

Abstract
The spatial localization of charge carriers to promote the formation of bound excitons and concomitantly enhance radiative recombination has long been a goal for luminescent semiconductors. Zero‐dimensional materials structurally impose carrier localization and result in the formation of localized Frenkel excitons. Now the fully inorganic, perovskite‐derived zero‐dimensional SnII material Cs4SnBr6 is presented that exhibits room‐temperature broad‐band photoluminescence centered at 540 nm with a quantum yield (QY) of 15±5 %. A series of analogous compositions following the general formula Cs4−xAxSn(Br1−yIy)6 (A=Rb, K; x≤1, y≤1) can be prepared. The emission of these materials ranges from 500 nm to 620 nm with the possibility to compositionally tune the Stokes shift and the self‐trapped exciton emission bands.
Keywords: luminescence, perovskites, self-trapped excitons, solid-state synthesis, tin
Interest in low‐dimensional metal halide semiconductors,1 and ultimately their zero‐dimensional (0D) counterparts,2 has been spurred by the increasing interest in 3D lead halide perovskites.3 In recent years, lead halide perovskites have risen to prominence in the field of optoelectronics with their use in full‐color imaging,4 photodetection,5 X‐ray imaging,6 hard‐radiation detection,7 solar cells,8 and light‐emitting diodes,9 owing to their defect‐tolerant photophysics and charge transport.10
As the dimensionality decreases, the metal halide octahedra become progressively less‐connected and the optical and electrical properties shift away from those of a delocalized, 3D network towards 0D, molecular‐like, isolated octahedra. In such structures, self‐trapped excitons (STEs) form owing to the local deformation of the crystal lattice upon photoexcitation. This strong spatial localization, and the absence of electronic trapping processes that are inherent in electronically extended (higher‐dimensionality) solids, favors radiative recombination. Previously, the spatial confinement of carriers in 3D perovskites has been attained through crystal size control at the nanoscale (that is, top‐down and bottom‐up synthesis of nanocrystals).11 In the case of 0D materials, such elaborate crystal size engineering is not required as the optical properties are instead governed by their structural dimensionality. Highly localized Frenkel‐like excitons are formed instead of Wannier–Mott type excitons.
The library of 0D metal halides with octahedral building units includes both lead‐based and lead‐free compounds: TiIV,12 HfIV,13 ZrIV,14 PdIV,15 PbII,16 SnIV,17 TeIV,18 SbIII,19 and BiIII.20 However, only several of these examples exhibit photoluminescence (PL) at room temperature (RT), and this emission is seldom characterized by a high PL quantum yield (QY). The first examples with high QYs in excess of 50 % were demonstrated only recently: (C4N2H14Br)4SnBr6 (QY=95 %±5 %) and (C4N2H14I)4SnI6 (QY=75 %±4 %).21 Both structures are constructed from disconnected [SnX6]4− octahedra, separated by large organic cations with a distance of more than 1 nm between Sn2+ centers.
Given the high PL QY of these hybrid materials and their novel approach towards exciton localization, herein we chose to pursue fully inorganic analogues such as Cs4SnBr6. While several studies have reported on the structure and basic properties of Cs4SnBr6, none have observed PL at RT.22 Recently, calculations have shown that the Cs4SnBr6 phase should have a band gap of 3.37 eV.23 We prepared Cs4−xAxSn(Br1−yIy)6 (A=Rb, K) materials using a simple solid‐state heat‐and‐beat approach, characterized them structurally, and found them to be luminescent at RT.
According to the CsBr–SnBr2 pseudo‐binary phase diagram (Figure 1 a), Cs4SnBr6 melts incongruently and competes with the decomposition into the black CsSnBr3 and CsBr. Therefore, phase‐pure Cs4SnBr6 cannot be obtained by cooling from a melt of this composition. Instead, solid pellets of a mixture of CsBr (4.5 equiv.) and SnBr2 (1 equiv.) were repeatedly heated to 350 °C for 60 h and reground in a glovebox between heating cycles. The highlighted green region in Figure 1 a represents the experimental conditions that were found to yield the purest materials, that is, at temperatures below the decomposition point of Cs4SnBr6 (Supporting Information, Figures S1, S2; Tables S1, S2). Single crystals of Cs4SnBr6 were obtained by tempering a solid pellet at 350 °C. Cs4SnBr6 crystallizes in a trigonal crystal system (R c space group; Figure 1 b; Supporting Information, Tables S3–S9), wherein [SnBr6]4− octahedra are separated by Cs+ cations. The Cs+ cations occupy two distinct crystallographic positions (Supporting Information, Figure S3).
Figure 1.
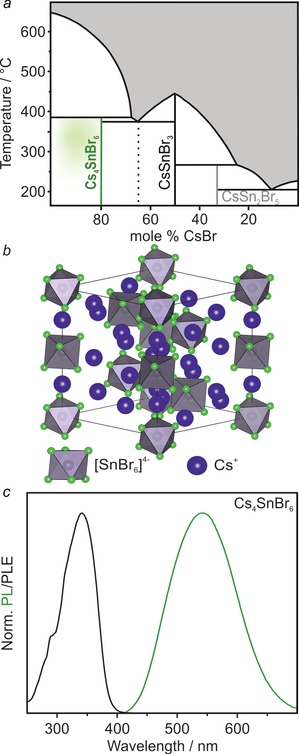
a) The pseudo‐binary CsBr–SnBr2 phase diagram.22a The highlighted green region represents the experimental conditions found to yield the purest Cs4SnBr6 material. b) The crystal structure of Cs4SnBr6 viewed along the (111) axis with [SnBr6]4− octahedra (gray with green bromine atoms) separated by Cs+ cations (blue). c) PL and PLE spectra for Cs4SnBr6 at RT.
Unlike the isostructural Cs4PbBr6, which shows narrow excitonic PL only at low temperatures and at wavelengths <400 nm,16 Cs4SnBr6 exhibits broad‐band green–yellow PL at RT from STEs (peak maximum at 540 nm, Figure 1 c). Upon measuring the PL excitation (PLE) and absorption spectra it was found that Cs4SnBr6 resembled a molecular material with a sharp excitation peak at 340 nm yielding a large Stokes shift of about 1.2 eV (Supporting Information, Figure S4). A PLQY of 15±5 % was measured at RT. PLE and PL spectra were found to be tunable through the partial substitution of both A‐site cations (Cs with Rb, K) and the halide anions (Br with I). Na was not observed to substitute Cs (Supporting Information, Figure S5).
In (C4N2H14X)4SnX6 (X=Br/I),21 the addition of iodide red‐shifts both the excitation and STE emission spectra. Similarly, the PL of Cs4SnX6 shifts to 620 nm (orange emission) at a Br:I ratio of ca. 1:1 (Figure 2 a). In agreement with Vegard's law, a linear change in both the a and c lattice parameters was observed over the range of compositions from Cs4SnBr6 to Cs4SnI6 (Supporting Information, Figures S6, S7; Tables S10, S11). Samples with greater than 50 % substitution by iodide, however, did not exhibit PL at RT and were not investigated further.
Figure 2.
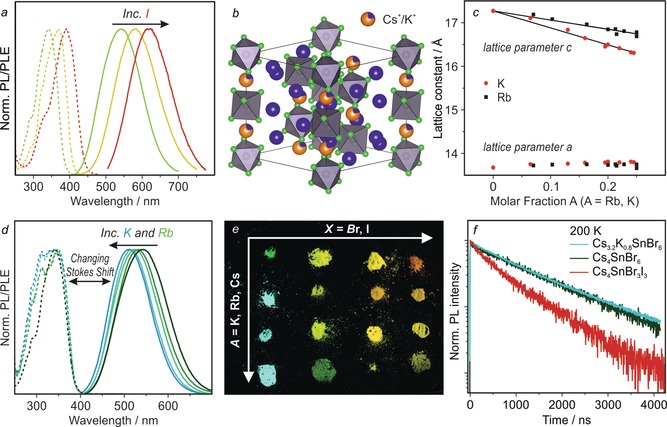
Structural and optical characterization of Cs4−xAxSn(Br,I)6 compounds (A=K, Rb). a) PL and PLE spectra of Cs4Sn(Br,I)6. b) Crystal structure of Cs3.2K0.8SnBr6 determined by Rietveld refinement. c) The change in the a and c lattice parameters upon Cs+ substitution by Rb+ and K+. d) PL and PLE spectra for Rb+ or K+ substituted compounds. e) Image of Cs4−xAxSn(Br,I)6 powders under 365 nm UV light. f) TRPL of Cs4SnBr6, Cs3.2K0.8SnBr6, and Cs4SnBr3I3 at 200 K.
While bromide and iodide occupy the same general position within the structure of Cs4SnBr6, Rb+ and K+ could potentially substitute two distinct crystallographic positions of Cs+ (Supporting Information, Figure S3). Substitution by Rb+ or K+ occurred only on the Cs2 site (1/4 of all Cs, six‐fold coordination, Figure 2 b; Supporting Information, Figure S8, Tables S12, S13), which explains the observed experimental 25 %‐limit for substitution (Supporting Information, Figure S9). Plotting the lattice constants against the molar fractions of K+ or Rb+ ions, estimated from Rietveld refinement and energy‐dispersive X‐ray spectroscopy (EDS), yields a seemingly linear trend for the c parameter, in agreement with Vegard's law, whereas the a parameter increases only slightly (Figure 2 c; Supporting Information, Figures S9–S11). The site affected is part of an infinite chain of [SnBr6]4− octahedra separated by Cs/Rb/K ions. Since the chain is oriented parallel to the c‐axis, substitution by Rb or K will cause the largest effect along this axis resulting in a decrease in the c parameter.
The substitution of Cs+ by Rb+ or K+ results in a blue‐shift of the PL maximum. This shift is dependent on both the degree of substitution and on the identity of the substituting cation. For 25 % substitution, Rb+ shifts the PL peak to 519 nm, whereas K+ yields a PL peak at 500 nm (Figure 2 d). Strikingly, the PLE remains unaffected. The same scenario was observed for the other examined Br:I ratios (ca. 5:1, 2:1, 1:1; Figure 2 e; Figure S12, Tables S14, S15).
Temperature‐dependent PL spectra were measured down to 6 K (Supporting Information, Figures S13, S14). It was found that the PL intensity increased with decreasing temperature and, in the case of Cs4SnBr6, the emission intensity reached a maximum at about 200 K. Given a RT QY of 15±5 %, the QY of Cs4SnBr6 is estimated to be near‐unity at 200 K (Supporting Information, Figure S15a). At 6 K, no higher‐energy emission, that is, from free excitons, could be observed in Cs4SnBr6 or Cs4SnBr3I3 (Supporting Information, Figures S14, S15b).
The time‐resolved PL (TRPL) of Cs4SnBr6 at RT exhibits a monoexponential decay with an average lifetime of 540 ns (Supporting Information, Figure S16). TRPL decays were then recorded at 200 K (the temperature that corresponds to highest QY, Figure 2 f; Supporting Information, Figure S15), and it was observed that the lifetime increased to 1381 ns (1424 ns for K‐substitution), while remaining monoexponential (Figure 2 f). Substitution with iodide accelerated the average lifetime (600 ns at 200 K for Cs4SnBr3I3, Figure 2 f; see the Supporting Information, Figure S16b for RT comparison). Additionally, the radiative lifetime was found to be insensitive towards dilution with CsBr, excitation intensity, crystallinity, and encapsulation within a UV‐curable epoxy (Supporting Information, Figure S16c,d), indicating a lack of surface effects on PL properties in these 0D materials.
Broad‐band, strongly Stokes‐shifted emission with relatively long radiative lifetimes indicates the formation of STEs (Figure 3 a). As in molecular complexes and similarly small entities, structural changes occur between the ground state and the excited state. Emission from STEs is then somewhat similar to an indirect‐gap optical transition since coupling to phonons is required.24 Besides 0D‐tin halides,21b emission from STEs has also been observed in 1D‐ and 2D‐layered materials.25
Figure 3.
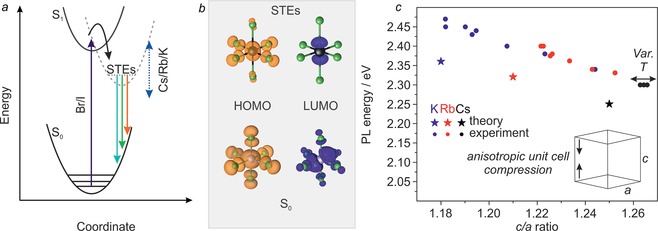
a) Generic configurational coordinate diagram illustrating the origin of STE PL in Cs4−xAxSn(Br1−yIy)6. b) Ground‐state and excited‐state (STE) HOMOs and LUMOs. c) Experimental and theoretical results demonstrating that PL energy varies linearly with the ratio of c‐axis to a‐axis length. Variable‐temperature measurements (three data points; black circles) are plotted for Cs4SnBr6 at 100 K, 200 K, and 273 K.
To rationalize the effect of A‐site substitution observed in these materials, calculations of the partial density of states (DOS) by density functional theory (DFT) were utilized (Figure 3 b; Supporting Information, Figure S17, Table S16).
A 1×1×1 unit cell (66 atoms) was used as a model system for Cs4SnBr6, Cs3RbSnBr6, and Cs3KSnBr6 materials. The electronic states were found to be highly localized in all three compositions and the band gap was found to consist of Sn 5s, Br 5p (conduction band/LUMO) and Sn 5p, Br 5p orbitals (valence band/HOMO). A‐cation orbitals do not significantly contribute to HOMO and LUMO states (Supporting Information, Figure S17). This is corroborated by the experiment given that A‐site substitution did not substantially affect the PLE spectra.
It was previously shown that both tin‐ and lead‐halide octahedra distort significantly upon photoexcitation in low‐dimensional materials.26 By modeling the excited state and ground state geometries, we find that the extent of distortion was the principle reason for the Stokes shift (Figure 3 b). A pseudo‐Jahn–Teller distortion was observed with elongation of up to 17 % in the axial Sn−Br bonds, and a contraction of up to 7 % in the equatorial bonds. This distortion was found to be greatest in Cs4SnBr6 and decreased from Cs+ to Rb+ to K+ (Supporting Information, Table S17). In other words, the energy required for distortion correlates well with the measured Stokes shift. Larger distortion is allowed by greater distance between the octahedra; primarily along the c‐axis. The ratio between the lengths of the c‐ and a‐axes was found to correlate most strongly with PL energy, experimentally and in calculations (Figure 3 c). On the other hand, isotropic changes to the unit cell (that is, contraction during cooling) do not affect the PL (Supporting Information, Table S18).
In summary, several new 0D tin halides with the general formula Cs4−xAxSn(Br1−yIy)6 have been found to exhibit broad‐band RT PL that is tunable from 500 nm to 620 nm. The PL peak position and Stokes shifts can be concomitantly adjusted by the substitution of Cs+ with either Rb+ or K+ as well as Br− by I−. PL properties were rationalized by the DFT analysis of STEs. Future studies might concern applications of such materials in luminescent solar concentrators and light emitting devices, harnessing their structural tunability and fully inorganic compositions.
Experimental Section
Materials and methods are included in the Supporting Information. Further details on the crystal structure investigation may be obtained from the Fachinformationszentrum Karlsruhe, 76344 Eggenstein‐Leopoldshafen, Germany (fax: (+49) 7247‐808‐666; e‐mail: crysdata@fiz‐karlsruhe.de), on quoting the depository numbers CSD‐434641, 434642, 434643, 434644, 434645, 434646, and 434647.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Dr. F. Krumeich for EDS measurements and acknowledge the support of the Scientific Center for Optical and Electron Microscopy (ScopeM) of the Swiss Federal Institute of Technology ETHZ. We thank Michael Solar for single‐crystal X‐ray diffraction measurements. This work was financially supported by the European Union through the FP7 (ERC Starting Grant NANOSOLID, GA No. 306733) and through the Horizon‐2020 (Marie‐Skłodowska Curie ITN network PHONSI, H2020‐MSCA‐ITN‐642656). Authors thank IBM‐Zurich Research, in particular Dr. T. Stöferle and Dr. R. F. Mahrt, for support with low‐temperature PL experiments. I.I. would like to thank The Netherlands Organization of Scientific Research (NWO) for providing financial support within the Innovational Research Incentive (Vidi) Scheme (Grant no. 723.013.002). DFT calculations were carried out on the Dutch national e‐infrastructure with the support of SURF Cooperative.
B. M. Benin, D. N. Dirin, V. Morad, M. Wörle, S. Yakunin, G. Rainò, O. Nazarenko, M. Fischer, I. Infante, M. V. Kovalenko, Angew. Chem. Int. Ed. 2018, 57, 11329.
References
- 1.
- 1a. Mitzi D. B., Felid C. A., Harrison W. T. A., Guloy A. M., Nature 1994, 369, 467–469; [Google Scholar]
- 1b. Mitzi D. B., Liang K., Wang S., Inorg. Chem. 1998, 37, 321–327; [Google Scholar]
- 1c. Xu Z., Mitzi D. B., Inorg. Chem. 2003, 42, 6589–6591; [DOI] [PubMed] [Google Scholar]
- 1d. Pedesseau L., Sapori D., Traore B., Robles R., Fang H. H., Loi M. A., Tsai H., Nie W., Blancon J. C., Neukirch A., Tretiak S., Mohite A. D., Katan C., Even J., Kepenekian M., ACS Nano 2016, 10, 9776–9786; [DOI] [PubMed] [Google Scholar]
- 1e. Mao L., Tsai H., Nie W., Ma L., Im J., Stoumpos C. C., Malliakas C. D., Hao F., Wasielewski M. R., Mohite A. D., Kanatzidis M. G., Chem. Mater. 2016, 28, 7781–7792; [Google Scholar]
- 1f. Stoumpos C. C., Cao D. H., Clark D. J., Young J., Rondinelli J. M., Jang J. I., Hupp J. T., Kanatzidis M. G., Chem. Mater. 2016, 28, 2852–2867; [Google Scholar]
- 1g. Tsai H., Nie W., Blancon J. C., Stoumpos C. C., Asadpour R., Harutyunyan B., Neukirch A. J., Verduzco R., Crochet J. J., Tretiak S., Pedesseau L., Even J., Alam M. A., Gupta G., Lou J., Ajayan P. M., Bedzyk M. J., Kanatzidis M. G., Nature 2016, 536, 312–316; [DOI] [PubMed] [Google Scholar]
- 1h. Mao L., Wu Y., Stoumpos C. C., Wasielewski M. R., Kanatzidis M. G., J. Am. Chem. Soc. 2017, 139, 5210–5215; [DOI] [PubMed] [Google Scholar]
- 1i. Mao L., Wu Y., Stoumpos C. C., Traore B., Katan C., Even J., Wasielewski M. R., Kanatzidis M. G., J. Am. Chem. Soc. 2017, 139, 11956–11963; [DOI] [PubMed] [Google Scholar]
- 1j. Cao D. H., Stoumpos C. C., Yokoyama T., Logsdon J. L., Song T.-B., Farha O. K., Wasielewski M. R., Hupp J. T., Kanatzidis M. G., ACS Energy Lett. 2017, 2, 982–990; [Google Scholar]
- 1k. Soe C. M. M., Stoumpos C. C., Kepenekian M., Traore B., Tsai H., Nie W., Wang B., Katan C., Seshadri R., Mohite A. D., Even J., Marks T. J., Kanatzidis M. G., J. Am. Chem. Soc. 2017, 139, 16297–16309; [DOI] [PubMed] [Google Scholar]
- 1l. Nazarenko O., Kotyrba M. R., Worle M., Cuervo-Reyes E., Yakunin S., Kovalenko M. V., Inorg. Chem. 2017, 56, 11552–11564; [DOI] [PubMed] [Google Scholar]
- 1m. Stoumpos C. C., Mao L., Malliakas C. D., Kanatzidis M. G., Inorg. Chem. 2017, 56, 56–73; [DOI] [PubMed] [Google Scholar]
- 1n. Nazarenko O., Kotyrba M. R., Yakunin S., Aebli M., Raino G., Benin B. M., Worle M., Kovalenko M. V., J. Am. Chem. Soc. 2018, 140, 3850–3853; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1o. Smith M. D., Karunadasa H. I., Acc. Chem. Res. 2018, 51, 619–627. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Saidaminov M. I., Mohammed O. F., Bakr O. M., ACS Energy Lett. 2017, 2, 889–896; [Google Scholar]
- 2b. Lin H., Zhou C., Tian Y., Siegrist T., Ma B., ACS Energy Lett. 2018, 3, 54–62; [Google Scholar]
- 2c. Akkerman Q. A., Abdelhady A. L., Manna L., J. Phys. Chem. Lett. 2018, 9, 2326–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Green M. A., Ho-Baillie A., Snaith H. J., Nat. Photonics 2014, 8, 506–514; [Google Scholar]
- 3b. Manser J. S., Christians J. A., Kamat P. V., Chem. Rev. 2016, 116, 12956–13008; [DOI] [PubMed] [Google Scholar]
- 3c. Akkerman Q. A., Raino G., Kovalenko M. V., Manna L., Nat. Mater. 2018, 17, 394–405. [DOI] [PubMed] [Google Scholar]
- 4. Yakunin S., Shynkarenko Y., Dirin D. N., Cherniukh I., Kovalenko M. V., NPG Asia Mater. 2017, 9, e431. [Google Scholar]
- 5.
- 5a. Hu X., Zhang X., Liang L., Bao J., Li S., Yang W., Xie Y., Adv. Funct. Mater. 2014, 24, 7373–7380; [Google Scholar]
- 5b. Dou L., Yang Y. M., You J., Hong Z., Chang W. H., Li G., Yang Y., Nat. Commun. 2014, 5, 1–6. [DOI] [PubMed] [Google Scholar]
- 6. Yakunin S., Sytnyk M., Kriegner D., Shrestha S., Richter M., Matt G. J., Azimi H., Brabec C. J., Stangl J., Kovalenko M. V., Heiss W., Nat. Photonics 2015, 9, 444–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Stoumpos C. C., Malliakas C. D., Peters J. A., Liu Z., Sebastian M., Im J., Chasapis T. C., Wibowo A. C., Chung D. Y., Freeman A. J., Wessels B. W., Kanatzidis M. G., Cryst. Growth Des. 2013, 13, 2722–2727; [Google Scholar]
- 7b. Yakunin S., Dirin D. N., Shynkarenko Y., Morad V., Cherniukh I., Nazarenko O., Kreil D., Nauser T., Kovalenko M. V., Nat. Photonics 2016, 10, 585–589. [Google Scholar]
- 8.
- 8a. Lee M. M., Teuscher J., Miyasaka T., Murakami T. N., Snaith H. J., Science 2012, 338, 643–647; [DOI] [PubMed] [Google Scholar]
- 8b. Burschka J., Pellet N., Moon S. J., Humphry-Baker R., Gao P., Nazeeruddin M. K., Gratzel M., Nature 2013, 499, 316–319. [DOI] [PubMed] [Google Scholar]
- 9. Tan Z.-K., Moghaddam R. S., Lai M. L., Docampo P., Higler R., Deschler F., Price M., Sadhanala A., Pazos L. M., Credgington D., Hanusch F., Bein T., Snaith H. J., Friend R. H., Nat. Nanotechnol. 2014, 9, 687–692. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Brandt R. E., Stevanović V., Ginley D. S., Buonassisi T., MRS Commun. 2015, 5, 265–275; [Google Scholar]
- 10b. Yaffe O., Guo Y., Tan L. Z., Egger D. A., Hull T., Stoumpos C. C., Zheng F., Heinz T. F., Kronik L., Kanatzidis M. G., Owen J. S., Rappe A. M., Pimenta M. A., Brus L. E., Phys. Rev. Lett. 2017, 118, 136001; [DOI] [PubMed] [Google Scholar]
- 10c. Miyata K., Atallah T. L., Zhu X.-Y., Sci. Adv. 2017, 3, e1701469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a. Schmidt L. C., Pertegas A., Gonzalez-Carrero S., Malinkiewicz O., Agouram S., Minguez Espallargas G., Bolink H. J., Galian R. E., Perez-Prieto J., J. Am. Chem. Soc. 2014, 136, 850–853; [DOI] [PubMed] [Google Scholar]
- 11b. Protesescu L., Yakunin S., Bodnarchuk M. I., Krieg F., Caputo R., Hendon C. H., Yang R. X., Walsh A., Kovalenko M. V., Nano Lett. 2015, 15, 3692–3696; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11c. Dirin D. N., Protesescu L., Trummer D., Kochetygov I. V., Yakunin S., Krumeich F., Stadie N. P., Kovalenko M. V., Nano Lett. 2016, 16, 5866–5874; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11d. Akkerman Q. A., Motti S. G., Srimath Kandada A. R., Mosconi E., D'Innocenzo V., Bertoni G., Marras S., Kamino B. A., Miranda L., De Angelis F., Petrozza A., Prato M., Manna L., J. Am. Chem. Soc. 2016, 138, 1010–1016; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11e. Huang H., Xue Q., Chen B., Xiong Y., Schneider J., Zhi C., Zhong H., Rogach A. L., Angew. Chem. Int. Ed. 2017, 56, 9571–9576; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 9699–9704; [Google Scholar]
- 11f. Protesescu L., Yakunin S., Nazarenko O., Dirin D. N., Kovalenko M. V., ACS Appl. Nano Mater. 2018, 1, 1300–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen M., Ju M.-G., Carl A. D., Zong Y., Grimm R. L., Gu J., Zeng X. C., Zhou Y., Padture N. P., Joule 2018, 2, 558–570. [Google Scholar]
- 13.
- 13a. Kang B., Biswas K., J. Phys. Chem. C 2016, 120, 12187–12195; [Google Scholar]
- 13b. Král R., Babin V., Mihóková E., Buryi M., Laguta V. V., Nitsch K., Nikl M., J. Phys. Chem. C 2017, 121, 12375–12382. [Google Scholar]
- 14. Saeki K., Fujimoto Y., Koshimizu M., Nakauchi D., Tanaka H., Yanagida T., Asai K., Jpn. J. Appl. Phys. 2018, 57, 030310. [Google Scholar]
- 15. Sakai N., Haghighirad A. A., Filip M. R., Nayak P. K., Nayak S., Ramadan A., Wang Z., Giustino F., Snaith H. J., J. Am. Chem. Soc. 2017, 139, 6030–6033. [DOI] [PubMed] [Google Scholar]
- 16. Nikl M., Mihokova E., Nitsch K., Somma F., Giampaolo C., Pazzi G. P., Fabeni P., Zazubovich S., Chem. Phys. Lett. 1999, 306, 280–284. [Google Scholar]
- 17.
- 17a. Lee B., Stoumpos C. C., Zhou N., Hao F., Malliakas C., Yeh C. Y., Marks T. J., Kanatzidis M. G., Chang R. P., J. Am. Chem. Soc. 2014, 136, 15379–15385; [DOI] [PubMed] [Google Scholar]
- 17b. Kaltzoglou A., Antoniadou M., Kontos A. G., Stoumpos C. C., Perganti D., Siranidi E., Raptis V., Trohidou K., Psycharis V., Kanatzidis M. G., Falaras P., J. Phys. Chem. C 2016, 120, 11777–11785. [Google Scholar]
- 18.
- 18a. Wernicke R., Kupka H., Ensslin W., Schmidtke H.-H., Chem. Phys. 1980, 47, 235–244; [Google Scholar]
- 18b. Blasse G., Dirksen G. J., Abriel W., Chem. Phys. Lett. 1987, 136, 460–464; [Google Scholar]
- 18c. Sedakova T. V., Mirochnik A. G., Karasev V. E., Opt. Spectrosc. 2011, 110, 755–761; [Google Scholar]
- 18d. Bukvetskii B. V., Sedakova T. V., Mirochnik A. G., Russ. J. Coord. Chem. 2012, 38, 106–110. [Google Scholar]
- 19.
- 19a. Sedakova T. V., Mirochnik A. G., Karasev V. E., Opt. Spectrosc. 2008, 105, 517–523; [Google Scholar]
- 19b. Vovna V. I., Dotsenko A. A., Korochentsev V. V., Shcheka O. L., Os'mushko I. S., Mirochnik A. G., Sedakova T. V., Sergienko V. I., J. Mol. Struct. 2015, 1091, 138–146; [Google Scholar]
- 19c. Zhou C., Worku M., Neu J., Lin H., Tian Y., Lee S., Zhou Y., Han D., Chen S., Hao A., Djurovich P. I., Siegrist T., Du M.-H., Ma B., Chem. Mater. 2018, 30, 2374–2378. [Google Scholar]
- 20. Elfaleh N., Kamoun S., J. Organomet. Chem. 2016, 819, 95–102. [Google Scholar]
- 21.
- 21a. Zhou C., Tian Y., Yuan Z., Lin H., Chen B., Clark R., Dilbeck T., Zhou Y., Hurley J., Neu J., Besara T., Siegrist T., Djurovich P., Ma B., ACS Appl. Mater. Interfaces 2017, 9, 44579–44583; [DOI] [PubMed] [Google Scholar]
- 21b. Zhou C., Lin H., Tian Y., Yuan Z., Clark R., Chen B., van de Burgt L. J., Wang J. C., Zhou Y., Hanson K., Meisner Q. J., Neu J., Besara T., Siegrist T., Lambers E., Djurovich P., Ma B., Chem. Sci. 2018, 9, 586–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.
- 22a. Andrews R. H., Clark S. J., Donaldson J. D., J. Chem. Soc. Dalton Trans. 1983, 767–770; [Google Scholar]
- 22b. Myagkota S. V., Savchin P. V., Voloshinovskiĭ A. S., Demkiv T. M., Boĭko Y. V., Vus R. S., Demkiv L. S., Phys. Solid State 2008, 50, 1473–1476. [Google Scholar]
- 23. Hu M., Ge C., Yu J., Feng J., J. Phys. Chem. C 2017, 121, 27053–27058. [Google Scholar]
- 24. Thirumal K., Chong W. K., Xie W., Ganguly R., Muduli S. K., Sherburne M., Asta M., Mhaisalkar S., Sum T. C., Soo H. S., Mathews N., Chem. Mater. 2017, 29, 3947–3953. [Google Scholar]
- 25.
- 25a. Dohner E. R., Jaffe A., Bradshaw L. R., Karunadasa H. I., J. Am. Chem. Soc. 2014, 136, 13154–13157; [DOI] [PubMed] [Google Scholar]
- 25b. Yuan Z., Zhou C., Tian Y., Shu Y., Messier J., Wang J. C., van de Burgt L. J., Kountouriotis K., Xin Y., Holt E., Schanze K., Clark R., Siegrist T., Ma B., Nat. Commun. 2017, 8, 1–7; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25c. Smith M. D., Watson B. L., Dauskardt R. H., Karunadasa H. I., Chem. Mater. 2017, 29, 7083–7087. [Google Scholar]
- 26.
- 26a. Zhou C., Lin H., Shi H., Tian Y., Pak C., Shatruk M., Zhou Y., Djurovich P., Du M. H., Ma B., Angew. Chem. Int. Ed. 2018, 57, 1021–1024; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 1033–1036; [Google Scholar]
- 26b. Kang B., Biswas K., J. Phys. Chem. Lett. 2018, 9, 830–836. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary