

Abstract
Accumulation of bile salts (BSs) during cholestasis leads to hepatic and biliary injury, driving inflammatory and fibrotic processes. The Na+‐Taurocholate Cotransporting Polypeptide (NTCP) is the major hepatic uptake transporter of BSs, and can be specifically inhibited by myrcludex B. We hypothesized that inhibition of NTCP dampens cholestatic liver injury. Acute cholestasis was induced in mice by a 3.5‐diethoxycarbonyl‐1.4‐dihydrocollidine (DDC) diet or by bile duct ligation (BDL). Chronic cholestasis was investigated in Atp8b1‐G308V and Abcb4/Mdr2 deficient mice. Mice were injected daily with myrcludex B or vehicle. Myrcludex B reduced plasma alkaline phosphatase (ALP) levels in DDC‐fed, Atp8b1‐G308V and BDL mice by 39%, 27% and 48% respectively. Expression of genes involved in fibrosis, proliferation and inflammation was reduced by myrcludex B treatment in DDC‐fed and Atp8b1‐G308V mice. NTCP‐inhibition increased plasma BS levels from 604±277 to 1746±719 μm in DDC‐fed mice, 432±280 to 762±288 μm in Atp8b1‐G308V mice and from 522±130 to 3625±378 μm in BDL mice. NTCP‐inhibition strongly aggravated weight loss in BDL mice, but not in other cholestatic models studied. NTCP‐inhibition reduced biliary BS output in DDC‐fed and Atp8b1‐G308V mice by ∼50% while phospholipid (PL) output was maintained, resulting in a higher PL/BS ratio. Conversely, liver injury in Abcb4 deficient mice, lacking biliary phospholipid output, was aggravated after myrcludex B treatment. Conclusion: NTCP‐inhibition by myrcludex B has hepatoprotective effects, by reducing BS load in hepatocytes and increasing the biliary PL/BS ratio. High micromolar plasma BS levels after NTCP‐inhibition were well tolerated. NTCP‐inhibition may be beneficial in selected forms of cholestasis. (Hepatology 2018).
Abbreviations
- ALP
alkaline phosphatase
- ALT
alanine amino transferase
- ASBT
apical sodium dependent bile salt transporter
- Atp8b1
p‐type ATPase 8b1
- BDL
bile duct ligation
- BS
bile salt
- BSEP
bile salt export pump
- BW
body weight
- C4
4‐cholesten‐3‐one
- CA
cholic acid
- Cdc25b
cell division control protein 25 b
- CK7
cytokeratin 7
- Col1a1
collagen 1a1
- CYP7A1
cytochrome p450 7A1
- DDC
3.5‐diethoxycarbonyl‐1.4‐dihydrocollidine
- F4/80
EGF‐like module‐containing mucin‐like hormone receptor‐like 1
- FGF15/19
fibroblast growth factor 15/19
- FXR
Farnesoid X receptor
- H&E
hematoxylin and eosin
- IL‐6
interleukin 6
- LW
liver weight
- Mcp1
monocyte chemotactic protein 1
- Mdr2
multidrug resistance protein 2
- MRP2‐4
multidrug resistance associated protein 2‐4
- NTCP
Na+‐taurocholate cotransporting polypeptide
- OATP
organic anion transporting protein
- PBC
primary biliary cholangitis
- PFIC
progressive familial intrahepatic cholestasis
- SHP
small heterodimer partner
- TβMCA
Tauro‐β‐muricholic acid
- TCA
tauro‐cholic acid
- TNF‐α
tumor necrosis factor‐α
- α‐SMA
α‐smooth muscle actin
Cholestatic liver diseases are characterized by impairment of bile formation and/or bile flow. Primary causes are (genetic) defects in bile formation at the canalicular membrane of hepatocytes, such as impaired phospholipid or bile salt excretion,1, 2 fibrosing inflammation of small and/or large bile ducts and mechanical obstruction of bile flow (e.g., cholelithiasis or obstruction by a tumor). As a consequence of impaired bile flow, (toxic) hydrophobic bile salts and other bile constituents damage cholangiocytes and accumulate in the hepatocyte, driving inflammatory reactions and disrupting cell membrane integrity.3, 4 Subsequent liver damage can progress to cirrhosis and liver failure, ultimately requiring liver transplantation.
The basolateral hepatic bile salt uptake machinery is downregulated in cholestatic conditions.5 Simultaneously, efflux transporters (e.g., bile salt export pump [BSEP]) and multi‐drug resistance‐associated protein 3‐4 (MRP3‐4) are upregulated.6 Furthermore, synthesis of new bile salts is repressed via farnesoid X receptor/short heterodimer partner (FXR/SHP)‐mediated downregulation of cytochrome P450 7A1 (CYP7A1).7 Such adaptations are attempts to lower intracellular concentrations of cytotoxic bile salts and other bile components to protect against further liver injury. Nevertheless, despite these physiological adjustments, cholestatic liver injury is often progressive and additional treatment strategies are employed. Endoscopic or surgical strategies include biliary tract stenting or balloon dilatation of fibrotic strictures aimed at the restoration of bile flow or, in rare cases, biliary diversion to lower the bile salt pool size. Furthermore, limited pharmacological treatments are available which slow the progression of cholestatic liver injury. So far ursodeoxycholic acid (UDCA), either alone or in combination with obeticholic acid (OCA) for primary biliary cholangitis (PBC), is the only clinically approved treatment for chronic cholestatic liver disease.8 Recently, pharmacological interruption of the enterohepatic transport of bile salts has been suggested as a means to attenuate cholestatic liver injury by reducing bile salt load on hepatocytes9 Indeed, apical sodium dependent bile salt transporter (ASBT) inhibitors reduce liver injury and fibrosis in Abcb4 deficient mice, a model of progressive familial intrahepatic cholestasis type 3 (progressive familial intrahepatic cholestasis [PFIC] 3) and sclerosing cholangitis,10, 11 and displayed a beneficial safety profile and reduced pruritus in PBC patients in a phase II trial.12 The effect of disruption of the enterohepatic circulation at the level of the basolateral membrane of the hepatocyte has not been tested as an anti‐cholestatic treatment strategy. Here, we hypothesize that reduction of hepatic bile salt uptake can dampen cholestatic liver injury by reducing hepatic BS load.
The Na+‐taurocholate cotransporting polypeptide (NTCP) is the major hepatic uptake transporter of conjugated bile salts, and deficiency of NTCP results in reduced clearance of bile salts from the circulation and increased systemic bile salt levels.13, 14 Administration of myrcludex B, a selective NTCP‐binding peptide currently used as hepatitis B and D virus entry inhibitor, also causes high systemic bile salt levels, which are tolerated very well in humans and rodents.15, 16, 17 We investigated whether myrcludex B reduces cholestatic liver injury in mice. Cholestasis was induced by 3,5‐diethoxycarbonyl‐1,4‐dihydrocollidine (DDC)‐feeding, bile duct ligation (BDL) or a consequence of genetic deficiencies associated with PFIC type 1 and 3 in humans. We demonstrate hepatoprotective effects of myrcludex B in certain models of experimental cholestasis which depend on a shift of conjugated BSs from hepatocytes to the circulation, in combination with preserved bile flow and Mdr2‐mediated phospholipid excretion in bile.
Materials and Methods
Detailed description of biochemical, histological and molecular measurements is provided in the Supporting material and methods.
ANIMAL STUDIES
Two‐month old male wild‐type mice in a C57Bl6/J background were purchased from Envigo (Venray, the Netherlands). Four mouse models for cholestasis were used to investigate possible hepatoprotective effects of NTCP‐inhibition. In all cholestasis models, subcutaneous injections of myrcludex B (2.5 μg/g BW) or placebo were given once daily. One cohort of mice was placed on a chow diet (D12450B1, Open Source Diets, USA), supplemented with 0.1% 3,5‐diethoxycarbonyl‐1,4‐dihydrocollidine (DDC, Sigma).18 After seven days DDC‐fed mice were sacrificed to investigate effects of myrcludex B treatment on the development of liver injury in this acute setting. A second cohort of mice was subjected to common bile duct ligation (BDL) and cholecystectomy, as described previously.19 BDL mice were sacrificed after five days because of animal welfare regulations (body weight loss>15%). Atp8b1‐G308V mutant mice (C57Bl6/J background) and Abcb4 knockout mice (FvB background), mouse models for PFIC type 1 and 3, were treated with myrcludex B to investigate effects of NTCP‐inhibition in a more chronic setting. Atp8b1‐G308V mice were fed a diet containing 0.1% cholate for 28 days to induce intrahepatic cholestasis, simultaneously mice were treated with placebo or myrcludex B. The cholestatic phenotype in Abcb4 knockout mice develops from early age. At the age of 6 weeks these mice were treated with placebo of myrcludex B for a period of 14 days. Mice were randomized to treatment using online randomization software and investigators were blinded for the treatments. Mice were sacrificed under anaesthesia with ketamine (Nimatek, 100 mg/kg) and xylazine (Sedamun, 10 mg/kg) 3 hours after the last Myrcludex B or placebo administration and fasted for 4‐6 hours in total. Blood was collected by cardiac puncture and plasma was separated by centrifugation at 800g for 10 minutes. Organs were fixed overnight in 4% paraformaldehyde or snap frozen in liquid N2 and stored at ‐80°C for further analysis. The study design and all protocols for animal care and handling were approved by the Institutional Animal Care and Use Committee of the University of Amsterdam. Experiments with the Abcb4 knockout mice were performed and approved by the local Animal Care and Use Committee in Vienna.
BILE DUCT CANNULATIONS
To investigate bile flow during cholestasis, gall bladder cannulation and bile collection was performed using a PE‐10 catheter. Bile was collected in aliquots every 10 minutes for 30 minutes after distal ligation of the common bile duct, as described by Slijepcevic, et al.14 Bile flow was determined gravimetrically assuming a density of 1 g/mL for bile. A heating pad maintained body temperature at 37°C.
STATISTICAL ANALYSIS
Data are provided as the mean ± standard error of the mean. Differences between groups were analyzed using Mann‐Whitney U. Statistical significance was considered when p< 0.05 and calculations and graphs were generated using GraphPad Prism 7.0 (GraphPad Software Inc., La Jolla, USA).
Results
NTCP‐INHIBITOR MYRCLUDEX B REDUCES CHOLESTATIC LIVER INJURY IN DDC‐TREATED AND ATP8B1‐G308V MICE
Myrcludex B treatment was well tolerated in DDC‐fed mice and Atp8b1‐G308V mice. DDC‐feeding induced gradual weight loss in all mice http://onlinelibrary.wiley.com/doi/10.1002/hep.29888/suppinfo. In liver sections of DDC‐fed mice luminal porphyrin plugs were observed, together with mild periportal ductular reaction and infiltration of inflammatory cells. These features were observed in both treatment groups (Fig. 1A). Marked elevation of hepatocellular damage marker alanine amino transferase ([ALT] Fig. 1B) and cholestatic marker alkaline phosphatase (ALP) was notable in vehicle‐treated mice (Fig. 1C), and both damage markers were clearly attenuated by myrcludex B treatment. Plasma bilirubin levels were not changed after myrcludex B treatment (Fig. 1D). In line with the biochemical findings, liver weight to BW ratio was lower in myrcludex B treated DDC‐fed mice (Fig. 1E).
Figure 1.
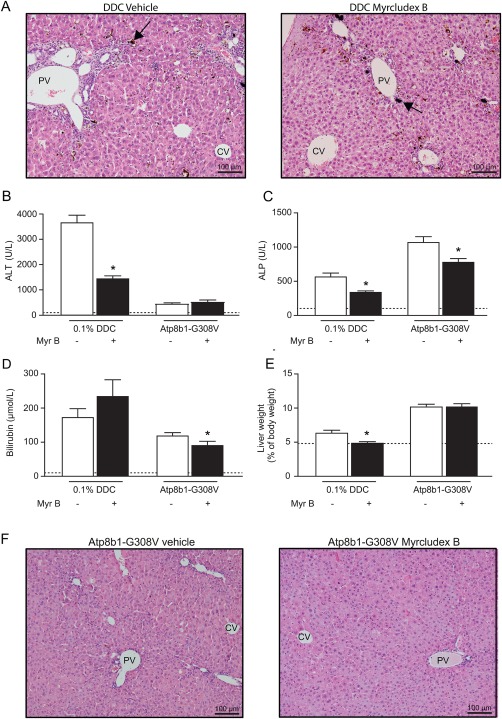
Reduced cholestatic liver injury by myrcludex B in DDC and PFIC1 models. (A) Liver H&E staining showing vehicle and myrcludex B treatment in DDC‐fed wild‐type mice. Black arrows indicate porphyrin plugs. (B‐C). Plasma biochemistry displaying degree of parenchymal liver injury (ALT in U/L) and cholestasis (ALP in U/L) in DDC‐fed mice and Atp8b1‐G308V mice. Dotted lines indicate physiological levels of the indicated enzymes. (D) Total bilirubin levels in plasma in DDC‐fed mice and Atp8b1‐G308V mutant mice. (E) Liver to BW ratio in DDC‐fed mice and Atp8b1‐G308V mice. (F) Liver H&E staining of Atp8b1‐G308V mice treated with vehicle or myrcludex B, compaction of hepatocytes is noted around periportal areas. Asterisk indicates significant changes (n=8‐14 mice/group). Abbreviations: CV, central vein; PV, portal vein.
Atp8b1‐G308V mice develop intrahepatic cholestasis upon cholate feeding.20 Histological evaluation by hematoxylin and eosin (H&E) staining of livers of Atp8b1‐G308V mice after 4 weeks of a diet supplemented with 0.1% cholate showed compaction of hepatocytes around periportal areas and mild ductular reaction, but no signs of fibrosis (Fig. 1F). Plasma ALT levels were only moderately increased compared to DDC‐induced cholestasis and not changed by myrcludex B treatment (Fig. 1B). However, there was a clear reduction of ALP and bilirubin levels after myrcludex B treatment (Fig. 1C,D). Food intake by Atp8b1‐G308V mice was comparable between groups, also liver weights and BWs were similar between both treatment groups (Fig. 1E and http://onlinelibrary.wiley.com/doi/10.1002/hep.29888/suppinfo.
NTCP‐INHIBITION DAMPENS PROLIFERATIVE, INFLAMMATORY AND FIBROTIC PROCESSES IN DDC‐FED MICE AND (TO LESSER EXTENT IN) ATP8B1‐G308V MICE
We investigated markers of inflammation, fibrosis and proliferation in more detail and assessed whether these were affected by myrcludex B treatment. We first analyzed expression of inflammatory markers in DDC‐fed mice. Myrcludex B treatment reduced Tnf‐α, Mcp1 and Il‐6 mRNA levels (Fig. 2A). Hepatic F4/80 mRNA level was reduced as well, however neither macrophage influx nor markers for stellate cell activation were altered after myrcludex B treatment on histological examination http://onlinelibrary.wiley.com/doi/10.1002/hep.29888/suppinfo. Investigation of ductular proliferation by CK7‐staining suggested that periportal ductular reaction was less pronounced in myrcludex B treated mice, although this did not reach significance (p=0.28; http://onlinelibrary.wiley.com/doi/10.1002/hep.29888/suppinfo). Only early signs of fibrotic changes were present in livers of DDC‐fed mice based on Sirius Red staining, but mRNA levels of Col1a1 and other pro‐fibrotic genes clearly demonstrated reduced fibrotic processes after myrcludex B treatment (Fig. 2B,C). In line with the attenuated inflammatory and fibrotic state, expression of proliferative genes was reduced and less Ki67 positive hepatocytes were present in livers of myrcludex B treated DDC‐fed mice (Fig. 2D,E).
Figure 2.
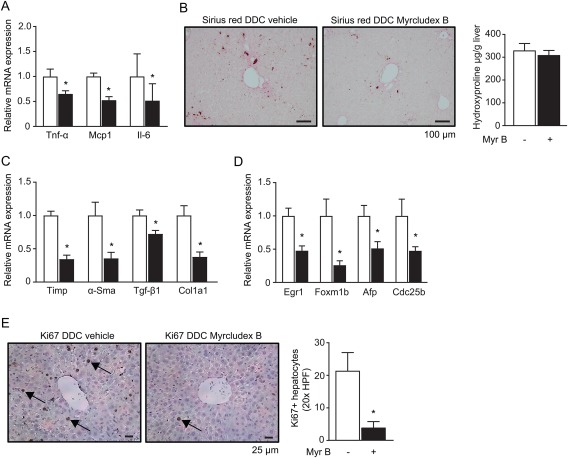
Specific immunohistochemistry and quantification of cholestatic liver injury and proliferation upon Myrcludex B. (A) Hepatic mRNA levels of inflammatory cytokines in DDC‐fed mice: Tnf‐α, Mcp‐1 and Il‐6. White bars represent vehicle treated mice, black bars represent myrcludex B treated mice. (B) Sirius red staining of livers from DDC‐fed mice treated with vehicle (left) or myrcludex B (right). (C) mRNA expression of pro‐fibrotic genes in the livers of DDC‐fed mice. (D) mRNA expression of genes involved in proliferation in livers of DDC‐fed mice. (E) Ki‐67+ staining on liver sections of DDC‐fed mice. 20x High‐power field (HPF), arrows indicate Ki67+ hepatocytes. (Right panel) Quantification of Ki67+ hepatocytes around central areas in the liver. Values are normalized to vehicle‐treated DDC‐fed. Asterisk indicates significant changes (n=8 mice/group).
In Atp8b1‐G308V mutant mice the number of CK7‐positive cells and mRNA levels of Ck7 as well as hepatic inflammatory markers Tnf‐α and Mcp1 all tended to decrease by myrcludex B treatment http://onlinelibrary.wiley.com/doi/10.1002/hep.29888/suppinfo. However, hepatic inflammation was hardly present in this model, and macrophage or hepatic stellate cell activation were unaltered by myrcludex B treatment http://onlinelibrary.wiley.com/doi/10.1002/hep.29888/suppinfo.
MYRCLUDEX B TREATMENT FURTHER RAISES PLASMA BILE SALT CONCENTRATIONS AND REDUCES INTRACELLULAR BILE SALT ACCUMULATION DURING CHOLESTASIS
The induction of cholestasis resulted in elevated plasma BS levels in DDC‐fed mice. Myrcludex B treatment further increased total BS levels by ∼3‐fold in DDC‐fed mice (p<0.01) and ∼2‐fold in Atp8b1‐G308V mutant mice (p<0.01; Fig. 3A), demonstrating potent inhibition of hepatic BS uptake. Plasma BSs mostly consisted of TCA after myrcludex B treatment, which was relatively more abundant than TβMCA. Myrcludex B treatment also led to higher plasma levels of the unconjugated BS CA in DDC‐fed mice (Fig. 3B). Fecal bile salt output was unaffected by myrcludex B treatment in both DDC‐fed and Atp8b1‐G308V mutant mice http://onlinelibrary.wiley.com/doi/10.1002/hep.29888/suppinfo. Renal Asbt was reduced and urinary bile salts showed a trend to increase directly after myrcludex B treatment only in DDC‐fed mice, without changes in renal function http://onlinelibrary.wiley.com/doi/10.1002/hep.29888/suppinfo.
Figure 3.
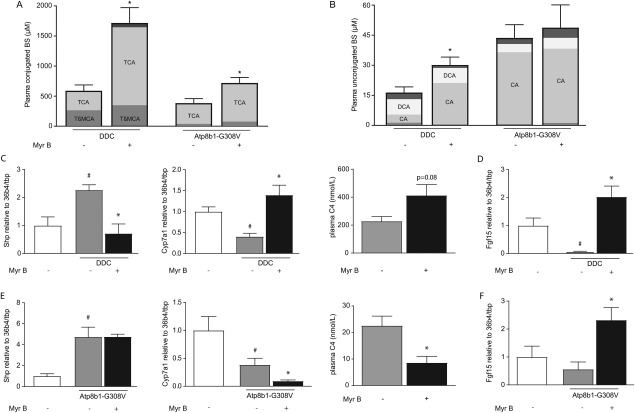
Myrcludex B treatment further raises plasma bile salt concentrations and reduces intracellular bile salt accumulation during cholestasis. Conjugated (A) and unconjugated (B) BS concentrations in DDC‐fed mice and Atp8b1‐G308V mutant mice. (C) mRNA expression levels of Shp and Cyp7a1 compared to untreated non‐cholestatic mice, and plasma levels of C4 in DDC‐fed mice. (D) mRNA expression level of Fgf15 in the terminal ileum of DDC‐fed mice compared to untreated non‐cholestatic mice. (E) mRNA expression levels of Shp and Cyp7a1 compared to untreated non‐cholestatic mice, and plasma levels of C4 in Atp8b1‐G308V mutant mice. (F) mRNA expression level of Fgf15 in the terminal ileum of Atp8b1‐G308V mutant mice compared to untreated non‐cholestatic mice. Asterisk indicates significant change compared to the cholestatic mouse group, hashtag indicates significant change compared to the non‐cholestatic mouse group (n=8‐14 mice/group).
To further evaluate whether myrcludex B treatment reduces intracellular bile salt accumulation during cholestasis, we analyzed hepatic mRNA levels of Fxr‐target genes. Myrcludex B treatment resulted in increased mRNA expression of Ntcp in both cholestasis models http://onlinelibrary.wiley.com/doi/10.1002/hep.29888/suppinfo, whereas mRNA levels of Oatps were unaffected, except for Oatp1a4 expression in DDC‐fed mice, which was increased http://onlinelibrary.wiley.com/doi/10.1002/hep.29888/suppinfo. We then assessed genes involved in bile salt synthesis, a process which is regulated via Fxr‐Shp and Fxr‐fibroblast growth factor 15 (Fgf15) mediated feedback loops in respectively the liver and intestine.21, 22 bile salt synthesis was restored to non‐cholestatic levels upon myrcludex B treatment in DDC‐fed mice, illustrated by the clear repression of Shp expression, induced Cyp7a1 expression and slightly elevated plasma 4‐cholesten‐3‐one (C4) levels compared to vehicle‐treated cholestatic mice (Fig. 3C; plasma C4 in non‐cholestatic mice: 91 ± 36 nmol/L). Similar to Cyp7a1, intestinal Fgf15 expression was completely repressed in vehicle‐treated DDC‐fed mice, whereas it was restored by myrcludex B to non‐cholestatic levels (Fig. 3D). In the Atp8b1‐G308V mutant mice, cholate feeding increased hepatic Shp in both treatment groups and reduced Cyp7a1 expression and plasma C4 levels (Fig. 3E). Myrcludex B treatment further repressed BS synthesis, likely through increased intestinal Fgf15 expression (Fig. 3F).
MYRCLUDEX B TREATMENT IN BDL‐INDUCED CHOLESTASIS
Considering the hepatoprotective effects of NTCP‐inhibition in the DDC model and Atp8b1‐G308V mice, we wondered whether myrcludex B injections would also be beneficial in complete obstructive cholestasis. To study this, we performed BDL in mice, which inhibits bile flow and blocks removal of (toxic) bile constituents. After 5 days of BDL, plasma ALP and ALT levels were significantly lower in myrcludex B treated mice compared to vehicle treated cholestatic mice (Fig. 4A,B). Also, liver weight to BW ratio significantly improved after myrcludex B treatment (Fig. 4C). However, biliary infarcts were observed in the periportal areas in both groups and tended to be aggravated in myrcludex B treated mice (Fig. 4D). In addition, substantial BW loss was observed in myrcludex B treated mice (Fig. 4E). Plasma bile salt levels increased to ∼3.5 mM after myrcludex B treatment in this model, and mainly consisted of TCA (79%) (Fig. 4F; left panel). Plasma bilirubin levels were elevated after myrcludex B treatment during BDL (Fig. 4F; right panel) and both Cyp7a1 mRNA levels and plasma C4 levels were reduced after myrcludex B treatment http://onlinelibrary.wiley.com/doi/10.1002/hep.29888/suppinfo. Expression of Egr1, Foxm1b and Cdc25, genes involved in cell proliferation, was reduced http://onlinelibrary.wiley.com/doi/10.1002/hep.29888/suppinfo, CK7 and Sirius red staining indicated early signs of ductular proliferation and fibrosis, which was not affected by myrcludex B treatment http://onlinelibrary.wiley.com/doi/10.1002/hep.29888/suppinfo. Histological markers α‐SMA and F4/80 showed periductular hepatic stellate cell activation and minor macrophage infiltration, which was not changed after NTCP‐inhibition http://onlinelibrary.wiley.com/doi/10.1002/hep.29888/suppinfo. A summary of the myrcludex effects in the described models is provided as http://onlinelibrary.wiley.com/doi/10.1002/hep.29888/suppinfo.
Figure 4.
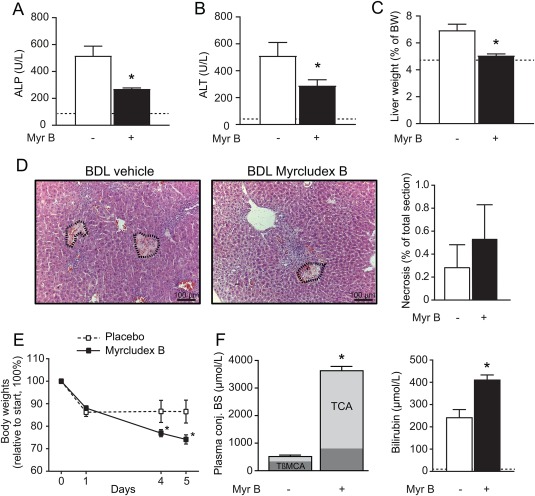
Myrcludex B improves plasma biochemistry during BDL‐cholestasis, at the expense of significant body weight loss. (A‐B) Plasma biochemistry displaying degree of parenchymal liver injury (ALT in U/L) and cholestasis (ALP in U/L) in BDL mice. White bars depict vehicle‐treated mice, black bars depict myrcludex B treated mice. (C) Liver to BW ratio in BDL mice. (D) H&E staining of the liver of vehicle and myrcludex B treated BDL mice. Areas of necrosis are indicated and quantified (right panel). (E) BW changes after start of BDL and daily vehicle or myrcludex B treatment. (F) Left panel showing conjugated BS levels in plasma of BDL mice treated with vehicle or myrcludex B. Right panel showing plasma total bilirubin levels in BDL mice. Asterisk indicates significant changes (n=6 mice/group).
BILIARY BILE SALT OUTPUT IS REDUCED BY MYRCLUDEX B TREATMENT, WHILE PHOSPHOLIPID OUTPUT IS STIMULATED
The ambiguous results of the BDL model suggested that the hepatoprotective effects of myrcludex B do not only depend on the reduction of intracellular bile salt accumulation. This prompted us to investigate the effects of NTCP‐inhibition on bile kinetics and composition. Myrcludex B treatment reduced bile flow in Atp8b1‐G308V mice and slightly lowered bile flow in DDC‐fed mice (Fig. 5A,B), with a concomitant lowering of biliary BS output, predominantly in the Atp8b1‐G308V mice (Fig. 5C,D). Similar to the plasma bile salt composition, myrcludex B rendered the biliary bile salts towards slightly less hydrophilic species, i.e., ratio TCA/TβMCA increased in both cholestasis models and tauro‐chenodeoxycholic acid (TCDCA) and tauro‐deoxycholic acid (TDCA) increased in DDC‐fed mice treated with myrcludex B (Table 1). Interestingly, biliary phospholipid output was increased in both DDC‐fed and Atp8b1‐G308V mice after myrcludex B treatment, resulting in a favorable ratio of biliary phospholipid to bile salts (Fig. 5E,F). The biliary bicarbonate output was unaffected by myrcludex B treatment in the DDC‐fed mice http://onlinelibrary.wiley.com/doi/10.1002/hep.29888/suppinfo.
Figure 5.
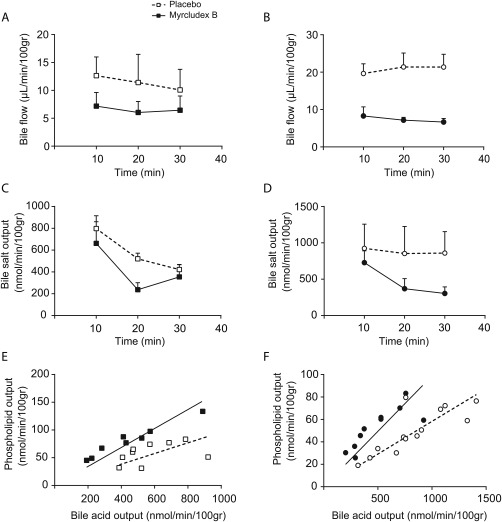
Bile flow and biliary BS output are reduced upon NTCP‐inhibition while phospholipid output is increased. (A‐B) Hepatic bile flow (μL/min/100g BW) determined at 10‐minute intervals during a 30‐minute period after gall bladder cannulation in DDC‐fed mice (A) and Atp8b1‐G308V mutant mice (B). (C‐D) Biliary bile salt output (nmol/min/100g BW) determined at 10‐minute intervals during a 30‐minute collection period in DDC‐fed mice (C) and Atp8b1‐G308V mutant mice (D). (E‐F) Correlation plot between biliary bile salt and phospholipid output in DDC‐fed mice (E) and Atp8b1‐G308V mutant mice (F) treated with vehicle (white squares) or myrcludex B (black squares). Data is from 3‐5 mice/group.
Table 1.
Biliary bile salt composition. The percentage of specific conjugated bile salt species of the total amount of biliary bile salts is indicated. n = 5‐14 mice per group. n.d. = not detectable. Asterisk indicates p‐value below 0.05.
| DDC‐fed | Atp8b1‐G308V | Abcb4 ‐/‐ | ||||
|---|---|---|---|---|---|---|
| vehicle | myrcludex B | vehicle | myrcludex B | vehicle | myrcludex B | |
| TαMCA | 0.96 ± 0.43 | 2.65 ± 0.13* | 2.09 ± 0.73 | 0.86 ± 0.33 | 1.34 ± 0.20 | 1.07 ± 0.21 |
| TβMCA | 43.63 ± 3.82 | 29.85 ± 2.53* | 4.13 ± 0.55 | 1.84 ± 0.24* | 33.98 ± 2.14 | 39.43 ± 0.31 |
| TUDCA | n.d. | n.d. | n.d. | n.d. | 6.90 ± 0.4 | 6.64 ± 0.23 |
| TCA | 54.68 ± 3.00 | 64.20 ± 2.23* | 93.77 ± 0.92 | 97.31 ± 0.27* | 56.85 ± 2.62 | 52.06 ± 0.81 |
| GUDCA | 0.01 ± 0.01 | 0.02 ± 0.01 | n.d. | n.d. | n.d. | n.d. |
| GCA | 0.04 ± 0.01 | 0.03 ± 0.01 | n.d. | n.d. | n.d. | n.d. |
| TCDCA | 0.65 ± 0.49 | 2.90 ± 0.19* | n.d. | n.d. | 0.71 ± 0.16 | 0.53 ± 0.08 |
| TDCA | 0.03 ± 0.10 | 0.35 ± 0.08* | n.d. | n.d. | 0.17 ± 0.06 | 0.25 ± 0.05 |
| GCDCA | n.d. | n.d. | n.d. | n.d. | 0.05 ± 0.03 | 0.02 ± 0.01 |
| GDCA | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
MDR2(ABCB4)‐DEPENDENT BILIARY PHOSPHOLIPID SECRETION IS REQUIRED FOR HEPATOPROTECTIVE EFFECTS OF MYRCLUDEX B TREATMENT
To explore whether the increased biliary phospholipid to bile salt ratio is involved in the myrcludex B‐mediated hepatoprotection, we tested the effects of myrcludex B in Abcb4 knockout mice. The transport of phospholipids from the hepatocyte to bile is exclusively mediated by Mdr2/Abcb4, and deficiency of this protein is the cause of PFIC type 3.23 Plasma ALP and ALT levels were not changed after myrcludex B treatment in Abcb4 knockout mice, but increased liver weight to BW ratio was observed (Fig. 6A,B). Sirius Red analysis of the liver showed accelerated fibrotic processes after 2 weeks of myrcludex B treatment compared to vehicle treatment, although this was not confirmed by hydroxyproline quantification (Fig. 6C). Col1a1 and F4/80 mRNA levels were elevated and ductular reaction was more pronounced, as quantified by Cytokeratin 19 (CK19) staining (Fig. 6D,E). This indicates that myrcludex B treatment is not effective in absence of biliary phospholipid secretion and appears to exacerbate liver injury. Immunohistochemical staining of F4/80 and αSMA both showed a similar trend towards induced inflammatory and fibrotic processes after myrcludex B administration http://onlinelibrary.wiley.com/doi/10.1002/hep.29888/suppinfo. Bile flow, biliary bile salt, cholesterol and bicarbonate output in Abcb4 knockout mice were not affected by myrcludex B treatment http://onlinelibrary.wiley.com/doi/10.1002/hep.29888/suppinfo.
Figure 6.
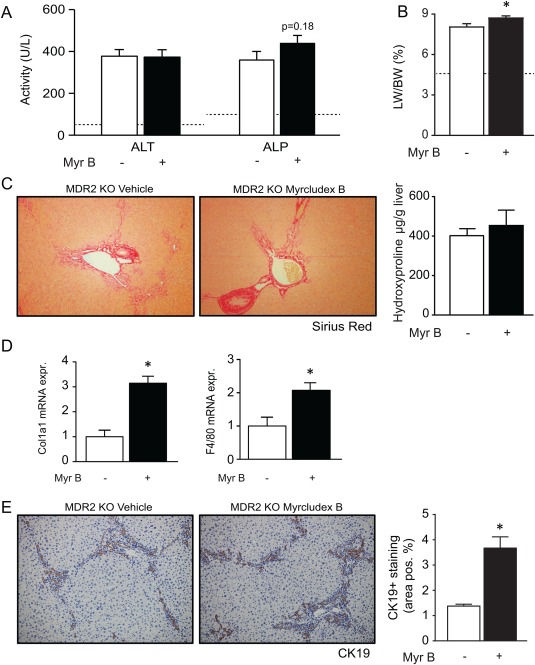
Absence of phospholipid excretion (Abcb4/MDR2‐deficiency) abrogates beneficial effects of myrcludex B. (A) Plasma ALT and ALP upon myrcludex B for 14 days. Dotted lines indicate physiological levels of the indicated enzymes. (B) Liver to BW ratio in Mdr2 knockout mice. (C) Sirius Red staining and hydroxyproline quantification in vehicle or myrcludex B treated Mdr2 knockout mice. (D) Hepatic mRNA expression of pro‐fibrotic gene Col1a1 and macrophage marker F4/80. (E) Images of liver stained with bile duct marker CK19, quantified in vehicle or myrcludex B treated Mdr2 knockout mice. Asterisk indicates significant changes (n=5 mice/group).
Discussion
This study demonstrates that the highly selective NTCP‐inhibitor myrcludex B can dampen cholestatic liver injury. The most prominent results were observed in the DDC‐fed mouse model, where myrcludex B treatment strongly reduced plasma ALP and ALT levels, markers of cholestatic liver damage. Simultaneously, inflammatory, proliferative and fibrotic responses were repressed. Prolonged NTCP‐inhibition also led to a reduction in cholestatic biochemical parameters in a mouse model for PFIC type 1 and in bile‐duct ligated mice. However, the latter also displayed highly elevated plasma bile salt levels and rapid weight loss. Finally, myrcludex B administration was not protective in mice deficient in biliary phospholipid secretion.
Comparing the effects of myrcludex B treatment in the various cholestatic models and to effects of interruption of the enterohepatic circulation of bile salts using ASBT inhibitors provided important insights into the hepatoprotective mechanisms at work upon inhibition of hepatic bile salt uptake.
NTCP‐inhibition leads to reduced hepatocellular accumulation of bile salts. This is similar to ASBT‐inhibitors, which target bile salt uptake at the luminal side of the enterocyte. Hepatocytes intrinsically counteract accumulation of bile salts by multiple protective mechanisms that reduce bile salt synthesis, reduce bile salt uptake and stimulate bile salt elimination, primarily regulated via FXR.24 Although NTCP expression and activity are already reduced during cholestasis,5 we show here that the residual activity can effectively be targeted by myrcludex B administration. This is comparable to the situation for ASBT inhibition, as this bile acid transporter is also downregulated during cholestasis,25 and further pharmacological inhibition still ameliorates cholangitis in mice10, 11 and reduces pruritus severity in PBC patients.12 One of the main differences between effects of ASBT and NTCP inhibition is the induction of diarrhea. This is a common side‐effect of ASBT‐inhibitors but was neither observed in this study nor in myrcludex B‐treated patients.
A second difference between ASBT and NTCP inhibition lies in the effect on bile salt synthesis. ASBT‐inhibition leads to elevated bile salt synthesis because FGF15‐mediated Cyp7a1 repression is lost.22 This is in contrast to NTCP inhibition in BDL and PFIC1 mice where bile salt synthesis is reduced, likely caused by an increase in Fgf15 expression in the gut after basolateral uptake of the bile salts present in blood.17 However, reduction of bile salt synthesis after NTCP‐inhibition was not observed in DDC‐fed mice treated with myrcludex B, which have normalized levels of Cyp7a1 expression, in line with reduced hepatic FXR activation.
A third and essential difference between the consequences of ASBT and NTCP inhibition is the change in plasma bile salt levels. ASBT inhibition results in lowering of plasma bile salt levels in contrast to NTCP inhibition leading to increased bile salt levels.
Although the hepatoprotective effects of NTCP‐inhibition in certain models are clear, the question arises whether the elevated systemic bile salt levels would be harmful in a clinical setting. The absolute increase in bile salts in the DDC and PFIC1 models was not as high compared to the BDL model and did not exert adverse effects. This indicates that increased systemic bile salt levels are well tolerated up to relatively high levels (1.5 millimolar in the DDC model). Similarly, in humans with NTCP‐deficiency elevated systemic bile salt levels are tolerated very well and do not lead to clinical problems, even at concentrations of 1,500 μm.13 Notably, long‐term myrcludex B treatment in humans does not lead to pruritus or diarrhea, despite increased plasma bile salt levels to ∼200 μm.16, 26 Prolonged use of myrcludex B and tenofovir in HBV/HDV coinfected patients in a phase IIb trial resulted in a decrease of ALT levels compared to tenofovir alone.27 This study supports further investigation of NTCP‐inhibition in cholestasis with defined etiologies. For example, in PBC/primary sclerosing cholangitis (PSC) plasma bile salt concentrations are moderately elevated in early stages of disease,28 allowing for an innocuous further increase in bile salt levels after myrcludex B treatment.
In the various cholestasis models described here, hepatic bile salt uptake was not inhibited to the same degree, as reflected by different increases in plasma bile salts, reduction in activation of FXR target genes in the liver and decreased biliary bile salt output. The DDC and Atp8b1‐G308V models showed reduced hepatic bile salt flux after NTCP‐inhibition, which contributed to the improved biochemical and molecular markers of cholestatic liver injury. In contrast, Mdr2 ‐/‐ mice treated with myrcludex B did not show alterations in bile salt flux. This discrepancy is probably due to differential expression of OATP‐isoforms. Particularly, Oatp1a1 contributes to hepatic bile salt uptake17 and Oatp1a1 expression was highest in MDR2 mice, absent in Atp8b1‐mutant and relatively low in DDC mice. Humans have different hepatic OATP‐orthologues and the role of OATP‐mediated BS transport in humans is far less prominent.17, 29 Important to note is that the human bile salt pool contains more hydrophobic CDCA than CA and mainly glycine conjugates compared to mice.30 These bile salts are more toxic when entering hepatocytes. This indicates that NTCP‐inhibition in humans may result in more pronounced reduction of hepatic bile salt accumulation and toxicity.
An important effect of NTCP‐inhibition in cholestasis is the alteration in bile composition. In both the DDC and Atp8b1‐G308V model we observed an increased phospholipid output and PL/BS ratio, making the bile less toxic. This protective effect of NTCP‐inhibition was absent in Mdr2 ‐/‐ mice, which lack phospholipid excretion in bile, and some parameters of liver damage even increased. We examined other biliary components that influence bile toxicity i.e., bile salt hydrophobicity, bicarbonate and cholesterol,31, 32 but these were not changed after myrcludex B administration. Therefore, these results indicate that myrcludex B treatment is beneficial when phospholipids secretion into bile is not functionally impaired.
Furthermore, despite improving levels of ALT and ALP in plasma, NTCP inhibition did not improve the overall phenotype in BDL mice, as presence of hepatic necrosis continued and weight loss increased upon myrcludex B treatment. Plasma bile salt levels were higher than any other model in this study (up to 4 millimolar). Additionally, expression of genes involved in hepatocyte proliferation were reduced in myrcludex B treated BDL mice, which may further contribute to the discrepancy in biochemical parameters and overall phenotype. A possible slower regeneration of the liver in this specific experimental model may be explained by decreased FXR‐activation33 visible upon inhibition of NTCP‐mediated bile salt accumulation in hepatocytes.
Myrcludex B treatment also lowered inflammation and proliferation in DDC‐fed cholestatic mice, in line with a recent observation of Cai, et al.34 that NTCP‐mediated bile salt uptake is required for hepatocellular cytokine release. Increased plasma bile salt levels, seen after NTCP inhibition, may stimulate TGR5 signalling on Kupffer cells which may further reduce the inflammatory response. Absence of TGR5 has been shown to be detrimental during BDL, while activation of the TGR5 receptor reduces hepatic inflammation.35, 36, 37
In summary, pharmacological NTCP‐inhibition with myrcludex B protects against cholestatic liver injury in conditions with residual bile flow and MDR2/3 functionality mainly by temporarily blocking entrance of conjugated bile salts into hepatocytes and increasing the biliary PL/BS ratio, thereby chaperoning potentially toxic biliary bile salts.
Supporting information
Additional Supporting Information may be found at http://onlinelibrary.wiley.com/doi/10.1002/hep.29888/suppinfo.
Supporting Information 1
Acknowledgment
We thank Chrystal Croes, Jonathan van de Meer and Suzanne Duijst (animal experiments), Dirk R de Waart (bile salt measurements) and Joanne Verheij (liver pathology).
Potential conflict of interest: M. Trauner consults, is on the speakers' bureau and received grants from Falk, MSD and Gilead. He consults and received grants from Albireo and Intercept. He is on the speakers' bureau and received grants from Roche. He consults for Phenex, Novartis and Bristol‐Myers Squibb. He received grants from Takeda.
Supported by the Netherlands Organization for Scientific Research (Vidi; 91713319 to SFJvdG), the European Research Council (starting grant 337479 to SFJvdG), and the Austrian Science Foundation (grant F3517‐B20 MT).
Disclosure: M. Trauner served as a consultant for Albireo, Falk, Genfit, Gilead, Intercept, MSD, Novartis and Phenex and is a member of the speakers' bureau of Falk, Gilead, MSD and Roche. He further received travel grants from Falk, Roche and Gilead and unrestricted research grants from Albireo, Falk, Gilead, Intercept and MSD. He is also co‐inventor of a patent on the medical use of norUDCA. Claudia Fuchs received travel grants from Gilead, Roche, Falk, Merck, Vifor, Abbvie and Böhringer Ingelheim. Ulrich Beuers received lecture fees from Abbvie, Falk Foundation, Gilead, Intercept, Roche, Shire, Zambon; consultant fees from Intercept and Novartis; support for investigator‐initiated studies from Dr. Falk and Intercept.
REFERENCES
- 1. Trauner M, Meier PJ, Boyer JL. Molecular pathogenesis of cholestasis. N Engl J Med 1998;339:1217‐1227. [DOI] [PubMed] [Google Scholar]
- 2. van der Woerd WL, Houwen RH, van de Graaf SF. Current and future therapies for inherited cholestatic liver diseases. World J Gastroenterol 2017;23:763‐775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Saito JM, Maher JJ. Bile duct ligation in rats induces biliary expression of cytokine‐induced neutrophil chemoattractant. Gastroenterology 2000;118:1157‐1168. [DOI] [PubMed] [Google Scholar]
- 4. Allen K, Jaeschke H, Copple BL. Bile acids induce inflammatory genes in hepatocytes: a novel mechanism of inflammation during obstructive cholestasis. Am J Pathol 2011;178:175‐186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gartung C, Ananthanarayanan M, Rahman MA, Schuele S, Nundy S, Soroka CJ, et al. Down‐regulation of expression and function of the rat liver Na+/bile acid cotransporter in extrahepatic cholestasis. Gastroenterology 1996;110:199‐209. [DOI] [PubMed] [Google Scholar]
- 6. Slitt AL, Allen K, Morrone J, Aleksunes LM, Chen C, Maher JM, et al. Regulation of transporter expression in mouse liver, kidney, and intestine during extrahepatic cholestasis. Biochim Biophys Acta 2007;1768:637‐647. [DOI] [PubMed] [Google Scholar]
- 7. Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, et al. A regulatory cascade of the nuclear receptors FXR, SHP‐1, and LRH‐1 represses bile acid biosynthesis. Mol Cell 2000;6:517‐526. [DOI] [PubMed] [Google Scholar]
- 8. Beuers U, Trauner M, Jansen P, Poupon R. New paradigms in the treatment of hepatic cholestasis: from UDCA to FXR, PXR and beyond. J Hepatol 2015;62:S25‐37. [DOI] [PubMed] [Google Scholar]
- 9. Trauner M, Fuchs CD, Halilbasic E, Paumgartner G. New therapeutic concepts in bile acid transport and signaling for management of cholestasis. Hepatology 2017;65:1393‐1404. [DOI] [PubMed] [Google Scholar]
- 10. Baghdasaryan A, Fuchs CD, Osterreicher CH, Lemberger UJ, Halilbasic E, Pahlman I, et al. Inhibition of intestinal bile acid absorption improves cholestatic liver and bile duct injury in a mouse model of sclerosing cholangitis. J Hepatol 2016;64:674‐681. [DOI] [PubMed] [Google Scholar]
- 11. Miethke AG, Zhang W, Simmons J, Taylor AE, Shi T, Shanmukhappa SK, et al. Pharmacological inhibition of apical sodium‐dependent bile acid transporter changes bile composition and blocks progression of sclerosing cholangitis in multidrug resistance 2 knockout mice. Hepatology 2016;63:512‐523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hegade VS, Kendrick SF, Dobbins RL, Miller SR, Thompson D, Richards D, et al. Effect of ileal bile acid transporter inhibitor GSK2330672 on pruritus in primary biliary cholangitis: a double‐blind, randomised, placebo‐controlled, crossover, phase 2a study. Lancet 2017;389:1114‐1123. [DOI] [PubMed] [Google Scholar]
- 13. Vaz FM, Paulusma CC, Huidekoper H, de Ru M, Lim C, Koster J, et al. Sodium taurocholate cotransporting polypeptide (SLC10A1) deficiency: conjugated hypercholanemia without a clear clinical phenotype. Hepatology 2015;61:260‐267. [DOI] [PubMed] [Google Scholar]
- 14. Slijepcevic D, Kaufman C, Wichers CG, Gilglioni EH, Lempp FA, Duijst S, et al. Impaired uptake of conjugated bile acids and hepatitis b virus pres1‐binding in na(+) ‐taurocholate cotransporting polypeptide knockout mice. Hepatology 2015;62:207‐219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Blank A, Eidam A, Haag M, Hohmann N, Burhenne J, Schwab M, et al. The NTCP ‐ inhibitor myrcludex B: Effects on bile acid disposition and tenofovir pharmacokinetics. Clin Pharmacol Ther 2018;103: 341‐348. [DOI] [PubMed] [Google Scholar]
- 16. Blank A, Markert C, Hohmann N, Carls A, Mikus G, Lehr T, et al. First‐in‐human application of the novel hepatitis B and hepatitis D virus entry inhibitor myrcludex B. J Hepatol 2016;65:483‐489. [DOI] [PubMed] [Google Scholar]
- 17. Slijepcevic D, Roscam Abbing RLP, Katafuchi T, Blank A, Donkers JM, van Hoppe S, et al. Hepatic uptake of conjugated bile acids is mediated by both sodium taurocholate cotransporting polypeptide and organic anion transporting polypeptides and modulated by intestinal sensing of plasma bile acid levels in mice. Hepatology 2017;66:1631‐1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fickert P, Trauner M, Fuchsbichler A, Stumptner C, Zatloukal K, Denk H. Bile acid‐induced Mallory body formation in drug‐primed mouse liver. Am J Pathol 2002;161:2019‐2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fickert P, Zollner G, Fuchsbichler A, Stumptner C, Weiglein AH, Lammert F, et al. Ursodeoxycholic acid aggravates bile infarcts in bile duct‐ligated and Mdr2 knockout mice via disruption of cholangioles. Gastroenterology 2002;123:1238‐1251. [DOI] [PubMed] [Google Scholar]
- 20. Pawlikowska L, Groen A, Eppens EF, Kunne C, Ottenhoff R, Looije N, et al. A mouse genetic model for familial cholestasis caused by ATP8B1 mutations reveals perturbed bile salt homeostasis but no impairment in bile secretion. Hum Mol Genet 2004;13:881‐892. [DOI] [PubMed] [Google Scholar]
- 21. Lu TT, Makishima M, Repa JJ, Schoonjans K, Kerr TA, Auwerx J, et al. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol Cell 2000;6:507‐515. [DOI] [PubMed] [Google Scholar]
- 22. Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab 2005;2:217‐225. [DOI] [PubMed] [Google Scholar]
- 23. Smit JJ, Schinkel AH, Oude Elferink RP, Groen AK, Wagenaar E, van Deemter L, et al. Homozygous disruption of the murine mdr2 P‐glycoprotein gene leads to a complete absence of phospholipid from bile and to liver disease. Cell 1993;75:451‐462. [DOI] [PubMed] [Google Scholar]
- 24. Wagner M, Zollner G, Trauner M. Nuclear receptor regulation of the adaptive response of bile acid transporters in cholestasis. Semin Liver Dis 2010;30:160‐177. [DOI] [PubMed] [Google Scholar]
- 25. Hruz P, Zimmermann C, Gutmann H, Degen L, Beuers U, Terracciano L, et al. Adaptive regulation of the ileal apical sodium dependent bile acid transporter (ASBT) in patients with obstructive cholestasis. Gut 2006;55:395‐402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bogomolov P, Alexandrov A, Voronkova N, Macievich M, Kokina K, Petrachenkova M, et al. Treatment of chronic hepatitis D with the entry inhibitor myrcludex B: First results of a phase Ib/IIa study. J Hepatol 2016;65:490‐498. [DOI] [PubMed] [Google Scholar]
- 27. Wedemeyer H, Alexandrov A, Bogomolov P, Blank A, Bremer B, Voronkova N, et al. Interim results of a multicenter, open‐label phase 2b clinical trial to assess safety and efficacy Myrcludex B in combination with Tenofovir in patients with chronic HBV/HDV co‐infection. Hepatology 2017;66:S1;20–21. [Google Scholar]
- 28. Trottier J, Bialek A, Caron P, Straka RJ, Heathcote J, Milkiewicz P, et al. Metabolomic profiling of 17 bile acids in serum from patients with primary biliary cirrhosis and primary sclerosing cholangitis: a pilot study. Dig Liver Dis 2012;44:303‐310. [DOI] [PubMed] [Google Scholar]
- 29. Klaassen CD, Aleksunes LM. Xenobiotic, bile acid, and cholesterol transporters: function and regulation. Pharmacol Rev 2010;62:1‐96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Woolbright BL, Jaeschke H. Novel insight into mechanisms of cholestatic liver injury. World J Gastroenterol 2012;18:4985‐4993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lammert F, Wang DQ, Hillebrandt S, Geier A, Fickert P, Trauner M, et al. Spontaneous cholecysto‐ and hepatolithiasis in Mdr2‐/‐ mice: a model for low phospholipid‐associated cholelithiasis. Hepatology 2004;39:117‐128. [DOI] [PubMed] [Google Scholar]
- 32. Hohenester S, Wenniger LM, Paulusma CC, van Vliet SJ, Jefferson DM, Elferink RP, et al. A biliary HCO3‐ umbrella constitutes a protective mechanism against bile acid‐induced injury in human cholangiocytes. Hepatology 2012;55:173‐183. [DOI] [PubMed] [Google Scholar]
- 33. Huang W, Ma K, Zhang J, Qatanani M, Cuvillier J, Liu J, Dong B, et al. Nuclear receptor‐dependent bile acid signaling is required for normal liver regeneration. Science 2006;312:233‐236. [DOI] [PubMed] [Google Scholar]
- 34. Cai SY, Ouyang X, Chen Y, Soroka CJ, Wang J, Mennone A, et al. Bile acids initiate cholestatic liver injury by triggering a hepatocyte‐specific inflammatory response. JCI Insight 2017;2:e90780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pean N, Doignon I, Garcin I, Besnard A, Julien B, Liu B, et al. The receptor TGR5 protects the liver from bile acid overload during liver regeneration in mice. Hepatology 2013;58:1451‐1460. [DOI] [PubMed] [Google Scholar]
- 36. Wang YD, Chen WD, Yu D, Forman BM, Huang W. The G‐protein‐coupled bile acid receptor, Gpbar1 (TGR5), negatively regulates hepatic inflammatory response through antagonizing nuclear factor kappa light‐chain enhancer of activated B cells (NF‐kappaB) in mice. Hepatology 2011;54:1421‐1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Keitel V, Donner M, Winandy S, Kubitz R, Haussinger D. Expression and function of the bile acid receptor TGR5 in Kupffer cells. Biochem Biophys Res Commun 2008;372:78‐84. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at http://onlinelibrary.wiley.com/doi/10.1002/hep.29888/suppinfo.
Supporting Information 1