

Abstract
Genetic alterations in the complement system have been linked to a variety of diseases, including atypical hemolytic uremic syndrome (aHUS), C3 glomerulopathy (C3G), and age‐related macular degeneration (AMD). We performed sequence analysis of the complement genes complement factor H (CFH), complement factor I (CFI), and complement C3 (C3) in 866 aHUS/C3G and 697 AMD patients. In total, we identified 505 low‐frequency alleles, representing 121 unique variants, of which 51 are novel. CFH contained the largest number of unique low‐frequency variants (n = 64; 53%), followed by C3 (n = 32; 26%) and CFI (n = 25; 21%). A substantial number of variants were found in both patients groups (n = 48; 40%), while 41 (34%) variants were found only in aHUS/C3G and 32 (26%) variants were AMD specific. Genotype‐phenotype correlations between the disease groups identified a higher frequency of protein altering alleles in short consensus repeat 20 (SCR20) of factor H (FH), and in the serine protease domain of factor I (FI) in aHUS/C3G patients. In AMD, a higher frequency of protein‐altering alleles was observed in SCR3, SCR5, and SCR7 of FH, the SRCR domain of FI, and in the MG3 domain of C3. In conclusion, we observed a substantial overlap of variants between aHUS/C3G and AMD; however, there is a distinct clustering of variants within specific domains.
Keywords: age‐related macular degeneration, alternative pathway, atypical hemolytic uremic syndrome, C3 glomerulopathy, complement system
1. INTRODUCTION
The complement system is part of the innate immune system, which balances host protection and immune defense. The complement system can be activated via 3 pathways: the classical, lectin, and alternative pathways, which converge at the step of cleavage of the central component C3 into C3a and C3b. C3b bound to activated factor B (Bb) forms the alternative pathway C3 convertase (C3bBb), which cleaves and activates more C3 molecules, thereby amplifying the cascade. When the alternative pathway C3 convertase binds additional C3b molecules, the C5 convertase is formed (C3bBbC3b), which cleaves C5 into C5a and C5b. C5b interacts with C6, C7, C8, and multiple C9 molecules to form the membrane attack complex (C5b‐9). Regulators, such as factor H (FH) and membrane cofactor protein (MCP; CD46), can inhibit complement activation by accelerating the decay of the C3 convertases or by acting as a cofactor for factor I (FI).1
Deregulation of the complement system, specifically the alternative pathway, has been implicated in a variety of diseases.1 Remarkably, protein‐altering variants in genes of the complement system have been associated with very different clinical outcomes: atypical hemolytic uremic syndrome (aHUS; MIM# 235400), C3 glomerulopathy (C3G; or dense deposit disease), and age‐related macular degeneration (AMD; MIM# 603075).2, 3
aHUS is a rare disorder characterized by acute renal failure, thrombocytopenia, and microangiopathic hemolytic anemia. It is estimated that genetic variants in the complement factor H (CFH), complement factor I (CFI), complement C3 (C3), complement factor B (CFB), CD46 molecule (CD46), thrombomodulin, and diacylglycerol kinase epsilon genes, the presence of genomic rearrangement in the CFH/CFH‐related (CFHR) gene cluster, and autoantibodies to FH account for 60% of all aHUS cases.4, 5, 6
C3G is another rare renal illness, which is characterized by C3 deposition in the glomeruli of the kidney and can lead to renal failure. C3G pathogenesis is linked to the presence of autoantibodies that stabilize the alternative and classical pathway C3 convertases (C3Nef and C4Nef). Genetic aberrations in the CFH, CFI, C3, CD46, CFHR5 genes, genomic rearrangements in the CFH/CFHR gene cluster, and anti‐FH autoantibodies genes have been described in 20% of C3G patients.6, 7, 8
AMD, in contrast to aHUS and C3G, is a common disease in which gradual visual impairment occurs at older age due to degeneration of the central retina. The disease is characterized by the disruption of normal retinal pigment epithelium (RPE) function through the accumulation of waste products, called drusen, between Bruch's membrane and the RPE. A combination of multiple genetic and environmental factors contribute to the pathogenesis of AMD. More than one‐third of the disease‐associated genetic variants reside in or near genes of the complement system: CFH, CFI, C3, complement C2 (C2)/CFB, complement C9 (C9), and vitronectin.9 In addition, rare protein‐altering variants in the CFH, CFI, C3, and C9 genes have been associated with AMD.9, 10
A number of low frequency protein‐altering variants have been described to cause both aHUS/C3G and AMD, such as the p.Arg53His and p.Arg1210Cys variants in CFH, 11, 12 p.Gly119Arg and p.Gly287Arg in CFI, 13, 14 and p.Lys155Gln and p.Arg161Trp in C3.10, 15, 16 A pathophysiologic explanation on how mutation in the same gene, in different patients, can lead to a different disease is not available.1 It has been suggested that the final disease outcome is determined by the individual's overall genetic risk to each of these diseases, and is influenced by environmental factors.17
However, a clustering of variants in certain protein domains in the different diseases has been described, supporting the existence of a genotype‐phenotype correlation.18 In C3, the p.Arg161Trp variant is located in the MG2 domain and is present in 4% to 16% of aHUS patients.19, 20 FH protein consists of 20 short consensus repeats (SCRs), which are also known as complement control protein domains. A particularly prominent hot spot for aHUS mutations is located within the C‐terminal (SCR15‐20) domains of FH, and is considered typical for this disease. Such clustering of variants in FH was not observed for C3G patients.21 In AMD, an enrichment of protein‐altering variants has been reported in the SCR1‐4 and SCR16‐20 of FH, and in the serine protease (SP) domain of FI.1, 22, 23 However, a systematic evaluation and comparison of genetic aberrations between the renal diseases (aHUS/C3G) and AMD has not yet been performed. Such an analysis could provide a more in‐depth insight into genotype‐phenotype correlations for these diseases, and could considerably enhance our understanding of complement deregulation in human disease.
In this study, we describe genetic variants identified in the CFH, CFI, and C3 genes in large patient cohorts consisting of 866 renal disease (aHUS/C3G) patients and 697 AMD patients, and provide a comprehensive genotype‐phenotype correlation analysis between these disease groups.
2. MATERIALS AND METHODS
2.1. Cohort description
The AMD cohort consisted of 697 individuals who were recruited as part of the European Genetic Database (EUGENDA) between December 2005 and June 2014. Only individuals affected by AMD were included in this study. To diagnose AMD, retinal images were evaluated according to the Cologne Image Reading Center and Laboratory protocol. In short, AMD was characterized as the presence of pigmentary changes together with at least 10 small drusen (<63 μm) or the presence of intermediate (63‐124 μm) to large drusen (≥125 μm diameter) near the macula. Furthermore, late AMD was defined as either subfoveal geographic atrophy or choroidal neovascularization in at least one eye. Written informed consent was obtained from all participants. The study was approved by the local ethics committees on Research Involving Human Subjects, and conducted according to the Declaration of Helsinki.
The aHUS/C3G cohort consisted of 886 patients that were referred to the Radboud University Medical Center for genetic screening between 2007 and 2015 to confirm the clinical diagnosis of aHUS or C3G. The diagnosis of aHUS was defined as a presence of hemolytic anemia, thrombocytopenia, and acute renal failure that was not preceded with infection with Shiga toxin‐producing Escherichia coli.6 C3G diagnosis was defined as active glomerulonephritis combined with predominantly C3 absent or marginal immunoglobulin deposition in renal biopsy.24 Most of the patients (around 70%) in this cohort were diagnosed with aHUS. However, in some of the cases, the clinical diagnosis of aHUS or C3G was unclear, and therefore a genetic screening was requested as part of their clinical care.
2.2. Genetic screening
In this study, we focused on the complement genes associated with both aHUS/C3G and AMD, which includes the CFH (HGNC:4883, MIM# 134370), CFI (HGNC:5394, MIM# 217030), and C3 (HGNC:1318, MIM# 120700) genes.4, 5, 9, 22
Genetic analysis was performed for the aHUS/C3G and AMD cohorts using DNA isolated from peripheral blood leucocytes using standard procedures. For the AMD cohort, whole‐exome sequencing (WES) of 697 unrelated individuals was performed. WES capture and variant calling was obtained through the Nimblegen SeqCap EZ Exome v2 kit by paired‐end sequencing on a Illumina HiSeq sequencer using TruSeq V3 chemistry as described in detail elsewhere (Corominas et al, manuscript submitted). WES data of the AMD cohort were filtered to select coding non‐synonymous and canonical splice‐site variants in CFH (NM_00186.3), CFI (NM_00204.3), and C3 (NM_000064.3). The mean coverage (and minimum to maximum range) for CFH, CFI, and C3 were 135X (32‐495), 124X (44‐356), and 32X (2‐137), respectively (Figure S1 in Appendix S1, Supporting Information). Strict quality filters were set to obtain true positive hits including minimum read depth (n > 20) and variant reads (>10). Variants identified in CFH were confirmed by Sanger sequencing due to the high similarities between CFH and the CHFR1‐4 genes. For the aHUS/C3G cohort, genetic analysis was performed by amplification of the coding regions and splice junctions of the CFH (NM_00186.3), CFI (NM_00204.3), and C3 (NM_000064.3) genes by PCR, followed by Sanger sequencing or next‐generation sequencing on an Ion torrent semiconductor (minimum coverage 40X).
Only low‐frequency and rare variants (minor allele frequency < 5%) based on the ExAC database25 were taken into account. Thus, common variants previously described as associated with the disease (such as Tyr402His and Ile62Val in AMD and polymorphisms described by Caprioli et al in aHUS)26, 27 have not been taken into account. Variants were considered aHUS/C3G or AMD specific when found only in the aHUS/C3G or AMD cohorts analyzed in this study and in previously published reports. Reported odds ratios for genetic variants in AMD were recently published by the International AMD Genomics Consortium.9, 10 Annotation of the variants, including minor allele frequency of the ExAC database, were obtained using ANNOVAR.28
2.3. Statistical analysis
Statistical analyses were performed using SPSS Software for Windows version 22.0 (Fisher exact, 2‐sided, weighted by number). We calculated if there was a higher percentage of alleles within a specific protein domain of FH, FI, or C3 in aHUS/C3G compared to AMD, and vice versa. P values <.05 were considered as significant.
2.4. Protein structure analysis
Protein domains that carried a significantly higher percentage of alleles in aHUS/C3G or AMD were analyzed in more depth. The potential effect of the variants mapping to these domains on interactions between C3b, FH and FI and their possible effect on C3b regulation were studied. To this end, the variants were mapped on the available molecular structures of the C3b‐miniFH‐FI protein complex (protein data bank, PDB 5O3229). Protein structures were retrieved from the PDB (http://www.rcsb.org)30 using YASARA.31
2.5. Literature search
We performed a literature search to review low‐frequency coding non‐synonymous and canonical splice‐site variants in CFH, CFI, and C3 previously reported in aHUS, C3G, and AMD. Moreover, variants were extracted from the FH aHUS Mutation Database (http://www.fh-hus.org/), and the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/).
3. RESULTS
3.1. Genetic variants identified in aHUS/C3G and AMD patients
We screened 886 aHUS/C3G patients and 697 AMD patients for low‐frequency variants in the coding and splice‐site regions of CFH, CFI, and C3. In total, we identified 505 low‐frequency alleles in CFH, CFI, and C3, of which 379 were found in aHUS/C3G and 126 were found in AMD. In aHUS/C3G patients, we identified 225 heterozygous and 7 homozygous carriers of variants in CFH, 46 heterozygous and 2 homozygous carriers of variants in CFI, and 88 heterozygous carriers and 1 homozygous carrier of variants in C3. All 126 variants identified in AMD patients were heterozygous, of which 55 were found in CFH, 31 in CFI, and 40 in C3 (Table S1 in Appendix S1).
The 505 alleles represent 121 unique low‐frequency variants, of which 51 are novel and 70 were previously reported in literature (Table S2 in Appendix S1). Of the 51 novel variants identified, 8 variants in CFH (n = 4), CFI (n = 2), and C3 (n = 2) lead to a premature stop, frameshift or abolished splice site, thereby leading to the loss of protein function. The remaining 43 novel variants lead to missense alterations or to an in‐frame insertion (Table S3 in Appendix S1).
CFH contained the largest number of unique low‐frequency variants (n = 64). Two‐third of these variants were aHUS/C3G (24/64) or AMD‐specific (17/64), and one‐third was found in both patients groups (23/64) (Figure 1; Table S3 in Appendix S1). Three of the low‐frequency variants (p.Gln950His, p.Asn1050Tyr, p.Gln1143Glu) identified in CFH in aHUS/C3G patients, were previously reported to have a protective effect for AMD.10 The p.Gln950His variant was detected in 15 aHUS/C3G patients, the p.Asn1050Tyr variant was detected in 21 aHUS/C3G patients, and the p.Gln1143Glu variant was detected in 9 aHUS/C3G patients.
Figure 1.
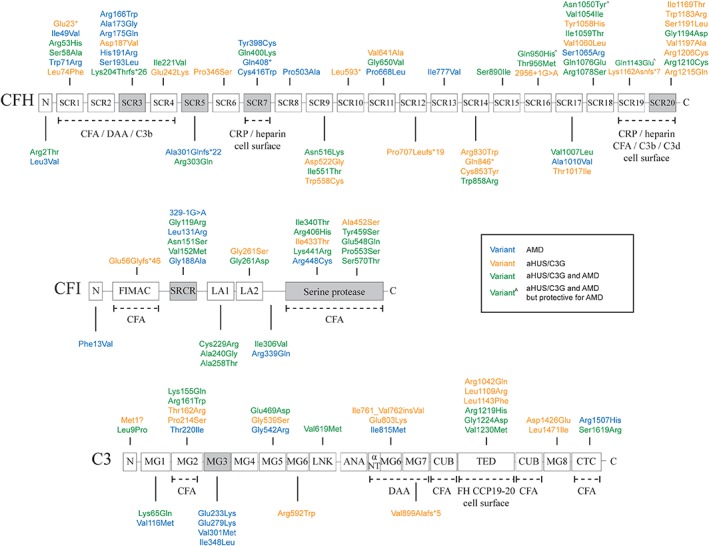
Genetic variants identified in 886 aHUS/C3G patients and 697 AMD patients. Colors represent the phenotype in which the variants were identified: green (both in aHUS/C3G and AMD), orange (aHUS/C3G only), blue (AMD only) or green^ (with circumflex) (protective for AMD but found recurrently in aHUS/C3G). Protein domains that carried a significantly higher percentage of alleles in aHUS/C3G or AMD are colored gray. Protein interaction sites are reported underneath the domains with dotted lines. Only low frequency variants (minor allele frequency <5%) based on the ExAC database are shown. CFA, cofactor activity; DAA, decay‐accelerating activity; CRP, C‐reactive protein
In CFI, 25 unique low‐frequency variants were identified of which 15 were found in both patient groups. Only 4 of the 25 variants were specific for aHUS/C3G and 6 variants were exclusively found in AMD (Figure 1; Table S3 in Appendix S1).
In C3, 32 unique low‐frequency variants were identified. These variants were distributed between aHUS/C3G‐specific variants (13/32), AMD‐specific variants (9/32), and both phenotypes (10/32) (Figure 1; Table S3 in Appendix S1).
3.2. Genotype‐phenotype correlations in aHUS/C3G and AMD
To determine genotype‐phenotype correlations, we calculated if there was a higher percentage of alleles within a specific protein domain of FH, FI, or C3 in aHUS/C3G compared to AMD, and vice versa. We observed a higher frequency of protein‐altering alleles in the SCR20 domain of FH (P = .010) and in the SP domain of FI (P = 2.96 × 10−3) in aHUS/C3G compared to AMD. In AMD, we observed a higher frequency of protein‐altering alleles in the SCR3, SCR5, and SCR7 domains of FH (P = 3.20 × 10−5, P = 1.97 × 10−4, and P = .003, respectively), the SRCR domain of FI (P = 1.60 × 10−3), and the MG3 domain of C3 (P = 2.30 × 10−3) compared to aHUS/C3G (Table 1 ).
Table 1.
Allelic distribution of CFH, CFI and C3 variants identified in 886 aHUS/C3G patients and 697 AMD patients
| Gene | Domain | aHUS/C3G | AMD | P‐value aHUS/C3G vs AMD |
|---|---|---|---|---|
| CFH | N‐terminus | 1 (0.4%) | 1 (1.8%) | 0.340 |
| SCR1 | 6 (2.5%) | 3 (5.5%) | 0.377 | |
| SCR2 | 0 (0%) | 0 (0%) | 1.000 | |
| SCR3 | 2 (0.8%) | 8 (14.5%) | 3.20E‐05 | |
| SCR4 | 3 (1.3%) | 0 (0%) | 1.000 | |
| SCR5 | 0 (0%) | 5 (9.1%) | 1.97E‐04 | |
| SCR6 | 1 (0.4%) | 0 (0%) | 1.000 | |
| SCR7 | 2 (0.8%) | 5 (9.1%) | 0.003 | |
| SCR8 | 0 (0%) | 1 (1.8%) | 0.187 | |
| SCR9 | 13 (5.4%) | 1 (1.8%) | 0.480 | |
| SCR10 | 1 (0.4%) | 0 (0%) | 1.000 | |
| SCR11 | 2 (0.8%) | 2 (3.6%) | 0.160 | |
| SCR12 | 2 (0.8%) | 0 (0%) | 1.000 | |
| SCR13 | 0 (0%) | 1 (1.8%) | 0.187 | |
| SCR14 | 8 (3.3%) | 1 (1.8%) | 1.000 | |
| SCR15 | 19 (7.9%) | 1 (1.8%) | 0.139 | |
| SCR16 | 20 (8.4%) | 8 (14.5%) | 0.199 | |
| SCR17 | 31 (13%) | 2 (3.6%) | 1.000 | |
| SCR18 | 77 (32.2%) | 14 (25.5%) | 0.419 | |
| SCR19 | 11 (4.6%) | 0 (0%) | 0.228 | |
| SCR20 | 40 (16.7%) | 2 (3.6%) | 0.010 | |
| N‐terminal (SCR1‐4) | 11 (4.6%) | 11 (20.0%) | 5.20E‐04 | |
| C‐terminal (SCR19‐20) | 51 (21.3%) | 2 (3.6%) | 1.44E‐03 | |
| Total CFH | 239 | 55 | ||
| CFI | N‐terminus | 0 (0%) | 1 (3.2%) | 0.383 |
| FIMAC | 1 (2.0%) | 0 (0%) | 1.000 | |
| SRCR | 7 (14.0%) | 15 (48.4%) | 1.60E‐03 | |
| LA1 | 3 (6.0%) | 3 (9.7%) | 0.670 | |
| LA2 | 6 (12.0%) | 2 (6.5%) | 0.704 | |
| Linker region | 1 (2.0%) | 1 (3.2%) | 1.000 | |
| SP | 32 (64.0%) | 9 (29.0%) | 2.96E‐03 | |
| Total CFI | 50 | 31 | ||
| C3 | MG1 | 4 (4.4%) | 3 (7.5%) | 0.675 |
| MG2 | 45 (50.0%) | 21 (52.5%) | 0.850 | |
| MG3 | 0 (0%) | 5 (12.5%) | 2.30E‐03 | |
| MG4 | 0 (0%) | 0 (0%) | 1.000 | |
| MG5 | 9 (10.0%) | 1 (2.5%) | 0.174 | |
| MG6a | 4 (4.4%) | 0 (0%) | 0.311 | |
| LNK | 4 (4.4%) | 1 (2.5%) | 1.000 | |
| ANA | 0 (0%) | 0 (0%) | 1.000 | |
| MG6b | 4 (4.4%) | 1 (2.5%) | 1.000 | |
| MG7 | 2 (2.2%) | 0 (0%) | 1.000 | |
| CUB | 0 (0%) | 0 (0%) | 1.000 | |
| TED | 8 (8.9%) | 0 (0%) | 0.106 | |
| MG8 | 2 (2.2%) | 0 (0%) | 1.000 | |
| CTC | 4 (4.4%) | 6 (15.0%) | 0.068 | |
| Total C3 | 90 | 40 |
Percentage of variant alleles identified with significant P‐values in bold.
For FH, we calculated the cumulative effect for the alleles found in the domains involved in cofactor activity, which include N‐terminal SCR1‐4 and C‐terminal SCR19‐20. We observed a burden of alleles in the SCR19‐20 domains at the C‐terminus for aHUS/C3G (21.3% vs 3.6% in AMD, P = 1.44 × 10−3), and a burden in the N‐terminal SCR1‐4 domains for AMD (20.0% vs 4.6% in aHUS/C3G, P = 5.20 × 10−4) (Table S3 in Appendix S1).
Next, we aimed to model the effect of specific mutated amino acids found in the domains that differ significantly in allele frequency between aHUS/C3G and AMD on interactions with ligands. The variants found in SCR3 and SCR20 domains of FH, the SRCR and SP domains of FI, and the MG3 domain of C3 were mapped on the structure of C3b‐FH‐FI complex29 to illustrate their effect on C3b regulation (Figure 2). FH SCR5 was not mapped as there are no structures available that illustrate possible interactions of this domain with ligands. A previous study illustrates an interaction of FH SCR7 with glycosaminoglycan (GAG) analogues; however, none of the residues that were altered in our cohorts were implicated in contact with GAGs (Table S3 in Appendix S1).32 Figure 3 shows a detailed view of the residues that are altered in the SCR3 domain of FH (Figure 3A,B) and the SP domain of FI (Figure 3C,D) in the FH‐C3b‐FI complex (PDB‐5O32), together with the amino acids of interacting protein partners that are in close proximity to the altered residues.
Figure 2.
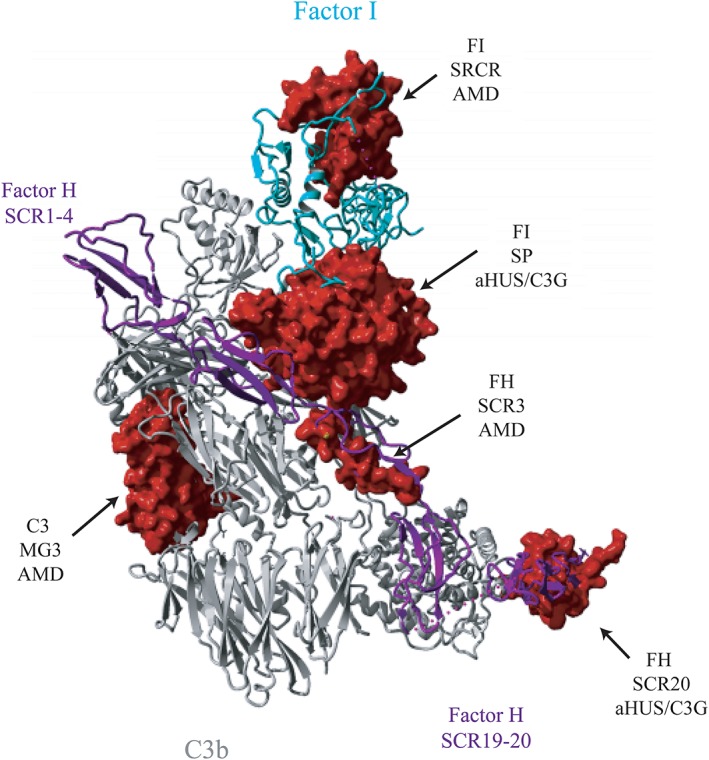
Location of protein domains containing a higher percentage of alleles in aHUS/C3G or AMD on the structure of the C3b‐FH‐FI complex. Three‐dimensional structure of C3b (gray) in complex with FI (cyan) and FH construct (purple) containing FH SCR1‐4 and SCR19‐20. Dotted line represents 12‐residue polyglycine linker that connects SCR1‐4 and SCR19‐20. Surfaces of fragments of C3b (233‐348), FI (119‐188, 340‐570) and FH (166‐193, 1169‐1215) carrying missense changes in domains, which contain a significantly higher percentage of alleles in aHUS/C3G or AMD, are shown in red. The figure is generated based on the PDB 5O32,29 using YASARA Version 17.8.1531
Figure 3.
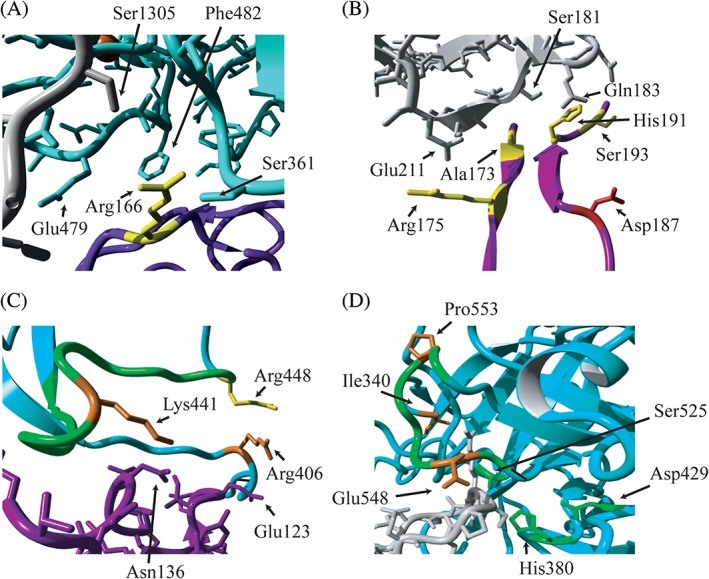
Localization of altered residues on the interface of C3b, FH and FI in proteins domains containing a higher percentage of alleles in aHUS/C3G or AMD. Fragments of the 3‐dimensional structure of C3b (gray), FI (cyan) and FH construct (purple) are shown. The residues altered in the SCR3 domain of FH (A and B) and the SP domain of FI (C and D) are shown. Residues that were found mutated in AMD only (yellow), aHUS/C3G (red) or both phenotypes (orange) are indicated, as well as amino acids of interacting partners that are in close proximity of the mutated residues (A, B, C). Important structural elements of FI are indicated in green: the charged loop 435‐448 (C); the activation loop 548‐553 and the catalytic triad (H380, D429, S525) (D). The figure is generated based on the PDB 5O32,29 using YASARA Version 17.8.1531
Based on the mapping of the variants on the C3b‐FH‐FI protein complex, several observations can be made. Amino acids affected by aHUS/C3G variants in the SCR20 domain of FH do not interact directly with C3b (Figure 2). Amino acids affected by AMD variants in the SCR3 domain are located at the interface of FH with C3b and with FI (Figure 2). The residue Arg166 of SCR3, mutated in AMD, is located at the interface of the SP domain of FI and the CUB domain of C3b (Figures 2 and 3A). Other amino acids altered in AMD (Arg175, Ala173, His191, Asp187, and Ser193) are located at the interface with the C3b molecule. (Figure 3B). Variants affecting the residues in the SP domain of FI appear to have different effects on FI binding and FI activity. Residues Arg406 and Lys441, affected by genetic variants identified in aHUS/C3G and AMD, make contacts with Glu123 and Asn136 of FH, respectively (Figure 3C). The residues Pro553 and Glu548, affected by genetic variants identified in aHUS/C3G and AMD, are located in one of the activation loops in proximity to the FI active site (Figure 3D). The AMD‐associated variants, affecting the SRCR domain of FI, are not located at the interface with C3b or FH (Figure 2).
3.3. aHUS/C3G and AMD variants found in literature
To compare the results of our study with previously reported variants, we compiled a list of low‐frequency variants found in literature for aHUS, C3G, and AMD (Table S4 in Appendix S1). We identified an additional 441 unique variants resulting in an amino acid change or splice site in CFH (n = 236), CFI (n = 104), and C3 (n = 101). These variants were found in aHUS/C3G (n = 212), AMD (n = 189), and both phenotypes (n = 40) (Table S5 in Appendix S1).
4. DISCUSSION
In aHUS/C3G patients, we observed a higher frequency of protein‐altering alleles in the C‐terminal SCR20 domain of FH and in the SP domain of FI (Figure 1). In AMD patients, an increased frequency of protein‐altering alleles was observed in the N‐terminal SCR domains of FH, specifically domains SCR3, SCR5, and SCR7, in addition to the SRCR domain of FI, and the MG3 domain of C3.
4.1. Complement FH
The SCR20 domain of FH, which is thought to interact with both C3b, C‐reactive protein, and endothelial cells, harbors significantly more aHUS‐/C3G‐associated genetic variants. Amino acids affected by genetic variants in this domain do not interact directly with C3b (Figure 2), but are probably to mediate FH attachment to the cell surface. Reduced binding to C3b and C3d was previously observed for variants p.Val1197Ala and p.Arg1210Cys in CFH (Table S3 in Appendix S1), which may be explained by the role of these residues in the overall structure of the FH C‐terminal domains.
Interestingly, like SCR20, SCR19 of FH is also described to be important for interaction with the thioester‐containing domain (TED) domain of C3b and C3d, and for cell‐surface interactions. However, for SCR 19, we observed only 11 protein altering alleles for aHUS/C3G and none for AMD. Only variant CFH p.Gln1143Glu, protective for AMD,9, 10 was detected (Table S3 in Appendix S1), indicating the importance of genetic changes in this domain for the pathogenesis of renal disease. Since phenotype‐specific prevalence of genetic variants in the TED domain of C3 for renal disease was not discovered by us or described in literature (Tables S3 and S4 in Appendix S1), the disease specificity of C‐terminal FH variants may lie within the interactions with the endothelium.
In AMD patients, genetic variants were more prevalent in the N‐terminal region of the protein. Available co‐crystallization structures show that SCR3 is located at the interface of FH with C3b and with FI (Figure 2). The residue Arg166 of SCR3, mutated in AMD, is located at the interface with the SP domain of FI as well as with the CUB domain of C3b (Figures 2 and 3A). Other amino acids altered in AMD (Arg175, Ala173, His191, Asp187, and Ser193) are located at the interface with C3b molecule (Figure 3B).
Our results are comparable with the variants found in literature. The variants more prevalent in aHUS/C3G are grouped at the C‐terminal SCR19‐20 domains of FH (52/148), while the AMD variants grouped in the first 4 SCR domains of CFH (28/74) (Table S4 in Appendix S1). Previous studies in densely affected AMD families detected several AMD‐specific variants in the first 4 SCR domains of FH (eg, p.Asp90Glu, p.Arg175Pro, p.Arg175Gln, p.Cys192Phe, and p.Ser193Leu), underscoring the importance of these domains in the disease pathogenesis of AMD.12, 22, 33 Previously, more than 60% of the aHUS‐associated FH mutations were reported in the C‐terminal domains.21 Variants in this region interfere with heparin, C3b, and C3d binding, and result in reduced cell‐surface interaction.34, 35 The low‐frequency variants reported in AMD are located at the N‐terminal domains of FH and decrease cofactor activity for the FI‐mediated cleavage of C3b.12, 22
The C‐terminal domains of FH are a hot spot for genetic variants in renal diseases, but not AMD, and are essential in endothelial binding of FH.36 Interestingly, the retinal Bruch's membrane differs from the renal glomerular basement membrane in GAG‐binding sites.37 It is hypothesized that not FH but Factor H‐like 1 (FHL‐1), the short form of FH (SCR1‐7), is involved in the pathogenesis of AMD. FHL‐1 has the same regulatory functions as FH, but due to its size only this FH form can diffuse into Bruch's membrane and drusen in the eye.38, 39 This may explain why the genetic alterations at the C‐terminal domains of FH, which are not present in FHL‐1, are less prevalent in AMD than in renal disease. Consistently, the SCR19‐20 region of FH localizes to GAGs in the glomeruli.37 Importance of specific GAG binding in AMD is further underscored by the effect of the common polymorphism Tyr402His located in SCR7 on binding of heparan sulfates.32 Moreover, our data show a higher allelic frequency in SCR7. The residues found altered in our cohorts do not make direct contact with GAGs; however, genetic variants may still influence interactions by introducing conformational changes to make patients more prone to AMD.
Overall, C3b interactions with glomerular endothelium in the kidney may be more important than the interaction with the endothelium in the retina for maintaining the tissue homeostasis. Furthermore, our data underscore the importance of N‐terminal region of FH in C3b regulation in AMD.
Three variants in CFH (c.2850G > T, p.Gln950His; c.3148A > T, p.Asn1050Tyr; c.3427C > G, p.Gln1143Glu) were reported as protective for AMD10. CFH p.Gln950His, residing in SCR16, is associated with disease risk for aHUS with moderate effect on cofactor function.40 CFH p.Asn1050Tyr, in SCR18, is a polymorphism reported various times in aHUS without functional analyses.41, 42, 43 No information is available on CFH p.Gln1143Glu and its association with aHUS/C3G. The protective mechanisms of these variants in AMD remain to be elucidated.
4.2. Complement factor I
In this study, we observed that genetic changes in the SP domain of FI are more prevalent in aHUS/C3G than in AMD patients. The SP domain, together with the FI membrane attack complex (FIMAC) domain, contains binding sites that are important for C3b and C4b degradation.44, 45 Variants found in the SP domain could interfere with proper cofactor activity. Residues Arg406 and Lys441 in the SP domain make contacts with Glu123 and Asn136 of FH, respectively (Figure 3C). Interestingly, a CFI variant affecting Arg448, found in AMD, is located in close proximity to these residues, within the charged loop (435‐448) that is important in interaction with various C3b regulators (Figure 3C).29 The CFI residues Pro553 and Glu548 are located in one of the activation loops in proximity to the FI active site (Figure 3D),46 while Ile340 at the N‐terminus of the light chain plays a role in stabilizing of the oxyanion hole.29 The residue Tyr459 is a part of the hydrophobic patch, which is important in heavy chain/light chain contact of FI.29
Furthermore, we observed a clustering of alleles in the SRCR domain in AMD patients (Tables S3 and S4 in Appendix S1). None of the variants are located at the interface with C3b or FH. However, FI serum levels of AMD individuals carrying low‐frequency variants in the SRCR domain were reported as reduced,14, 22, 47 thus leading to overall impaired ability to degrade and inactivate C3b.
In literature, the majority of variants were identified in the FIMAC (4/24 and 10/62), SRCR (5/24 and 12/62) and SP (10/24 and 23/62) domains for both aHUS/C3G and AMD. Previously, a burden of genetic variants was found in the light catalytic chain, the SP domain of FI for aHUS/C3G patients,44, 45 and for AMD.1, 23 However, we observe an enrichment of genetic variants in the SRCR domain of FI in AMD. It should be noted that rare genetic variant p.Gly119Arg, which confers high risk for AMD, and the rare genetic variant p.Leu131Arg that was identified in a densely affected AMD family, also reside in SRCR of FI.14, 22 FI genetic changes in AMD, which are more prevalent in the SRCR domain, are probably to affect the FI expression and thus overall activity,14, 22 while FI changes more prevalent in renal disease are mostly involved in structural elements around the active site.
4.3. Complement component 3
In this study, an increased number of variants were found in the MG3 domain of C3 in AMD patients (Tables S3 and S4 in Appendix S1). There are no interactions known for the MG3 domain in C3b, including that with FB or complement inhibitors.48, 49 However, MG3 is located at the predicted interface between C3b and C3 in the C3bBb‐C3 enzyme‐substrate complex. Genetic alterations in MG3 may thus result in changes at this interface and in more efficient C3 activation by C3 convertase.50 Therefore, AMD variants affecting the residues in the MG3 domain may affect the overall domain structure and the rates of C3 secretion and/or activation into C3b.
In literature, the majority of variants were identified in the TED domain for both aHUS/C3G (22/40) and AMD (14/53) (Table S4 in Appendix S1). For aHUS, it was previously reported that the TED, MG2, and MG5 domains of C3 exhibited most of the missense mutations.16 Variant identified in aHUS/C3G were found in the TED (8.9%) and MG2 (50%) domains. For AMD, we did not identify any variants in the TED domain, but the majority of variants identified resided in MG2 (52.5%) as well.
4.4. Clinical relevance
Detailed analysis of rare genetic variants that are phenotype specific or that are not, and the variability in which certain domains have significant associations with renal or AMD phenotypes will generate valuable insights into disease mechanisms. A deeper understanding of domain‐specific effects in these diseases will allow for the development of novel therapeutic interventions. Targeting protein domains, rather than the entire protein, increases specificity and reduces any potential side effects. A combination of genetic and functional analyses will probably become instrumental in diagnostics as well as stratifying patients for highly specific personalized treatment.
In conclusion, our data underscore the importance of the N‐terminal domains of FH in AMD, and of the FH C‐terminal domains in renal disease. In AMD, alterations in the SRCR domain of FI are prevalent, while FI changes related to renal disease are predominantly found in structural elements around the active site. For C3, AMD alterations were located in the MG3 domain. We observed a substantial overlap in variants between aHUS/C3G and AMD; however, there is a distinct clustering of variants within specific domains.
Depending on the location of the variant, the genetic variants follow a distinctive genotype‐phenotype correlation. Some genetic variants are associated with aHUS, C3G, and AMD but individuals carrying these risk variants only present phenotypic characteristics of one disorder.17, 51 It is probably not always one specific variant that results in the manifestation of aHUS, C3G, or AMD, but a combination of rare and common disease specific risk variants and environmental factors will lead to the manifestation of either an eye or renal phenotype.
Supporting information
Appendix S1. Supporting Information
Table S1 Number of alleles found in this study. (combined PDF)
Table S2 Number of genetic variants found in this study. (combined PDF)
Table S3 Variants identified in this study. (Excel file)
Table S4 Variants identified in literature. (Excel file)
Table S5 Number of unique variants identified in literature. (combined PDF)
Figure S1 Mean depth of coverage per gene of the AMD cohort. (combined PDF)
Appendix S2. Supporting Information
ACKNOWLEDGEMENTS
The research leading to these results has received funding from the European Research Council under the European Union's Seventh Framework Programme (FP/2007‐2013)/ERC Grant Agreement no. 310644 (MACULA). This study received funding from the Dutch Kidney Foundation (CP 14.27 COMBAT consortium, 13OI116, KFB 11.007, IP 10.22)
Conflict of interest
The authors declare no potential conflict of interests.
Geerlings MJ, Volokhina EB, de Jong EK, et al. Genotype‐phenotype correlations of low‐frequency variants in the complement system in renal disease and age‐related macular degeneration. Clin Genet. 2018;94:330–338. 10.1111/cge.13392
Maartje J. Geerlings and Elena B. Volokhina contributed equally to this study.
REFERENCES
- 1. Liszewski MK, Java A, Schramm EC, Atkinson JP. Complement dysregulation and disease: insights from contemporary genetics. Annu Rev Pathol. 2017;12:25‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Klein RJ, Zeiss C, Chew EY, et al. Complement factor H polymorphism in age‐related macular degeneration. Science. 2005;308:385‐389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Warwicker P, Goodship TH, Donne RL, et al. Genetic studies into inherited and sporadic hemolytic uremic syndrome. Kidney Int. 1998;53:836‐844. [DOI] [PubMed] [Google Scholar]
- 4. Caprioli J, Noris M, Brioschi S, et al. Genetics of HUS: the impact of MCP, CFH, and IF mutations on clinical presentation, response to treatment, and outcome. Blood. 2006;108:1267‐1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Malina M, Roumenina LT, Seeman T, et al. Genetics of hemolytic uremic syndromes. Presse Med. 2012;41:e105‐e114. [DOI] [PubMed] [Google Scholar]
- 6. Noris M, Remuzzi G. Glomerular diseases dependent on complement activation, including atypical hemolytic uremic syndrome, Membranoproliferative glomerulonephritis, and C3 glomerulopathy: Core curriculum 2015. Am J Kidney Dis. 2015;66:359‐375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Servais AA. Acquired and genetic complement abnormalities play a critical role in dense deposit disease and other C3 glomerulopathies. Kidney Int. 2012;82:454‐464. [DOI] [PubMed] [Google Scholar]
- 8. Zipfel PF, Skerka C, Chen Q, et al. The role of complement in C3 glomerulopathy. Mol Immunol. 2015;67:21‐30. [DOI] [PubMed] [Google Scholar]
- 9. Fritsche LG, Igl W, Bailey JN, et al. A large genome‐wide association study of age‐related macular degeneration highlights contributions of rare and common variants. Nat Genet. 2016;48:134‐143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Geerlings MJ, de Jong EK, den Hollander AI. The complement system in age‐related macular degeneration: a review of rare genetic variants and implications for personalized treatment. Mol Immunol. 2017;84:65‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kavanagh D, Goodship TH, Richards A. Atypical hemolytic uremic syndrome. Semin Nephrol. 2013;33:508‐530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yu Y, Triebwasser MP, Wong EK, et al. Whole‐exome sequencing identifies rare, functional CFH variants in families with macular degeneration. Hum Mol Genet. 2014;23:5283‐5293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Maga TK, Nishimura CJ, Weaver AE, Frees KL, Smith RJH. Mutations in alternative pathway complement proteins in American patients with atypical hemolytic uremic syndrome. Hum Mutat. 2010;31:E1445‐E1460. [DOI] [PubMed] [Google Scholar]
- 14. van de Ven JP, Nilsson SC, Tan PL, et al. A functional variant in the CFI gene confers a high risk of age‐related macular degeneration. Nat Genet. 2013;45:813‐817. [DOI] [PubMed] [Google Scholar]
- 15. Martinez‐Barricarte R, Heurich M, Lopez‐Perrote A, et al. The molecular and structural bases for the association of complement C3 mutations with atypical hemolytic uremic syndrome. Mol Immunol. 2015;66:263‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rodriguez E, Rallapalli PM, Osborne AJ, Perkins SJ. New functional and structural insights from updated mutational databases for complement factor H, factor I, membrane cofactor protein and C3. Biosci Rep. 2014;34:635‐649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Recalde S, Tortajada A, Subias M, et al. Molecular basis of factor H R1210C association with ocular and renal diseases. J Am Soc Nephrol. 2016;27:1305‐1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Westra D, Volokhina E, van der Heijden E, et al. Genetic disorders in complement (regulating) genes in patients with atypical haemolytic uraemic syndrome (aHUS). Nephrol Dial Transplant. 2010;25:2195‐2202. [DOI] [PubMed] [Google Scholar]
- 19. Roumenina LT, Frimat M, Miller EC, et al. A prevalent C3 mutation in aHUS patients causes a direct C3 convertase gain of function. Blood. 2012;119:4182‐4191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Volokhina E, Westra D, Xue X, et al. Novel C3 mutation p.Lys65Gln in aHUS affects complement factor H binding. Pediatr Nephrol. 2012;27:1519‐1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. de Cordoba SR, Tortajada A, Harris CL, Morgan BP. Complement dysregulation and disease: from genes and proteins to diagnostics and drugs. Immunobiology. 2012;217:1034‐1046. [DOI] [PubMed] [Google Scholar]
- 22. Geerlings MJ, Kremlitzka M, Bakker B, et al. The functional effect of rare variants in complement genes on C3b degradation in patients with age‐related macular degeneration. JAMA Ophthalmol. 2017;135:39‐46. [DOI] [PubMed] [Google Scholar]
- 23. Seddon JM, Yu Y, Miller EC, et al. Rare variants in CFI, C3 and C9 are associated with high risk of advanced age‐related macular degeneration. Nat Genet. 2013;45:1366‐1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pickering MC, D'Agati VD, Nester CM, et al. C3 glomerulopathy: consensus report. Kidney Int. 2013;84:1079‐1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein‐coding genetic variation in 60,706 humans. Nature. 2016;536:285‐291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hageman GS, Anderson DH, Johnson LV, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age‐related macular degeneration. Proc Natl Acad Sci USA. 2005;102:7227‐7232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Caprioli J, Castelletti F, Bucchioni S, et al. Complement factor H mutations and gene polymorphisms in haemolytic uraemic syndrome: the C‐257T, the A2089G and the G2881T polymorphisms are strongly associated with the disease. Hum Mol Genet. 2003;12:3385‐3395. [DOI] [PubMed] [Google Scholar]
- 28. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Res. 2010;38:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xue X, Wu J, Ricklin D, et al. Regulator‐dependent mechanisms of C3b processing by factor I allow differentiation of immune responses. Nat Struct Mol Biol. 2017;24:643‐651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Berman HM, Westbrook J, Feng Z, et al. The protein data Bank. Nucleic Acids Res. 2000;28:235‐242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Krieger E. Vriend G. YASARA view—molecular graphics for all devices—from smartphones to workstations. Bioinformatics. 2014;30:2981‐2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Prosser BE, Johnson S, Roversi P, et al. Structural basis for complement factor H linked age‐related macular degeneration. J Exp Med. 2007;204:2277‐2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wagner EK, Raychaudhuri S, Villalonga MB, et al. Mapping rare, deleterious mutations in factor H: association with early onset, drusen burden, and lower antigenic levels in familial AMD. Sci Rep. 2016;6:31531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jozsi M, Heinen S, Hartmann A, et al. Factor H and atypical hemolytic uremic syndrome: mutations in the C‐terminus cause structural changes and defective recognition functions. J Am Soc Nephrol. 2006;17:170‐177. [DOI] [PubMed] [Google Scholar]
- 35. Ferreira VP, Herbert AP, Cortes C, et al. The binding of factor H to a complex of physiological polyanions and C3b on cells is impaired in atypical hemolytic uremic syndrome. J Immunol. 2009;182:7009‐7018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jokiranta TS, Cheng ZZ, Seeberger H, et al. Binding of complement factor H to endothelial cells is mediated by the carboxy‐terminal glycosaminoglycan binding site. Am J Pathol. 2005;167:1173‐1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Clark SJ, Ridge LA, Herbert AP, et al. Tissue‐specific host recognition by complement factor H is mediated by differential activities of its glycosaminoglycan‐binding regions. J Immunol. 2013;190:2049‐2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Clark SJ, Schmidt CQ, White AM, et al. Identification of factor H‐like protein 1 as the predominant complement regulator in Bruch's membrane: implications for age‐related macular degeneration. J Immunol. 2014;193:4962‐4970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Clark SJ, Bishop PN. Role of factor H and related proteins in regulating complement activation in the macula, and relevance to age‐related macular degeneration. J Clin Med. 2015;4:18‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mohlin FC, Nilsson SC, Levart TK, et al. Functional characterization of two novel non‐synonymous alterations in CD46 and a Q950H change in factor H found in atypical hemolytic uremic syndrome patients. Mol Immunol. 2015;65:367‐376. [DOI] [PubMed] [Google Scholar]
- 41. Stahl AL, Vaziri‐Sani F, Heinen S, et al. Factor H dysfunction in patients with atypical hemolytic uremic syndrome contributes to complement deposition on platelets and their activation. Blood. 2008;111:5307‐5315. [DOI] [PubMed] [Google Scholar]
- 42. Neumann HP, Salzmann M, Bohnert‐Iwan B, et al. Haemolytic uraemic syndrome and mutations of the factor H gene: a registry‐based study of German speaking countries. J Med Genet. 2003;40:676‐681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Richards A, Buddles MR, Donne RL, et al. Factor H mutations in hemolytic uremic syndrome cluster in exons 18–20, a domain important for host cell recognition. Am J Hum Genet. 2001;68:485‐490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nilsson SC, Nita I, Mansson L, et al. Analysis of binding sites on complement factor I that are required for its activity. J Biol Chem. 2010;285:6235‐6245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sanchez‐Gallego JI, Groeneveld TW, Krentz S, et al. Analysis of binding sites on complement factor I using artificial N‐linked glycosylation. J Biol Chem. 2012;287:13572‐13583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Roversi P, Johnson S, Caesar JJ, et al. Structural basis for complement factor I control and its disease‐associated sequence polymorphisms. Proc Natl Acad Sci USA. 2011;108:12839‐12844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kavanagh D, Pappworth IY, Anderson H, et al. Factor I autoantibodies in patients with atypical hemolytic uremic syndrome: disease‐associated or an epiphenomenon? Clin J Am Soc Nephrol. 2012;7:417‐426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Forneris F, Ricklin D, Wu J, et al. Structures of C3b in complex with factors B and D give insight into complement convertase formation. Science. 2010;330:1816‐1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Forneris F, Wu J, Xue X, et al. Regulators of complement activity mediate inhibitory mechanisms through a common C3b‐binding mode. EMBO J. 2016;35:1133‐1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rooijakkers SH, Wu J, Ruyken M, et al. Structural and functional implications of the alternative complement pathway C3 convertase stabilized by a staphylococcal inhibitor. Nat Immunol. 2009;10:721‐727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Martinez‐Barricarte R, Pianetti G, Gautard R, et al. The complement factor H R1210C mutation is associated with atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2008;19:639‐646. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supporting Information
Table S1 Number of alleles found in this study. (combined PDF)
Table S2 Number of genetic variants found in this study. (combined PDF)
Table S3 Variants identified in this study. (Excel file)
Table S4 Variants identified in literature. (Excel file)
Table S5 Number of unique variants identified in literature. (combined PDF)
Figure S1 Mean depth of coverage per gene of the AMD cohort. (combined PDF)
Appendix S2. Supporting Information