

Abstract
Central nervous system (CNS) infection by Mycobacterium tuberculosis is one of the most devastating complications of tuberculosis, in particular in early childhood. In order to induce CNS infection, M. tuberculosis needs to cross specialised barriers protecting the brain. How M. tuberculosis crosses the blood–brain barrier (BBB) and enters the CNS is not well understood. Here, we use transparent zebrafish larvae and the closely related pathogen Mycobacterium marinum to answer this question. We show that in the early stages of development, mycobacteria rapidly infect brain tissue, either as free mycobacteria or within circulating macrophages. After the formation of a functionally intact BBB, the infiltration of brain tissue by infected macrophages is delayed, but not blocked, suggesting that crossing the BBB via phagocytic cells is one of the mechanisms used by mycobacteria to invade the CNS. Interestingly, depletion of phagocytic cells did not prevent M. marinum from infecting the brain tissue, indicating that free mycobacteria can independently cause brain infection. Detailed analysis showed that mycobacteria are able to cause vasculitis by extracellular outgrowth in the smaller blood vessels and by infecting endothelial cells. Importantly, we could show that this second mechanism is an active process that depends on an intact ESX‐1 secretion system, which extends the role of ESX‐1 secretion beyond the macrophage infection cycle.
Keywords: blood–brain barrier, ESX‐1 secretion, Trojan horse mechanism, tuberculosis, tuberculous meningitis, zebrafish
1. INTRODUCTION
Tuberculous meningitis (TBM) is one of the most severe extra‐pulmonary manifestations of tuberculosis (TB) and significantly contributes to mycobacterial disease burden (WHO, 2017). Invasion of Mycobacterium tuberculosis, the causative agent of TB, into the central nervous system (CNS) occurs in 1% of all cases (Thwaites, van Toorn, & Schoeman, 2013; Wilkinson et al., 2017). Major risk groups for developing TBM include young children and HIV‐positive individuals in TB endemic areas (van Well et al., 2009; Wilkinson et al., 2017). Despite extensive research efforts, the diagnosis and treatment of TBM is often delayed because of its insidious onset (Wilkinson et al., 2017). Consequently, half of the patients are diagnosed in the most advanced stage of disease, resulting in a high mortality rate of nearly 20% and neurological sequelae in more than half of the survivors (Chiang et al., 2014). These poor odds of (full) recovery for TBM patients can be mostly attributed to the severe neuro‐inflammation at the base of the brain, on‐going neural ischaemia, and vasculitis (Donald & Van Toorn, 2016).
The histological hallmark of TB is the granuloma, a cluster of immune cells that shields off the infected macrophages from the surrounding tissue. In 1933, it was established that in TBM, granulomas are present in brain parenchyma and meninges. This important observation led to the hypothesis that granulomas were the main aetiology of TBM and these infectious foci were later called Rich foci (Rich & McCordock, 1933; Rich & Thomas, 1946). Today, the concept of the Rich focus still stands; however, the question remains how the first mycobacterium enters the brain to seed the Rich focus.
To induce granuloma formation and subsequently meningitis, M. tuberculosis must traverse the blood–brain barrier (BBB), a selectively permeable layer that separates brain tissue from the blood circulation. The BBB consists of specialised endothelial cells connected by tight junctions, closely surrounded and monitored by several cell types, including astrocytes, pericytes, and microglia. The BBB regulates the passage of molecules and effectively protects the brain from circulating toxins and microorganisms (Abbott, Patabendige, Dolman, Yusof, & Begley, 2010; Abbott, Rönnbäck, & Hansson, 2006; Obermeier, Daneman, & Ransohoff, 2013). Little is known about the steps preceding granuloma formation, in particular how M. tuberculosis manages to traverse the BBB.
Only a small subset of bacterial pathogens is able to cause meningitis or CNS infections. Thus far, three different BBB traversal strategies have been described for these pathogens. The most commonly utilised route is transcellular migration. This receptor‐mediated process results in endocytosis of the pathogen by endothelial cells that line the blood vessels and is used by Streptococcus pneumoniae, Haemophilus influenzae, and Neisseria meningitidis (Bencurova, Mlynarcik, & Bhide, 2011; Kim, 2008; Orihuela et al., 2009; van Sorge & Doran, 2013). A second route is paracellular migration, which usually occurs when BBB integrity is disrupted by direct contact with the pathogen or as a result of secreted bacterial toxins. A third mechanism of BBB crossing is the Trojan horse mechanism; the pathogen infects a macrophage that subsequently traverses the BBB. Based on the fact that M. tuberculosis is an intracellular pathogen capable of surviving and replicating within the macrophage, the latter mechanism seems logical for BBB traversal (Nguyen & Pieters, 2005). In line with this hypothesis, M. tuberculosis was found to cross an epithelial barrier with significantly higher efficiency when phagocytosed by monocytes than when mycobacteria alone were introduced in an in vitro system (Bermudez, Sangari, Kolonoski, Petrofsky, & Goodman, 2002). Furthermore, macrophages played an essential role in early dissemination and establishment of extra‐pulmonary foci (Clay et al., 2011; Polena et al., 2016). However, the ability of M. tuberculosis to invade brain endothelial cells in vitro has been described as well (Be, Bishai, & Jain, 2012; Chen, Sakamoto, Quinn, Chen, & Fu, 2015; Jain, Paul‐Satyaseela, Lamichhane, Kim, & Bishai, 2006; Mehta, Karls, White, Ades, & Quinn, 2006). Consequently, the exact mechanisms involved in mycobacterial invasion into brain tissue are still not completely understood. We reasoned that, in order to study such a detailed sequence of events, it is essential to observe the interplay between host and pathogen in vivo.
Several in vivo models to study mycobacterial pathogenesis exist, such as rabbits, guinea pig, and mice (Be et al., 2012; Be, Klinkenberg, Bishai, Karakousis, & Jain, 2011; Skerry et al., 2013; Tsenova et al., 2005; Tucker et al., 2016; van Well et al., 2007; Zucchi et al., 2012). However, none of these models could demonstrate the course of events during mycobacterial trafficking across the BBB in a living host. Another in vivo model that has proven to mimic human mycobacterial disease well is the zebrafish—Mycobacterium marinum infection model (Berg & Ramakrishnan, 2012; Lesley & Ramakrishnan, 2011; van der Sar, Appelmelk, Vandenbroucke‐Grauls, & Bitter, 2004). The translucency of the Danio rerio larvae in combination with many fluorescent tools offers unique possibilities to study host–pathogen interaction in real life (Kuipers, Kalicharan, Wolters, van Ham, & Giepmans, 2016; Tobin, May, & Wheeler, 2012). Moreover, the anatomy of the zebrafish BBB is highly similar to the human BBB. Already after 3‐days postfertilisation (dpf), the zebrafish BBB functionally prevents exchange of large molecules (Fleming, Diekmann, & Goldsmith, 2013; Xie, Farage, Sugimoto, & Anand‐Apte, 2010). Importantly, upon infection with M. marinum, TBM does occur in adult zebrafish, with granuloma formation in the meninges and brain parenchyma in 20% of the cases (van Leeuwen et al., 2014). Therefore, the zebrafish model allows us to specifically address questions regarding mycobacterial invasion into the brain in an in vivo model (Bernut et al., 2014; Tenor, Oehlers, Yang, Tobin, & Perfect, 2015; van Leeuwen et al., 2014)
This study provides in vivo evidence that mycobacteria utilise phagocytic cells to cross the BBB. Additionally, by using in vivo macrophage depletion and correlated light and electron microscopy (CLEM), we show that mycobacteria also employ transcellular migration by infecting and damaging brain endothelial cells in an ESX‐1‐dependent manner.
2. RESULTS
2.1. M. marinum crosses a functionally intact BBB within phagocytic cells
To examine the importance of an intact BBB in mycobacterial trafficking to the brain, we used larvae at different developmental stages and followed infection progression daily (Figure 1a). BBB biogenesis in zebrafish starts at 3 dpf and can be determined by systemic injection of fluorescent tracers. A 3‐kDa fluorescent dye was excluded from the larval brain from 3 dpf onwards, indicating BBB maturation (compare Figure 1b with 1c). Please notice that the blood vessels in close proximity to the eyes and gills do not possess a BBB and therefore do not restrict diffusion of the dye into the surrounding tissue (Figure 1c; van Leeuwen et al., 2017).
Figure 1.
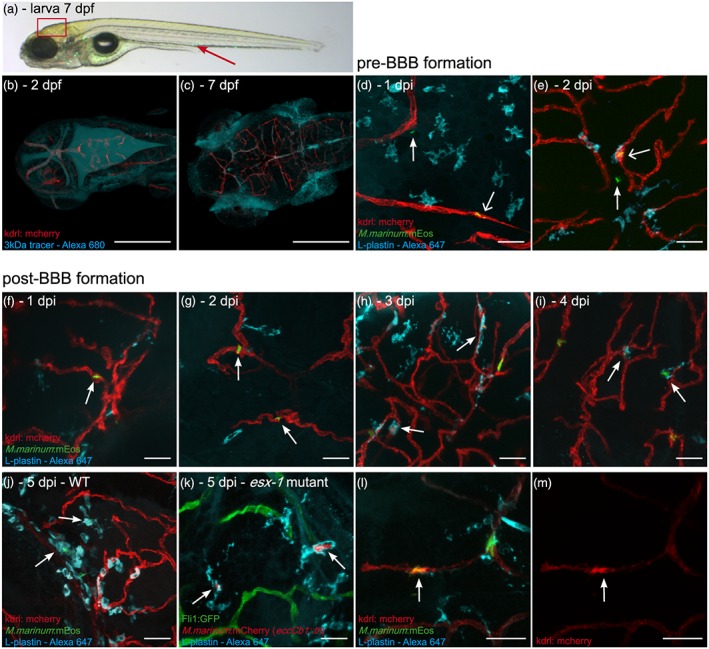
M. marinum WT and esx‐1 mutant traverse across an intact blood–brain barrier within macrophages. (a) Lateral view of a casper zebrafish larva at 3 dpi (corresponds to 7 dpf), infected with M. marinum E11:mEos3.1 (green). Red arrow marks the caudal vein injection spot. Boxed area represents the brain region of which representative images are shown in this figure. (b) Tg(kdrl:mCherry) is5 zebrafish larva, uninfected, 1 hr after 3‐kDa Alexa 680 tracer injection at 2 dpf, showing massive leakage of tracer to the ventricles, confirming the immaturity of the BBB. (c) Injection of tracer at 7 dpf, showing localisation in brain blood vessels, indicating that the BBB is functionally intact at this moment. Blood vessels in close proximity to the eyes and gills do not possess a BBB and do not restrict diffusion of the dye, leading to extravasation of the dye at these locations. Scale bars bc = 250 μm. (d) Tg(kdrl:mCherry) is5 zebrafish larvae infected at 2 dpf (before formation of a functional BBB), 1 dpi single nonphagocytosed bacteria (green) can be found in blood vessel (red; open arrow) and in brain tissue (arrow), showing that M. marinum can enter brain tissue at this time point. (e) Also at 2 dpi, phagocytosed bacteria (colocalisation of green bacteria and cyan phagocytes labelled with anti‐L‐plastin) in blood vessel (open arrow) and single nonphagocytosed bacteria in brain tissue (arrow) are found. (f) Systemic infection at 4 dpf (larvae with a functional BBB), M. marinum (green) is solely found in brain blood vessels (red) at 1 dpi and (g) 2 dpi (arrows). (h–j) From 3 dpi onwards, phagocytosed mycobacteria (colocalisation of green bacteria and cyan phagocytes) are able to leave the bloodstream (arrows). (k) Representative image of section of brain in Tg(Fli1:GFP) y1 zebrafish larvae, systemically infected with the esx‐1 mutant (red) at 4 dpf, showing that mutant bacteria also cross the BBB within phagocytic cells (anti‐L‐plastin, cyan). (l,m) Examples of spot with high expression of mCherry tagged VEGFr2 in Tg(kdrl:mCherry) is5 larva, colocalising with phagocytosed (cyan) M. marinum E11 (green) in blood vessel (red). (l) Merged image, (m) corresponding single red channels. Scale bars d–m = 25 μm
Infection experiments performed at 2 dpf, that is, before BBB biogenesis, showed that mycobacteria readily crossed blood vessel walls in the brain at this time point (Figure 1d,e). In these larvae, mycobacterial migration was observed as early as 1‐day postinfection (dpi; Figure 1d). Examination of larvae infected at 4 dpf, that is, after the formation of the BBB, showed that mycobacteria are present in brain blood vessels at 1 and 2 dpi (Figure 1f,g; n = 5 larvae, 0/12 bacteria in parenchyma) but only entered brain tissue from 3 dpi onwards, indicating a significant delay (Figure 1h–j; 3 dpi: 6/22 bacteria in parenchyma, n = 5 larvae; 4 dpi: 9/17 bacteria in parenchyma, n = 3 larvae). Notably, upon visualisation of phagocytic cells with the L‐plastin marker, we always observed colocalisation of bacteria with phagocytic cells (Figures 1h–j and S1). This strongly suggests that phagocytes act as carriers (Trojan horse mechanism) to transfer mycobacteria to brain tissue once the protective function of the BBB is in place.
Because BBB crossing was only observed after 3 days after intravenous infection, we hypothesised that possibly at this stage, the BBB might be compromised due to the infection and a concomitant inflammatory response. In order to study the integrity of the BBB during initial mycobacterial migration to brain tissue, we injected a 3‐kDa fluorescent dye at 3 and 5 dpi and monitored dye distribution 60–180 min postinjection. At all time points, we found many single bacteria associated with the blood vessel wall, possibly in the process of migration, with fluorescent dye restricted to the vessel lumen (Figure S2A,B,D open arrow). The lack of leakage of dye in the parenchyma suggests an intact BBB at these spots. However, injection of dye at 3 and 5 dpi resulted in accumulation and colocalisation of dye within these clusters (Figure S2A,C,E,F closed arrow). This indicates that once an inflammatory focus is formed, the local integrity of the BBB is reduced. Because we did not observe accumulation of dye in the ventricles, we reasoned that there is no increased overall leakage and no substantial breakdown of the BBB in this inflammatory setting.
To further study the Trojan horse as migration mechanism, we compared M. marinum WT with a mutant strain (eccCb1::tn) deficient in ESX‐1 secretion (esx‐1 mutant). ESX‐1 secretion mutants are severely attenuated (Davis & Ramakrishnan, 2009; Stoop et al., 2011; Volkman et al., 2004), but most importantly for our purposes, these mutants are unable to complete the macrophage infection cycle and are therefore predominantly located inside phagocytic cells (Houben et al., 2012; Simeone et al., 2012; van der Wel et al., 2007).
As expected, we observed significantly lower numbers of mycobacteria in zebrafish larvae infected with our esx‐1 mutant (compare Figure 1j [WT: 28 single infected phagocytes and 14 early clusters in six larvae] with 1k [esx‐1 mutant: 16 single infected phagocytes and three small clusters in eight larvae]; see also Figure 5a,b). The observed bacteria were always associated with phagocytes (Figure 1k). Despite these lower numbers, esx‐1 mutants were still able to infect brain tissue and in both WT and esx‐1 mutant infected larvae, approximately half of the infected macrophages were found in brain parenchyma (WT: 13/28, esx‐1 mutant: 8/16). Collectively, our findings confirm the protective function of the BBB against M. marinum infection of brain tissue in developing zebrafish larvae and indicate that M. marinum also uses phagocytes to cross the BBB. In addition, we have indications that local BBB integrity seems to be reduced once an inflammatory focus is established.
Figure 5.
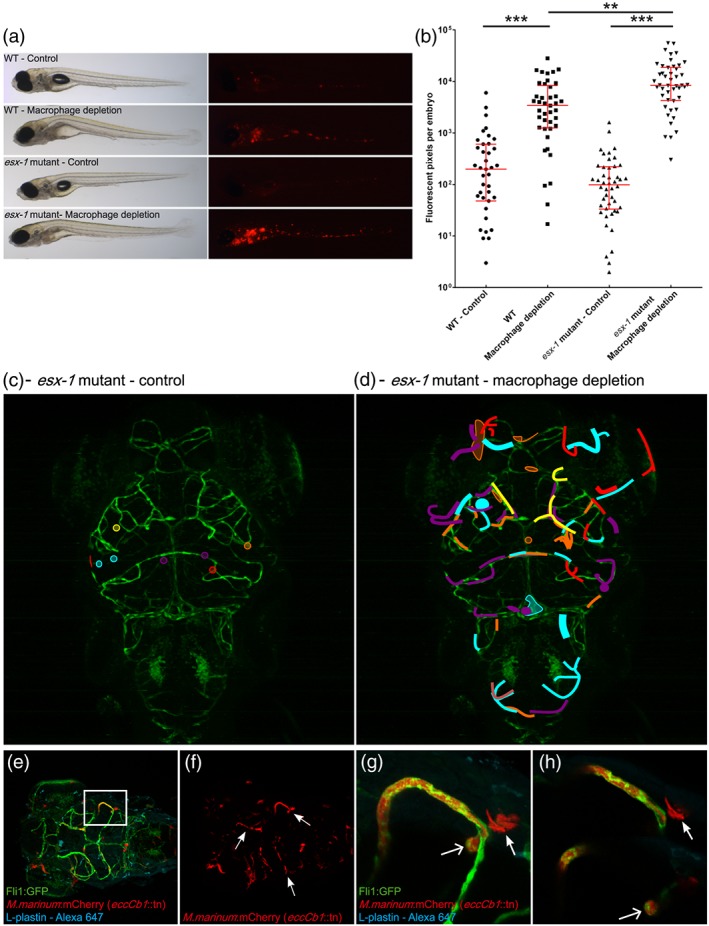
ESX‐1‐deficient mycobacteria are found predominantly in blood vessels. (a) Representative bright field and corresponding fluorescent image of infected larvae at 4 dpf with either M. marinum E11 or M. marinum esx‐1 mutant with or without phagocyte depletion. Images clearly illustrate increased fluorescent intensity in phagocyte‐depleted groups. (b) Corresponding fluorescent pixel counts of infected larvae of three pooled biological independent infection experiments. Graphs show mean and SEM. ** = <.05, *** = <.005. (c) Schematic representation of esx‐1 mutant infection pattern in head of control larvae, pooled data from five larvae. (d) Schematic representation of esx‐1 mutant infection distribution found in five phagocyte‐depleted larva, showing that high amounts of mycobacteria are found predominantly in blood vessels. (e) Z‐stack, dorsal view of Tg(Fli1:GFP) y1 larvae (green), depleted of its phagocytic pool and systemically infected with the esx‐1 mutant (red) at 4 dpf, stained with L‐plastin (cyan) to visualise remaining phagocytes, showing high infection load in brain area. (f) Shows single red fluorescent signal, demonstrating that bacteria are strictly localised in the vasculature (arrows). Boxed area is enlarged in (g) z‐stack, and (h) two single Z‐slices, evidently showing an abundant amount of bacteria clogging the blood vessel, with subsequent bacterial overgrowth, protrusion (open arrow) and (very rare) bursting of the blood vessel wall (closed arrow)
2.2. Intensified VEGFR2 signal colocalises with infected phagocytes
Previously, it has been shown that upregulation of vascular endothelial growth factor (VEGF) had a promoting effect on macrophage‐mediated extra pulmonary dissemination of M. tuberculosis (Polena et al., 2016). To examine the role of VEGF in our zebrafish model, we systemically infected Tg(kdrl:mCherry) is5 embryos with M. marinum. These embryos are modified to express mCherry under control of the promoter regulating kdrl/vegfr2 gene expression, which allowed us to determine the effect of M. marinum on vergfr2 expression in blood vessels of the brain. We observed that, from 3 dpi, the VEGFR2 signal was more intense at 54% of the spots in which phagocytosed mycobacteria colocalised with blood vessels (Figure 1l,m, 13 out of 24 spots in 20 larvae). This time course corresponds with the observed migration of mycobacteria into brain tissue. Nonphagocytosed bacteria found in brain blood vessels showed significantly lower colocalisation with an intensified VEGFR2 signal (16% of the cases, data not shown). This observation is indicative for local upregulation of vegfr2 in endothelial cells by phagocytes carrying mycobacteria.
We hypothesised that alterations in general VEGF levels could affect the percentage of migrated infected phagocytes. Therefore, we manipulated VEGF levels using two different approaches, (a) inducing VEGF levels with GS4012 (Wu et al., 2015) or (b) blocking the VEGF receptor with SU5416 (Oehlers et al., 2014; Wu et al., 2015). Although differences in overall infection levels were found with an increase in the absolute number of infected cells after inducing VEGFR2 signalling (Figure S3), the proportion of infected macrophages crossing the BBB at 3 dpi was similar for all groups (GS4012: 48%; control: 52%; SU5416: 52%; Table S1).
Taken together, although we do observe a local upregulation of VEGFR2 at the site of BBB crossing by infected phagocytes, generic manipulation of the VEGF levels does not alter the percentage of migrated phagocytosed mycobacteria in zebrafish embryos.
2.3. Wild type M. marinum can still infect brain tissue when phagocytes are depleted
To examine whether the Trojan horse mechanism forms the only transport route to cross the BBB, we studied mycobacterial invasion in parenchyma of zebrafish larvae that were depleted of phagocytes. Successful depletion was achieved by injecting both pu.1 morpholinos at the single cell stage, to prevent macrophage development (Clay et al., 2011) and clodronate‐filled liposomes at 3 dpf to kill the remaining circulating phagocytic cells (Figure 2a,b; Bernut et al., 2014; Pagán et al., 2015; van Rooijen, Sanders, & van den Berg, 1996).
Figure 2.
Wild type M. marinum can still infect brain tissue when macrophages are depleted. Left panel shows untreated zebrafish larvae (control), whereas right panel shows larvae depleted of phagocytes by treatment with pu.1 and clodronate filled liposomes at 3 dpf, to kill circulating phagocytes. (a) Control casper larva at 5 dpf, stained with anti‐L‐plastin to visualise normal phagocyte distribution. (b) Five dpf phagocyte‐depleted casper larva. Anti‐L‐plastin is used in (a) and (b) to stain phagocytes. (c) Dorsal view of wild type Tg(Fli1:GFP) y1 larvae (green) at 5 dpi, systemically infected at 4 dpf with M. marinum (red) and stained with anti‐L‐plastin (cyan), showing formation of early granuloma in brain tissue. (d) Z‐stack of boxed area in (c), allowing for a more precise analysis of the position of M. marinum and phagocytes. (e) Corresponding red fluorescent channel to clearly show infection pattern. (f) In the presence of macrophages, M. marinum leaves the bloodstream within phagocytes (arrow) and forms early granulomas in brain tissue. Scale bar c = 100 μm. Scale bar d–f = 25 μm. (g) Z‐stack of head of Tg(Fli1:GFP) y1 larva, phagocyte‐depleted, and systemically infected with M. marinum (red) at 4 dpf and stained with L‐plastin (cyan). Boxed area is enlarged in h–j. (h) Shows mycobacteria outside blood vessels in brain tissue in the absence of phagocytic cells (*). (i) Shows the corresponding red fluorescent channel, depicting that tissue infiltration follows the shape of the blood vessels. (j) Single Z‐slice, which shows the intracellular phenotype of M. marinum (arrow), colocalising with a blood vessel, but in the absence of L‐plastin labelling. Scale bar g = 100 μm; Scale bar h–j = 25 μm. (k) Schematic representation of pooled data of all early granulomas found in nine wild type larvae showing a random distribution in the brain. (l) Schematic representation of pooled data of infection distribution found in nine phagocyte‐depleted larvae, showing that mycobacteria are found in brain tissue, but do not migrate into deeper tissue
As expected, infection with wild type M. marinum in control larvae with normal phagocyte counts resulted in clusters of infected macrophages in the brain of zebrafish larvae (Figure 2c–f). In these zebrafish, we even identified mycobacteria‐loaded phagocytes that appear to be in the process of crossing the BBB (Figure 2d,f, arrow). In contrast, infection in larvae without phagocytes resulted in a huge expansion of mycobacteria in blood vessels without the formation of early granulomas in brain tissue (Figure 2g–j). Surprisingly, however, mycobacteria were also still present in brain tissue in all cases (Figure 2h,i, indicated with *). This observation suggests that mycobacteria can utilise another, phagocyte‐independent, route to cross the BBB. Closer analysis of the bacterial aggregates showed colocalisation with Fli1 labelling, which labels endothelial cells (Lawson & Weinstein, 2002; Figure 2h,j, arrow), suggesting mycobacterial outgrowth in other cell types than phagocytes. In addition, the normal vascular architecture seemed to be disrupted at these heavily infected spots, which indicates major changes in endothelial cells. Although mycobacteria were still located in brain tissue in these larvae, we observed a completely different pattern and distribution of infection (compare Figure 2k with 2l). In phagocyte‐depleted larvae, M. marinum was always found in close vicinity of blood vessels in the brain that were highly loaded with bacteria, whereas in untreated zebrafish, granulomas were located at more distant locations indicating that phagocytes are essential for transport of bacteria into deeper tissue.
In conclusion, M. marinum has the capability to migrate into brain tissue in the absence of phagocytes, which means that an alternative migration route is present.
2.4. Wild type M. marinum causes damage to blood vessels and surrounding tissue
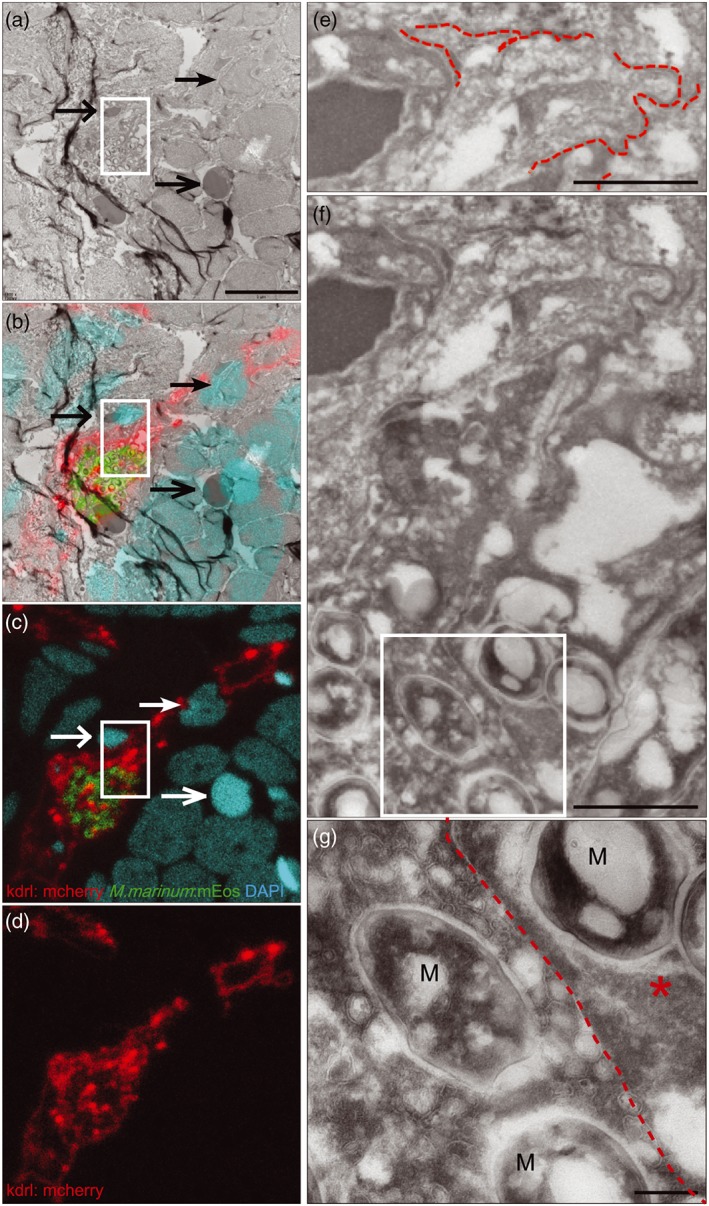
To be able to understand the phagocyte‐independent interaction with the BBB in more detail, we used CLEM, which facilitates the direct correlation of fluorescent confocal microscopy with electron microscopy of consecutive slides of the same tissue (Figures 3a–c and 4a–c).
Figure 3.
Correlative light and electron microscopy of M. marinum infected blood vessels showing irregular blood vessel walls and invasion of endothelial cells. Tg(kdrl:mCherry) is5 larva (9 dpf) with red fluorescent blood vessels infected with green fluorescent M. marinum after phagocyte depletion. To aid correlation of confocal and EM imaging, nuclei were stained with DAPI after fixation (cyan). (a) Electron microscopy and (b) correlative light and electron microscopy, and (c) confocal microscopy. Arrows indicate landmarks used to merge images obtained from consecutive slices in the same area of the zebrafish brain. Boxed area is enlarged in (f), scale bar a = 5 μm (applies to b–d). (d) Single red channel illustrating the irregularity of the infected blood vessel and the more regular shape of the non‐infected blood vessel (right upper corner). (e,f) High magnification EM image showing the irregular shaped infected blood vessels and basal lamina. Red dotted lines represent basal lamina found in this area. Boxed area is enlarged in (g). Scale bar = 1 μm. (g) High magnification of area where mycobacteria can both be found intravascular as intracellular. Vesicles, indicative for intravascular localisation, can only be found left of the red dotted line. This suggest that mycobacteria right of the line are localised in an endothelial cell (*). Scale bar = 200 nm. M = M. marinum
Figure 4.
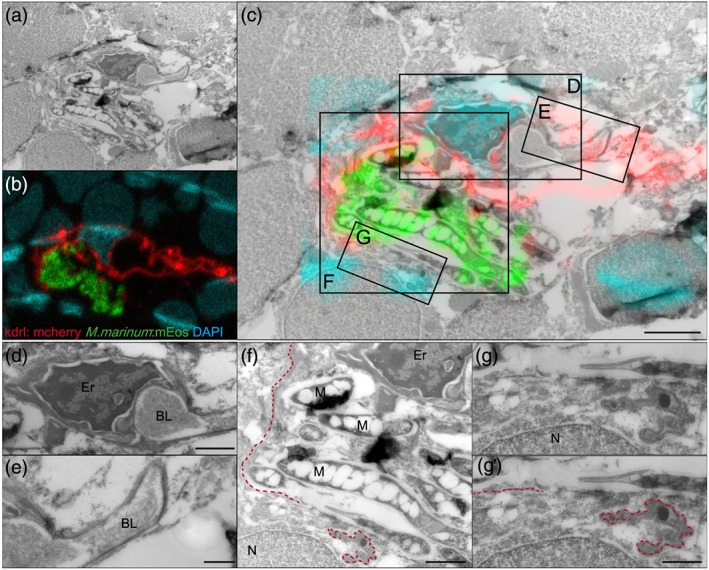
M. marinum causes damage to blood vessels and surrounding tissue. (a) Electron microscopy and (b) confocal imaging merged into (c) correlative light and electron microscopy of 9 dpf Tg(kdrl:mCherry) is5 larva with red fluorescent blood vessels infected with green fluorescent M. marinum E11, after phagocyte depletion. Nuclei were stained with DAPI after fixation (cyan). Boxed areas are enlarged in (d)–(g). Scale bar in c = 2 μm. (d) High magnification of infected blood vessel in brain of infected zebrafish with erythrocyte, vessel lumen, and intact basal lamina on one side of the blood vessel visible. Scale bar = 1 μm. (e) Intact basal lamina at uninfected part of this infected blood vessel. Scale bar = 500 nm. (f) As a consequence of bacterial replication and invasion of endothelial cells, the basal lamina is disrupted (red dotted line). Scale bar = 1 μm. (g,g') higher magnification of the disrupted basal lamina. Scale bar = 500 nm. Er = Erythrocyte, BL = blood vessel lumen, M = M. marinum, N = nucleus
In the wild type situation, M. marinum is found to cross the BBB and invade brain tissue, apparently without disrupting the integrity of the blood vessel. Bacteria‐loaded phagocytes are clearly detected outside of the intact vessels (Figure S4A‐C). In contrast, the phenotype found in phagocyte‐depleted larvae is completely different. Notably, individual infected blood vessels were shaped irregularly (Figure 3d–f), whereas non‐infected blood vessels appear intact (Figure 3d, upper‐right corner). Furthermore, several blood vessels seemed to be segmented, visible within a cross section of a vessel (Figure 3d–f). Higher magnifications of these infected blood vessels showed that bacteria can be found both intravascular and intracellular. For example, in Figure 3g, the bacteria are located in an endothelial cell (specified with *), as the red dotted line indicates the membrane separating an endothelial cell from the vascular lumen. Furthermore, we observed that infection with wild type M. marinum disrupts the vessel wall (Figure 4c,f) and consequently the basal lamina (Figure 4f,g).
Taken together, we show that, in the absence of phagocytes, mycobacteria are capable of invading surrounding tissue, presumably the endothelial cells, and induce damage to the basal lamina and local distribution in the surrounding brain tissue.
2.5. ESX‐1 secretion is essential for macrophage independent BBB crossing
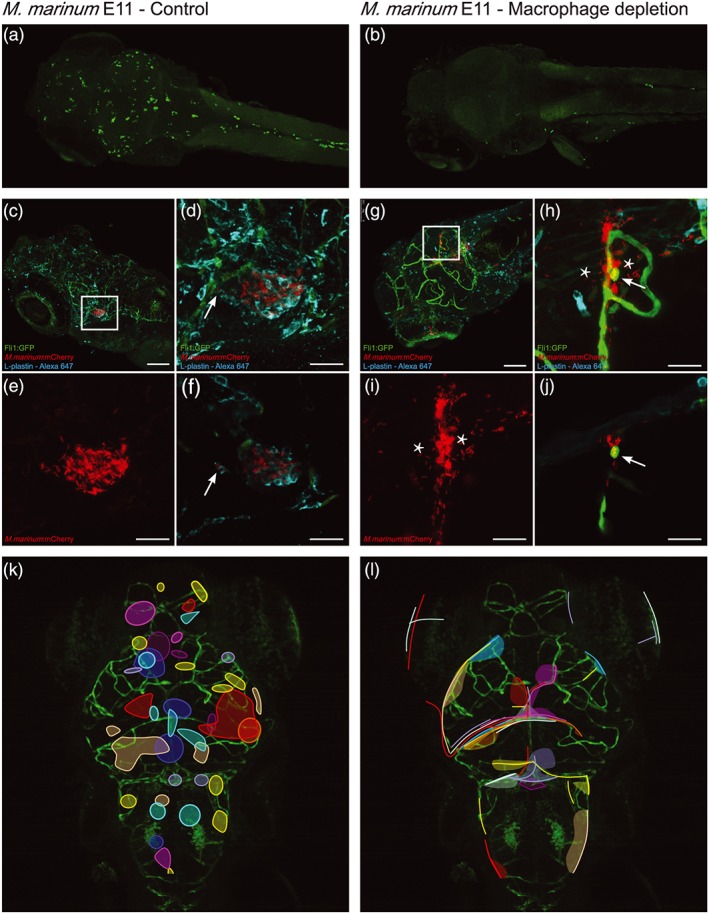
Next, we examined the fate of our esx‐1 mutant in macrophage‐depleted zebrafish larvae. In line with previous findings (Clay et al., 2011), we observed significantly higher outgrowth of the esx‐1 mutant under these conditions, as compared with outgrowth in normal zebrafish larvae (Figure 5a,b), confirming that the absence of phagocytes compensates for the attenuation of this strain. Closer examination of phagocyte‐depleted larvae revealed an important difference with WT M. marinum infection: Although blood vessels in the brain were filled and clogged with ESX‐1‐deficient mycobacteria (Figure 5c,d), esx‐1 mutants were only rarely found outside the blood vessels. (Figure 5e,f, 16/61 cases in five larvae). Therefore, esx‐1 mutants seem unable to cross the BBB efficiently under these conditions, with subsequent bacterial outgrowth in the vessel lumen and protrusion of vessels (Figure 5g,h, open arrow). Only occasionally, we observed bursting of blood vessels (Figure 5g,h, closed arrow). Therefore, for macrophage‐independent crossing, ESX‐1 secretion seems to be an important factor.
Also for the esx‐1 mutant infections, CLEM analysis was performed in phagocyte‐depleted larvae (Figure 6a–c), which confirmed that high amounts of extracellular bacteria were present in brain blood vessels. The esx‐1 mutant mycobacteria were predominantly found in the lumen of the blood vessels and were never found to cross or disrupt the basal lamina and blood vessel wall (Figure 6d, red dotted line), which is in contrast with WT infections. Only sporadically, bacteria were endocytosed by an endothelial cell (Figure 6e, *). In addition, no segmentation of blood vessels was observed, although the infected vessels were often enlarged in diameter (Figure 6f; lumen diameter WT infection: average 6.2 μm, range 4–9 μm, n = 15; lumen diameter esx‐1 mutant infection: average 18.4 μm, range 7.5–37 μm, n = 15).
Figure 6.
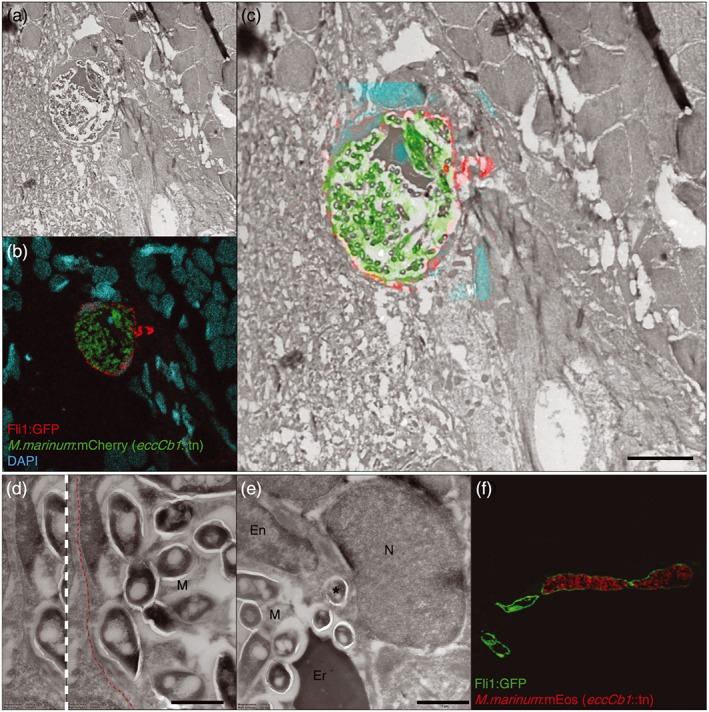
ESX‐1 secretion is required for macrophage independent BBB crossing in vivo. Infected blood vessel (depicted in red to facilitate comparison with Figures 3 and 4) found in brain tissue of 9 dpf phagocyte‐depleted Tg(Fli1:GFP) y1 larva, systemically infected with M. marinum eccCb1::tn (green), nuclei are stained with DAPI (cyan). (a) Electron microscopy, (b) confocal microscopy, and (c) correlative light and electron microscopy, showing an abundant amount of extracellular ESX‐1‐deficient M. marinum restricted to a blood vessel. Scale bar = 5 μm. Also notice the regular shape of the blood vessel. (d) Higher magnification of the highly infected blood vessel depicted in (c), showing that bacteria did not cross or disrupt the basal lamina (red dotted line). (e) Although esx‐1 mutant bacteria were mainly found in the lumen of the vessel, sporadically one was found to invade endothelial cell (*). (f) Cross section of infected (red fluorescent esx‐1 mutant) and non‐infected blood vessels (green) of Tg(Fli1:GFP) y1 larva, showing high bacterial load in a single blood vessel with subsequent enlargement of the vessel lumen. En = Endothelial cell (nucleus), Er = Erythrocyte, M = M. marinum, N = nucleus, Scale bar e and f = 1 μm
Collectively, CLEM analysis confirmed that mycobacteria require ESX‐1 secretion for macrophage‐independent crossing of the BBB.
2.6. M. marinum invasion of brain endothelial cells is dependent on ESX‐1 secretion
To study the interaction of M. marinum with brain endothelial cells (BECs) in more detail and to examine the role of ESX‐1 secretion in this process, human BECs were infected with live or heat‐killed M. marinum WT, the esx‐1 mutant or the complemented esx‐1 mutant. Bacterial levels at 24‐hr postinfection (hpi) were analysed with FACS and compared with uptake of these bacteria by RAW macrophages. RAW macrophages phagocytosed both strains with similar efficiency, both at 2 and 24 hpi (Figure 7a), confirming previous reports that the level of phagocytosis of M. marinum WT and our esx‐1 mutant are similar (Houben et al., 2012; Simeone et al., 2012; van der Wel et al., 2007).
Figure 7.
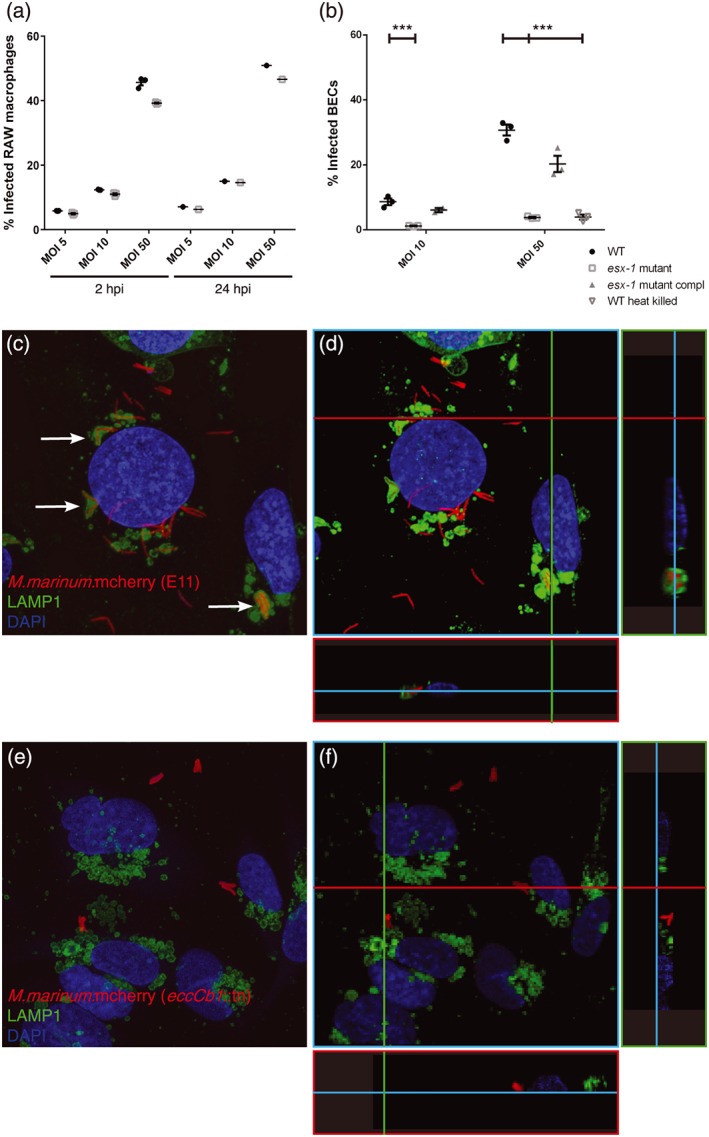
ESX‐1 secretion required for brain endothelial cell invasion. (a) FACS experiment showing uptake of M. marinum E11 and ESX‐1‐deficient M. marinum in RAW macrophages. No significant differences can be found in phagocytosis at 2 and 24 hpi. (b) Infection of brain endothelial cells show significant differences for both concentrations in uptake between M. marinum WT, the esx‐1 mutant, complemented esx‐1 mutant, and heat‐killed M. marinum WT, graph shows one out of three experiments with representative data, performed in triplo. *** = p < .005. (c) M. marinum WT (red) infects BECs (nuclei, cyan) and is transferred to the lysosome, shown by colocalisation of mycobacteria and LAMP1 (green; arrows). (d) 3D model of the same stack provides more evidence for the colocalisation of M. marinum with LAMP1, illustrated with two cross sections in this stack, visualised with green and red line. (e) Few esx‐1 mutant bacteria are found associated with BECs and clearly show no colocalisation with LAMP1. (f) 3D modelling of the same stack shows the probable extracellular localisation of the esx‐1 mutant
When compared with macrophages, infection of BECs with M. marinum is less efficient. To observe infection, we needed a higher multiplicity of infection (MOI 50 instead of MOI 10) and even then the number of infected cells was lower compared with the RAW macrophages (compare 45% infected RAW macrophages in Figure 7a with 30% infected BECs in Figure 7b). However, if we just look at BECs, we could observe significant differences between M. marinum WT and the esx‐1 mutant (Figure 7b). The esx‐1 mutant showed almost eight times lower uptake at 24 hpi, for both experiments performed with an MOI of 10 and an MOI of 50. Importantly, the infection defect was restored when cells were infected with the complemented strain. Interestingly, infection with heat‐killed M. marinum WT also resulted in reduced uptake, at a level comparable with the phenotype seen for the esx‐1 mutant. This indicates that invasion of BECs by M. marinum WT is an active process that depends on an active ESX‐1 secretion system.
Confocal analysis of infected BECs showed colocalisation of WT M. marinum with LAMP1, a late endosomal marker, demonstrating that M. marinum E11 is transported to the phagolysosome (Figure 7c,d). In contrast, the few esx‐1 mutant bacteria that were associated with BECs did not colocalise with LAMP1 and based on the 3D modelling appeared to be located at the cell periphery, probably even at the cell surface (Figure 7e,f).
In conclusion, these experiments show that M. marinum is capable of actively invading and infecting brain endothelial cells. In addition, the ESX‐1 secretion system is essential for active invasion of BECs, extending the role of ESX‐1 secretion beyond the macrophage infection cycle.
3. DISCUSSION
In this study, we show that M. marinum employs two different strategies to cross the BBB: the Trojan horse mechanism and ESX‐1‐dependent invasion and damaging of endothelial cells (Figure 8, graphical abstract).
Figure 8.
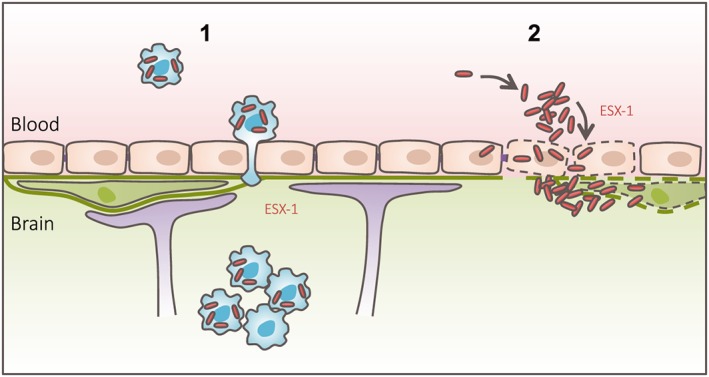
Graphical abstract. Graphical abstract summarising the main findings of this study, in which we show that mycobacteria are able to employ two different migration routes to cross the BBB: (1) Trojan horse mechanism, in which mycobacteria can cross a functionally intact BBB within phagocytic cells; (2) ESX‐1‐dependent invasion and infection of endothelial cells, with subsequent damage of the basal lamina
Our observation that, under normal conditions, all bacteria that cross the BBB colocalise with a phagocyte is a strong indication that this is one of the mechanisms for M. marinum. There was a clear difference in M. marinum trafficking before and after the presence of an intact BBB. Infection experiments at early time points, that is, performed before BBB formation, showed that mycobacteria could readily traverse into brain tissue. These results are comparable with mycobacterial invasion into tissue outside the CNS (Lesley & Ramakrishnan, 2011). After the formation of the BBB, mycobacterial crossing was delayed by several days. This shows that the BBB is an obstacle for entering the brain, even when using the Trojan horse mechanism. This also means that in vivo experiments performed to study dissemination of mycobacteria at early time points, that is, 2 dpf, cannot be used to study natural infection of brain tissue.
For extra‐cellular pathogens causing meningitis, such as S. pneumoniae, N. meningitides, and H. influenza, it has been shown that haematogenous dissemination directly results in acute meningitis (Panackal, Williamson, van de Beek, Boulwared, & Williamson, 2016; van Sorge & Doran, 2013). However, it seems likely that the initial seeding of M. tuberculosis into brain tissue only induces a mild inflammatory response, because massive inflammation seen during clinical presentation of TBM is only observed when a pre‐existing granuloma in the brain progresses into meningitis (Donald, Schaaf, & Schoeman, 2005; Donald & Van Toorn, 2016; Panackal et al., 2016). This might be explained by the Trojan horse mechanism but also by a sporadic bacteraemia with subsequent non‐inflammatory deposition of free mycobacteria on the endothelium. Using a macrophage as protective niche instead of endothelial cell infection, M. tuberculosis avoids interaction with the endothelial cells and a subsequent inflammatory response. Our fluorescent dye experiments support that this initial non‐inflammatory event is not accompanied with substantial breakdown of the BBB. However, we cannot exclude the possibility of local, perhaps transient, areas of compromised BBB integrity, serving as entry point for infected phagocytes. Additional evidence for a Trojan horse mechanism was obtained by studying esx‐1‐deficient bacteria. Infection with this attenuated mutant strain that is not able to escape the phagocyte resulted in reduced infection levels and inflammation, but nevertheless, phagocytes filled with these bacteria did infect the brain. Extensive breakdown of the BBB by a massive (local) inflammatory response is unlikely in this case, because we have relatively low levels of infection by this attenuated mutant. Both concepts fit with the generally accepted Rich focus theory about TBM pathogenesis, describing that meningitis only occurs when a granuloma in brain tissue or meninges discharges its content in the subarachnoid space. However, more recently, it has been debated that haematogenous dissemination in the form of miliary TB in young children, with a high bacteraemia, has a high likelihood to lead to TBM soon after infection and may have a stronger correlation with the onset of TBM in children than initially thought (Donald et al., 2005). Therefore, it has been suggested that M. tuberculosis might be able to use other entry routes into the brain when high bacterial levels in blood are present. Moreover, mycobacteria associated with endothelium and in close proximity of blood vessels could explain endovasculitis as major pathological hallmark of TBM seen in historical studies and might suggest a specific role of these bacteria in Rich focus formation (Rich & McCordock, 1933).
Our new data uncover the presence of such an alternative route, where M. marinum is able to enter the CNS without the help of phagocytes. We used CLEM to show the localisation of mycobacteria in the blood vessel lumen, in endothelial cells, and in the surrounding tissue. These experiments suggested that these bacteria cross the BBB transcellular. Our in vivo studies are consistent with in vitro studies demonstrating the ability of M. tuberculosis to invade endothelial cells (Chen et al., 2015; Jain et al., 2006; Mehta et al., 2006). Remarkably, this transcellular migration by M. marinum seems to differ from the “classical” transcellular migration route, in which changes in endothelial barrier integrity are generally not observed (Kim, 2008). However, strategies that affect BBB integrity to gain direct entry have been demonstrated for other pathogens (van Sorge & Doran, 2013), and it is not unlikely that mycobacteria developed similar strategies. The perception that M. tuberculosis is also invading other host cells than only the phagocytic cells is not new. Recently, lymphatic endothelial cells were suggested as a replicative niche for M. tuberculosis (Lerner et al., 2016), and also the importance of the interaction of M. tuberculosis with epithelial cells for early dissemination has been noted previously (Menozzi et al., 2006). Here, we show the role of differential host cells for dissemination across the BBB.
We used an ESX‐1‐deficient mutant to study the effect of a well‐known virulence factor on BBB trafficking. Interestingly, while our results confirmed the importance of ESX‐1 secretion in dissemination and virulence (Stoop et al., 2011; Volkman et al., 2004), it also revealed a completely new and different phenotype. In phagocyte‐depleted larvae infected with the ESX‐1‐deficient strain, a high bacterial load is found in the zebrafish brain. However, in contrast to WT bacteria, these bacteria are almost exclusively restricted to the blood vessel lumen and do not invade endothelial cells. Previously, the work of Jain et al. showed a difference between Mycobacterium bovis BCG and M. tuberculosis isolates in invasion efficiency of brain endothelial cells, but they did not attribute this difference to ESX‐1 (Jain et al., 2006). Our novel findings now show that invasion and infection of endothelial cells by M. marinum, both in vivo and in vitro, are dependent on ESX‐1 secretion. Our data suggest that ESX‐1 substrates mediate an active process in which M. marinum is able to invade endothelial cells. This attributes a novel function to ESX‐1 secretion, in addition to phagosomal escape (Abdallah et al., 2011; Houben et al., 2012; Simeone et al., 2012; van der Wel et al., 2007). Furthermore, the damaging effect of mycobacteria on endothelial cells might be part of the explanation for the extensive vasculopathy, including stroke and vasculitis, found in clinical TBM presentation and autopsy material (Donald & Van Toorn, 2016; van der Flier et al., 2004; Zaharie et al, unpublished).
Another factor linked to cerebral vasculopathy is VEGF. In the case of TBM, increased VEGF levels have been found in CSF and are associated with cerebral oedema formation, hydrocephalus, and basal meningeal enhancement (van der Flier et al., 2004; Visser et al., 2014). The observed colocalisation of intensified kdrl/vegfr2 signal with M. marinum‐loaded phagocytes in larval brain blood vessels in our study suggests an interaction between infected macrophages and the blood vessel wall and a role for VEGF in this process. It has been shown that macrophages secrete VEGF after infection with M. tuberculosis in an ESX‐1‐dependent manner, which in turn interacts with VEGFR2 specifically (Polena et al., 2016). This mechanism is essential for angiogenetic vessel growth and bacterial expansion and dissemination. Our data suggest a similar mechanism involved in BBB crossing. The intensified VEGFR2 signal phenotype is less frequently seen when extracellular bacteria colocalise with blood vessels, suggesting that infected macrophages play an important role in this process.
Although it is clear that VEGF is not the only factor involved in mycobacterial CNS infection, the importance of VEGF is of great interest in the context of clinical TBM presentation and therapeutic approaches. Various animal studies suggest that anti‐VEGF treatment, such as the widely prescribed anti‐VEGF antibody bevacizumab, can resolve vascular leakiness and improve outcome of TB (Datta et al., 2015; Oehlers et al., 2014; Oehlers et al., 2016). With the rising awareness that host‐directed therapy could be a valuable addition to antibiotics, VEGF seems to be an interesting target. However, our study failed to show any effect of VEGF level manipulation on phagocyte migration. The absence of a link might be obscured by many factors, such as VEGF interacting with VEGFR1 in addition to VEGFR2 or TNFα interacting with VEGFR2 (Jeltsch, Leppänen, Saharinen, & Alitalo, 2013; Zielonka et al., 2011). Hypothetically, these pathways interact with each other and may alter the effect seen in our experiments. This implies that anti‐VEGF treatment might not be as straightforward as hoped for or this might suggest that VEGF levels are not essential for mycobacterial CNS invasion.
In summary, our results support the longstanding idea that mycobacteria employ macrophages as Trojan horse as migration mechanism across the BBB and suggest that VEGF might play a role in this process, but we also show that the Trojan horse is not the only route this pathogen uses. We demonstrate an additional type of migration whereby virulent M. marinum actively infect and disrupt the endothelium to gain excess to brain tissue in an ESX‐1‐dependent manner. The next step would be to determine if the same mechanisms apply for M. tuberculosis. As such, this study reveals early steps of TBM pathogenesis and might help us to explain variation in pathological and clinical presentation.
4. EXPERIMENTAL PROCEDURES
4.1. Bacterial strains and growth conditions
Mycobacterium marinum E11 WT, a sea bass isolate of M. marinum, and the ESX‐1 secretion mutant eccCb1::tn, derivative of this strain (esx‐1 mutant), were used in this study (Stoop et al., 2011). M. marinum E11 and eccCb1::tn were transformed by electroporation with pSMT3‐hsp60‐mCherry, to express mCherry, or pSMT3‐hsp60‐mEos3.1 and pSMT3‐hsp60‐mEos3.2, to express mEos3.1 or mEos3.2 respectively. All three plasmids were used to visualise infection in zebrafish larvae and human Cerebral Microvascular Endothelium Cells (hCMEC/D3) infection experiments. Complementation of eccCb1::tn was done by introduction of plasmid pMV‐hsp60‐eccCb1. Transformants were selected on plates with the appropriate antibiotic selection markers (25‐μg/ml kanamycin [Sigma] and/or 50‐μg/ml hygromycin [Roche]). All constructs were confirmed by sequencing of plasmid inserts.
Strains were routinely grown at 30 °C on Middlebrook 7H10 agar plates (Difco) supplemented with 10% oleic acid‐albumin‐dextrose‐catalase (BD Bioscience) or in Middlebrook 7H9 broth (Difco) with 10% Middlebrook albumin‐dextrose‐catalase (BD Bioscience), 0.05% Tween‐80, and the appropriate antibiotic selection marker.
4.2. Construction of plasmids
To visualise bacterial infection in zebrafish embryo, infection experiments pSMT3‐hsp60‐mEos3.1 and pSMT3‐hsp60‐mEos3.2 were created. Both mEos3.1 and mEos3.2 were obtained from Zhang et al. (2012). mEos3.1 and mEos3.2 were amplified by PCR with 15 base pair flanking regions corresponding to the target vector using mEos3‐FW and mEos3‐RV (Table S2). The target vector pSMT3‐hsp60‐mCherry (Meijer et al., 2008) was digested with NheI and BamHI. Subsequently, the insert was introduced into the digested vector by In‐Fusion cloning (Clontech) to produce the plasmids pSMT3‐hsp60‐mEos3.1 and pSMT3‐hsp60‐mEos3.2. A complementation vector for the eccCb1::tn mutant was constructed by amplification of the eccCb1 gene from genomic DNA of M. marinum E11 using primers EccCb1‐comp‐FW and EccCb1‐comp‐RV (Table S2). The primers introduced a 15 base pair overlap with the target vector on both 5′ and 3′ sides of the insert to allow In‐Fusion cloning. The target vector, pMV‐hsp60‐mEos3.1 (Van De Weerd et al., 2016) was digested with NheI and ClaI. Subsequently, the insert was introduced into the digested vector by In‐Fusion cloning to produce pMV‐hsp60‐eccCb1. The plasmids were subsequently transformed by electroporation into M. marinum E11 or the respective M. marinum E11 eccCb1::tn mutant.
4.3. Injection stocks
Injection stocks were prepared by growing bacteria until the logarithmic phase (OD600 of 0.7–1). Bacteria were briefly spun down and washed with 0.3% Tween‐80 in phosphate buffered saline (PBS) for declumping and resuspended in PBS with 20% glycerol and stored at −80 °C. Before use, bacteria were resuspended in PBS containing 0.17% (v/v) phenol red (Sigma) to aid visualisation of the injection process.
4.4. Zebrafish
Zebrafish lines used in this study: transparent casper zebrafish (White et al., 2008), Tg(Fli1:GFP) y1 casper zebrafish, with green fluorescent endothelial cells (Lawson & Weinstein, 2002), and Tg(kdrl:mCherry) is5, with red fluorescent endothelial cells (Jin, Beis, Mitchell, Chen, & Stainier, 2005).
All procedures involving D. rerio (zebrafish) larvae were performed in compliance with local animal welfare laws and maintained according to standard protocols (http://zfin.org). The breeding of adult fish and infection of embryos older than 5–7 dpf was approved by the institutional animal welfare committee (Animal Experimental licensing Committee, DEC). All protocols adhered to the international guidelines specified by the EU Animal Protection Directive 86/609/EEC, which allows zebrafish embryos to be used up to the moment of free‐living (approximately 5–7 dpf). Infection of older embryos was approved under DEC number MMI10‐02 by the institutional animal welfare committee (Animal Experimental licensing Committee, DEC) of the VU University medical centre.
4.5. Infection procedure
At 2 or 4 dpf, embryos were infected with M. marinum E11 or eccCb1::tn by microinjection in the caudal vein (E11‐2dpf: 250–400 CFU; E11‐4dpf: 250–900; eccCb1::tn‐4dpf: 325–1,040 CFU). Injection was performed as described previously (Benard et al., 2012). At 1–5 dpi bacterial infection was monitored daily with a Leica MZ16FA fluorescence microscope. Following analysis, larvae were fixated on indicated time points overnight in 4% (v/v) paraformaldehyde (EMS, 100122) in PBS, and stored in 100% methanol at −20 °C for immune‐histochemical staining and confocal imaging. To determine the exact number of bacteria injected, the injection volume was plated on 7H10 plates containing the proper antibiotic selection. During injection and microscopic examining, larvae were anaesthetised in egg water with 0.02% (W/V) buffered 3‐aminobenzoic acid (Tricaine; Sigma‐Aldrich, A‐5040).
4.6. Phagocyte depletion
To deplete the phagocytic pool in larvae, pu.1 morpholino (Rhodes et al., 2005) was injected at the 1–4 cellular stage. At 3 dpf clodronate‐filled liposomes, (1:1 [v/v] diluted in phenol red, 10 nl end volume) were injected into the caudal vein, to deplete the larvae of all residual systemic phagocytes (Bernut et al., 2014; Pagán et al., 2015; Van Rooijen & Sanders, 1994).
4.7. BBB functionality assay with fluorescent tracer
Uninfected Tg(kdrl:mCherry) is5 larvae were injected with a 3‐kDa fluorescent dye (Dextran, Alexa Fluor 647, Thermofisher) into the caudal vein at 2–9 dpf. Leakage of tracer into brain tissue, as a measure for BBB integrity, was subsequently monitored on a confocal microscope every 10 min between 30‐ and 120‐min postinjection.
4.8. VEGF modulation experiments
To study the effect of VEGF on infection levels and bacterial BBB crossing, either 250 nM SU5416 (VEGF receptor blocker, Sigma S8442; Oehlers et al., 2014; Wu et al., 2015) or 2.5 μM GS4012 (VEGF inducer, Santa Cruz Biotech sc‐222411; Wu et al., 2015) was used. Compounds were added directly after infection and refreshed every second day as described previously (Oehlers et al., 2014). Six to eight larvae per group were fixated in 4% (v/v) paraformaldehyde (EMS, 100122) in PBS at 3 and 5 dpi for further analysis with confocal microscopy.
4.9. Immunohistochemical stain
Larvae were fixated on indicated time points overnight in 4% (v/v) paraformaldehyde (EMS, 100122) in PBS and stored in 100% methanol at −20 °C for immune‐histochemical staining and confocal imaging. Larvae were labelled with anti‐L‐plastin, which stains phagocytic cells according to established protocols (Bennett et al., 2001; Herbomel, Thisse, & Thisse, 1999). In short, larvae were rinsed with 1% PBTx, (1% Triton X‐100 in PBS), permeated in 0.24% trypsin in PBS and blocked for 3 hr in block buffer (10% normal goat serum [NGS] in 1% PBTx). Samples were incubated with anti‐L‐plastin (1:500 [v/v] dilution) in antibody buffer (PBTx containing 1% [v/v] NGS and 1% [w/v] BSA) overnight at RT. Samples were washed with PBTx, incubated for 1 hr in block buffer and stained with an Alexa‐Fluor‐647 goat‐anti‐rabbit as secondary antibody (Invitrogen A21070, 1:400), overnight at 4 °C.
4.10. Microscopy
Bacterial infection was monitored initially with a Leica MZ16FA fluorescence microscope. Bright field and fluorescence images were generated with a Leica DFC420C camera. Early granuloma formation was analysed visually and quantified with custom‐made pixel‐counting software (http://www.elaborant.com). Confocal analysis was performed on hCMEC/D3 cells and larvae, embedded in 1% low melting‐point agarose (Boehringer Mannheim, 12841221‐01) in an eight‐well microscopy μ‐slide (ibidi). Analysis was performed with a confocal laser‐scanning microscope (Leica TCS SP8 X Confocal Microscope). Leica Application Suite X software and ImageJ software were used to adjust brightness and contrast and to create overlay images and 3D models.
4.11. CLEM
Representative embryos were selected to study infection in the zebrafish brain using CLEM. Larvae were fixated overnight in 4% (v/v) paraformaldehyde (EMS, 100122) dissolved in PBS, and stored in 0.1‐M PHEM and 0.5% PFA. 0.4‐M PHEM was made with 240‐mM PIPES, dissolved in 0.3‐M NaOH. Following this, 40‐mM EGTA, 100‐mM HEPES, and 8‐mM MgCl2 were added in this order. pH was adjusted to 6.9 with NaOH, and PHEM buffer was stored at −20 °C until use. Larvae were incubated overnight in 2‐M sucrose and snap‐frozen on a pin in liquid nitrogen. Semithin and ultrathin EM sections were cut as described by Bedussi et al. (2016). Semithin sections (300–400 nm) were stained with DAPI for counterstaining of nuclei in the tissue and analysed with confocal microscopy. When fluorescent vessels were found to colocalise with fluorescent M. marinum, 70‐ to 100‐nm thin sections were cut and transferred to a 150‐mesh cupper grid and stained with Uranyl Acetate for TEM analysis, grids with ultrathin sections were washed and stained with a Uranyl acetate/Tylose mixture and imaged using Tecnai T12 at 120 kV. The position of the nuclei, which is visible in both FM and EM, was used to align and overlay the images (Adobe Photoshop CS6).
4.12. Cell infection
The human Cerebral Microvascular Endothelium Cells line hCMEC/D3 (or BECs; Weksler et al., 2005), kindly provided by Dr. P.‐O. Couraud (Institut Cochin, Universite Paris Descartes, Paris, France), and mouse RAW264.7 macrophages (American Type Culture Collection) were used for cell infection experiments. The hCMEC/D3 cells were grown in EBM−2 medium supplemented with hEGF, hydrocortisone, GA‐1000, fetal bovine serum, VEGF, hFGF‐B, R3‐IGF‐1, ascorbic acid, and 2.5% fetal calf serum (Lonza, Basel, Switzerland). Before use, cells were washed with PBS and human endothelial SFM was added (Invitrogen, ca. no 11111‐044). RAW264.7 cells were cultured in RPMI 1640 with Glutamax‐1 (Gibco) supplemented with 10% FBS (Gibco), 100 U of penicillin/ml, 100 μg of streptomycin/ml at 37 °C, 5% CO2.
4.13. Cell infection–flow cytometry
hCMEC/D3 cells were seeded until confluent in 24‐well plates. For RAW macrophages, a total of 3 × 107 cells were seeded in T175 flasks (Corning). M. marinum E11, M. marinum eccCb1::tn, and M. marinum eccCb1::tn‐comp were grown until the exponential growth phase, spun down, and resuspended in specialised medium. Brain endothelial cells were infected with a multiplicity of infection of 10 or 50, incubated for 2 or 24 hr at 30 °C, 5% CO2, washed with PBS and detached with trypsin. RAW macrophages were infected with an MOI of 5, 10, or 50 for 2 or 24 hr and incubated at 30 °C, 5% CO2. Uptake of both strains was quantified for both cell lines with a BD Accuri C6 flow cytometer (BD Biosciences) with a 488‐nm laser and 585‐/40‐nm filter to detect mEos3.1. A minimum of 10,000 gated events were collected per sample per time point; data were analysed using BD CFlow software.
4.14. Cell infection–confocal microscopy
hCMEC/D3 cells were seeded until confluent in eight‐well microscopy μ‐slide (ibidi, cat no. 80826). M. marinum E11 and M. marinum eccCb1::tn were grown until the exponential growth phase, spun down, and resuspended in specialised medium. Brain endothelial cells were infected with an MOI of 10 or 50, incubated for 2 or 24 hr, washed with PBS and fixated with 4% PFA dissolved in PBS for 20 min. For labelling, cells were blocked for 60 min in block buffer (5% NGS in 0.3% Triton X100). Samples were incubated with anti‐LAMP1 (Cell Signalling, cat. no 9091P, 1:100) in antibody buffer (1% BSA and 0.3% Triton X100 in PBS) overnight at 4 °C. Samples were washed with PBS and incubated with Alexa‐Fluor‐488 goat‐anti‐rabbit (Molecular Probes, cat. no A‐11008, 1:400) in antibody buffer for 90 min at RT. Cells were washed with PBS, incubated with Hoechst (1:1000, Molecular Probes, cat. no 33258) for 1 min, washed with PBS, and stored in PBS at 4 °C until further analysis with confocal microscopy (Leica TCS SP8 X Confocal Microscope). Leica Application Suite X software was used for 3D analysis.
4.15. Graphs and statistical analysis
Graphs were made using Graph Pad Prism 6.0. Pixel counts obtained with eLaborant were logarithmic transformed; error bars represent mean and standard error of the mean. A one‐way ANOVA was performed followed by a Bonferroni multiple comparison test to analyse statistical significance. Graphs with results for D3 and RAW cell infection show percentage infected cells of total cells; error bars represent mean and standard error of the mean. A one‐way ANOVA was performed for statistical analysis.
Supporting information
Figure S1: Single channel images corresponding to Figure 1. (A) Merged confocal image of systemically infected zebrafish larvae, 3 days post infection (dpi), corresponding to Figure 1H, with single red channel depicting blood vessels, single green channel showing M. marinum::mEos and single cyan channel with L‐plastin labeled phagocytic cells. (B) depicts single channels corresponding to Figure 1I, (C) corresponds to Figure 1J, (D) corresponds to Figure 1K.
Figure S2: Fluorescent dye leakage experiments in infected larvae. Tg(kdrl:mCherry) is5 zebrafish larvae (red blood vessels), infected with M. marinum::mEos (green) at 4 dpf, 1 hour after 3 kDa Alexa 680 tracer injection (cyan) at 3 dpi (A‐C) and 5 dpi (D‐F). Figure shows merged confocal images (A, B, C, D, E, F) and single cyan channel (A', B', C', D', E', F'). Open arrows point at single bacteria associated with the blood vessel wall, without leakage of the dye in the adjacent tissue. Closed arrows show localization of tracer in brain parenchyma, which was always concentrated in small spots.
Figure S3: Intensified VEGFr signal co‐localizing with infected phagocytes. (A – C) Z‐stack of head of 5dpi/9dpf Tg(kdrl:mCherry) zebrafish larva with red fluorescent blood vessels, systemically infected with M. marinum E11 (green) at 4 dpf, treated directly after infection with GS4012 to induce VEGF levels (A), nothing (B) or SU5416 to block the VEGFr (C). Representative image per group is shown. Clear difference in infection level can be seen, in particular in GS4012‐treatment compared to control. Scale bar = 250 μm.
Figure S4: Correlative light and electron microscopy. (A) Correlative light and electron microscopy of (B) Confocal Microscopy and (C) Electron microscopy, of a 9 dpf Tg(kdrl:mCherry) control larva with red fluorescent blood vessels, infected at 4 dpf with green fluorescent M. marinum E11, nuclei counter stain after fixation with DAPI (cyan). (B) clearly illustrates the limitations of confocal microscopy in terms of resolution.
Table S1: Intensified VEGFr signal co‐localizing with infected phagocytes. Table showing total number of phagocytosed M. marinum found in brain blood vessels or brain tissue in all larvae included per group: GS4012‐treated vs. control vs. SU5416‐treated. Per topic an absolute number and percentage of total is shown. (D) Time point: 3 dpi, (E) time point: 5 dpi.
Table S2: Primers used in this study to create bacterial constructs
ACKNOWLEDGEMENTS
We thank Kin Ki Jim, Roy Ummels, Theo Verboom, and Wim Schouten for technical assistance; the cellular imaging facility at the AMC for the use of their confocal microscope; Wikky Tigchelaar‐Gutter for technical assistance with EM experiments; Nanne Paauw for help with confocal imaging at VUmc, and Prof. P. Martin (Bristol University, UK) for the L‐plastin antibody.
This research is partially funded by ESPID/Wyeth fellowship 2010–2012 (awarded to M.v.d.K.) and Innovative Medicines Initiative Joint Undertaking Grant Agreement 115337 (to W.B.).
van Leeuwen LM, Boot M, Kuijl C, et al. Mycobacteria employ two different mechanisms to cross the blood–brain barrier. Cellular Microbiology. 2018;20:e12858 10.1111/cmi.12858
REFERENCES
- Abbott, N. J. , Patabendige, A. A. , Dolman, D. E. , Yusof, S. R. , & Begley, D. J. (2010). Structure and function of the blood‐brain barrier. Neurobiology of Disease, 37(1), 13–25. Available at: doi: 10.1016/j.nbd.2009.07.030 [DOI] [PubMed] [Google Scholar]
- Abbott, N. J. , Rönnbäck, L. , & Hansson, E. (2006). Astrocyte‐endothelial interactions at the blood‐brain barrier. Nature Reviews. Neuroscience, 7(1), 41–53. Available at: http://www.ncbi.nlm.nih.gov/pubmed/16371949. Accessed May 22, 2013 [DOI] [PubMed] [Google Scholar]
- Abdallah, A. M. , Bestebroer, J. , Savage, N. D. , de Punder, K. , van Zon, M. , Wilson, L. , … Peters, P. J. (2011). Mycobacterial secretion systems ESX‐1 and ESX‐5 play distinct roles in host cell death and inflammasome activation. Journal of Immunology, 187(9), 4744–4753. Available at: http://www.ncbi.nlm.nih.gov/pubmed/21957139. Accessed December 31, 2013 [DOI] [PubMed] [Google Scholar]
- Be, N. a. , Bishai, W. R. , & Jain, S. K. (2012). Role of Mycobacterium tuberculosis pknD in the pathogenesis of central nervous system tuberculosis. BMC Microbiology, 12, 7 Available at: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3322341&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- Be, N. A. , Klinkenberg, L. G. , Bishai, W. R. , Karakousis, P. C. , & Jain, S. K. (2011). Strain‐dependent CNS dissemination in guinea pigs after Mycobacterium tuberculosis aerosol challenge. Tuberculosis (Edinburgh, Scotland), 91(5), 386–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedussi, B. , van der Wel, N. N. , de Vos, J. , van Veen, H. , Siebes, M. , Van Bavel, E. , & Bakker, E. N. (2016). Paravascular channels, cisterns, and the subarachnoid space in the rat brain: A single compartment with preferential pathways. Jounal of Cerebral Blood Flow & Metabolism, 0(0), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benard, E. L. , van der Sar, A. M. , Ellett, F. , Lieschke, G. J. , Spaink, H. P. , & Meijer, A. H. (2012). Infection of zebrafish embryos with intracellular bacterial pathogens. Journal of Visualized Experiments, 61, 1–9. Available at: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3415172&tool=pmcentrez&rendertype=abstract. Accessed January 3, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bencurova, E. , Mlynarcik, P. , & Bhide, M. (2011). An insight into the ligand‐receptor interactions involved in the translocation of pathogens across blood‐brain barrier. FEMS Immunology and Medical Microbiology, 63(3), 297–318. [DOI] [PubMed] [Google Scholar]
- Bennett, C. M. , Kanki, J. P. , Rhodes, J. , Liu, T. X. , Paw, B. H. , Kieran, M. W. , … Look, A. T. (2001). Myelopoiesis in the zebrafish, Danio rerio. Blood, 98(3), 643–651. Available at: doi: 10.1182/blood.V98.3.643. Accessed October 24, 2012 [DOI] [PubMed] [Google Scholar]
- Berg, R. D. , & Ramakrishnan, L. (2012). Insights into tuberculosis from the zebrafish model. Trends in Molecular Medicine, 18(12), 689–690. Available at: http://www.ncbi.nlm.nih.gov/pubmed/23084762. Accessed June 4, 2013 [DOI] [PubMed] [Google Scholar]
- Bermudez, L. E. , Sangari, F. J. , Kolonoski, P. , Petrofsky, M. , & Goodman, J. (2002). The efficiency of the translocation of Mycobacterium tuberculosis across a bilayer of epithelial and endothelial cells as a model of the alveolar wall is a consequence of transport within mononuclear phagocytes and invasion of alveolar epithelial cells. Infection and Immunity, 70(1), 140–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernut, A. , Herrmann, J. L. , Kissa, K. , Dubremetz, J. F. , Gaillard, J. L. , Lutfalla, G. , & Kremer, L. (2014). Mycobacterium abscessus cording prevents phagocytosis and promotes abscess formation. Proceedings of the National Academy of Sciences of the United States of America, 111(10), E943–E952. Available at: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3956181&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, X. , Sakamoto, K. , Quinn, F. D. , Chen, H. , & Fu, Z. (2015). Lack of intracellular replication of M. tuberculosis and M. bovis BCG caused by delivering bacilli to lysosomes in murine brain microvascular endothelial cells. Oncotarget, 6(32), 32456–32467. Available at: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4741705&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang, S. S. , Khan, F. A. , Milstein, M. B. , Tolman, A. W. , Benedetti, A. , Starke, J. R. , & Becerra, M. C. (2014). Treatment outcomes of childhood tuberculous meningitis: a systematic review and meta‐analysis. The Lancet Infectious Diseases, 14(10), 947–957. [DOI] [PubMed] [Google Scholar]
- Clay, H. , Davis, J. M. , Beery, D. , Huttenlocher, A. , Lyons, S. E. , & Ramakrishnan, L. (2011). Dichotous role of the macrophage in early mycobacterium marinum Infection of the Zebrafish. Cell Host & Microbe, 2(1), 29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta, M. , Via, L. E. , Kamoun, W. S. , Liu, C. , Chen, W. , Seano, G. , … Jain, R. K. (2015). Anti‐vascular endothelial growth factor treatment normalizes tuberculosis granuloma vasculature and improves small molecule delivery. Proceedings of the National Academy of Sciences, 112(6), 1827–1832. Available at: doi: 10.1073/pnas.1424563112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, J. M. , & Ramakrishnan, L. (2009). The role of the granuloma in expansion and dissemination of early turberculous infection. Cell, 136(1), 37–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donald, P. R. , Schaaf, H. S. , & Schoeman, J. F. (2005). Tuberculous meningitis and miliary tuberculosis: the Rich focus revisited. The Journal of Infection, 50(3), 193–195. Available at: http://www.ncbi.nlm.nih.gov/pubmed/15780412. Accessed October 24, 2012 [DOI] [PubMed] [Google Scholar]
- Donald, P. R. , & Van Toorn, R. (2016). Use of corticosteroids in tuberculous meningitis. The Lancet, 387(10038), 2585–2587. Available at: http://linkinghub.elsevier.com/retrieve/pii/S014067361630770X [DOI] [PubMed] [Google Scholar]
- Fleming, A. , Diekmann, H. , & Goldsmith, P. (2013). Functional characterisation of the maturation of the blood‐brain barrier in larval zebrafish. PLoS One, 8(10), e77548 Available at: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3797749&tool=pmcentrez&rendertype=abstract. Accessed January 3, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbomel, P. , Thisse, B. , & Thisse, C. (1999). Ontogeny and behaviour of early macrophages in the zebrafish embryo. Development, 126(17), 3735–3745. Available at: http://www.ncbi.nlm.nih.gov/pubmed/10433904 [DOI] [PubMed] [Google Scholar]
- Houben, D. , Demangel, C. , van Ingen, J. , Perez, J. , Baldeón, L. , Abdallah, A. M. , … Peters, P. J. (2012). ESX‐1‐mediated translocation to the cytosol controls virulence of mycobacteria. Cellular Microbiology, 14(8), 1287–1298. Available at: http://www.ncbi.nlm.nih.gov/pubmed/22524898. Accessed October 29, 2012 [DOI] [PubMed] [Google Scholar]
- Jain, S. K. , Paul‐Satyaseela, M. , Lamichhane, G. , Kim, K. S. , & Bishai, W. R. (2006). Mycobacterium tuberculosis invasion and traversal across an in vitro human blood‐brain barrier as a pathogenic mechanism for central nervous system tuberculosis. The Journal of Infectious Diseases, 193(9), 1287–1295. [DOI] [PubMed] [Google Scholar]
- Jeltsch, M. , Leppänen, V. M. , Saharinen, P. , & Alitalo, K. (2013). Receptor tyrosine kinase‐mediated. Cold Spring Harbor Perspectives in Biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, S.‐W. , Beis, D. , Mitchell, T. , Chen, J. N. , & Stainier, D. Y. (2005). Cellular and molecular analyses of vascular tube and lumen formation in zebrafish. Development (Cambridge, England), 132(23), 5199–5209. Available at: http://www.ncbi.nlm.nih.gov/pubmed/16251212. Accessed January 6, 2014 [DOI] [PubMed] [Google Scholar]
- Kim, K. S. (2008). Mechanisms of microbial traversal of the blood‐brain barrier. Nature Reviews. Microbiology, 6(8), 625–634. Available at: http://www.ncbi.nlm.nih.gov/pubmed/18604221. Accessed July 14, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuipers, J. , Kalicharan, R. D. , Wolters, A. H. , van Ham, T. J. , & Giepmans, B. N. (2016). Large‐scale scanning transmission electron microscopy (nanotomy) of healthy and injured zebrafish Brain. Journal of Visualized Experiments, 14(111), 12–14. Available at: http://www.jove.com/video/53635/large-scale-scanning-transmission-electron-microscopy-nanotomy [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson, N. D. , & Weinstein, B. M. (2002). In vivo imaging of embryonic vascular development using transgenic zebrafish. Developmental Biology, 248(2), 307–318. Available at: http://linkinghub.elsevier.com/retrieve/pii/S0012160602907116. Accessed October 24, 2012 [DOI] [PubMed] [Google Scholar]
- Lerner, T. R. , de Souza Carvalho‐Wodarz, C. , Repnik, U. , Russell, M. R. , Borel, S. , Diedrich, C. R. , … Gutierrez, M. G. (2016). Lymphatic endothelial cells are a replicative niche for Mycobacterium tuberculosis. Journal of Clinical Investigation, 126(3), 1093–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesley, R. , & Ramakrishnan, L. (2011). Insights into early mycobacterial pathogenesis from the zebrafish. Current Opinion in Microbiology, 11(3), 277–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta, P. K. , Karls, R. K. , White, E. H. , Ades, E. W. , & Quinn, F. D. (2006). Entry and intracellular replication of Mycobacterium tuberculosis in cultured human microvascular endothelial cells. Microbial Pathogenesis, 41(2–3), 119–124. [DOI] [PubMed] [Google Scholar]
- Meijer, A. H. , van der Sar, A. M. , Cunha, C. , Lamers, G. E. , Laplante, M. A. , Kikuta, H. , … Spaink, H. P. (2008). Identification and real‐time imaging of a myc‐expressing neutrophil population involved in inflammation and mycobacterial granuloma formation in zebrafish. Developmental and Comparative Immunology, 32(1), 36–49. Available at: http://www.ncbi.nlm.nih.gov/pubmed/17553562. Accessed September 19, 2013 [DOI] [PubMed] [Google Scholar]
- Menozzi, F. D. , Reddy, V. M. , Cayet, D. , Raze, D. , Debrie, A. S. , Dehouck, M. P. , … Locht, C. (2006). Mycobacterium tuberculosis heparin‐binding haemagglutinin adhesin (HBHA) triggers receptor‐mediated transcytosis without altering the integrity of tight junctions. Microbes and Infection, 8(1), 1–9. [DOI] [PubMed] [Google Scholar]
- Nguyen, L. , & Pieters, J. (2005). The Trojan horse: survival tactics of pathogenic mycobacteria in macrophages. Trends in Cell Biology, 15(5), 269–276. [DOI] [PubMed] [Google Scholar]
- Obermeier, B. , Daneman, R. , & Ransohoff, R. M. (2013). Development, maintenance and disruption of the blood‐brain barrier. Nature Medicine, 19(12), 1584–1596. Available at: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4080800&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oehlers, S. H. , Cronan, M. R. , Scott, N. R. , Thomas, M. I. , Okuda, K. S. , Walton, E. M. , … Tobin, D. M. (2014). Interception of host angiogenic signalling limits mycobacterial growth. Nature, 517(7536), 612–615. Available at: doi: 10.1038/nature13967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oehlers, S. H. , et al. (2016). Infection‐induced vascular permeability aids mycobacterial growth. Journal of Infectious Diseases, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orihuela, C. J. , Mahdavi, J. , Thornton, J. , Mann, B. , Wooldridge, K. G. , Abouseada, N. , … Tuomanen, E. I. (2009). Laminin receptor initiates bacterial contact with the blood brain barrier in experimental meningitis models. The Journal of Clinical Investigation, 119(6), 1638–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagán, A. J. , Yang, C. T. , Cameron, J. , Swaim, L. E. , Ellett, F. , Lieschke, G. J. , & Ramakrishnan, L. (2015). Myeloid growth factors promote resistance to mycobacterial infection by curtailing granuloma necrosis through macrophage replenishment. Cell Host & Microbe, 18(1), 15–26. Available at: http://linkinghub.elsevier.com/retrieve/pii/S1931312815002565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panackal, A. A. , Williamson, K. C. , van de Beek, D. , Boulwared, D. R. , & Williamson, P. R. (2016). Fighting the monster: applying the host damage framework to human central nervous system infections. MBio, 7(1), 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polena, H. , Boudou, F. , Tilleul, S. , Dubois‐Colas, N. , Lecointe, C. , Rakotosamimanana, N. , … Tailleux, L. (2016). Mycobacterium tuberculosis exploits the formation of new blood vessels for its dissemination. Scientific Reports, 6(September, 33162 Available at: http://www.nature.com/articles/srep33162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes, J. Hagen A, Hsu K, Deng M, Liu TX, Look AT, Kanki JP, 2005. Interplay of pu.1 and gata1 determines myelo‐erythroid progenitor cell fate in zebrafish. Developmental Cell, 8(1), pp.97–108. Available at: http://www.ncbi.nlm.nih.gov/pubmed/15621533 [Accessed May 24, 2013]. [DOI] [PubMed] [Google Scholar]
- Rich, A. , & Thomas, C. (1946). The pathogenesis of meningeal tuberculosis In The pathogenesis of tuberculosis (pp. 882–896). [Google Scholar]
- Rich, A. R. , & McCordock, H. A. (1933). The pathogenesis of Tuberculous Meningitis. Bulletin of the Johns Hopkins Hospital, 52, 2–33. [Google Scholar]
- Simeone, R. , Bobard, A. , Lippmann, J. , Bitter, W. , Majlessi, L. , Brosch, R. , & Enninga, J. (2012). Phagosomal rupture by Mycobacterium tuberculosis results in toxicity and host cell death. PLoS Pathogens, 8(2), e1002507 Available at: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3271072&tool=pmcentrez&rendertype=abstract. Accessed November 14, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skerry, C. , Pokkali, S. , Pinn, M. , Be, N. A. , Harper, J. , Karakousis, P. C. , & Jain, S. K. (2013). Vaccination with recombinant Mycobacterium tuberculosis PknD attenuates bacterial dissemination to the brain in guinea pigs. PLoS One, 8(6), e66310 Available at: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3679071&tool=pmcentrez&rendertype=abstract. Accessed November 15, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoop, E. J. M. , Schipper, T. , Rosendahl Huber, S. K. , Nezhinsky, A. E. , Verbeek, F. J. , Gurcha, S. S. , … van der Sar, A. M. (2011). Zebrafish embryo screen for mycobacterial genes involved in the initiation of granuloma formation reveals a newly identified ESX‐1 component. Disease Models & Mechanisms, 4(4), 526–536. Available at: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3124061&tool=pmcentrez&rendertype=abstract. Accessed October 24, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenor, J. , Oehlers, S. H. , Yang, J. L. , Tobin, D. M. , & Perfect, J. R. (2015). Live imaging of host‐parasite interactions in a zebrafish infection model reveals cryptococcal determinants of virulence and central nervous system invasion. MBio, 6(5), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thwaites, G. E. , van Toorn, R. , & Schoeman, J. (2013). Tuberculous meningitis: More questions, still too few answers. The Lancet Neurology, 12(10), 999–1010. Available at: doi: 10.1016/S1474-4422(13)70168-6 [DOI] [PubMed] [Google Scholar]
- Tobin, D. M. , May, R. C. , & Wheeler, R. T. (2012). Zebrafish: a see‐through host and a fluorescent toolbox to probe host‐pathogen interaction. PLoS Pathogens, 8(1), e1002349 Available at: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3252360&tool=pmcentrez&rendertype=abstract. Accessed April 29, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsenova, L. , Ellison, E. , Harbacheuski, R. , Moreira, A. L. , Kurepina, N. , Reed, M. B. , … Kaplan, G. (2005). Virulence of selected Mycobacterium tuberculosis clinical isolates in the rabbit model of meningitis is dependent on phenolic glycolipid produced by the bacilli. The Journal of Infectious Diseases, 192(1), 98–106. [DOI] [PubMed] [Google Scholar]
- Tucker, E. W. , Pokkali, S. , Zhang, Z. , DeMarco, V. P. , Klunk, M. , Smith, E. S. , … Kannan, S. (2016). Microglia activation in a pediatric rabbit model of tuberculous meningitis. Disease Models & Mechanisms, 9(12), 1497–1506. Available at: doi: 10.1242/dmm.027326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van De Weerd, R. , Boot, M. , Maaskant, J. , Sparrius, M. , Verboom, T. , van Leeuwen, L. M. , … Geurtsen, J. (2016). Inorganic phosphate limitation modulates capsular polysaccharide composition in mycobacteria. Journal of Biological Chemistry, 291(22), 11787–11799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Flier, M. , Hoppenreijs, S. , van Rensburg, A. J. , Ruyken, M. , Kolk, A. H. , Springer, P. , … Schoeman, J. F. (2004). Vascular endothelial growth factor and blood‐brain barrier disruption in Tuberculous Meningitis. The Pediatric Infectious Disease Journal, 23(7), 608–613. Available at: http://content.wkhealth.com/linkback/openurl?sid=WKPTLP:landingpage&an=00006454-200407000-00004. Accessed January 6, 2014 [DOI] [PubMed] [Google Scholar]
- van der Sar, A. M. , Appelmelk, B. J. , Vandenbroucke‐Grauls, C. M. , & Bitter, W. (2004). A star with stripes: zebrafish as an infection model. Trends in Microbiology, 12(10), 451–457. Available at: http://www.ncbi.nlm.nih.gov/pubmed/15381194. Accessed October 24, 2012 [DOI] [PubMed] [Google Scholar]
- van der Wel, N. , Hava, D. , Houben, D. , Fluitsma, D. , van Zon, M. , Pierson, J. , … Peters, P. J. (2007). M. tuberculosis and M. leprae translocate from the phagolysosome to the cytosol in myeloid cells. Cell, 129(7), 1287–1298. Available at: http://www.ncbi.nlm.nih.gov/pubmed/17604718. Accessed October 7, 2012 [DOI] [PubMed] [Google Scholar]
- van Leeuwen, L. M. , Evans, R. J. , Jim, K. K. , Verboom, T. , Fang, X. , Bojarczuk, A. , … van der Sar, A. M. (2017). A transgenic zebrafish model for the in vivo study of the blood and choroid plexus brain barriers using claudin 5. Biology Open, 1–12. Available at: doi: 10.1101/180653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leeuwen, L. M. , van der Kuip, M. , Youssef, S. A. , de Bruin, A. , Bitter, W. , van Furth, A. M. , & van der Sar, A. M. (2014). Modeling tuberculous meningitis in zebrafish using Mycobacterium marinum. Disease Models & Mechanisms, (July), 1111–1122. Available at: http://www.ncbi.nlm.nih.gov/pubmed/24997190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Rooijen, N. , & Sanders, A. (1994). Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. Journal of Immunological Methods, 174(1–2), 83–93. Available at: http://linkinghub.elsevier.com/retrieve/pii/0022175994900124 [DOI] [PubMed] [Google Scholar]
- van Rooijen, N. , Sanders, A. , & van den Berg, T. K. (1996). Apoptosis of macrophages induced by liposome‐mediated intracellular delivery of clodronate and propamidine. Journal of Immunological Methods, 193(1), 93–99. Available at: http://www.ncbi.nlm.nih.gov/pubmed/8690935 [DOI] [PubMed] [Google Scholar]
- van Sorge, N. M. , & Doran, K. (2013). Defense at the border: the blood‐brain barrier versus bacterial foreigners. 7(3), 383–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Well, G. T. J. , Paes, B. F. , Terwee, C. B. , Springer, P. , Roord, J. J. , Donald, P. R. , … Schoeman, J. F. (2009). Twenty years of pediatric tuberculous meningitis: a retrospective cohort study in the western cape of South Africa. Pediatrics, 123(1), e1–e8. Available at: doi: 10.1542/peds.2008-1353. Accessed October 24, 2012 [DOI] [PubMed] [Google Scholar]
- van Well, G. T. J. , Wieland, C. W. , Florquin, S. , Roord, J. J. , van der Poll, T. , & van Furth, A. M. (2007). A new murine model to study the pathogenesis of tuberculous meningitis. The Journal of Infectious Diseases, 195(5), 694–697. Available at: http://www.ncbi.nlm.nih.gov/pubmed/17262711. Accessed October 24, 2012 [DOI] [PubMed] [Google Scholar]
- Visser, D. H. , Solomons, R. S. , Ronacher, K. , van Well, G. T. , Heymans, M. W. , Walzl, G. , … van Furth, A. M. (2014). Host immune response to Tuberculous Meningitis. Clinical Infectious Diseases, 60(2), 177–187. Available at: doi: 10.1093/cid/ciu781 [DOI] [PubMed] [Google Scholar]
- Volkman, H. E. , Volkman, H. E. , Clay, H. , Beery, D. , Chang, J. C. W. , Sherman, D. R. , & Ramakrishnan, L. (2004). Tuberculous granuloma formation is enhanced by a mycobacterium virulence determinant. PLoS Biology, 2(11), 1946–1956. Available at: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=524251&tool=pmcentrez&rendertype=abstract. Accessed October 25, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weksler, B. B. , Subileau, E. A. , Perrière, N. , Charneau, P. , Holloway, K. , Leveque, M. , … Couraud, P. O. (2005). Blood‐brain barrier‐specific properties of a human adult brain endothelial cell line. The FASEB Journal, 19(13), 1872–1874. Available at: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16141364 [DOI] [PubMed] [Google Scholar]
- White, R. M. , Sessa, A. , Burke, C. , Bowman, T. , LeBlanc, J. , Ceol, C. , … Zon, L. I. (2008). Transparent adult zebrafish as a tool for in vivo transplantation analysis. Cell Stem Cell, 2(2), 183–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO (2017). Global Tuberculosis Report, 2017 Available at: http://apps.who.int/iris/bitstream/10665/259366/1/9789241565516-eng.pdf?ua=1 [Google Scholar]
- Wilkinson, R. J. , Rohlwink, U. , Misra, U. K. , van Crevel, R. , Mai, N. T. H. , Dooley, K. E. , … Tuberculous Meningitis International Research Consortium (2017). Tuberculous meningitis. Nature Reviews Neurology, 13(10), 581–598. Available at: doi: 10.1038/nrneurol.2017.120 [DOI] [PubMed] [Google Scholar]
- Wu, Y. C. , Chang, C. Y. , Kao, A. , Hsi, B. , Lee, S. H. , Chen, Y. H. , & Wang, I. J. (2015). Hypoxia‐induced retinal neovascularization in zebrafish embryos: a potential model of retinopathy of prematurity. PLoS One, 10(5), 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, J. , Farage, E. , Sugimoto, M. , & Anand‐Apte, B. (2010). A novel transgenic zebrafish model for blood‐brain and blood‐retinal barrier development. BMC Developmental Biology, 10(76), 1–14. Available at: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2914679&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, M. , Chang, H. , Zhang, Y. , Yu, J. , Wu, L. , Ji, W. , … Xu, T. (2012). Rational design of true monomeric and bright photoactivatable fluorescent proteins. Nature Methods, 9(7), 727–729. Available at: 10.1038/nmeth.2021/nhttp://www.nature.com/nmeth/journal/v9/n7/abs/nmeth.2021.html/nhttp://www.nature.com/nmeth/journal/v9/n7/pdf/nmeth.2021.pdf [DOI] [PubMed] [Google Scholar]
- Zielonka, T. M. , Demkow, U. , Michalowska‐Mitczuk, D. , Filewska, M. , Bialas, B. , Zycinska, K. , … Skopinska‐Rozewska, E. (2011). Angiogenic activity of sera from pulmonary tuberculosis patients in relation to IL‐12p40 and TNFα serum levels. Lung, 189(4), 351–357. Available at: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3140944&tool=pmcentrez&rendertype=abstract. Accessed December 19, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucchi, F. C. R. , Pelegrini‐da‐Silva, A. , Neder, L. , Silva, C. L. , Tsanaclis, A. M. , & Takayanagui, O. M. (2012). The contribution of a murine CNS‐TB model for the understanding of the host‐pathogen interactions in the formation of granulomas. Journal of Neuroscience Methods, 206(1), 88–93. Available at: http://www.ncbi.nlm.nih.gov/pubmed/22387263. Accessed October 24, 2012 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Single channel images corresponding to Figure 1. (A) Merged confocal image of systemically infected zebrafish larvae, 3 days post infection (dpi), corresponding to Figure 1H, with single red channel depicting blood vessels, single green channel showing M. marinum::mEos and single cyan channel with L‐plastin labeled phagocytic cells. (B) depicts single channels corresponding to Figure 1I, (C) corresponds to Figure 1J, (D) corresponds to Figure 1K.
Figure S2: Fluorescent dye leakage experiments in infected larvae. Tg(kdrl:mCherry) is5 zebrafish larvae (red blood vessels), infected with M. marinum::mEos (green) at 4 dpf, 1 hour after 3 kDa Alexa 680 tracer injection (cyan) at 3 dpi (A‐C) and 5 dpi (D‐F). Figure shows merged confocal images (A, B, C, D, E, F) and single cyan channel (A', B', C', D', E', F'). Open arrows point at single bacteria associated with the blood vessel wall, without leakage of the dye in the adjacent tissue. Closed arrows show localization of tracer in brain parenchyma, which was always concentrated in small spots.
Figure S3: Intensified VEGFr signal co‐localizing with infected phagocytes. (A – C) Z‐stack of head of 5dpi/9dpf Tg(kdrl:mCherry) zebrafish larva with red fluorescent blood vessels, systemically infected with M. marinum E11 (green) at 4 dpf, treated directly after infection with GS4012 to induce VEGF levels (A), nothing (B) or SU5416 to block the VEGFr (C). Representative image per group is shown. Clear difference in infection level can be seen, in particular in GS4012‐treatment compared to control. Scale bar = 250 μm.
Figure S4: Correlative light and electron microscopy. (A) Correlative light and electron microscopy of (B) Confocal Microscopy and (C) Electron microscopy, of a 9 dpf Tg(kdrl:mCherry) control larva with red fluorescent blood vessels, infected at 4 dpf with green fluorescent M. marinum E11, nuclei counter stain after fixation with DAPI (cyan). (B) clearly illustrates the limitations of confocal microscopy in terms of resolution.
Table S1: Intensified VEGFr signal co‐localizing with infected phagocytes. Table showing total number of phagocytosed M. marinum found in brain blood vessels or brain tissue in all larvae included per group: GS4012‐treated vs. control vs. SU5416‐treated. Per topic an absolute number and percentage of total is shown. (D) Time point: 3 dpi, (E) time point: 5 dpi.
Table S2: Primers used in this study to create bacterial constructs