

Abstract
Aim
Diabetes is a result of an interplay between genetic, environmental and lifestyle factors. Keratin intermediate filaments are stress proteins in epithelial cells, and keratin mutations predispose to several human diseases. However, the involvement of keratins in diabetes is not well known. K8 and its partner K18 are the main β‐cell keratins, and knockout of K8 (K8−/−) in mice causes mislocalization of glucose transporter 2, mitochondrial defects, reduced insulin content and altered systemic glucose/insulin control. We hypothesize that K8/K18 offer protection during β‐cell stress and that decreased K8 levels contribute to diabetes susceptibility.
Methods
K8‐heterozygous knockout (K8+/−) and wild‐type (K8+/+) mice were used to evaluate the influence of keratin levels on endocrine pancreatic function and diabetes development under basal conditions and after T1D streptozotocin (STZ)‐induced β‐cell stress and T2D high‐fat diet (HFD).
Results
Murine K8+/− endocrine islets express ~50% less K8/K18 compared with K8+/+. The decreased keratin levels have little impact on basal systemic glucose/insulin regulation, β‐cell health or insulin levels. Diabetes incidence and blood glucose levels are significantly higher in K8+/− mice after low‐dose/chronic STZ treatment, and STZ causes more β‐cell damage and polyuria in K8+/− compared with K8+/+. K8 appears upregulated 5 weeks after STZ treatment in K8+/+ islets but not in K8+/−. K8+/− mice showed no major susceptibility risk to HFD compared to K8+/+.
Conclusion
Partial K8 deficiency reduces β‐cell stress tolerance and aggravates diabetes development in response to STZ, while there is no major susceptibility to HFD.
Keywords: β‐cell, cytoskeleton, diabetes, glucose‐stimulated insulin secretion, high‐fat diet, keratin intermediate filaments, streptozotocin
1. INTRODUCTION
Diabetes development is commonly described to result from an interplay between genetic and environmental factors, and several susceptibility genes and environmental or lifestyle factors are known to contribute to both type 1 (T1D) and type 2 diabetes (T2D).1, 2 In T1D, genetic mutations of human leucocyte antigen (HLA) are the most common variants,3 while also virus infections have been implicated in T1D development.3 The application of genome‐wide association studies (GWAS) has identified over 50 gene variants associated with T2D.2, 4 The high number of genetic alleles with low odds ratios for disease predictability in T2D confirms that genetic and environmental factors interlink in the development of the disease.5, 6 Keratins (K) constitute the intermediate filament proteins of epithelial cells.7 They make up a part of the cytoskeletal cell reinforcement and assist in essential cellular functions such as protein targeting, cell stress and cell polarity.8, 11 Keratin mutations in humans cause or predispose to several diseases, for example in skin and liver.10, 12, 13 In the liver, keratin mutations do not cause disease per se but clearly contribute or predispose to disease when liver cells are exposed to additional stress, emphasizing the role of keratins during cell stress or cell injury.10, 14 In mouse models, knock‐down or expression of mutant simple epithelial K8 or K18, phenocopy human conditions and cause or predispose to a range of pathologies and abnormalities in the organs where these keratins are expressed, such as in the intestine, liver and thymus.10, 15, 16 K8 knockout (K8−/−) in mice, for example, causes 50% embryonic lethality and leads to an ulcerative‐colitis‐like phenotype in the surviving mice.17, 18 Moreover, liver fragility is observed in both K8−/− and K18−/− as well as in K18 mutant mice, while K8 knockdown disrupts cell morphology and increases apoptosis in the thymus.7, 15 Keratins are upregulated in epithelial cells after organ‐specific stress or disease conditions, as documented in several organs such as colon, liver, kidney, β‐cells and gall bladder.10, 19, 20
The loss of K8 also leads to altered glucose metabolism seen as decreased fasting blood glucose levels,20 as well as increased glucose uptake and glycogen synthesis in hepatocytes.21 In β‐cells, K8 loss causes disturbances in the plasma membrane targeting of glucose transporter 2 (GLUT2) resulting in a delayed sensitivity to streptozotocin (STZ)‐induced T1D.20 K8 loss is moreover associated with a decrease in insulin vesicle size and insulin secretion, as well as mitochondrial defects, including smaller sized mitochondria, decreased mitochondrial membrane potential and ATP production in β‐cells.20, 22 Interestingly, K8 has been reported to be upregulated in cultured β‐cells upon stimulation with glucose,23 and ectopic expression of epidermal keratins K1 and K10 in the pancreas leads to diabetes due to loss of β‐cell insulin vesicles and decreased insulin secretion.24 Taken together, these findings suggest that keratins are intricately involved in the regulation of β‐cell processes. Given that keratins have well‐described cell stress functions in many organs and are associated with glucose/insulin regulation in β‐cells and liver,21 we herein investigated whether a transgenic partial downregulation of K8 by deletion of one K8 allele would render mice more susceptible to β‐cell stress induced by STZ or metabolic stress induced by high‐fat diet (HFD). Our results indicate that K8+/− mice, expressing less K8 than K8+/+ mice, are more vulnerable to diabetes induced by chronic β‐cell injury caused through STZ exposure but not by HFD.
2. RESULTS
2.1. K8+/− mice express decreased levels of β‐cell K8 and K18 but maintain normal blood glucose and insulin regulation under basal conditions
To determine the levels of the main β‐cell keratins, K8 and K18, in K8+/+ and K8+/− mice, isolated hand‐picked islets and pancreatic tissues were analysed by Western blotting and immunofluorescence microscopy respectively. K8 and K18 are expressed in both K8+/+ and K8+/− mouse pancreatic islets (Figure 1A‐C). K8+/− mouse islets, however, express roughly 50%‐80% less of both K8 and K18 (Figure 1A‐C) reflecting the loss of one K8 allele. The decreased islet levels of K8 and K18 in K8+/− compared to K8+/+ were confirmed by immunostaining (Figure 1D). Type II K7 protein levels are high in exocrine ducts, but very low in islets.20 Thus, K7 was detectable only by immunofluorescence staining and K7 appeared slightly decreased in K8+/− compared to K8+/+ islets (Figure S1). Type I K19, which is expressed in the exocrine pancreas, was not detected in pancreatic islets by Western blotting or immunostaining analysis (not shown). K8+/− islets have a normal appearance similar to islets in K8+/+ mice (Figure 1D).
Figure 1.
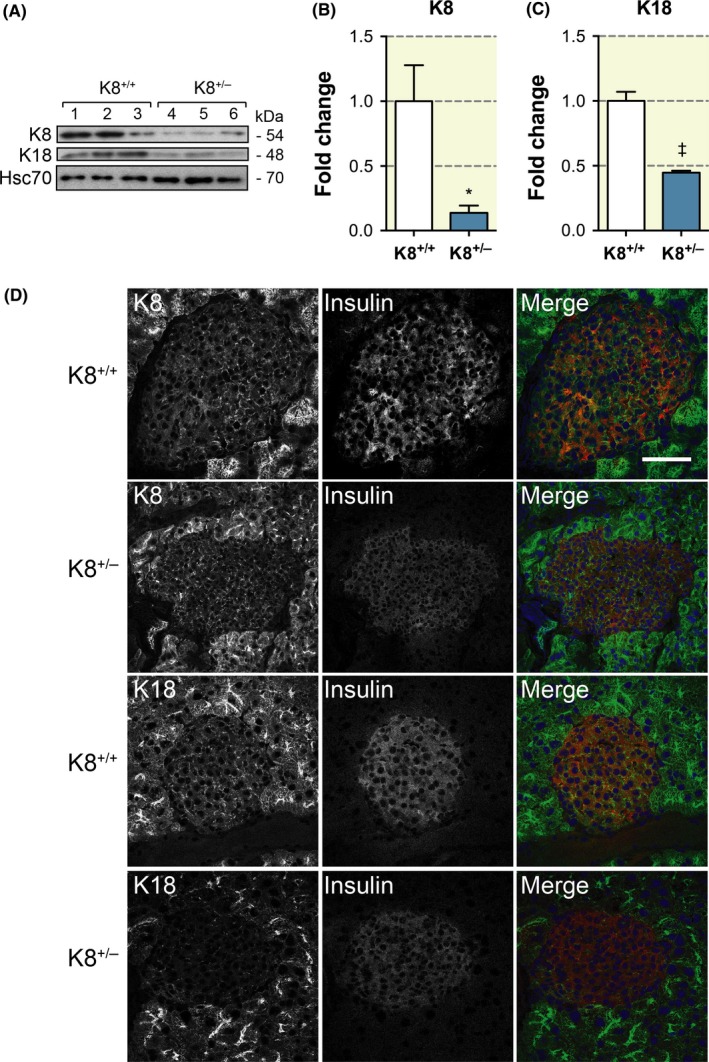
K8+/− mouse islet cells express less K8 and K18 than K8+/+ islets. A, Western blot for K8, K18 and Hsc70 (loading control) of protein lysate from pancreatic islets isolated from K8+/+ and K8+/− mice (lane 1‐3, K8+/+; lane 4‐6, K8+/−). B, Quantification of immunoblotting showed a statistically significant fivefold decrease in K8 protein levels in K8+/−‐isolated pancreatic islets compared to K8+/+ islets when normalized to Hsc70 levels. *P < .05, error bars represent mean ± SEM. C, Quantification of immunoblotting showed a significant twofold decrease in K18 protein levels in K8+/−‐isolated pancreatic islets compared to K8+/+ islets when normalized to Hsc70 levels. †P < .001, error bars represent mean ± SEM. D, Immunostaining of K8 or K18 (green), insulin (red, used as a marker for islets) and nuclei (blue, colours seen in the merged images) of pancreatic sections from K8+/+ and K8+/− mice shows a less dense islet keratin network in K8+/−.mice compared to K8+/+ mice. Scale bar: 100 μm
To test whether the decreased keratin levels in the K8+/− β‐cells affect blood glucose regulation, blood glucose levels were measured following overnight fasting in untreated, young (3‐4 months old) K8+/− and K8+/+ mice (Figure 2A). Likewise, the change in blood glucose levels after intraperitoneal (i.p.) glucose or insulin administration, and insulin levels in serum after glucose administration, were measured (Figure 2B‐D). The results show that under normal physiological circumstances, or after a short‐term glucose or insulin challenge, K8+/− mice are adept at regulating blood glucose levels despite the reduction in K8/K18 in the K8+/− mice, as no significant differences between K8+/− and K8+/+ could be observed in these measured metabolic parameters (Figure 2A‐D). GSIS was elevated as expected in K8+/+, but not in K8+/− mice (Figure 2D). Pancreatic insulin levels, analysed both from ex vivo pancreatic extracts using enzyme‐linked immunosorbent assay (ELISA) (Figure 2E) and by Western blotting (Figure 2F), of isolated islets showed similar hormone levels in K8+/+ and K8+/− mice.
Figure 2.
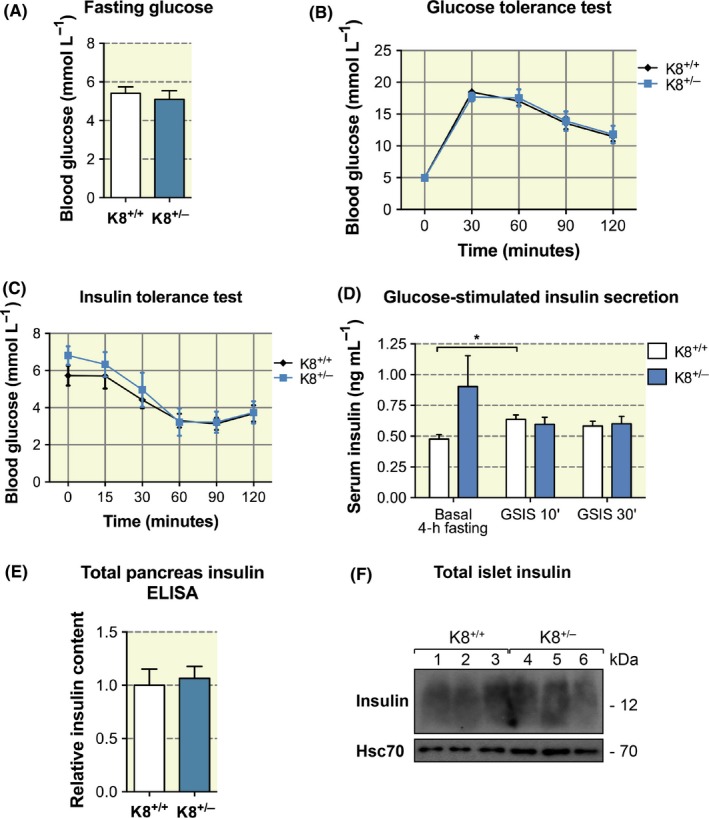
K8+/− mice exhibit no significant changes in blood glucose and insulin regulation compared to K8+/+ mice. A, Blood glucose levels after overnight fasting in 3‐ to 4‐mo‐old K8+/+ mice and K8+/− mice showed no significant differences using t test. Bars represent mean ± SEM, n = 10 for K8+/+ and n = 8 for K8+/− mice. B, K8+/+ and K8+/− mice were subjected to a glucose tolerance test. Mice were fasted overnight and then challenged with 2 g/kg d‐glucose administered i.p. No changes in blood glucose levels were observed between K8+/+ and K8+/− mice as evaluated using repeated measures two‐way ANOVA. Bars represent mean ± SEM, n = 10 for K8+/+ and n = 8 for K8+/− mice. C, K8+/+ and K8+/− mice showed no significant changes in blood glucose levels, using repeated measures two‐way ANOVA, when subjected to an insulin tolerance test by 0.75 U/kg i.p. insulin after 4‐h fasting. Bars represent mean ± SEM, n = 7 for K8+/+ and n = 9 for K8+/− mice. D, Glucose‐stimulated insulin secretion (GSIS) was measured 10 and 30 min (') after i.p. glucose 2 g/kg of body weight in K8+/+ mice and K8+/− mice. A significant increase in fasting versus 10 min GSIS can be seen for K8+/+ mice, analysed by one‐way ANOVA and Bonferroni's correction, but no overall differences in GSIS responses between the genotypes were observed when compared using two‐way ANOVA. Plot shows mean ± SEM, n = 8‐10 mice/group for fasting insulin and 4‐5 mice/group for glucose‐stimulated insulin secretion. E, Relative pancreatic insulin content as determined by enzyme‐linked immunosorbent assay showed no differences between K8+/+ mice and K8−/− mice. Bars indicate mean ± SEM, n = 5 for K8+/+ mice and n = 7 for K8+/− mice. F, Immunoblotting of K8+/+ (n = 3, lanes 1‐3) and K8+/− (n = 3, lanes 4‐6) isolated pancreatic islets showed no changes in the protein amount of insulin between the genotypes
2.2. K8+/− mice are more sensitive to T1D after streptozotocin treatment and develop a more severe T1D than K8+/+ mice
In addition to providing structural support, keratins are known stress proteins that undergo rapid and dynamic changes in response to cell stress or injury.9 In order to investigate whether a reduction in keratin levels, due to the knockout of one K8 allele in K8+/− mice, increases sensitivity to β‐cell injury and diabetes, the STZ model for T1D development was used. A low STZ dose (40 mg/kg, i.p.) was administered to mice once per day for 5 consecutive days, after which blood glucose levels were monitored for up to 5 weeks. A significantly (P = .016) higher proportion of K8+/− than K8+/+ mice became diabetic in response to the STZ treatment, and the onset of diabetes occurred sooner in K8+/− mice (Figure 3A). Six weeks after the start of the experiment, ~90% of K8+/− mice had diabetic blood glucose levels, compared to ~60% in K8+/+ mice (Figure 3A). K8+/− mouse blood glucose levels were also higher than in K8+/+ mice after STZ treatment at the two‐week time point (Figure 3B) and remained at a higher level in K8+/− mice until the end of the experiment (not shown). Moreover, polyuria, which is an indication of the severity of diabetes,25 was significantly increased in STZ‐treated diabetic K8+/− compared with K8+/+ mice at 34 days post‐STZ treatments (Figure 3c), confirming the more severe diabetic condition in these mice. In untreated control mice, no differences in urine volume were seen between the genotypes (Figure 3C). Histological examination showed that the pancreatic islets in K8+/− mice were significantly more damaged 2 weeks as well as 5 weeks after STZ treatments (Figure 4A). These results show that even if a reduction in K8 levels does not markedly alter glucose/insulin metabolism or β‐cell function under basal conditions, it reduces the capacity to endure the prolonged cell stress caused by the β‐cell toxin and diabetes‐inducing agent, STZ.
Figure 3.
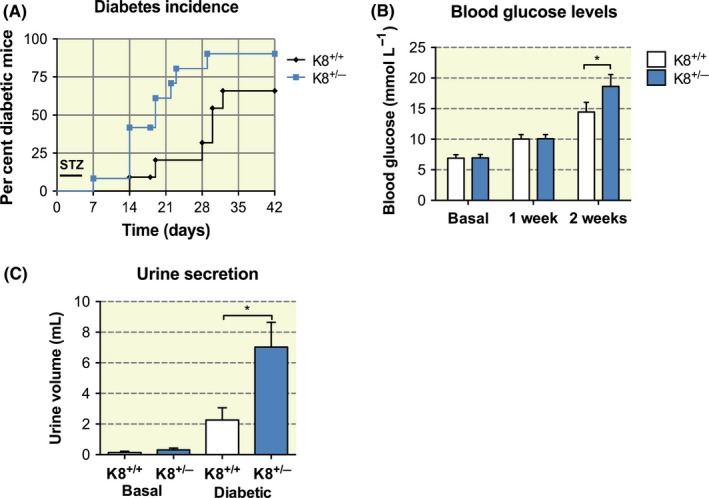
K8+/− mice exhibit increased diabetes incidence and aggravated diabetes after chronic low‐dose STZ treatment when compared to K8+/+ mice. A, A significantly higher diabetes incidence was seen for K8+/− compared to K8+/+ mice after daily low‐dose STZ i.p. injections during the first 5 d. n = 13 for K8+/+ and n = 11 for K8+/− mice. P = .016, calculated with log‐rank test. B, K8+/− mice showed higher blood glucose levels 2 wk after low‐dose STZ injections compared to K8+/+ mice. Bars show mean ± SEM, n = 17 for K8+/+ mice and n = 18 for K8+/− mice. *P < .05 calculated with two‐way ANOVA and Bonferroni's post hoc test. C, Urine secretion for basal untreated and STZ‐treated diabetic (collected 34 d after treatment) K8+/+ and K8+/− mice showed significantly larger urine volume for K8+/− mice compared to K8+/+ mice. *P < .05 calculated with two‐way ANOVA
Figure 4.
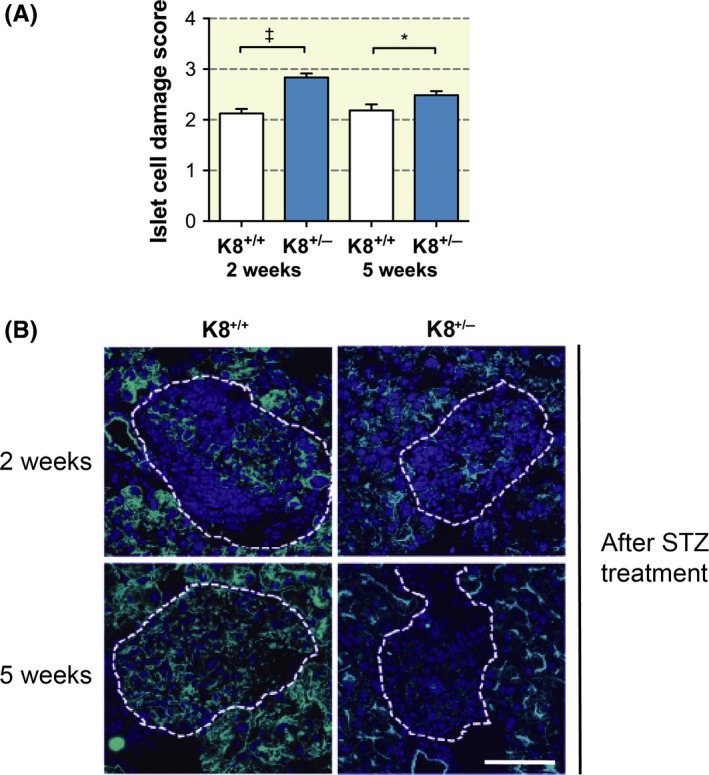
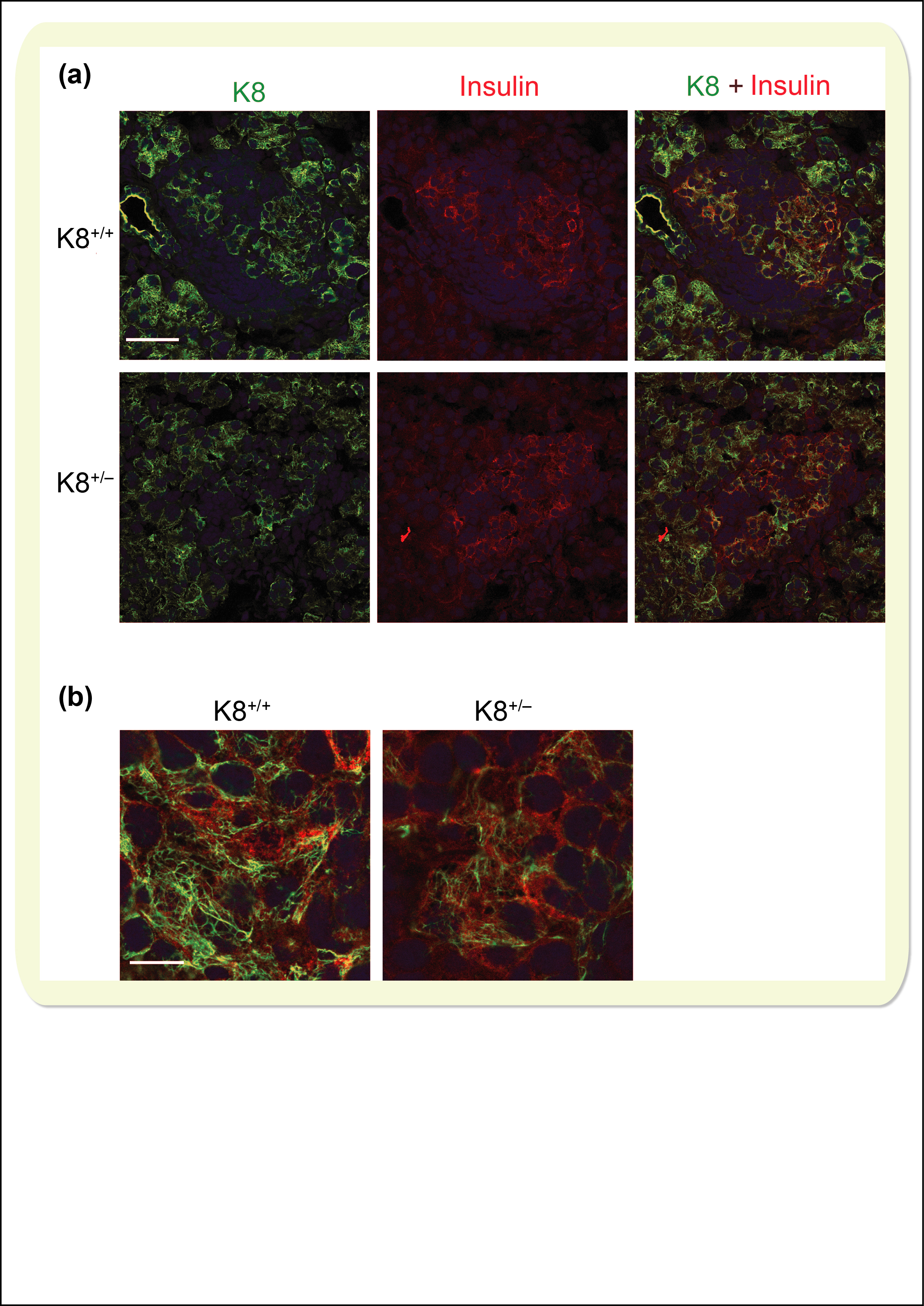
Increased islet damage in K8+/− mice and more persistent keratin upregulation in K8+/+ than in K8+/− mice after STZ treatment. A, Scoring of islet cell damage in STZ‐treated (2 or 5 wk) K8+/+ and K8+/− haematoxylin‐eosin–stained pancreata showed more islet damage in K8+/− mice compared to K8+/+ mice. n = 6 for K8+/+ and K8+/− mice (2 wk STZ treatment); n = 3 for K8+/+ and n = 4 for K8+/− mice (5‐wk STZ treatment). *P < .05, † P < .001. B, Immunostaining for K8 (green) and nuclei (blue) of pancreatic sections from STZ‐induced diabetic K8+/+ and K8+/− mice 2 wk and 5 wk after start of the STZ treatment shows increased K8 levels after 2 wk of STZ treatment in both genotypes. At 5 wk, K8 was increased in islet cells compared to untreated control mice (see Figure S2 for keratin/insulin staining of these samples). Islets are indicated with white broken lines. Scale bar: 100 μm
Keratins undergo dynamic changes in response to cell injury, and these responses often include a rapid upregulation of keratins, which is thought to help the cell withstand injury.9 We have previously shown that K8 is upregulated in remaining β‐cells in K8+/+ mice exposed to STZ and in non‐obese diabetic mice during the development of diabetes.20 Despite the reduced basal K8 levels in K8+/− mice, K8 also appears upregulated in K8+/− mouse islets in insulin‐positive β‐cells (Figure S2) 2 weeks after STZ treatment (Figure 4B), when compared to exocrine keratin staining and controls (Figure 1D). Importantly, 5 weeks after STZ, K8+/+ mouse islets still retained some K8 staining, while K8+/− islets had little or no K8 expression left (Figure 4B), supporting our observations of increased β‐cell damage in these mice.
As complete K8 loss in β‐cells leads to decreased mitochondrial function and MFN2 levels, as well as GLUT2 mislocalization,20, 22 we were interested to investigate whether decreased keratin amount affects these proteins. However, both MFN2 and GLUT2 protein levels (Figure 5A, B, D, E) as well as GLUT2 cellular localization (Figure 5C) were unaltered in K8+/− β‐cells. This suggests that mitochondria and GLUT2 targeting can be maintained, at least under basal circumstances, even after a 50% reduction in keratin levels.
Figure 5.
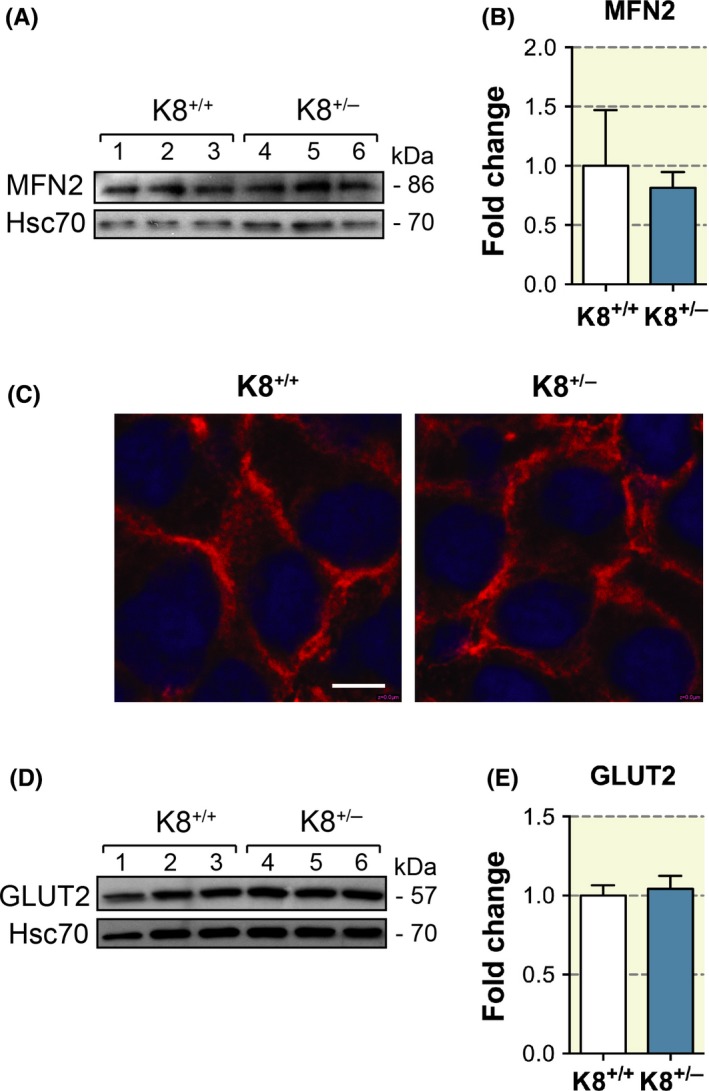
MFN2 protein levels and GLUT2 protein levels and localization are unaltered in K8+/− mice. A, Western blot analysis for MFN2 and Hsc70 (loading control) of protein lysate from pancreatic islets isolated from K8+/+ and K8+/− mice (lanes 1‐3, K8+/+; lanes 4‐6, K8+/−) showed no changes in the protein levels of MFN2 between the genotypes. B, Quantification of MFN2 immunoblotting from (A) using Hsc70 as loading control showed no significant changes in protein levels of MFN2 between K8+/+ and K8+/− mice. C, Immunostaining of pancreatic sections from K8+/+ and K8+/− mice for GLUT2 (red) and nuclei (blue) showed equal intensities of GLUT2 staining on the β‐cell membranes. Scale bar: 5 μm. D, Western blot for GLUT2 and Hsc70 (loading control) of isolated islet lysates showed no differences in GLUT2 amounts in total lysates of K8+/+ and K8+/− pancreas (lanes 1‐3, K8+/+; lanes 4‐6, K8+/−). (E) Quantification of GLUT2 immunoblotting from (D) balanced to loading control Hsc70 showed no significant changes between the genotypes
2.3. High‐fat diet induces no increased T2D susceptibility risk for K8+/− mice
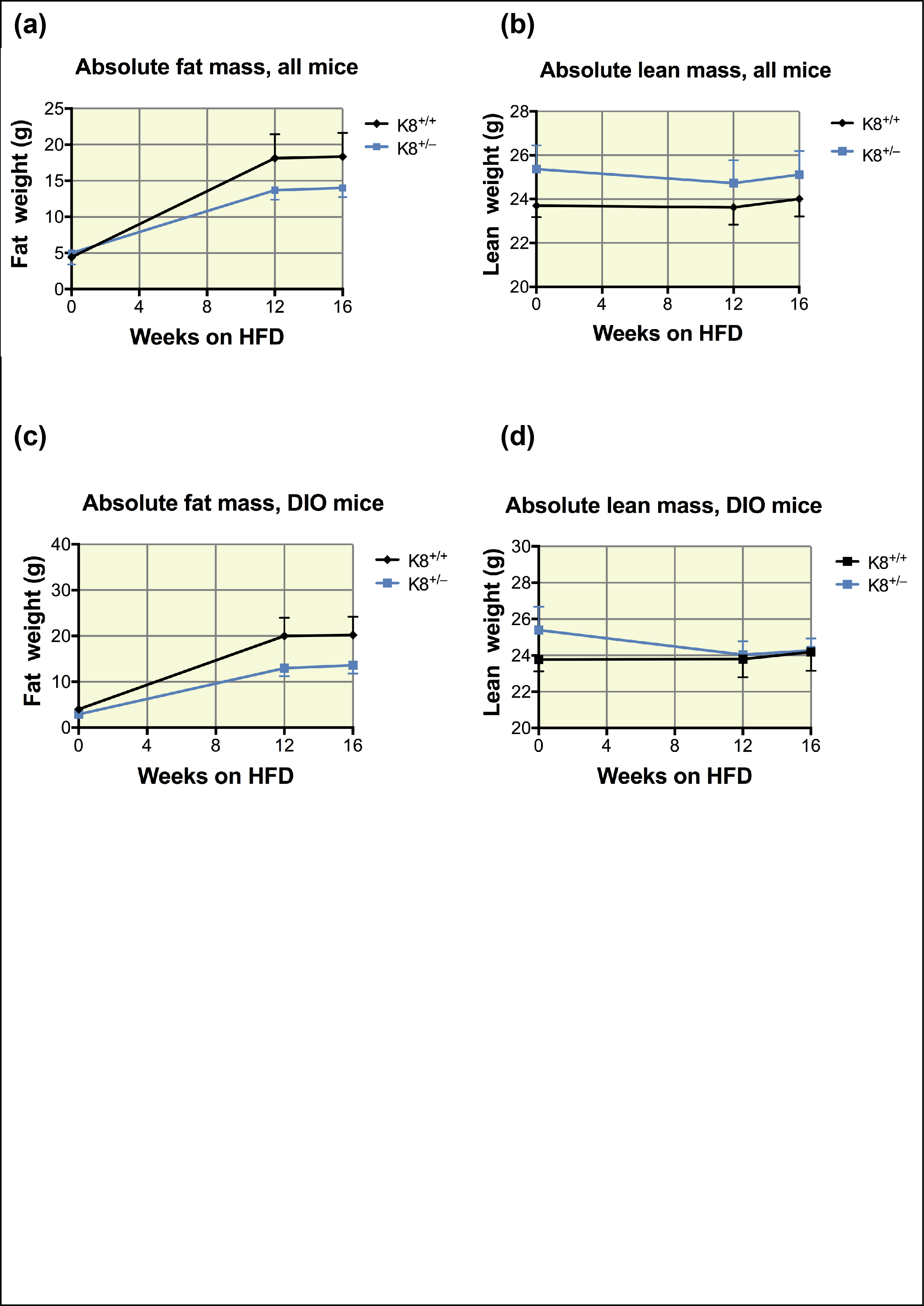
To further test the stress sensitivity of K8+/−, HFD was used as an experimental T2D model. While STZ‐induced diabetes develops due to the direct toxicity of STZ to cells, HFD provides a different type of “stress situation” caused by the metabolic imbalance, insulin resistance and indirectly β‐cell stress.26 While the body weight of mice steadily increased on average 20%‐25% during the 16‐week HFD treatment period as expected, there were no differences in body weight (Figure 6A) or body weight change (not shown) between K8+/+ and K8+/− mice. The fat mass index measured by EchoMRI also increased similarly on average 55% in both strains during the experiment (Figure 6B). Absolute fat mass and absolute lean mass analyses did not show any differences between the genotypes, when both all mice and diet‐induced obesity (DIO) mice were analysed (Figure S3). Blood glucose levels did not increase dramatically over the course of the HFD studies, and they were comparable between the genotypes over the 16‐week HFD treatment period. K8+/− mice showed no difference in blood glucose levels during HFD (week 1‐7, P = .32‐.91; week 8, P = .06; week 9, P = .15; week 10, P = .06; week 11‐16, P = .24‐.96) compared to their K8+/+ littermates (Figure 6C; consistent in two different sets of experiments, not shown). During 2 of these 3 weeks (weeks 8‐10) on HFD, 71% of K8+/− mice compared to 33% of K8+/+ had blood glucose levels higher than 10 mmol/L per L. Glucose tolerance analysis after 14 weeks of HFD revealed no differences between the genotypes (Figure 6D), and islet histology of K8+/− mice was unaltered compared to their K8+/+ littermates (Figure S4). Taken together, HFD elicits no major increased T2D susceptibility for K8+/− mice.
Figure 6.
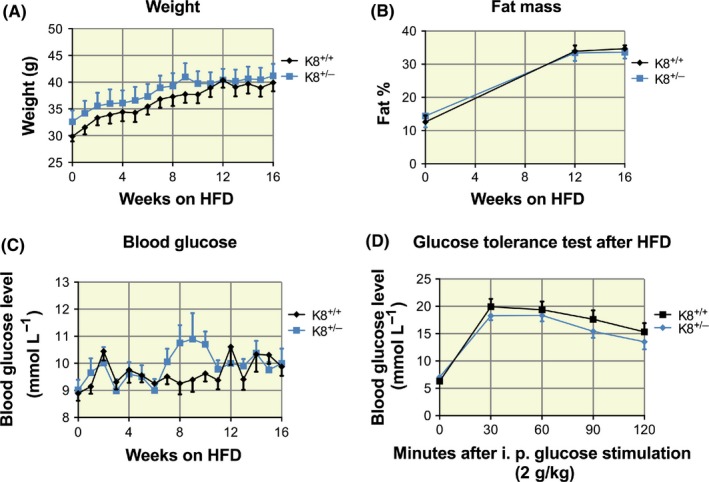
High‐fat diet induces no increased T2D susceptibility risk for K8+/− mice. Four‐ to 6‐mo‐old male K8+/+ (n = 9) and K8+/− mice (n = 7) were treated for a HFD regimen for 16 wk. A, K8+/+ and K8+/− mice show similarly increased body weight in response to HFD, calculated by two‐way ANOVA. B, Fat % of K8+/+ and K8+/− mice (measured by EchoMRI) similarly increased in response to HFD (two‐way ANOVA). C, Analysed by two‐way ANOVA, K8+/− mice showed no statistical difference in non‐fasting blood glucose values in response to 8‐10 wk of HFD (wk 8, P = .06; wk 9, P = .15; wk 10, P = .06) compared to K8+/+. D, Glucose tolerance test of K8+/+ and K8+/− mice after HFD showed no changes in fasting glucose levels before and after 2 g/kg i. p. glucose stimulation analysed by two‐way ANOVA
3. DISCUSSION
The present study shows that decreased levels of intermediate filament keratins predispose pancreatic endocrine islet cells to stress‐induced injury and diabetes. The K8+/− mouse model used in this study has an over 50% reduction in endocrine pancreatic K8 and its dimerization partner K18. In colonic epithelial cells of K8+/− mice that exhibit a similar 50% keratin reduction, the K8 loss is partially compensated by an increased expression of the only other type II simple epithelial keratin, K7.27 In pancreatic islets, however, no such compensatory keratin expression could be detected in K8+/− mice. Moreover, despite the reduction in islet keratin levels in K8+/− mice, normal islet functions, including mitochondrial MFN2 and membrane‐bound GLUT2, as well as normal glucose/insulin regulation can still be maintained under basal conditions. Our findings thus show that K8 is the most important type II simple epithelial keratin in islets, but that a partial reduction in K8 does not jeopardize β‐cell functions at basal conditions.
Although K8‐compromised K8+/− mice showed no aberrancies under basal circumstances, they were significantly more sensitive to the T1D model STZ. After STZ exposure, K8+/− mice suffered more injury to the pancreatic islets, displayed earlier and higher diabetes incidence and displayed a more severe disease profile, including increased blood glucose levels and increased polyuria compared with diabetic STZ‐treated K8+/+ mice. Increased disease susceptibility to colitis has previously been described in K8+/−,27 as well as increased sensitivity to liver toxicity in mice overexpressing the human K8 G62C liver disease mutant.28 These findings support the now well‐accepted notion that simple epithelial keratin mutations do not, per se, cause but predispose to disease.10, 29 K8‐compromised K8+/− mice should, thus, be a useful model to study functional keratin‐level–related disease mechanisms.
In contrast to the increased STZ sensitivity, K8+/− mice showed no clear susceptibility to the HFD model. Supportive of that only a mild keratin‐related phenotype occurs after a high‐fat regimen, is that K8+/− and K8+/+ gall bladders and livers responded similarly to a high‐fat lithogenic gallstone‐inducing diet,19 while HFD induced hepatocellular injury in susceptible K8−/− mice.30 HFD, which has a systemic metabolic effect on the organism, is likely to primarily reflect liver health and not pancreatic islet health. As blood glucose levels were only moderately increased after HFD in our study, the stress on the endocrine pancreas was probably minor, as can also be extrapolated from the unchanged pancreatic histology in both K8+/+ and K8+/− mice in our study.
In a recent study, K8 was suggested to be a negative regulator of TLR‐mediated signalling, as it was shown that K8 downregulation in mice leads to increased TLR responses, higher inflammatory cytokine levels and more inflammatory tissue injury in response to lipopolysaccharide‐induced sepsis.31 It is possible that the aggravated diabetes in K8+/− mice in the present study also, in part, may stem from upregulated inflammatory responses due to increased TLR signalling in the K8‐compromised mice, and this potential association warrants further study. The present study shows that the genetically downregulated K8 expression causes increased sensitivity to STZ‐induced diabetes. We have previously shown that total K8 ablation in K8−/− mice leads to GLUT2 mislocalization from the membrane to the cytoplasm correlating with a delayed response to acute STZ toxicity,20 as STZ is taken up into the β‐cells by GLUT2. K8+/− islets, however, have normal membrane‐bound distribution of GLUT2 readily allowing import of STZ and hence causing aggravated injury in the K8+/− β‐cells. This is in contrast to K8−/− mice, where mistargeted GLUT2 correlates with delayed sensitivity to STZ.20 These data support that GLUT2 must be correctly located at the membrane for STZ to have effect and that the relatively low keratin levels in K8+/− islet cells are still sufficient for GLUT2 membrane targeting.
Keratins are well‐known stress proteins that are rapidly regulated both through changes in expression levels and through post‐translational modifications, such as phosphorylation and glycosylation, in response to cell stress or injury.32, 33 However, also during normal physiological processes including cell movement and division, keratins are actively assembled, disassembled and remodelled in the cells.34 We have previously reported that K8 is increased in the pancreatic β‐cells during the early stages of diabetes development in non‐obese diabetic mice as well as in STZ‐induced diabetes.20 In this study, although K8+/− mice also appeared to be able to upregulate K8 in response to STZ exposure, K8+/− mice still sustained more injury to the pancreatic islet cells and developed more severe diabetes compared to K8+/+ mice, indicating that the K8 stress response to sustained β‐cell injury is inadequate in these mice. A recent theory connecting the genetic predisposition in diabetes with environmental triggers is the “β‐cell fragility model.”35 This model suggests that an increased susceptibility to β‐cell stress factors underlies diabetes development both in T1D and T2D. According to the model, β‐cells have differential sensitivity to β‐cell stress factors, depending on the genetic variations in the cells. Consistent with this idea, Dooley et al36 reported that defects in two different genes that regulate the apoptosis or DNA damage repair pathways are linked with both T1D and T2D. These genetic defects render the β‐cells more vulnerable to stress‐induced injury, thus accelerating diabetes development.36 Our results suggest that a reduced synthesis of K8 in K8+/− mice interferes with epithelial cell stress responses and renders these mice more susceptible to diabetes development in response to pancreatic stress caused by STZ. K8, thus, protects from β‐cell fragility and may compose a novel factor that interlinks the genetics and environmental triggers for diabetes.
4. MATERIALS AND METHODS
4.1. Experimental animals
Sex‐ and age‐matched K8+/+ and K8+/− mice in the FVB/n mouse background were used throughout this study. All animals were bred and raised at the central animal laboratory at the University of Turku, Turku, Finland. All animals used for experiments were 4‐6 months of age (HFD mice were 4‐6 months at the start of the experiment). At the end of the experiments, mice were sacrificed by CO2 inhalation. Animal experiments were approved by the National Animal Experiment Board and conformed to the regulations set by The Finnish Act on Animal Experimentation.
4.2. Fasting glucose measurement, glucose tolerance and insulin tolerance tests
Fasting blood glucose was measured in K8+/+ and K8+/− mice after 16 hours (overnight) of fasting, using a hand‐held glucose monitor (Contour XT, Bayer, Leverkusen, Germany). Glucose tolerance tests in these mice were performed after 18 hours (overnight) fasting followed by 2 g/kg, i.p. glucose (Sigma‐Aldrich, Saint Louis, MI, USA) administration. Blood glucose was measured before and at 30‐minute intervals up to 150 minutes after glucose administration. Insulin tolerance tests were performed by fasting mice for 4 hours, then administering 0.75 U/kg insulin by i.p. (Humalog, Lilly, Indianapolis, IN, USA), after which blood glucose was monitored before and at indicated intervals up to 2 hours after insulin challenge.
4.3. Insulin enzyme‐linked immunosorbent assay
Blood was collected from the submandibular vein with Goldenrod lancets (MEDIpoint International, New York, NY, USA) from K8+/+ and K8+/− mice fasted for 4 hours. The blood was allowed to clot, after which the serum was separated from the blood by centrifugation. The insulin concentration was determined according to manufacturer's recommendations, using a Mouse Ultrasensitive Insulin ELISA kit (Alpco, Salem, NH, USA).
4.4. Insulin extraction from pancreatic tissue
For measurement of insulin levels from pancreas samples, the acid‐ethanol method was used to extract insulin from the pancreas. Mice were sacrificed by CO2 inhalation, and pancreata were collected in acid‐ethanol, containing 1.5% vol/col concentrated HCl (Sigma‐Aldrich, Saint Louis, MI, USA) in 70% ethanol (20 μL solvent/mg pancreas tissue) and homogenized using a TissueRuptor homogenizer (Qiagen, Hilden, Germany). The homogenate was incubated under constant stirring at 4°C overnight to extract the insulin. Samples were then centrifuged at 13 000 g for 30 minutes, and supernatants were collected for insulin measurements. The insulin concentrations in the serum and in pancreas samples were determined with ELISA as described above.
4.5. STZ treatments and urine collection
STZ (Sigma‐Aldrich) was administered i.p. to mice in daily doses of 40 mg/kg/d for 5 consecutive days. The animals were fasted 4 hours before the first injection. Blood glucose levels were measured from the tail vein before STZ administration, then daily between day 7 and 14 and thereafter once a week. Animals were considered diabetic when 2 consecutive blood glucose measurements exceeded 14 mmol/L per L. Urine was collected from control mice (basal condition) and from diabetic mice treated with STZ on day 34 after the start of the treatment. Urine was collected for a 12‐hour period when mice were housed in individual metabolic cages, after which the urine volume in mL was recorded. STZ‐treated animals were sacrificed by CO2 inhalation, and samples were collected 15 or 35 days after the start of the experiment.
4.6. Pancreas histology
Pancreata from STZ‐treated K8+/+ and K8+/− mice were dissected and fixed in paraformaldehyde (4% [vol./vol.] in PBS, pH 7.4) for preparation of haematoxylin‐eosin–stained paraffin‐embedded sections. Islet damage was scored according to Ref. (20) from 0 to 3, where 0 = no apparent damage, 1 = loss of ≤10% of islet cells, 2 = 10%‐25%, 3 = 25%‐50% and 4 = ≥50% islet cell loss. A minimum of 20 islets per mouse were scored, and a mean islet damage score was calculated for the K8+/+ and K8+/− mice. Pancreata from K8+/+ and K8+/− mice subjected to HFD were frozen in tissue embedding compound (O.C.T. Compound, Tissue‐Tek, Alphen aan den Rijn, the Netherlands). Cryosections (7 μm thick) were cut and further fixed in acetone for 10 minutes at −20°C. Sections were stained with haematoxylin and counterstained in 0.5% eosin, after which sections were dehydrated and mounted with a xylene‐based mounting media.
4.7. Immunofluorescence staining
Pancreata were dissected from K8+/+ and K8+/− mice and frozen in O.C.T, sectioned as described above and stained using specific antibodies as described in Alam et al.20 Mouse anti‐K7 (RCK 105; Progen, Heidelberg, Germany), rat anti‐K8 (Troma I; Developmental Studies Hybridoma Bank, NIH, USA), rabbit anti‐K18 (275, kind gift from Professor J.E. Eriksson), goat anti‐insulin (polyclonal; Santa Cruz Biotechnology, Dallas, TX, USA), rabbit anti‐insulin (polyclonal; Santa Cruz Biotechnology) and rabbit anti‐GLUT2 (Polyclonal; Millipore, Temecula, CA, USA) antibodies were used to stain the pancreas tissue. Cell nuclei were counterstained with DRAQ5 (Sigma‐Aldrich, Saint Louis, MI, USA). Secondary antibodies used were as follows: donkey anti‐rabbit Alexa 546, donkey anti‐goat Alexa 488, donkey anti‐rat Alexa 488 and goat anti‐mouse Alexa 488 (Molecular Probes, Eugene, OR, USA). The sections were mounted with ProLong Gold antifade reagent (Invitrogen, Carlsbad, CA, USA). Images were acquired under strict comparable conditions between genotypes and treatments using a SP5 confocal microscope (Leica, Wetzlar, Germany).
4.8. Islet isolation
Isolation of murine islets of Langerhans was performed according to a modified protocol Zmuda et al, 201137 except that 1.5 mg/mL collagenase p (Roche, Mannheim, Germany) was injected through the common bile duct to bloat and digest the pancreas. Islets were hand‐picked, on ice under a dissection microscope, for lysate preparation.
4.9. SDS‐PAGE and western blotting
Isolated and hand‐picked islets from K8+/+ and K8−/− mice were homogenized with a 1‐mL syringe (BD, Franklin Lakes, NJ, USA) and 30‐G needle (Henke Sass Wolf, Tuttlingen, Germany), and samples were prepared for SDS‐PAGE and Western blotting as described.20 Primary antibodies used were as follows: rabbit anti‐insulin (Santa Cruz Biotechnologies), rat anti‐K8 (Troma I; Developmental Studies Hybridoma Bank), rabbit anti‐K18 (275, kind gift from Professor J.E. Eriksson), rat anti‐Hsc70 (Stressgen Bioreagents, Ann Arbor, MI, USA), rabbit anti‐GLUT2 (Polyclonal; Millipore) and rabbit anti‐MFN2 (Sigma‐Aldrich, St. Louis, MO, USA). Anti‐rabbit HRP (Promega Biosciences, San Luis Obispo, CA, USA) and anti‐rat HRP (GE Healthcare, Little Chalfont, UK) secondary antibodies were used. The signal on PVDF‐membranes was developed with ECL developing solution (GE Healthcare) and further exposed to X‐ray films (Fuji, Tokyo, Japan). The Western blot films were then analysed with IMAGE J software (NIH) for individual band quantification.
4.10. High‐fat diet
Four‐ to 6‐month‐old male K8+/+ (n = 9) and K8+/− mice (n = 7) were fed with high‐fat diet (HFD; D12492, Research diets, New Brunswick, NJ, USA) for 16 weeks with unlimited access to water. The macronutrient composition of the diet by kcal% was 20% protein, 20% carbohydrate and 60% fat, with an energy content of 5.24 kcal/g. Food, water, cages and bedding were changed every 7 days. Weight and blood glucose levels were measured every 7 days at fixed time points. Blood glucose levels (non‐fasting) were measured using a glucose monitor (Contour, Bayer, Leverkusen, Germany) from blood obtained from the tail vein. Fat and lean mass were measured at weeks 0, 12 and 16 with EchoMRI (EchoMRI LLC, Houston, TX, USA). Fat % was calculated according to: (fat mass/mouse weight)*100, and lean % was calculated according to (lean mass/mouse weight)*100. Absolute values for fat/lean masses were obtained from the EchoMRI device. Similarly, individuals that developed diet‐induced obesity (DIO) were analysed, excluding non‐responders. For this analysis, mice that increased their absolute fat weight more than 3 times from the beginning of the experiment until 16 weeks of HFD were included (n = 7 for K8+/+ and n = 5 for K8+/‐). After 14 weeks of HFD, glucose tolerance test was carried out as previously described and mice were fasted 17 hours before the test. After 16 weeks, the experiment was ended and the mice were sacrificed by CO2 inhalation.
4.11. Statistics
Statistical calculations were performed using GRAPHPAD PRISM and Student's t test, one‐way and regular or repeated measures two‐way ANOVA, log‐rank test and Bonferroni post hoc tests as appropriate. Statistical analysis for diabetes incidence was performed using the Kaplan–Meier test.
CONFLICT OF INTEREST
The authors have no conflict of interests to declare.
AUTHOR CONTRIBUTION
C.M.A. designed and performed experiments, analysed data and wrote the manuscript. J.S.G.S. designed experiments, performed experiments, analysed data and edited the manuscript. T.O.H designed experiments, performed experiments, analysed data, wrote and edited the manuscript. D.M.T. designed the study, analysed data and edited the manuscript.
Supporting information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGEMENTS
We thank Aruna Ghimire, Keshav Thapa, Ciaran Butler‐Hallissey, Anup Shrestha, Taina Heikkilä (Åbo Akademi University, ÅAU), the personnel at the Central Animal Laboratory (University of Turku) and members of the Toivola laboratory (Biosciences, Faculty of Science and Engineering, ÅAU) for skilful assistance and joint maintenance of the mouse colony. This work was funded by the Swedish Cultural Foundation (to J.S.G.S. and T.O.H.) and Doctoral Network in Molecular Biosciences at ÅAU (to J.S.G.S.) and The Novo Nordisk Foundation, The Finnish Diabetes Research Foundation, Diabetes Wellness, The Sigrid Juselius Foundation and The Academy of Finland (to D.M.T.).
Alam CM, Silvander JSG, Helenius TO, Toivola DM. Decreased levels of keratin 8 sensitize mice to streptozotocin‐induced diabetes. Acta Physiol. 2018;224:e13085 10.1111/apha.13085
J. S. G. Silvander and T. O. Helenius equally contributed.
REFERENCES
- 1. Murea M, Ma L, Freedman BI. Genetic and environmental factors associated with type 2 diabetes and diabetic vascular complications. Rev Diabet Stud. 2012;9:6‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vaarala O, Atkinson MA, Neu J. The, “perfect storm” for type 1 diabetes: the complex interplay between intestinal microbiota, gut permeability, and mucosal immunity. Diabetes. 2008;57:2555‐2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Atkinson MA, Eisenbarth GS, Michels AW. Type 1 diabetes. Lancet. 2014;383:69‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Prasad RB, Groop L. Genetics of type 2 diabetes‐pitfalls and possibilities. Genes (Basel). 2015;6:87‐123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Clayton DG. Prediction and interaction in complex disease genetics: experience in type 1 diabetes. PLoS Genet. 2009;5:e1000540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pitkaniemi J, Onkamo P, Tuomilehto J, Arjas E. Increasing incidence of Type 1 diabetes–role for genes? BMC Genet. 2004;5:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Omary MB, Ku NO, Strnad P, Hanada S. Toward unraveling the complexity of simple epithelial keratins in human disease. J Clin Invest. 2009;119:1794‐1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Omary MB. Intermediate filament proteins of digestive organs: physiology and pathophysiology. Am J Physiol Gastrointest Liver Physiol. 2017;312:G628‐G634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Toivola DM, Strnad P, Habtezion A, Omary MB. Intermediate filaments take the heat as stress proteins. Trends Cell Biol. 2010;20:79‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Toivola DM, Boor P, Alam C, Strnad P. Keratins in health and disease. Curr Opin Cell Biol. 2015;32:73‐81. [DOI] [PubMed] [Google Scholar]
- 11. Moll R, Divo M, Langbein L. The human keratins: biology and pathology. Histochem Cell Biol. 2008;129:705‐733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chamcheu JC, Siddiqui IA, Syed DN, Adhami VM, Liovic M, Mukhtar H. Keratin gene mutations in disorders of human skin and its appendages. Arch Biochem Biophys. 2011;508:123‐137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Szeverenyi I, Cassidy AJ, Chung CW, et al. The human intermediate filament database: comprehensive information on a gene family involved in many human diseases. Hum Mutat. 2008;29:351‐360. [DOI] [PubMed] [Google Scholar]
- 14. Strnad P, Paschke S, Jang KH, Ku NO. Keratins: markers and modulators of liver disease. Curr Opin Gastroenterol. 2012;28:209‐216. [DOI] [PubMed] [Google Scholar]
- 15. Odaka C, Loranger A, Takizawa K, et al. Keratin 8 is required for the maintenance of architectural structure in thymus epithelium. PLoS One. 2013;8:e75101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Omary MB. “IF‐pathies”: a broad spectrum of intermediate filament‐associated diseases. J Clin Invest. 2009;119:1756‐1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Baribault H, Penner J, Iozzo RV, Wilson‐Heiner M. Colorectal hyperplasia and inflammation in keratin 8‐deficient FVB/N mice. Genes Dev. 1994;8:2964‐2973. [DOI] [PubMed] [Google Scholar]
- 18. Baribault H, Price J, Miyai K, Oshima RG. Mid‐gestational lethality in mice lacking keratin 8. Genes Dev. 1993;7:1191‐1202. [DOI] [PubMed] [Google Scholar]
- 19. Tao GZ, Toivola DM, Zhong B, et al. Keratin‐8 null mice have different gallbladder and liver susceptibility to lithogenic diet‐induced injury. J Cell Sci. 2003;116(Pt 22):4629‐4638. [DOI] [PubMed] [Google Scholar]
- 20. Alam CM, Silvander JS, Daniel EN, et al. Keratin 8 modulates beta‐cell stress responses and normoglycaemia. J Cell Sci. 2013;126(Pt 24):5635‐5644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mathew J, Loranger A, Gilbert S, Faure R, Marceau N. Keratin 8/18 regulation of glucose metabolism in normal versus cancerous hepatic cells through differential modulation of hexokinase status and insulin signaling. Exp Cell Res. 2013;319:474‐486. [DOI] [PubMed] [Google Scholar]
- 22. Silvander JSG, Kvarnstrom SM, Kumari‐Ilieva A, Shrestha A, Alam CM, Toivola DM. Keratins regulate beta‐cell mitochondrial morphology, motility, and homeostasis. FASEB J. 2017;31:4578‐4587. [DOI] [PubMed] [Google Scholar]
- 23. Ahmed M, Bergsten P. Glucose‐induced changes of multiple mouse islet proteins analysed by two‐dimensional gel electrophoresis and mass spectrometry. Diabetologia. 2005;48:477‐485. [DOI] [PubMed] [Google Scholar]
- 24. Blessing M, Ruther U, Franke WW. Ectopic synthesis of epidermal cytokeratins in pancreatic islet cells of transgenic mice interferes with cytoskeletal order and insulin production. J Cell Biol. 1993;120:743‐755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Katsarou A, Gudbjornsdottir S, Rawshani A, et al. Type 1 diabetes mellitus. Nat Rev Dis Primers. 2017;3:17016. [DOI] [PubMed] [Google Scholar]
- 26. Graham ML, Janecek JL, Kittredge JA, Hering BJ, Schuurman HJ. The streptozotocin‐induced diabetic nude mouse model: differences between animals from different sources. Comp Med. 2011;61:356‐360. [PMC free article] [PubMed] [Google Scholar]
- 27. Asghar MN, Silvander JS, Helenius TO, et al. The amount of keratins matters for stress protection of the colonic epithelium. PLoS One. 2015;10:e0127436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ku NO, Omary MB. A disease‐ and phosphorylation‐related nonmechanical function for keratin 8. J Cell Biol. 2006;174:115‐125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Strnad P, Stumptner C, Zatloukal K, Denk H. Intermediate filament cytoskeleton of the liver in health and disease. Histochem Cell Biol. 2008;129:735‐749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kucukoglu O, Guldiken N, Chen Y, et al. High‐fat diet triggers Mallory‐Denk body formation through misfolding and crosslinking of excess keratin 8. Hepatology. 2014;60:169‐178. [DOI] [PubMed] [Google Scholar]
- 31. Dong XM, Liu ED, Meng YX, et al. Keratin 8 limits TLR‐triggered inflammatory responses through inhibiting TRAF6 polyubiquitination. Sci Rep. 2016;6:32710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ku NO, Toivola DM, Strnad P, Omary MB. Cytoskeletal keratin glycosylation protects epithelial tissue from injury. Nat Cell Biol. 2010;12:876‐885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kirfel J, Magin TM, Reichelt J. Keratins: a structural scaffold with emerging functions. Cell Mol Life Sci. 2003;60:56‐71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Windoffer R, Beil M, Magin TM, Leube RE. Cytoskeleton in motion: the dynamics of keratin intermediate filaments in epithelia. J Cell Biol. 2011;194:669‐678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liston A, Todd JA, Lagou V. Beta‐cell fragility as a common underlying risk factor in type 1 and type 2 diabetes. Trends Mol Med. 2017;23:181‐194. [DOI] [PubMed] [Google Scholar]
- 36. Dooley J, Tian L, Schonefeldt S, et al. Genetic predisposition for beta cell fragility underlies type 1 and type 2 diabetes. Nat Genet. 2016;48:519‐527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zmuda EJ, Powell CA, Hai T. A Method for Murine Islet Isolation and Subcapsular Kidney Transplantation. J. Vis. Exp. 2011;50:e2096, 10.3791/2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials