

Abstract
Aberrant activation of the classical complement pathway is the common underlying pathophysiology of orphan diseases such as bullous pemphigoid, antibody‐mediated rejection of organ transplants, cold agglutinin disease, and warm autoimmune hemolytic anemia. Therapeutic options for these complement‐mediated disorders are limited and sutimlimab, a humanized monoclonal antibody directed against complement factor C1s, may be potentially useful for inhibition of the classical complement pathway. A phase I, first‐in‐human, double‐blind, randomized, placebo‐controlled, dose‐escalation trial of single and multiple doses of sutimlimab or placebo was conducted in 64 volunteers to evaluate safety, tolerability, pharmacokinetic, and pharmacodynamic profiles. Single and multiple infusions of sutimlimab were well tolerated without any safety concerns. sutimlimab exhibited a steep concentration–effect relationship with a Hill coefficient of 2.4, and an IC90 of 15.5 μg/mL. This study establishes the foundation for using sutimlimab as a highly selective inhibitor of the classical complement pathway in different diseases.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Therapeutic options for complement‐mediated disorders, such as bullous pemphigoid, antibody‐mediated rejection of organ transplants, cold agglutinin disease, and warm autoimmune hemolytic anemia are limited and sutimlimab, a humanized monoclonal antibody directed against complement factor C1s, may be potentially useful for inhibition of the classical complement pathway.
WHAT QUESTION DID THIS STUDY ADDRESS?
This study assessed the safety, PK, and PD of sutimlimab after intravenously infused single and multiple ascending doses in healthy volunteers.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
Single and multiple infusions of sutimlimab, a novel, first‐in‐class, classical complement pathway‐specific inhibitor, had a good safety profile and predictable and consistent PK and PD.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
Targeting complement factor C1s by the humanized monoclonal antibody sutimlimab provides highly selective inhibition of the classical complement pathway. Subsequent testing of sutimlimab in safety and efficacy studies in the target population of patients with complement‐mediated disorders is promising.
sutimlimab is a humanized monoclonal IgG4 antibody directed against human complement factor C1s, a serine protease responsible for the enzymatic propagation of the classical complement pathway (CP).1 C1s is part of the C1 complex, a multimeric protein assembly containing the pattern recognition receptor C1q and the C1s‐activating serine protease, C1r. By binding C1s, sutimlimab specifically inhibits the classical pathway, preventing the enzymatic action of the C1 complex on its substrates (C4 and C2), thereby blocking formation of the pivotal enzyme, C3‐convertase. Importantly, sutimlimab preserves the function of both the alternative and lectin complement pathways to mediate humoral surveillance of pathogens. sutimlimab also leaves the opsonic function of C1q (i.e., phagocytic clearance of apoptotic cellular debris) intact.2 Target indications for sutimlimab are classical complement pathway‐mediated disorders including the hematologic disorders cold agglutinin disease (CAD) and warm autoimmune hemolytic anemia (WAIHA), the autoimmune blistering skin disease bullous pemphigoid (BP), and antibody‐mediated transplant rejection (AMR).3 Cold agglutinin disease and WAIHA are two clinical manifestations of autoimmune hemolytic anemia (AIHA), a rare autoimmune disease characterized by hemolytic anemia caused by autoantibodies against red blood cells.4 First‐line therapy for WAIHAs is glucocorticoids, but steroid‐dependency and refractory/relapsed cases may require second‐line treatment such as splenectomy or rituximab. While no therapeutic agent has been approved for the treatment of CAD, rituximab is considered first‐line therapy. However, ∼40% of patients do not respond to rituximab and only 21% of patients have a complete hematologic response, and sustained remissions are uncommon with rituximab therapy.5
Bullous pemphigoid is an autoimmune blistering disease characterized by autoantibody deposition at the dermal–epidermal junction and a subsequent activation of the complement system.6 First‐line treatment includes topical and systemic glucocorticoids, followed by immunosuppressive agents. Because BP occurs most commonly in patients aged over 70 years, much of the morbidity and mortality arises from comorbidities and drug‐related side effects.
Antibody‐mediated rejection is a serious medical condition and the major cause of late graft failure in kidney transplant recipients. Complement is activated by antibodies reactive to donor‐specific HLA expressed within the graft,7 leading to endothelial injury and microvascular inflammation.8, 9 Current standard of care includes plasmapheresis and intravenous (i.v.) immune globulin; however, it is unclear whether such therapeutic approaches are effective.7
Therapeutic options for these complement‐mediated disorders are limited and drugs selectively inhibiting the CP are currently not approved for these diseases. sutimlimab and its murine parental monoclonal antibody, TNT003, have demonstrated in vitro activity in preventing antibody‐mediated complement activation on the target cell or tissue in various preclinical disease models.1, 10, 11, 12 Therefore, sutimlimab may be potentially useful for treatment of patients with complement‐mediated disorders. This first‐in‐human trial evaluated the safety, tolerability, pharmacokinetic, and pharmacodynamic profiles of single and multiple doses of sutimlimab in healthy volunteers.
RESULTS
Healthy female and male subjects aged 19 to 59 years (median age part A, placebo: 31, sutimlimab: 32; part B, placebo: 30, sutimlimab: 27) were included between June 29, and December 10, 2015. Demographic and baseline characteristics are provided in Table 1. A flow diagram of the progress through the phase I trial is summarized in Figure 1. In part A, all randomized subjects received a single sutimlimab infusion, while four subjects were not randomized. One subject was not randomized for personal reasons, one subject was a screening failure due to withdrawal of informed consent by the subject, and two subjects were screened but held in reserve as backup. Subjects randomized in part B received four weekly doses of sutimlimab. One subject did not receive the second infusion in part B (60 mg/kg of sutimlimab) due to gastroenteritis. Because there was only minimal variation in multiple infusion pharmacokinetic (PK) data after adjustment (Table 2), the subject was not excluded from final analysis.
Table 1.
Summary statistics of demographic and baseline characteristics of part A (single dose study) and part B (multiple dose study)
| Single sutimlimab (i.v., mg/kg) | Multiple sutimlimab (i.v., mg/kg) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Active group | Placebo (N = 12) | 0.3 (N = 3) | 1 (N = 3) | 3 (N = 6) | 10 (N = 6) | 30 (N = 6) | 60 (N = 6) | 100 (N = 6) | Total (N = 36) | Placebo (N = 4) | 30 (N = 6) | 60 (N = 6) | Total (N = 12) |
| Placebo group | — | (N = 1) | (N = 1) | (N = 2) | (N = 2) | (N = 2) | (N = 2) | (N = 2) | — | — | (N = 2) | (N = 2) | — |
| Age (years) | |||||||||||||
| Median (range) | 31 (24–52) | 38 (28–45) | 33 (27–40) | 33 (28–59) | 33 (24–46) | 27 (22–48) | 34 (27–48) | 26 (19–37) | 32 (19–59) | 30 (23–35) | 26 (24–30) | 27 (22–41) | 27 (22–41) |
| Gender | |||||||||||||
| Female, n (%) | 3 (25) | 2 (66.7) | 1 (33.3) | 4 (66.7) | 3 (50) | 1 (16.7) | 2 (33.3) | 6 (100) | 19 (52.8) | 1 (25) | 2 (33.3) | 1 (16.7) | 3 (25) |
| Male, n (%) | 9 (75) | 1 (33.3) | 2 (66.7) | 2 (33.3) | 3 (50) | 5 (83.3) | 4 (66.7) | 0 | 17 (47.2) | 3 (75) | 4 (66.7) | 5 (83.3) | 9 (75) |
| Ethnic group; n (%) | |||||||||||||
| Caucasian | 11 (91.7) | 3 (100) | 3 (100) | 6 (100) | 5 (83.3) | 6 (100) | 5 (83.3) | 6 (100) | 34 (94.4) | 4 (100) | 6 (100) | 6 (100) | 12 (100) |
| Asian | 1 (8.3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| African | 0 | 0 | 0 | 0 | 1 (16.7) | 0 | 1 (16.7) | 0 | 2 (5.6) | 0 | 0 | 0 | 0 |
| Weight (kg) | |||||||||||||
| Mean (SD) | 74 (15) | 77 (16) | 63 (5) | 64 (7) | 73 (12) | 75(12) | 78 (13) | 55 (3) | 69 (13) | 73 (15) | 79 (4) | 78 (16) | 79 (11) |
| Height (cm) | |||||||||||||
| Mean (SD) | 176 (12) | 171 (15) | 172 (5) | 168 (12) | 173 (10) | 178 (4) | 180 (12) | 170 (3) | 174 (10) | 176 (8) | 180 (9) | 184 (8) | 182 (9) |
Figure 1.
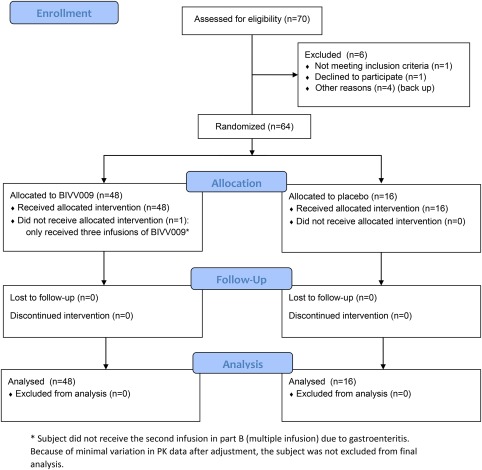
Flow diagram. [Color figure can be viewed at http://cpt-journal.com]
Table 2.
Summary statistics for sutimlimab serum pharmacokinetic parameters by treatment
| Cmax (µg/mL) | tmax (h) | AUC0‐∞ (µg·h/mL) | AUC0‐168 (µg·h/mL) | Half‐life (h) | |
|---|---|---|---|---|---|
| Part A | |||||
| 3 (N = 6) | 40 (28) | 2.5 (1, 8) | NC | 521 (57) | NC |
| 10 (N = 6) | 211 (21) | 4 (1, 8) | 7330 (32) | 6368 (28) | 19.1 (37.8) |
| 30 (N = 6) | 602 (14) | 2.5 (1, 24) | 55168 (27) | 48795 (22) | 53.3 (19.8) |
| 60 (N = 6) | 1464 (16) | 1.0 (1.0, 8.0) | 162835 (13) | 124342 (12) | 65.1 (31.7) |
| 100 (N = 6) | 2036 (14) | 1.0 (1.0, 23.5) | 335927 (8) | 198026 (11) | 132 (18.5) |
| Part B | |||||
| single sutimlimab (mg/kg, 60 min infusion) | |||||
| 30 (N = 6) | 653 (16) | 2.5 (1, 4) | 52161 (15) | 46604 (12) | 51.2 (15.9) |
| 60 (N = 6) | 1252 (17) | 6 (1, 8) | 150570 (13) | 111480 (14) | 87.8 (14.1) |
| multiple sutimlimab (mg/kg, 60 min infusion) | |||||
| 30 (N = 6) | 832 (18) | 6.8 (4, 8) | 99015 (30) | 74064 (19) | 67 (47) |
| 60 (N = 6) | 2073 (10) | 4 (1, 8) | 557551 (23) | 235612 (16) | 210 (13) |
| 60 (N = 5)a | 2079 (11) | 4 (1, 8) | 568045 (25) | 237821 (17) | 212 (14) |
Values are represented as mean (CV%) for each parameter, except for tmax, for which the values are the median and (minimum–maximum). AUC, area under the concentration–time curve; Cmax, maximum serum concentration; tmax, time to maximum serum concentration; NC, not calculated.
Adjusted for one subject who did not receive the second infusion due to gastroenteritis.
Safety
A total of 48 subjects received i.v. infusions of sutimlimab, with single doses as high as 100 mg/kg and with four repeated doses given weekly as high as 60 mg/kg. No drug‐related serious adverse events, premature withdrawals due to adverse events, or severe drug‐related adverse events were observed. In part A, 11 subjects (31%) receiving sutimlimab had a total of 18 adverse events and six subjects (50%) in the placebo group had a total of eight adverse events. In part B, eight subjects (67%) receiving sutimlimab had a total of 19 adverse events and all four subjects in the placebo group had a total of 10 adverse events. Headache (6/48 subjects = 13%) and nasopharyngitis (4/48 subjects = 8%) were the most common adverse events reported in subjects receiving sutimlimab. No clinically significant changes were observed in ECG recordings.
Due to the reported association of genetic deficiencies in classical pathway proteins and an increased risk of systemic lupus erythematosus (SLE) or circulating immune complex disease,13 an SLE screening panel including antinuclear antibodies (ANA), anti‐ds‐DNA, anti‐Sm, anti‐phospholipid, anti‐Ro antibodies, and circulating immune complexes was performed throughout the course of the trial. There were no subjects dosed with sutimlimab who changed from negative at baseline to positive at the end of the study for any analyte measured in the SLE screening panel (Supplemental Tables S1, S2). Circulating immune complexes were below the limit of quantification in all subjects before dosing with sutimlimab and at the end of the study. Furthermore, there were no incident cases of SLE or systemic bacterial infection.
Pharmacokinetics
In part A, i.v. infusions of sutimlimab over 60 minutes were associated with similar median time to reach maximum concentration (tmax) values across doses of 3–30 mg/kg, with values ranging from 2.5–4 hours (Table 2). Peak serum sutimlimab concentrations after doses of 60 and 100 mg/kg were observed with the end of infusion (median tmax 1 hour). Mean maximum concentration (Cmax) increased dose proportionally, ranging from 40–2036 μg/mL. Over the 10–100 mg/kg dose range, mean half‐life (t1/2) ranged from 19–132 hours and increased with higher dose levels. From 3–10 mg/kg and from 10–30 mg/kg, the mean sutimlimab exposure area under the curve (AUC0‐168) increased in a greater than dose proportional manner (12.2‐ and 7.7‐fold, respectively). On the other hand, from 30–60 mg/kg and from 60–100 mg/kg, the mean sutimlimab exposure (AUC0‐168) increased in a dose‐proportional manner (2.5‐ and 1.6‐fold, respectively). Serum sutimlimab concentrations were below the limit of quantification with the two lowest doses (0.3 and 1 mg/kg). Mean concentration–time profiles of sutimlimab are provided in Figure 2 a. Based on visual investigation of the concentration–time profiles of sutimlimab, nonlinear elimination was clearly apparent at concentrations lower than ∼100 μg/mL.
Figure 2.
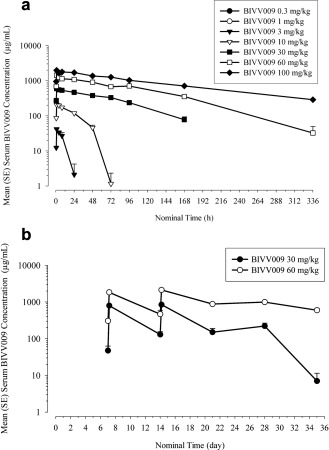
(a) Mean (+SE) serum concentrations of sutimlimab vs. time following a single 60‐minute i.v. infusion of sutimlimab in healthy volunteers (part A). (b) Mean (+SE) serum trough concentrations of sutimlimab vs. time following weekly 60‐minute i.v. infusions of sutimlimab (part B).
In part B, peak serum sutimlimab concentrations were observed at a median of 2.5 and 6 hours after a single infusion (30 or 60 mg/kg, respectively) and 6.8 and 4 hours (30 or 60 mg/kg, respectively) following multiple infusions (Table 2). For a 2‐fold increase in sutimlimab dose from 30–60 mg/kg, mean AUC0‐168 increased 2.4‐fold after a single infusion and 3.2‐fold after multiple infusions. Cmax increased 1.9‐fold after a single infusion and 2.5‐fold after multiple infusions. Mean t1/2 ranged from 51.2–87.8 hours (single 30 or 60 mg/kg dose, respectively) and from 67–212 hours (multiple 30 or 60 mg/kg doses, respectively). Predose concentrations increased over time, indicating some sutimlimab accumulation (Figure 2 b).
Figures of PK parameters vs. individual body weight from part A and part B (first dose) were plotted to explore any potential relationships (Figure 3). A negative slope was observed in the linear regression for AUClast, Cmax, and half‐life vs. weight plots, suggesting that body weight may play a role in sutimlimab exposure and disposition. However, a formal covariate analysis testing the effect of weight on exposure was not performed, since performing such an analysis using the limited number of subjects available in parts A and B (i.e., <50 subjects) would be deemed unreliable.14 These plots should thus be considered exploratory, and further investigation of the dose effect at extremes of the weight range may be warranted.
Figure 3.
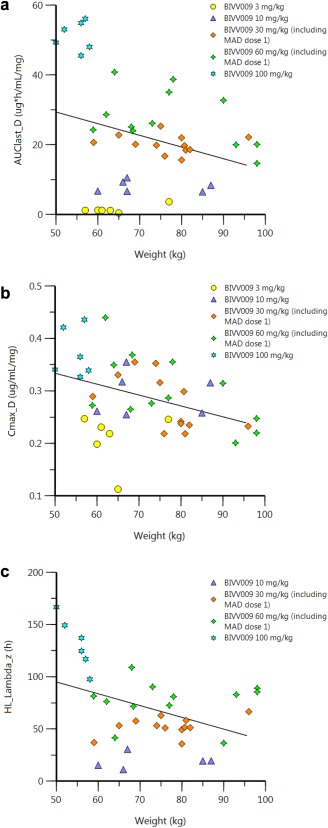
Individual body weight vs. PK parameters (parts A and B). AUC, area under the concentration–time curve; MAD, multiple ascending doses; Cmax, maximum serum concentration; HL, half‐life.
Pharmacodynamics
In part A, all subjects had normal CP activity at baseline (placebo 95% ± 10%; sutimlimab 97% ± 14%). A single infusion of 3, 10, 30, 60, and 100 mg/kg sutimlimab suppressed CP activity by >90% within 1 hour after the start of the infusion (Figure 4 a). The duration of suppression persisted dose‐proportionally from 8 hours (3 mg/kg) to up to 14 days (100 mg/kg). The CP activity returned to baseline levels within 2 weeks, whereas no reversal was observed in the 100 mg/kg dose group and only a partial return was observed in the 60 mg/kg group. No significant changes in CP activity were observed in subjects in the placebo group.
Figure 4.
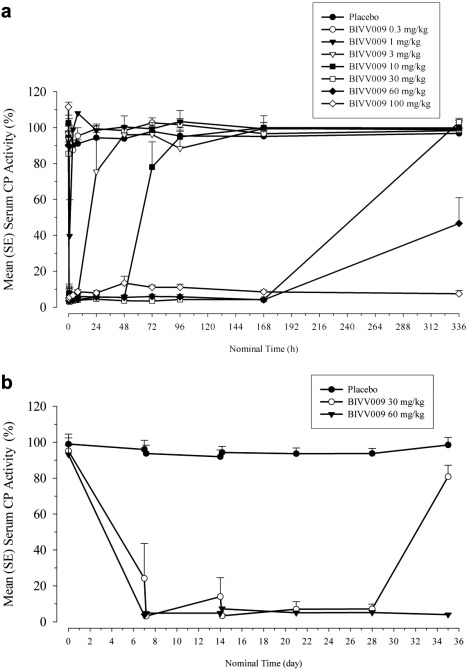
(a) Mean (+SE) serum classical complement pathway (CP) activity vs. time following a single 60‐minute i.v. infusion of sutimlimab in healthy volunteers (part A). (b) Mean (+SE) serum trough CP activity vs. time following single or weekly 60‐minute i.v. infusions of sutimlimab (part B).
In part B, all subjects had normal CP activity at baseline (placebo 99% ± 7%; sutimlimab 94% ± 18%). A single infusion of sutimlimab (30 and 60 mg/kg) profoundly suppressed CP activity by >95% in less than 1 hour after the start of the infusion. Multiple infusions suppressed CP activity by ≥90% in almost all individuals (Figure 4 b). Classical pathway activity did not completely return to baseline in the 30 mg/kg sutimlimab dose group 2 weeks after the last infusion (Figure 4 b). At the same time, mean CP activity was still <5% in the 60 mg/kg dose group. Among subjects receiving 30 mg/kg, one individual had predose activity levels of 102%, 67%, and 24% after 7, 14, and 21 days, respectively, indicating faster CP activity reversal than the other individuals. However, sutimlimab rapidly suppressed CP activity (<1 hour) by >95% compared with baseline in this subject and sustained suppression for at least 5 days (Figure S1).
Pharmacokinetic/pharmacodynamic (PD) correlations of sutimlimab and CP activity
Based on exploratory analyses, near‐maximal CP activity knockdown was observed for both dose levels in part B, much like the knockdown seen at similar dose levels in part A and B (day 0) and no delay was observed in CP activity (results retained on file). As a result, individual serum concentrations of sutimlimab and CP activity were time‐matched and the PK/PD relationship was modeled using an inhibitory Emax model, as shown in Figure S2. The relationship between serum sutimlimab concentrations and CP activity is presented in Figure 5, while parameters derived with the inhibitory Emax model are presented in Table S3. A steep concentration–effect relationship was observed for the knockdown of serum CP activity. Based on the inhibitory Emax model, the maximum percent inhibition (Imax) of sutimlimab on CP activity was 90.2%, with a 50% knockdown of CP activity (IC50) predicted at a sutimlimab concentration of 6.2 μg/mL. The sutimlimab concentration associated with a 90% reduction of CP activity (IC90) was 15.5 μg/mL. The very low IC50, combined with a Hill parameter of 2.4, suggests a very steep concentration–effect relationship and that sutimlimab concentrations above 100 μg/mL would be sufficient to maintain a near‐maximal knockdown of CP activity and avoid nonlinear PK.
Figure 5.
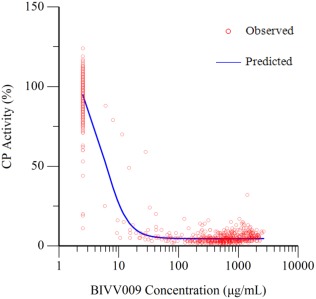
Relationship between concentrations of sutimlimab and CP activity: parts A and B.
Immunogenicity
In part A, eight subjects (17%), 10 subjects (21%), and 18 subjects (38%) had samples that tested positive in the screening assay for antidrug antibodies (ADA) at day 0, day 7, and at day 14, respectively. Confirmatory assays were performed and absolute ADA concentrations were determined for the subjects tested as reactive in the screening assays. One subject had a confirmed, reactive ADA result (42 ng/mL) from a sample collected on day 7, but also had a reactive but unconfirmed ADA result prior to the first dose of sutimlimab (predose sample). A sample from another subject collected on day 14 tested positive with a confirmed, reactive ADA result (28 ng/mL).
In part B, two subjects (13%), two subjects (13%), one subject (7%), and four subjects (27%) had positive ADA screening results from samples taken on day 0, day 7, day 21, and at day 35, respectively. Antidrug antibodies were positive in 1 of 4 subjects (25%) receiving placebo and in 4 of 12 subjects (33%) receiving sutimlimab, all of whom received 30 mg/kg sutimlimab. One of these subjects had a confirmed ADA result in their day 0 sample (0 ng/mL), before receiving the first dose of sutimlimab.
DISCUSSION
In this first‐in‐human trial, the primary objective was to characterize the safety/tolerability profile of sutimlimab, a classical complement pathway‐specific inhibitor, in healthy volunteers. Infusions of up to 100 mg/kg of sutimlimab to 48 subjects were well tolerated and no serious or severe adverse events occurred. Importantly, although complement inhibition increases the risk of invasive bacterial infection,15 no systemic bacterial infections were observed during the entire study period, presumably because the mode of action of sutimlimab leaves the alternative pathway and the lectin pathway function intact, and all participants were vaccinated against encapsulated bacterial pathogens prior to dosing. Another theoretical concern, extrapolated from rare human cases of deficiencies or mutations in classical pathway components including C1s, is the potential development of systemic autoimmune disease.16 In this study, we did not observe any incidental cases of SLE in subjects dosed with sutimlimab, consistent with the finding that levels of commonly associated serological markers of SLE were unchanged with sutimlimab treatment. Thus, short‐term pharmacologic C1s inhibition does not appear to increase the risk of developing SLE. Larger and longer clinical trials will be required to determine this risk under chronic treatment.
The Cmax of sutimlimab was dose proportional over the dose range from 10–100 mg/kg. However, the mean sutimlimab exposure (AUC) increased in a greater than dose‐proportional manner in the lower dose range (3–30 mg/kg) and in an approximately dose‐proportional manner at higher doses (60–100 mg/kg). Over the 10–100 mg/kg dose range, mean t1/2 ranged from 19–132 hours and increased with higher dose levels. Nonlinear elimination of sutimlimab was clearly apparent at concentrations lower than ∼100 μg/mL. This nonlinear behavior suggests potential target‐mediated elimination, which is usually apparent at lower concentrations, as previously reported for other monoclonal antibodies.17 As the linear component was more dominant at higher doses, prediction of therapeutic blood concentrations may be more accurate. Following repeated weekly dosing, mean total sutimlimab exposure increased in a slightly greater than dose‐proportional manner. The trough concentrations also increased with repeated dosing, suggesting some accumulation of sutimlimab in healthy volunteers. sutimlimab doses of 1 mg/kg or lower had little effect on CP activity, whereas complete inhibition, defined by CP activity <10%, was achieved in all subjects who received a sutimlimab dose of 3 mg/kg or higher. The complete inhibition of CP activity persisted <4 days, >7 days, and >14 days with low (3, 10 mg/kg), moderate (30, 60 mg/kg), and high (100 mg/kg) doses, respectively. As such, the duration of CP inhibition appears to be dose‐related. The data also show that sutimlimab inhibits CP activity at very low doses, although for a short time. An inhibitory Imax model confirmed a very steep concentration–effect relationship (Hill parameter of 2.4), while the sutimlimab concentration associated with a 90% reduction of CP activity (IC90) was 15.5 μg/mL. Multiple infusions of 60 mg/kg sutimlimab resulted in complete and consistent suppression of CP activity for more than 14 days after the last infusion. In contrast, CP activity was almost reversed in the 30 mg/kg dose group (81% of baseline) at the same time. Therefore, a 60 mg/kg or higher dose in combination with different dosing intervals may achieve long‐acting CP inhibition, possibly more suitable for clinical practice. The mean predose CP activity was slightly higher with weekly 30 mg/kg sutimlimab compared with the weekly 60 mg/kg dose. This was caused by considerably higher trough CP activity in one individual in the 30 mg/kg cohort, who also was reactive in the screening and confirmatory ADA assay at the beginning of part B before receiving sutimlimab. Although the preformed ADAs presumably had some effects on the individual's PK and PD profile, 30 mg/kg of sutimlimab was still sufficient to induce a rapid and sustained suppression of CP activity without any clinical manifestation. This may indicate that sutimlimab retains most of its inhibitory activity with no apparent side effects, even if ADAs are present. No other participants were confirmed to have ADAs in part B. In part A, two subjects developed ADAs in response to sutimlimab (10 mg/kg and 60 mg/kg dose group). While no specific tests have been performed to characterize the neutralizing activity of ADAs, both individuals had similar CP activity when compared to other subjects in their cohorts. Additionally, the measured ADA concentrations (42 ng/mL and 28 ng/mL) were ∼500–1,000‐fold lower than the drug levels required for a PD effect, suggesting no clinically relevant inhibition of sutimlimab functional activity. The ADA positive rate of 6% in part A and 8% in part B is comparable to the ADA frequency reported from other studies with therapeutic antibodies.18
The results presented here provide mandatory data on safety, PK, and PD of sutimlimab in healthy volunteers. It is important, however, to recognize that although patients with BP, AMR, WAIHA, or CAD may share the same underlying effector pathway, differences in safety, PK, and PD may exist. Since assessment of proof of concept should be made in the target population, we used an integrated protocol design that included an ongoing investigation of patients with BP, AMR, WAIHA, or CAD.3 Overall, sutimlimab had a good safety profile and predictable and consistent PK and PD in healthy volunteers. These results are the basis of subsequent testing of sutimlimab in safety and efficacy studies in the target population of patients with complement‐mediated disorders. Our data support this novel therapeutic principle to be successfully transferred into the clinical setting.
METHODS
We conducted a phase I, first‐in‐human, double‐blind, randomized, placebo‐controlled, dose‐escalation trial of sutimlimab at the Department of Clinical Pharmacology, at the Medical University of Vienna, Austria. The trial was approved by the Institutional Review Board at the Medical University of Vienna and registered at clinicaltrials.gov (NCT02502903) and was conducted in accordance with the Declaration of Helsinki and International Conference on Harmonization–Good Clinical Practice. While we used an integrated protocol design that included three subparts (part A, a single ascending dose trial in normal healthy volunteers, part B, a multiple ascending dose trial in normal healthy volunteers, and part C, an ongoing multiple dose trial in patients with a complement‐mediated disorder),3 this report is limited to part A and part B (normal healthy volunteers). All 64 volunteers gave written informed consent before enrollment. Randomization (open access randomization generator, http://www.randomization.com) was performed using sealed opaque envelopes, which were produced before the start of the study by staff members (secretaries) not otherwise involved the study.
Subjects
Healthy female and male subjects aged ≥18 years were eligible for enrollment. Subjects had to be either previously vaccinated against encapsulated bacterial pathogens (Neisseria meningitidis, Haemophilus influenzae, and Streptococcus pneumoniae) or willing to undergo vaccination (at least 14 days before study drug administration). Subjects with body weight >98 kg (for all subjects in all dose cohorts other than the 100 mg/kg dose cohort of part A, for which the body weight upper limit was 58 kg) were excluded.
Trial design
In part A, single doses of sutimlimab (0.3, 1, 3, 10, 30, 60, or 100 mg/kg) or placebo were infused i.v. over a period of ∼60 minutes in a 3:1 ratio (0.3 and 1 mg/kg: n = 4 per group; the remainder: n = 8 per group). The lowest dose of sutimlimab given was based on 1/300th of the No Observed Adverse Effect Level (NOAEL) in nonhuman primates (NHP), which was expected not to inhibit the classical pathway. In part B, four repeated doses of sutimlimab (30 or 60 mg/kg) or placebo were given once weekly to 16 subjects (8 per dose group) in a 3:1 ratio, with an additional observation period of 2 weeks. Infusion of sutimlimab or placebo followed a stepwise dose‐escalation procedure. Part B was initiated after confirming the tolerability and safety of the highest dose step of part A. In part A, safety (adverse events, vital signs), PK profiles and PD responses were monitored 1 hour before and 0.5, 1, 4, 8, and 24 hours after the start of the infusion and 2, 3, 4, 7, and 14 days after administration. In part B, safety, PK, and PD were monitored at the following timepoints: 1 hour before and 0.5, 1, 4, and 8 hours after the start of the first infusion, and daily on the next 4 days; 1 hour before and 4 hours after the start of the second and third infusion; 1 hour before and 0.5, 1, 4, and 8 hours after the start of the last/fourth infusion, and daily on the next 4 days; 1 week and 2 weeks after the last/fourth infusion.
Pharmacokinetics (PK)
PK variables of sutimlimab were determined from serum concentrations and included Cmax, t½, tmax, area under the concentration–time curve (AUC) up to the last timepoint with a concentration above the lower limit of quantification extrapolated to infinity (AUC∞), and up to the last timepoint with a concentration above the lower limit of quantification (AUClast). Serum concentrations of sutimlimab were measured with a validated immunoassay by a GLP‐certified laboratory (Vela Laboratories, Vienna, Austria).
Pharmacodynamics (PD)
Activity of the classical complement pathway was measured semiquantitatively in serum by the use of a commercially available enzyme immunoassay (Complement System Classical Pathway WIESLAB; Euro Diagnostica, Malmö, Sweden) as previously published.19
PK/PD
The relationship between concentrations of sutimlimab and CP activity was first explored to assess potential delay in response (i.e., hysteresis). Based on exploratory analyses, the concentration–effect relationship of sutimlimab and CP activity was explored using various PK/PD models. PK/PD modeling was performed with Phoenix NLME (v. 7, Certara, Princeton, NJ).
Safety
Safety measurements were assessed by adverse events, vital signs, physical examination, electrocardiogram, and laboratory tests. The severity of adverse events was graded using the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE, v. 4.03). An SLE screening panel consisting of ANA, anti‐ds‐DNA, anti‐Sm, anti‐phospholipid, anti‐Ro antibodies, and circulating immune complexes was performed with longitudinal samples taken throughout the course of the trial. Laboratory tests were determined in an accredited routine laboratory and consisted of hematology, blood chemistry, and coagulation tests, urinalysis, and immunoassays for SLE associated autoantibodies.
Immunogenicity
Anti‐sutimlimab antibodies (antidrug antibodies (ADAs)) were analyzed in a two‐step approach (screening assay, followed by a confirmatory assay and absolute ADA concentration determination) with validated immunoassays by a GLP‐certified laboratory (Vela Laboratories). ADAs were measured in serum before infusion and after 7 and 14 days in part A, and before infusion and after 7, 21, and 35 days in part B. Specific tests to characterize neutralizing activity of ADAs were not performed.
Sample size and statistical analysis
No formal sample size calculation was conducted, but the clinical trial followed the usual dose escalation design for first‐in‐human trials. The first two cohorts included only three subjects because we assumed only minimal PK/PD readouts could be obtained in those groups. No inferential statistical testing was performed because no formal hypothesis was tested. Data are presented descriptively, as appropriate.
FUNDING
This work was supported by Bioverativ Therapeutics Inc., South San Francisco, CA, USA.
CONFLICT OF INTEREST
The authors declare no competing interests for this work.
AUTHOR CONTRIBUTIONS
J.B., M.B., C.C., J.M., D.N., J.C.G., S.P., and B.J. wrote the article; J.C.G. and B.J. designed the research; J.B., C.S., M.S., C.F., and B.J. performed the research; J.B., M.B., C.C., J.M., D.N., J.C.G., S.P., and B.J. analyzed the data.
Supporting information
References
- 1. Shi, J. et al TNT009, a classical complement pathway specific inhibitor, prevents complement dependent hemolysis induced by cold agglutinin disease patient autoantibodies. Blood 122, 42 (2013). [Google Scholar]
- 2. Colonna, L. , Parry, G.C. , Panicker, S. & Elkon, K.B. Uncoupling complement C1s activation from C1q binding in apoptotic cell phagocytosis and immunosuppressive capacity. Clin. Immunol. 163, 84–90 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Derhaschnig, U. , Gilbert, J. , Jager, U. , Bohmig, G. , Stingl, G. & Jilma, B. Combined integrated protocol/basket trial design for a first‐in‐human trial. Orphanet J. Rare Dis. 11, 134 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Meulenbroek, E.M. , Wouters, D. & Zeerleder, S.S. Lyse or not to lyse: clinical significance of red blood cell autoantibodies. Blood Rev. 29, 369–376 (2015). [DOI] [PubMed] [Google Scholar]
- 5. Reynaud, Q. et al Efficacy and safety of rituximab in auto‐immune hemolytic anemia: a meta‐analysis of 21 studies. Autoimmun. Rev. 14, 304–313 (2015). [DOI] [PubMed] [Google Scholar]
- 6. Liu, Z. et al The role of complement in experimental bullous pemphigoid. J. Clin. Invest. 95, 1539–1544 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Stites, E. , Le Quintrec, M. & Thurman, J.M. The complement system and antibody‐mediated transplant rejection. J. Immunol. 195, 5525–5531 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Feucht, H.E. et al Vascular deposition of complement‐split products in kidney allografts with cell‐mediated rejection. Clin. Exp. Immunol. 86, 464–470 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Abe, T. et al Anti‐huCD20 antibody therapy for antibody‐mediated rejection of renal allografts in a mouse model. Am. J. Transplant. 15, 1192–1204 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Thomas, K.A. et al An anti‐C1s monoclonal, TNT003, inhibits complement activation induced by antibodies against HLA. Am. J. Transplant. 15, 2037–2049 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shi, J. et al TNT003, an inhibitor of the serine protease C1s, prevents complement activation induced by cold agglutinins. Blood 123, 4015–4022 (2014). [DOI] [PubMed] [Google Scholar]
- 12. Kasprick, A. et al The anti‐C1s antibody TNT003 prevents complement activation in the skin induced by bullous pemphigoid autoantibodies. J Invest Dermatol 138, 458–461 (2018). [DOI] [PubMed] [Google Scholar]
- 13. Walport, M.J. Complement and systemic lupus erythematosus. Arthritis Res. 4 Suppl 3, S279–293 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bonate, P.L. Pharmacokinetic‐Pharmacodynamic Modeling and Simulation. (New York, Springer, 2006). [Google Scholar]
- 15. Dmytrijuk, A. , Robie‐Suh, K. , Cohen, M.H. , Rieves, D. , Weiss, K. & Pazdur, R. FDA report: eculizumab (Soliris) for the treatment of patients with paroxysmal nocturnal hemoglobinuria. Oncologist 13, 993–1000 (2008). [DOI] [PubMed] [Google Scholar]
- 16. Lintner, K.E. et al Early components of the complement classical activation pathway in human systemic autoimmune diseases. Front. Immunol. 7, 36 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mould, D.R. & Sweeney, K.R. The pharmacokinetics and pharmacodynamics of monoclonal antibodies—mechanistic modeling applied to drug development. Curr. Opin. Drug Discov. Dev. 10, 84–96 (2007). [PubMed] [Google Scholar]
- 18. Baker, M.P. , Reynolds, H.M. , Lumicisi, B. & Bryson, C.J. Immunogenicity of protein therapeutics: the key causes, consequences and challenges. Self Nonself 1, 314–322 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Roos, A. & Wieslander, J. Evaluation of complement function by ELISA. Methods Mol. Biol. 1100, 11–23 (2014). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials