a study clinician indicated the low platelet count was not related to the drug. In total, data were collected from 20 volunteers, and each group had five volunteers (Figure 2).
Figure 2.
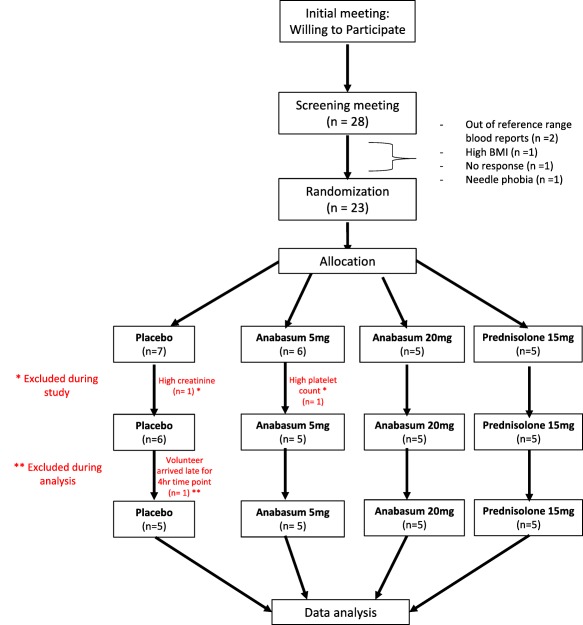
Consort diagram of the open label, randomized, parallel group, placebo‐controlled clinical study to test effect of 5 mg anabasum, 20 mg anabasum, and prednisolone on anti‐inflammatory and pro‐resolution biomarkers in UVkEc triggered self‐resolving dermal inflammation model.
Anabasum is anti‐inflammatory in a human model of acute inflammation
Injecting UVkEc into the dermis of healthy male volunteers elicited a robust local infiltration of neutrophils followed by phagocytosing monocyte‐derived macrophage populations with the response resolving within 48–72 h. Anabasum inhibited neutrophil numbers at the peak of inflammatory onset (4 h) by about 85% compared to the placebo control; this profound suppressive effect was seen with both 5 mg b.i.d. (P = 0.005) and 20 mg b.i.d. (P = 0.008) and was similar to the inhibition brought about by prednisolone (P = 0.01) (Figure 3 a). In contrast, neither doses of anabasum affected the numbers of macrophages (Figure 3 b), which increased from 4–10 h coincident with their established role in clearing dead/dying neutrophils and in affecting resolution. Interestingly, prednisolone exerted a trend towards reduction in macrophage numbers compared to placebo at 4 h that was significant at 10 h (P ≤ 0.05, Figure 3 b).
Figure 3.
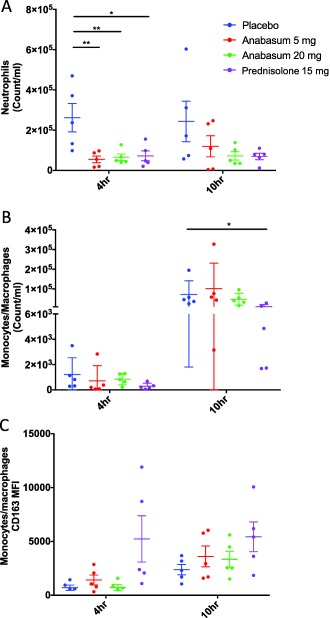
Anabasum inhibits neutrophil migration to the site of UVkEc triggered dermal inflammation. Healthy male volunteers were randomized to receive by oral route either placebo, 5 mg anabasum, 20 mg anabasum, twice daily, or 15 mg prednisolone once daily for 4 days. On the fourth day, acute inflammation was triggered by intradermal injection of 1.5 × 107 UV‐killed E. coli (UVkEc) suspended in 100 μL of saline in both forearms. Inflammatory exudate at the injection site was acquired into a suction blister raised after 4 h (onset phase) on one forearm and after 10 h (resolution phase) on the contralateral forearm. Cells in the exudate were phenoytped by multicolor flow cytometry. Neutrophil count/mL (a), monocyte/macrophage count/mL (b), and surface expression of CD163 on monocytes/macrophages at 4 h and 10 h is shown here (c). Data expressed as individual values with mean ± SD. n = 5/group. *P < 0.05, **< 0.01.
We have noted that as inflammation progresses towards resolution in this model, macrophages acquire increased expression of CD163 as a consequence of efferocytosing apoptotic neutrophils.13 Anabasum did not elevate CD163 level on infiltrating macrophages (Figure 3 c), while prednisolone exerted a trend towards an increase consistent with its role in enhancing efferocytosis.20, 21 These data provide some insight into the mechanisms of action of anabasum in this model, where inhibition of neutrophil infiltration is more obvious than enhancing neutrophil clearance/efferocytosis.
Unlike their inhibitory effect on neutrophil infiltration at the local site of UVkEc‐triggered acute inflammation, anabasum and prednisolone caused no significant changes in peripheral blood neutrophils or blood monocytes at 4 h or 10 h and was without effect on peripheral blood C‐reactive protein (CRP) levels (Supplemental Figure S1).
Effects of anabasum on inflammatory cytokines
Despite the striking effect of anabasum on neutrophil trafficking, it had only a modest effect on concentrations of typical cytokines/chemokines known to drive or inhibit acute inflammation including tumor necrosis factor alpha (TNFα) and interleukin (IL)‐1β (Figure 4 a–h). Most notable was a dose‐dependent, although statistically insignificant, reduction in concentrations of IL‐8 at the local site of inflammation. Indeed, prednisolone was equally unremarkable in this regard, exerting a significant effect on interferon gamma only at 4 h, P ≤ 0.05 (Figure 4 c).
Figure 4.
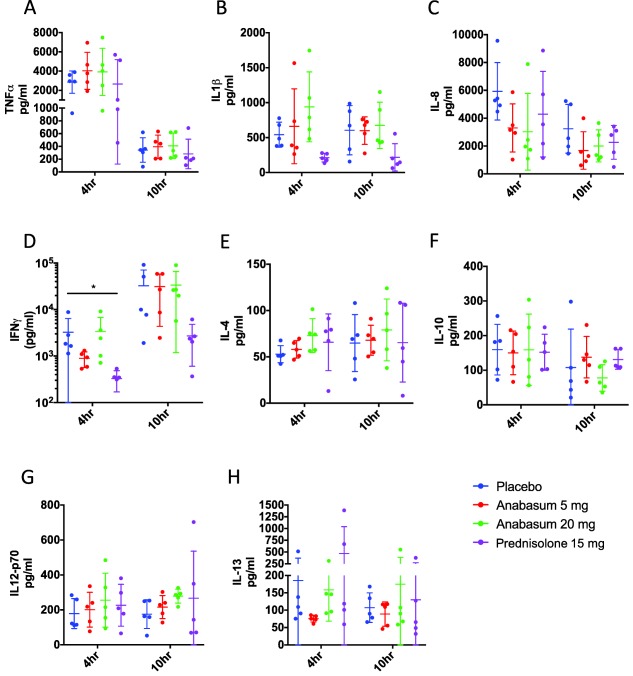
Effects of anabasum and prednisolone on cytokine and chemokine biosynthesis. Healthy male volunteers were randomized to receive by oral route either placebo, 5 mg anabasum, 20 mg anabasum, twice daily, or 15 mg prednisolone once daily for 4 days. On the fourth day, acute inflammation was triggered by intradermal injection of 1.5 × 107 UV‐killed E. coli (UVkEc) suspended in 100 μL of saline in both forearms. Inflammatory exudate at the injection site was acquired into a suction blister raised after 4 h (onset phase) on one forearm and after 10 h (resolution phase) on the contralateral forearm. Cytokines and chemokines in the inflammatory exudate were measured by multiplex enzyme‐linked immunosorbent assay (ELISA). The concentrations of cytokines and chemokines in the inflammatory exudate at 4 h and 10 h are shown. Data expressed as individual values with mean ± SD. n = 5/group. *P < 0.05, **< 0.01.
Effects of anabasum on lipid mediator biosynthesis
Targeted lipid mediator metabololipidomics was carried out on the inflammatory exudate obtained from the site of UVkEc‐induced inflammation at both 4 h and 10 h. The 4‐h timepoint represents the switch from onset to resolution as defined by the beginning of neutrophil clearance and cytokine/chemokine catabolism—established determinants of resolution. Indeed, in support of this we found a temporal switch in lipid mediator families from prostanoids to SPMs. At both 4 h and 10 h, anabasum‐treated volunteers gave a distinct lipid mediator profile characterized by a decrease in levels of proinflammatory prostaglandin PGE2, thromboxane TXB2, PGF2α and leukotriene LTB4 and a concomitant increase in levels of pro‐resolving mediators including LXA4, RvD1, and RvD3 (Figure S2).
The main effects of anabasum on SPM concentrations were at the highest dose of 20 mg b.i.d., where it elevated RvD1 and RvD3 at 4 h; an equivalent increase was also seen at 10 h. Prednisolone increased RvD1 at both timepoints but was without effect on RvD3. Anabasum caused a trend towards an increase in LXA4 and LXB4 at 20 mg b.i.d. only, being significant at 10 h for LXA4 (Figure 5 a–e).
Figure 5.
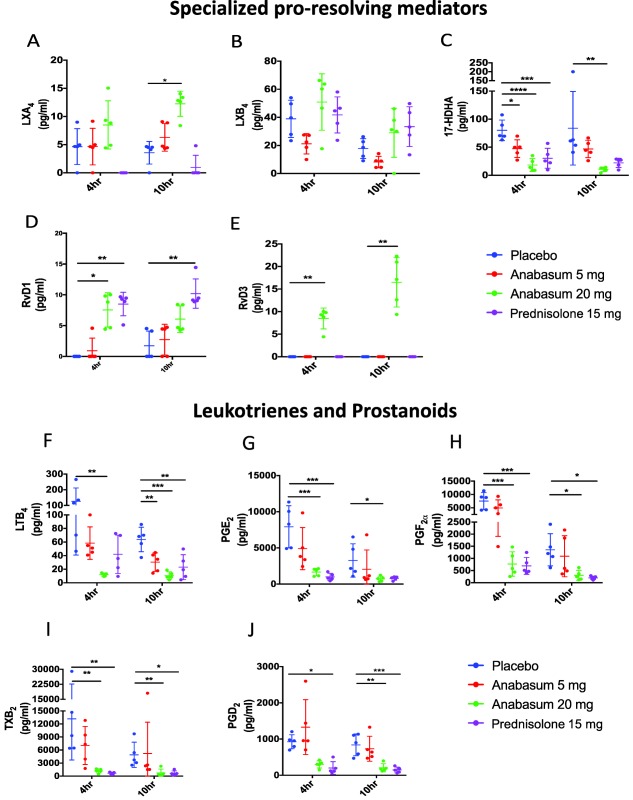
Anabasum differentially regulates specialized pro‐resolving mediators (SPMs) and prostanoids. Healthy male volunteers were randomized to receive by oral route either placebo, 5 mg anabasum, 20 mg anabasum, twice daily, or 15 mg prednisolone once daily for 4 days. On the fourth day, acute inflammation was triggered by intradermal injection of 1.5 × 107 UV‐killed E. coli (UVkEc) suspended in 100 μL of saline in both forearms. Inflammatory exudate at the injection site was acquired from the suction blister raised after 4 h (onset phase) on one forearm and after 10 h (resolution phase) on the contralateral forearm. Lipid mediators in the inflammatory exudate were analyzed by liquid chromatography mass spectrophotometry (LC‐MS) in Prof. Serhan's laboratory (Harvard University). The concentrations of SPMs and prostanoids in the inflammatory exudate at 4 h and 10 h are shown. Data expressed as individual values with mean ± SD. n = 5/group. *P < 0.05, **< 0.01, ***< 0.001, ****< 0.0001.
In contrast, anabasum brought about a dose‐dependent inhibition in concentrations of PGE2, TXB2, PGF2α, and LTB4 within the elicited blister fluid at both 4 h and 10 h, reaching statistical significance at the highest does of 20 mg b.i.d.. These suppressive effects on cyclooxygenase and lipoxygenase activity were seen also with prednisolone (Figure 5 f–j).
A post‐hoc analysis of these data revealed that when anabasum reduced LTB4 levels at 20 mg b.i.d. there was a corresponding significant reduction in neutrophils (Figure S3).
Anabasum increased vascular hyperemia around the site of inflammation
Using laser Doppler, we were able to determine changes in vascular hyperemia surrounding the site of inflammation. As shown previously, this was found to peak at 24 h and to decline thereafter up to 72 h.13 Of interest, this reduction in microvascular hyperemia always occurs once neutrophils have been cleared and macrophage populations have fully populated the inflamed site.13 Anabasum at 5 mg b.i.d. and 20 mg b.i.d. caused a dose‐dependent increase in microvascular hyperemia that nonetheless resolved at the same rate as in placebo controls (Figure 6 a) with representative laser Doppler and skin images shown in Figure 6 b. To ensure that this increase in redness at the site of inflammation following 20 mg b.i.d. anabasum was not indicative of enhanced localized or rebound inflammation, we elicited suction blister over these site in an additional group of volunteers at 24 h and 48 h and found that there were equivalent numbers of neutrophil in this group compared to placebo controls (Figure 6 c). Indeed, clinical examination of the inflamed site revealed that the increased vascular hyperemia trigger by anabasum was not edematous or painful and was therefore concluded not to be inflammatory in nature.
Figure 6.
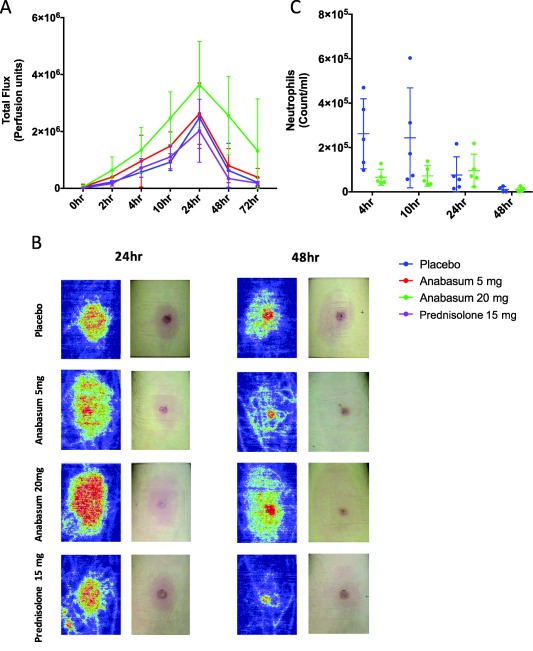
Anabasum alters vascular hyperemia at the site of UVkEc triggered self‐resolving dermal inflammation. Healthy male volunteers were randomized to receive by oral route either placebo, 5 mg anabasum, 20 mg anabasum, twice daily, or 15 mg prednisolone once daily for 4 days. On the fourth day, acute inflammation was triggered by intradermal injection of 1.5 × 107 UV‐killed E. coli (UVkEc) suspended in 100 μL of saline in both forearms. Vascular hyperemia at the injection site was assessed at specified intervals by a laser Doppler imager. The comparison of vascular hyperemia (mean ± SD) between the four treatment groups (a), the representative Doppler flux images at 24 h and 48 h in the four treatment groups (b) and the neutrophil count at 24 h and 48 h in the placebo and 20 mg anabasum (c) are shown. Data expressed as individual values with mean ± SD. n = 5/group.
Anabasum enhances bacterial clearance
One of the key determinants of inflammatory resolution is antigen clearance.4 Indeed, both 5 mg b.i.d. and 20 mg b.i.d. anabasum caused a profound reduction in levels of endotoxin within the elicited blister fluid, whereas prednisolone had no effect; in fact, there appeared to be an increase in endotoxin levels by prednisolone compared to placebo controls at 10 h (Figure 7 a). To understand how anabasum caused this reduction in endotoxin levels, we questioned whether there was a correlation between endogenous regulators of bacterial phagocytosis and levels of endotoxin. For instance, PGE2 is known to impair bacterial phagocytosis, while SPM has been shown to enhance bacterial clearance.22, 23, 24 Therefore, we carried out post‐hoc correlation analysis between levels of blister fluid endotoxin levels vs. blister fluid prostanoids and SPM levels (Figure S4). It transpired that there is a direct correlation between PGE2/endotoxin (P = 0.0167 and r = 1.0) as well as TxB2/endotoxin (P = 0.0167 and r = 1.0). No significant correlation was obtained with SPMs/endotoxin (Figure S4). To determine whether anabasum's inhibition of PGE2 and TxB2 at 4 h caused a reduction in blister endotoxin, we incubated whole blood with anabasum for 4 h followed by LPS for 15 min, after which time samples were spiked with FITC‐labeled E. coli bioparticles. However, anabasum did not directly enhance E. coli bioparticles uptake (Figure 7 b). Moreover, a combination of RvD3, LXB4, RvD1, and LXA4 were also without effect in this phagocytosis assay (Figure 7 c). We repeated these experiments, but stimulated with LPS for 4 h to represent conditions within the 4‐h blister site. Here, anabasum caused an increased uptake of E. coli bioparticles at 0.1 μM, which is equivalent to plasma levels of anabasum found in blood after dosing with 5 mg b.i.d. Importantly, this increased uptake was reversed by PGE2 and TXB2 at levels found in the blister fluid at 4 h post‐anabasum (Figure 7 d). These results support the hypothesis that reduced endotoxin levels brought about by anabasum at 4 h resulted from the inhibition of antiphagocytic prostanoids. An interesting observation was an inverse correlation between local blood flow as measured by laser Doppler vs. and blister endotoxin levels, where greater local blood flow was associated with lower endotoxin levels, (P = 0.0167 and r = 1) (Figure S4).
Figure 7.
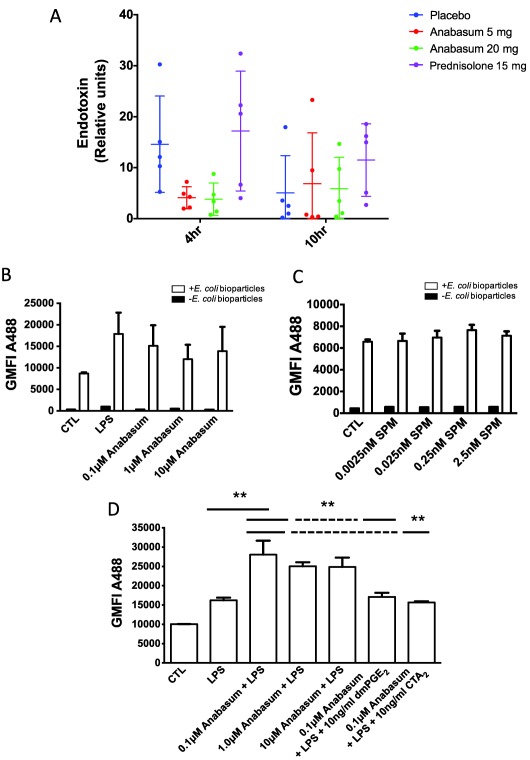
Anabasum actively clears UVkEc endotoxin from the site of inflammation. Healthy male volunteers were randomized to receive by oral route either placebo, 5 mg anabasum, 20 mg anabasum, twice daily, or 15 mg prednisolone once daily for 4 days. On the fourth day, acute inflammation was triggered by intradermal injection of 1.5 × 107 UV‐killed E. coli (UVkEc) suspended in 100 μL of saline in both forearms. Inflammatory exudate at the injection site was acquired into a suction blister raised after 4 h (onset phase) on one forearm and after 10 h (resolution phase) on the contralateral forearm. Endotoxin as a surrogate for inflammatory stimulus (UVkEc) was measured using the kinetic turbidimetric limulus ameobocyte lysate test. The relative concentration of endotoxin in the inflammatory exudate at 4 h and 10 h is shown (a). Whole blood from healthy volunteers was incubated with anabasum for 4 h before adding 0.1 μg/mL LPS for 15 min, after which time red blood cells were lysed and remaining leukocytes incubated with AF488‐labeled E. coli bioparticles and analyzed by flow cytometry to determine neutrophil phagocytosis (b). For comparison, lysed whole blood was stimulated with varying concentrations of SPMs (RvD3, LXB4, RvD1, and LXA4) for 15 min before incubating with AF488 E. coli bioparticles for 15 min and analyzed by flow cytometry (c). Experiments in panel b were repeated but incubated with 0.1 μg/mL LPS for 4 h with and without either 10 ng/mL dmPGE2 or a TxA2 receptor agonist (CTA2) (d); the idea was to replicate, as much as possible, the events occurring at the site of inflammation at 4 h where anabasum inhibited prophagocytic prostanoid biosynthesis. Data expressed as individual values with mean ± SD. n = 5/group. **P < 0.01.
DISCUSSION
In this study, we found that anabasum, a selective CB2 receptor agonist with minimal cannabimetic CNS activity, exerts potent anti‐inflammatory, anti‐infective, as well as a pro‐resolution effect in a model of self‐resolving acute inflammation in healthy human volunteers. The anti‐inflammatory properties of anabasum arose from the inhibition of the neutrophil chemoattractant LTB4, thereby reducing neutrophil infiltration and hence the potential for further neutrophil‐mediated tissue damage and propagation of inflammation. Moreover, in line with the classic definition of resolution, anabasum hastened antigen clearance, which alongside increased biosynthesis of SPMs at the 20 mg b.i.d. dose collectively resulted in effects that were superior to that of prednisolone. We propose, therefore, that anabasum is the first of a new generation of anti‐inflammatory/pro‐resolving drug that acts on diverse aspects of the immune response bringing back into homeostatic check multiple proinflammatory pathways that are activated by infection or injury. These unique pharmacological properties of anabasum appear to exert a more favorable effect on human pathophysiology compared to the properties of steroids, which as a consequence of their broad actions carry with them a burden of immunosuppressive side effects not seen with anabasum.
Treating neutrophils with anabasum for 4 h followed by LPS for 15 min did not alter their ability to engulf bacteria compared to untreated controls. If we extended the LPS‐stimulation time to 4 h then anabasum significantly enhanced the ability to neutrophils to phagocytose labeled E. coli bioparticles. These experimental conditions, which are akin to the 4 h blister setting and where anabasum significantly inhibits endotoxin levels, suggest that anabasum either triggered the release of prophagocytic factor/s or inhibited the synthesis of factor/s that impair bacterial uptake. While SPMs have been shown to enhance bacterial killing in various settings,25 anabasum did not alter levels of these lipid mediators at 4 h. However, it did dose‐dependently and significantly inhibit synthesis of both PGE2 and TxB2. Moreover, adding these prostanoids back to the ex vivo phagocytosis assays reversed anabasum's uptake of labeled bacteria. PGE2 has been shown to inhibit NADPH oxidase‐mediated bacterial killing via upregulation of cAMP26 and also inhibit FcγR‐mediated phagocytosis22, 27; indeed, we have shown that its elevation in cirrhosis patients underpins their susceptibility to infection that is typical of these individuals.28 Moreover, a post‐hoc analysis revealed a significant correlation between levels of blister fluid endotoxin at 4 h and PGE2 as well as TXB2 such that when levels of these prostanoids were inhibited, so also were levels of endotoxin. Thus, inhibiting prostanoid biosynthesis could explain how anabasum reduced endotoxin levels.
There was also a strong correlation between the inhibition of TxB2 by anabasum (5 mg b.i.d.) and endotoxin levels at 4 h in elicited blister fluid. While there is little evidence that thromboxane can directly affect neutrophil and/or macrophage bacterial phagocytosis, we did find that TxB2 blocked anabasum‐mediated uptake of E. coli bioparticles by neutrophils. However, there is also compelling evidence that TxA2‐mediated vasoconstriction may decrease the numbers of bacteria leaving the site of infection,29 supporting the hypothesis that this may be an important mechanism by which TxA2 may regulate immune responses to infection in vivo. Indeed, we found that levels of TxB2 after anabasum at 4 h (5 mg b.i.d.) correlated inversely (P = 0.0167 and r = 1) with local vascular blood flow as determined by laser Doppler. Thus, the mechanism by which anabasum lowered endotoxin levels could be due to increased phagocytosis and/or reverse migration to systemic circulation; however, the latter seems unlikely, as there was no spike in systemic CRP levels.
While there are many effective anti‐inflammatory treatment regimens on the market (nonsteroidal anti‐inflammatory drugs (NSAIDs), anticytokine therapies, steroids, etc.), switching off the underlying disease process is much more challenging. In terms of diseases driven by ongoing inflammation, the rationale is therefore to activate pro‐resolution pathways that might be pathologically silenced and/or activate functional pro‐resolution pathways to ameliorate persistent inflammation.4 Indeed, it may transpire that combining pro‐resolution with anti‐inflammatory technology may represent a superior strategy to existing approaches. Along these lines, it was originally feared that hastening resolution might lead to incomplete clearance of the original trigger, especially in the context of infections. Specialized pro‐resolution mediators, for instance, have to date been shown not just to augment resolution processes such as nonphlogistic monocyte recruitment and efferocytosis of neutrophils, but also antimicrobial properties.30 Indeed, while NSAIDs are clearly anti‐inflammatory, they also augment host defense. Therefore, it must be emphasized that dampening inflammation is not necessarily associated with compromised host defense.
While anabasum was effective at dampening neutrophil trafficking and hastening antigen clearance in a self‐resolving model of inflammation in healthy volunteers, its ability to exert similar actions on chronic inflammatory disease has been documented as well. In a rat adjuvant arthritis model, anabasum reduced inflammation, weight loss, pannus formation in the joints, and joint tissue injury over the entire 4‐week period of the study.31, 32 Likewise, in bleomycin‐induced lung and dermal inflammation and fibrosis models, anabasum, administered therapeutically after the start of bleomycin, inhibited organ fibrosis, collagen deposition, and myofibroblast differentiation.33, 34 In clinical studies anabasum treatment over a 12‐week period has been shown to reduce skin thickening in systemic sclerosis patients, and the rate and number of pulmonary exacerbations and inflammatory biomarkers in cystic fibrosis patients.35, 36
One interesting observation made during this study was that anabasum caused a dose‐dependent increase in local microvascular hyperemia around the injection site. While this suggested increased “inflammatory flare,” upon clinical examination the site was neither edematous nor painful. Moreover, we found no increase in neutrophils or macrophages in the affected tissue. In order to understand the mechanism underlying this observation, we measured levels of nitrite/nitrate in the blister fluid and found no evidence of increased NO biosynthesis (Figure S5). However, anabasum dose‐dependently inhibited TxB2, but was without effect on prostacyclin, measured as 6‐keto PGF1α (Figure S6). This inhibition of a potent vasoconstrictor while keeping levels of the vasodilator prostacyclin unaffected could result in a net increase in local microvascular hyperemia. Indeed, these data are very reminiscent of the beneficial effects of low‐dose aspirin on pathophysiology, namely, the inhibition of vasoconstrictive thromboxane while maintaining prostacyclin levels due to its preference for acetylating platelets in the portal circulation.
While both doses of anabasum inhibited inflammation, the mechanisms by which it exerted this potent effect may be dose‐specific. For instance, at 20 mg b.i.d. anabasum increased levels of LXA4, LXB4, RvD1, and RvD3, which is of interest because each one of these mediators carries potent pro‐resolving actions. For instance, LXA4 and RvD1 reduce human neutrophilic chemotaxis and infiltration,37 while RvD3 increases efferocytosis and phagocytosis, while, alongside aspirin‐triggered RvD3, are potent immunoresolvents.38, 39 In contrast, 5 mg b.i.d. anabasum had no effect on these SPMs. PGE2 and TxB2 inhibition occurred with both 5 mg b.i.d. and 20 mg b.i.d. doses. These findings suggest lower concentrations of anabasum or lower levels of CB2 receptor occupancy have metabolic effects on the arachidonic acid pathway, whereas higher concentrations are required to trigger SPM synthesis pathways. Thus, 20 mg b.i.d. may activate both effects of anabasum, whereas 5 mg b.i.d. only activates metabolic effects on the arachidonic acid pathway, at least in the setting of exposure limited to 4 days. These data suggest that 20 mg b.i.d. may be a more effective therapeutic dose of anabasum for treating chronic inflammatory diseases because it engages resolution pathways, with the acknowledgment that a longer duration of exposure to lower doses might also lead to activation of resolution pathways.
In summary, we used UV‐killed E. coli‐driven acute dermal inflammation to report the potent anti‐inflammatory properties of the CB2 receptor agonist, anabasum. Specifically, anabasum was as efficacious as prednisolone in inhibiting neutrophil infiltration and also accelerated the clearance of the injected bacteria, a key determinant for the resolution of inflammation. It transpires that its mode of action is, at least in part, through inhibition of chemoattractant LTB4, as well as through inhibition of antiphagocytic prostanoids, while maintaining levels of the vasodilator prostacyclin. Anabasum also triggered specialized pro‐resolving lipid mediators including lipoxins and resolvins. Therefore, anabasum exerts its effects on multiple aspects of the inflammatory and resolution cascade, resulting in both anti‐inflammatory and pro‐resolving properties in a manner that does not appear to compromise host defense or vascular homeostasis.
METHODS
Ethics statement
Study approval was obtained from UCL Institutional Ethics Committee (Project ID: 5051/002). All volunteers provided written informed consent.
Study design and volunteer recruitment
Healthy, young (18–45 years) male volunteers were recruited for this study. An open‐label, randomized, parallel group, placebo‐controlled study was conducted and the volunteers were randomized to one of four study groups: Group A: placebo, twice daily for 4 days, oral. Group B: 5 mg anabasum, twice daily for 4 days, oral. Group C: 20 mg anabasum, twice daily for 4 days, oral, Group D: 15 mg prednisolone, single morning dose for 4 days, oral (Figure 1). The study exclusion criteria are detailed in the Supplementary Methods.
Figure 1.
Study protocol.
Human model of self‐resolving acute dermal inflammation
The model was performed as described previously.13 Briefly, acute inflammation was triggered by intradermal injection of 1.5 × 107 UVkEc bacteria (Strain: NCTC 10418, Public Health England, UK) suspended in 100 μL of saline in both the forearm of healthy volunteers. Using negative pressure, a skin blister was created over the inflamed site to acquire inflammatory exudate at 4 h (onset phase) on one forearm and at 10 h (resolution phase) on the contralateral forearm after the UVkEc injection. Cells in the inflammatory exudate were processed immediately for flow cytometry (described in the Supplementary Methods) and the cell free exudate was frozen into aliquots at –80°C and was analyzed later for soluble mediators including cytokines and lipid mediators (described in the Supplementary Methods). Cell count in inflammatory exudate was obtained using an automatic cell counter (ADAM MC 2000, NanoEntek, South Korea). To assess local vascular hyperemia, the forearm was scanned by a laser Doppler imager (moorLDI, UK) at specific intervals after UVkEc injection.
Endotoxin measurement
Endotoxin in the blister exudate was measured using the Pyrogent‐5000 endotoxin measurement kit (Lonza, Basel, Switzerland) as per the manufacturer's instructions. For use with this kit, the cell‐free blister exudate was first treated, as recommended previously for human biological matrices,40 by diluting 1:20 in 0.1% tween 80 buffer (Sigma, St. Louis, MO) to a final volume of 100 μL, followed by heating at 70°C for 15 min.
Phagocytosis assay
One ml of ethylenediamine‐tetraacetic acid (EDTA) anticoagulated whole blood was mixed with 4 mL of RPMI. The mixture was then incubated with 0.1, 1, and 10 μM anabasum for 4 h at 37°C, and then 0.1 μg/mL of LPS was added for an additional 4 h with or without 10 ng/mL dmPGE2 and CTA2. One × 107 AF488 labeled E. coli (K‐12 strain) BioParticles were opsonized with autologous serum, and incubated with 500,000 lysed whole blood cells for 15 min at 37°C before stopping with 1% paraformaldehyde (PFA). Samples were quenched with 0.4 mg/mL Trypan blue before reading on an LSR II Fortessa flow cytometer. For SPM experiments, lysed whole blood was incubated for 15 min at 37°C with SPM (RvD3, LXB4, RvD1, and LXA4) before incubating with bioparticles as above.
Statistical analysis
GraphPad Prism software (v. 7, San Diego, CA) was used for statistical analysis. Data are presented as individual values with the mean ± standard deviation (SD) on a linear scale. For normally distributed data, differences between groups were tested for statistical significance by ordinary one‐way analysis of variance (ANOVA) followed by Bonferroni's test to correct for multiple comparisons. For non‐normal distributed data, differences were detected by the Kruskal–Wallis test (for unpaired data) followed by Dunn's test to correct for multiple comparisons. Spearman correlation was performed to test correlation between variables. P < 0.05 was taken as significant.
AUTHOR CONTRIBUTIONS
D.W.G. and M.P.M. wrote the article; D.W.G., M.T., B.W., M.P.M., R.M., A.A.M., and M.J.G. designed the research; M.P.M., P.C.N., F.B., A.H., J.N., and A.J.H. performed the research; M.P.M., P.C.N., F.B., A.H., J.N., A.J.H., C.N.S., and D.W.G. analyzed the data; P.C.N., C.N.S., and A.J.H. contributed new reagents/analytical tools.
CONFLICT OF INTEREST
The study was funded by Corbus Pharmaceuticals, Norwood, MA, USA in a form of investigator initiated mechanisitc study grant to DWG.
FUNDING
Corbus Pharmaceuticals, Norwood, MA, USA, in the form of investigator initiated mechanisitc study grant to D.W.G. (UCL Grant code: 534126)
Supporting information
Supporting Information
Figure S1: Effects of anabasum on peripheral blood neutrophils, monocytes and CRP
Figure S2: Anabasum (20 mg) decreases pro‐inflammatory prostanoids and increases specialized pro‐resolving mediators
Figure S3: Correlation between neutrophil count and soluble mediators of inflammation at 4hr and 10hr
Figure S4: Correlation between endotoxin and soluble mediators of inflammation at 4hr
Figure S5: Effect of Anabasum on nitric oxide synthesis at the site of UVkEc triggered acute resolving inflammation
Figure S6: Effect of Anabasum on generation of 6‐keto‐PGF1ɑ at the site of UVkEc triggered acute resolving inflammation
References
- 1. Vane, J.R. & Botting, R.M. Anti‐inflammatory drugs and their mechanism of action. Inflamm. Res. 47(suppl. 2), S78–87 (1998). [DOI] [PubMed] [Google Scholar]
- 2. Greaves, M.W. Anti‐inflammatory action of corticosteroids. Postgrad. Med. J. 52, 631–633 (1976). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Monaco, C. , Nanchahal, J. , Taylor, P. & Feldmann, M. Anti‐TNF therapy: past, present and future. Int. Immunol. 27, 55–62 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fullerton, J.N. & Gilroy, D.W. Resolution of inflammation: a new therapeutic frontier. Nat. Rev. Drug Discov. 15, 551–567 (2016). [DOI] [PubMed] [Google Scholar]
- 5. Serhan, C.N. et al Resolution of inflammation: state of the art, definitions and terms. FASEB J. 21, 325–332 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bannenberg, G.L. et al Molecular circuits of resolution: formation and actions of resolvins and protectins. J. Immunol. 174, 4345–4355 (2005). [DOI] [PubMed] [Google Scholar]
- 7. Morgenstern, D.E. , Gifford, M.A. , Li, L.L. , Doerschuk, C.M. & Dinauer, M.C. Absence of respiratory burst in X‐linked chronic granulomatous disease mice leads to abnormalities in both host defense and inflammatory response to Aspergillus fumigatus. J. Exp. Med. 185, 207–218 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Segal, A.W. & Peters, T.J. Characterisation of the enzyme defect in chronic granulomatous disease. Lancet 1, 1363–1365 (1976). [DOI] [PubMed] [Google Scholar]
- 9. Ariel, A. et al Apoptotic neutrophils and T cells sequester chemokines during immune response resolution through modulation of CCR5 expression. Nat. Immunol. 7, 1209–1216 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jamieson, T. et al The chemokine receptor D6 limits the inflammatory response in vivo. Nat. Immunol. 6, 403–411 (2005). [DOI] [PubMed] [Google Scholar]
- 11. Cohen, J.J. , Duke, R.C. , Fadok, V.A. & Sellins, K.S. Apoptosis and programmed cell death in immunity. Annu. Rev. Immunol. 10, 267–293 (1992). [DOI] [PubMed] [Google Scholar]
- 12. Savill, J. , Fadok, V. , Henson, P. & Haslett, C. Phagocyte recognition of cells undergoing apoptosis. Immunol. Today 14, 131–136 (1993). [DOI] [PubMed] [Google Scholar]
- 13. Motwani, M.P. et al Novel translational model of resolving inflammation triggered by UV‐killed E. coli. J. Pathol. Clin. Res. 2, 154–165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tepper, M.A. , Zurier, R.B. & Burstein, S.H. Ultrapure ajulemic acid has improved CB2 selectivity with reduced CB1 activity. Bioorg. Med. Chem. 22, 3245–3251 (2014). [DOI] [PubMed] [Google Scholar]
- 15. Burstein, S.H. et al Synthetic nonpsychotropic cannabinoids with potent anti‐inflammatory, analgesic, and leukocyte antiadhesion activities. J. Med. Chem. 35, 3135–3141 (1992). [DOI] [PubMed] [Google Scholar]
- 16. Zurier, R.B. et al Dimethylheptyl‐THC‐11 oic acid: a nonpsychoactive anti‐inflammatory agent with a cannabinoid template structure. Arthritis Rheum. 41, 163–170 (1998). [DOI] [PubMed] [Google Scholar]
- 17. Stebulis, J.A. et al Ajulemic acid, a synthetic cannabinoid acid, induces an anti‐inflammatory profile of eicosanoids in human synovial cells. Life Sci. 83, 666–670 (2008). [DOI] [PubMed] [Google Scholar]
- 18. Gilroy, D.W. et al Inducible cyclooxygenase may have anti‐inflammatory properties. Nat. Med. 5, 698–701 (1999). [DOI] [PubMed] [Google Scholar]
- 19. Zurier, R.B. et al Ajulemic acid, a synthetic cannabinoid, increases formation of the endogenous proresolving and anti‐inflammatory eicosanoid, lipoxin A4. FASEB J. 23, 1503–1509 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Heasman, S.J. et al Glucocorticoid‐mediated regulation of granulocyte apoptosis and macrophage phagocytosis of apoptotic cells: implications for the resolution of inflammation. J. Endocrinol. 178, 29–36 (2003). [DOI] [PubMed] [Google Scholar]
- 21. Giles, K.M. et al Glucocorticoid augmentation of macrophage capacity for phagocytosis of apoptotic cells is associated with reduced p130Cas expression, loss of paxillin/pyk2 phosphorylation, and high levels of active Rac. J. Immunol. 167, 976–986 (2001). [DOI] [PubMed] [Google Scholar]
- 22. Aronoff, D.M. , Canetti, C. & Peters‐Golden, M. Prostaglandin E2 inhibits alveolar macrophage phagocytosis through an E‐prostanoid 2 receptor‐mediated increase in intracellular cyclic AMP. J. Immunol. 173, 559–565 (2004). [DOI] [PubMed] [Google Scholar]
- 23. Spite, M. et al Resolvin D2 is a potent regulator of leukocytes and controls microbial sepsis. Nature 461, 1287–1291 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Abdulnour, R.E. et al Aspirin‐triggered resolvin D1 is produced during self‐resolving gram‐negative bacterial pneumonia and regulates host immune responses for the resolution of lung inflammation. Mucosal Immunol. 9, 1278–1287 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Basil, M.C. & Levy, B.D. Specialized pro‐resolving mediators: endogenous regulators of infection and inflammation. Nat. Rev. Immunol. 16, 51–67 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Serezani, C.H. , Ballinger, M.N. , Aronoff, D.M. & Peters‐Golden, M. Cyclic AMP: master regulator of innate immune cell function. Am. J. Respir. Cell Mol. Biol. 39, 127–132 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Medeiros, A.I. , Serezani, C.H. , Lee, S.P. & Peters‐Golden, M. Efferocytosis impairs pulmonary macrophage and lung antibacterial function via PGE2/EP2 signaling. J. Exp. Med. 206, 61–68 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. O'Brien, A.J. et al Immunosuppression in acutely decompensated cirrhosis is mediated by prostaglandin E2. Nat. Med. 20, 518–523 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tripp, C.S. , Needleman, P. & Unanue, E.R. Indomethacin in vivo increases the sensitivity to Listeria infection in mice. A possible role for macrophage thromboxane A2 synthesis. J. Clin. Invest. 79, 399–403 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chiang, N. et al Infection regulates pro‐resolving mediators that lower antibiotic requirements. Nature 484, 524–528 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zurier, R.B. et al Dimethylheptyl‐THC‐11 oic acid: a nonpsychoactive anti‐inflammatory agent with a cannabinoid template structure. Arthritis Rheum. 41, 163–170 (1998). [DOI] [PubMed] [Google Scholar]
- 32. Dajani, E.Z. et al 1',1'‐Dimethylheptyl‐Δ‐8‐tetrahydrocannabinol‐11‐oic acid: a novel, orally effective cannabinoid with analgesic and anti‐inflammatory properties. J. Pharmacol. Exp. Ther. 291, 31–38 (1999). [PubMed] [Google Scholar]
- 33. Lucattelli, M. et al Ajulemic acid exerts potent anti‐fibrotic effect during the fibrogenic phase of bleomycin lung. Respir. Res. 17, 49 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gonzalez, E.G. et al Synthetic cannabinoid ajulemic acid exerts potent antifibrotic effects in experimental models of systemic sclerosis. Ann. Rheum. Dis. 71, 1545–1551 (2012). [DOI] [PubMed] [Google Scholar]
- 35. Spiera, R.F. et al A phase 2 study of safety and efficacy of anabasum (JBT‐101), a cannabinoid receptor type 2 agonist, in diffuse cutaneous systemic sclerosis. In: ACR Meeting Abstracts [Internet]. <http://acrabstracts.org/abstract/a-phase-2-study-of-safety-and-efficacy-of-anabasum-jbt-101-a-cannabinoid-receptor-type-2-agonist-in-diffuse-cutaneous-systemic-sclerosis/>.
- 36. Chmiel, J.F. , Elborn, J.S. , Constantine, S. & White, B. WS01.5 A phase 2 study of the safety, pharmacokinetics, and efficacy of anabasum (JBT‐101) in cystic fibrosis (CF). J. Cyst. Fibros. 16, S2 (2017). 28986024 [Google Scholar]
- 37. Norling, L.V. et al Proresolving and cartilage‐protective actions of resolvin D1 in inflammatory arthritis. JCI Insight. 1, e85922 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dalli, J. et al Resolvin D3 and aspirin‐triggered resolvin D3 are potent immunoresolvents. Chem. Biol. 20, 188–201 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Norris, P.C. et al Resolvin D3 multi‐level proresolving actions are host protective during infection. Prostagland. Leukot. Essent. Fatty Acids (2016) [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wong, J. , Jeraj, H. , Vilar, E. , Viljoen, A. & Farrington, K. Endotoxin detection in end‐stage kidney disease. J. Clin. Pathol. 68, 73–78 (2015). [DOI] [PubMed] [Google Scholar]
- 41. English, J.T. , Norris, P.C. , Hodges, R.R. , Dartt, D.A. & Serhan, C.N. Identification and profiling of specialized pro‐resolving mediators in human tears by lipid mediator metabolomics. Prostagland. Leukot Essent. Fatty Acids 117, 17–27 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Figure S1: Effects of anabasum on peripheral blood neutrophils, monocytes and CRP
Figure S2: Anabasum (20 mg) decreases pro‐inflammatory prostanoids and increases specialized pro‐resolving mediators
Figure S3: Correlation between neutrophil count and soluble mediators of inflammation at 4hr and 10hr
Figure S4: Correlation between endotoxin and soluble mediators of inflammation at 4hr
Figure S5: Effect of Anabasum on nitric oxide synthesis at the site of UVkEc triggered acute resolving inflammation
Figure S6: Effect of Anabasum on generation of 6‐keto‐PGF1ɑ at the site of UVkEc triggered acute resolving inflammation