

Abstract
Semaglutide is a human glucagon‐like peptide‐1 analog that has been co‐formulated with the absorption enhancer, sodium N‐(8‐[2‐hydroxybenzoyl] amino) caprylate, for oral administration. This trial (NCT02016911) investigated whether hepatic impairment affects the pharmacokinetics, safety, and tolerability of oral semaglutide. Subjects were classified into groups: normal hepatic function (n = 24), and mild (n = 12), moderate (n = 12), or severe (n = 8) hepatic impairment according to Child‐Pugh criteria, and received once‐daily oral semaglutide (5 mg for 5 days followed by 10 mg for 5 days). Semaglutide plasma concentrations were measured during dosing and for up to 21 days post‐last dose. Area under the semaglutide plasma concentration–time curve from 0–24 hours after the 10th dose (primary end point) and maximum semaglutide concentration after the 10th dose appeared similar across hepatic function groups. Similarly, there was no apparent effect of hepatic impairment on time to maximum semaglutide concentration (median range 1.0–1.5 hours) or half‐life (geometric mean range 142–156 hours). No safety concerns were identified in subjects with hepatic impairment receiving semaglutide. Reported adverse events were in line with those observed for other glucagon‐like peptide‐1 receptor agonists. There was no apparent effect of hepatic impairment on the pharmacokinetics, safety, and tolerability of oral semaglutide. The results of this trial suggest that dose adjustment of oral semaglutide is not warranted in subjects with hepatic impairment.
Keywords: GLP‐1 receptor agonists, semaglutide, pharmacokinetics, hepatic impairment
Despite the availability of a broad range of pharmacological options for the treatment of type 2 diabetes, optimal glycemic control remains a challenge for many patients.1 Native glucagon‐like peptide 1 (GLP‐1) stimulates insulin secretion and suppresses glucagon secretion in a glucose‐dependent manner, thereby resulting in improved glucose homeostasis and reduced hyperglycemia.2, 3 GLP‐1 has also been shown to reduce appetite, with a subsequent decrease in energy intake and without an increase in energy expenditure.2, 4 These actions make the GLP‐1 receptor a therapeutic target for the management of type 2 diabetes. Several GLP‐1 receptor agonists are now available and are included in best‐practice guideline recommendations.5, 6 While currently available GLP‐1 receptor agonists must be administered by subcutaneous injection on a daily or weekly basis, oral administration of a GLP‐1 receptor agonist may lead to earlier initiation of treatment and may improve acceptance and adherence to this drug class for some patients.7
Oral delivery of peptides is, however, hindered by the limited permeability of the gastrointestinal (GI) tract and rapid enzymatic and pH‐induced degradation in the stomach. Semaglutide is a long‐acting human GLP‐1 analog that differs from native human GLP‐1 by 3 minor but important structural modifications. These confer increased resistance to degradation by dipeptidyl peptidase‐4 and improve binding to albumin, extending the half‐life to approximately 1 week.8, 9 Semaglutide for once‐daily oral administration is co‐formulated with the absorption enhancer, sodium N‐(8‐[2‐hydroxybenzoyl] amino) caprylate (SNAC), in a tablet for once‐daily administration. SNAC is a small fatty acid derivative that promotes absorption of semaglutide across the gastric mucosa via effects on transcellular pathways.10, 11
Hepatic impairment, a frequent comorbidity in patients with type 2 diabetes, may influence the pharmacokinetics (PK) of antihyperglycemic agents.12 Semaglutide is metabolized via proteolytic cleavage of the peptide backbone and sequential β‐oxidation of the fatty acid chain, with no single organ acting as the major route of elimination.13 However, degradation products of semaglutide are excreted via urine and feces, implying at least partial involvement of the liver in the elimination of semaglutide. Additionally, semaglutide binds to albumin, the concentration of which may be lower in subjects with hepatic impairment compared to individuals with normal hepatic function. Although semaglutide exposure did not appear to be affected by hepatic impairment when administered by subcutaneous injection in a previous trial,14 the effects of liver dysfunction on the PK of the oral formulation of semaglutide have not previously been assessed. In addition, there are no previously reported data on the possible effects of hepatic impairment on the PK of SNAC, which is extensively metabolized via β‐oxidation and glucoronidation and is also highly bound to albumin.
Understanding the effects of hepatic impairment on the PK and tolerability of oral semaglutide is important for the management of patients with type 2 diabetes and reduced liver function. This trial was conducted to investigate the PK of oral semaglutide in subjects with hepatic impairment to determine whether dose adjustment may be required in this population. The short‐term safety and tolerability of oral semaglutide in these subjects was also assessed.
Methods
Relevant Ethics Committees (IKEM FTNsP/University Hospital Královské Vinohrady, Prague, Czech Republic, and the Office of Regional Government, Bratislava, Slovakia) approved the trial protocol. All subjects provided written, informed consent. The trial was conducted in accordance with Good Clinical Practice, the Declaration of Helsinki, and US Food and Drug Administration/European Medicines Agency guidelines for studies in subjects with hepatic impairment.15, 16
Trial Population
Male or female subjects were eligible for inclusion in the trial if they were aged 18–85 years with a body mass index of 18.5–40.0 kg/m2. Subjects with normal and subjects with impaired hepatic function were comparable with respect to age, sex, and body weight to the extent possible, with subjects in the normal hepatic function group being recruited once a minimum number of subjects (9, 9, and 6 subjects with mild, moderate, and severe hepatic impairment, respectively) had initiated dosing. Subjects with hepatic impairment were included based on a diagnosis of cirrhosis due to parenchymal liver disease as assessed through medical history and physical examination and confirmed by hepatic ultrasound, computerized axial tomography scan or magnetic resonance imaging, and/or liver biopsy. Hepatic impairment had to be stable, defined as no clinically significant change in disease status in the 30 days before screening, according to recent medical history. Hepatic function was categorized by the investigator according to Child‐Pugh criteria as mild impairment (Child‐Pugh Grade A; 5–6 points), moderate impairment (Child‐Pugh Grade B; 7–9 points), or severe impairment (Child‐Pugh Grade C; 10–15 points). Subjects with type 2 diabetes could be included in the hepatic impairment groups. Exclusion criteria for hepatically impaired subjects included the use of dipeptidyl peptidase‐4 inhibitors or GLP‐1 receptor agonists (except the trial product), biliary obstruction, and/or other causes of hepatic impairment not related to parenchymal disorders and/or diseases. Subjects with severe hepatic encephalopathy (grade ≥3) were also excluded.
Subjects with normal hepatic function were eligible for inclusion if they were judged to be of general good health by the investigator, based on a medical examination (including physical examination, medical history, vital signs, and electrocardiogram) and had liver enzyme (alanine transaminase, aspartate aminotransferase, and gamma‐glutamyl transpeptidase) levels within prespecified limits (within lower normal range –100% and upper normal range +50%).
Key exclusion criteria for all subjects included liver transplantation, history of inflammatory bowel disease, previous GI surgery, history of chronic or idiopathic acute pancreatitis, serious cardiac disease (New York Heart Association heart failure functional class III–IV; myocardial infarction within 3 months; unstable angina pectoris), uncontrolled hypertension (diastolic blood pressure ≥100 mmHg or systolic blood pressure ≥180 mmHg), or clinically significant renal disease (creatinine clearance <60 mL/min as calculated by the Cockcroft‐Gault formula at screening).
Trial Design and Treatment
This was a multicenter, open‐label, multiple‐dose, parallel‐group trial (NCT02016911). All subjects were treated once daily with oral semaglutide (Novo Nordisk A/S, Denmark) for 10 consecutive days, from day 1 to day 10 (5 days of 5 mg followed by 5 days of 10 mg) (Figure 1). All oral semaglutide tablets included 300 mg of SNAC. Subjects received a single tablet of oral semaglutide with 120 mL of water in the morning after overnight fasting (≥6 hours), with no fluid intake for at least 2 hours before dosing. Subjects had no food or liquid intake for 30 minutes after dosing, after which time breakfast was started. Administration of oral concomitant medication was avoided 2 hours before and after dosing. A multiple‐dose trial design, with 10 consecutive days of dosing, was chosen to avoid subjects with all measured concentrations below the lower limit of quantification (LLOQ) and to reduce the variability observed in exposure after a single dose.17
Figure 1.

Trial design. PK, pharmacokinetics.
A total of 26 blood samples were collected from each subject for analysis of plasma concentrations of semaglutide. Blood samples were taken on day 1 at up to 30 minutes prior to the first administration of semaglutide and 1 hour after dosing; predose on days 2, 4, 6, 8, and 9; on day 10 at up to 30 minutes predose and postdose every 30 minutes until 3 hours, and at 4, 6, 12, 18, 24, 36, 48, 96, 168, 264, 336, and 504 hours (21 days) after the last oral semaglutide dose. Blood samples (n = 50) for analysis of plasma concentrations of SNAC were collected at additional time points within the first 24 hours after dosing on days 1 and 10 to account for the shorter half‐life of SNAC. On day 1, samples were taken at up to 30 minutes before the first administration and postdose every 10 minutes in the first hour and at 1.5, 2, 2.5, 3, 4, 5, 6, 7, 8, 10, 12, 18, and 24 hours (predose on day 2); predose on days 4, 6, 8, and 9; and on day 10 up to 30 minutes predose and postdose every 10 minutes in the first hour, then at 1.5, 2, 2.5, 3, 4, 5, 6, 7, 8, 10, 12, 18, 24, 36, 48, 96, 168, 336, and 504 hours (21 days) after the last oral semaglutide dose.
Semaglutide Plasma Bioanalysis
Venous blood samples were drawn in tripotassium ethylenediaminetetraacetic acid (K3‐EDTA) tubes and stored at −20°C until analyzed. A liquid chromatography tandem mass spectroscopy (LC‐MS/MS) assay was used following precipitation of the plasma proteins (Celerion Switzerland AG, Fehraltorf, Switzerland). The LC‐MS/MS assay was validated according to current guidelines for bioanalysis of plasma samples in the concentration range 0.729–60.8 nmol/L (3.00–250 ng/mL). A 5‐fold dilution of each sample was validated to extend the assay range above 60.8 nmol/L. A stable isotope–labeled analogue of semaglutide was used as an internal standard (IS). The analysis was carried out using an AB Sciex API QTrap 5500 mass spectrometer (AB Sciex LLC, Framingham, Massachusetts). Positive ions were monitored in the multiple reaction‐monitoring (MRM) mode, with mass transitions m/z 1029.1 → 136.0 Da (semaglutide) and m/z 1033.2 → 136.0 Da (IS). The LC system was a Waters Acquity UPLC system and the LC column an Acquity UPLC BEH300 C18, 2.1 50 mm (Waters Corporation, Milford, Massachusetts). Quantification was performed by peak area ratios of semaglutide versus IS. The calibration curve fitting was done by weighted linear regression (1/concentration2). The LLOQ for semaglutide was 0.729 nmol/L.
SNAC Plasma Bioanalysis
Venous blood samples were drawn in K3‐EDTA tubes and stored at −20°C until analyzed. A LC‐MS/MS assay was used following in‐line solid‐phase sample preparation (Celerion Switzerland AG, Fehraltorf, Switzerland). The LC‐MS/MS assay was validated according to current guidelines for bioanalysis of plasma samples in the concentration range 5.00–2000 ng/mL. A 5‐fold dilution of each sample was validated to extend the assay range above 2000 ng/mL. A structural analogue of SNAC was used as an IS. The analysis was carried out using an AB Sciex API 4000 Triple Quadrupole Mass Spectrometer (AB Sciex LLC). Negative ions were monitored in the MRM mode, with mass transitions m/z 278.1 → 118.0 Da (SNAC) and m/z 249.0 → 135.0 Da (IS). The LC system was a Cohesive Turbulent Flow system (Cohesive Technologies LLC, Alpharetta, Georgia) with LC loading column Turboflow Cyclone‐P, 50 × 0.5 mm (Thermo Scientific, San Jose, California) and analytical column Onyx Monolithic C18, 50 × 2.0 mm (Phenomenex, Inc., Torrance, California). Quantification was performed using the peak area ratios of SNAC versus IS. The calibration curve fitting was done by weighted linear regression (1/concentration2). The LLOQ for SNAC was 5.00 ng/mL.
Urine was sampled predose on day 1 and postdose on day 10 (fractionated collection in predefined intervals of 0–4, 4–8, 16–24, and 24–36 hours postdose) to assess renal clearance (CLR) of semaglutide and SNAC.
Semaglutide and SNAC Urine Bioanalysis
Urine samples were stored at −20°C until analyzed. To avoid nonspecific binding of semaglutide to urine collection containers, 1% Triton X‐100 (Sigma‐Aldrich, St. Louis, Missouri) was added to the urine samples in a ratio of 1 part per 9 parts urine. LC‐MS/MS assays identical to the above for plasma were used for analyzing urine samples for semaglutide and SNAC. The assay ranges and the LLOQ were identical to those in plasma but due to dilution with Triton X‐100, the assay ranges were corrected for dilution (correction factor 1.111).
Protein Binding
A blood sample was collected from each subject prior to dosing on day 1 to assess protein binding of semaglutide. Plasma protein binding of semaglutide was determined using surface plasmon resonance technology as described elsewhere.18 Plasma protein binding of SNAC was determined using the Rapid Equilibrium Dialysis (RED) system (Covance Laboratories Ltd, Harrogate, UK) with blood samples collected 30 minutes after dosing on day 10.
SNAC metabolites were also analyzed (data not reported).
Trial End Points and Statistical Analysis
The sample size was based on the precision of the ratio of area under the semaglutide plasma concentration–time curve from 0–24 hours after the 10th dose (AUC0–24h,Day10) between the group of subjects with moderate hepatic impairment and the group of subjects with normal hepatic function, and was done using SAS 9.3 (SAS Institute, Cary, North Carolina). The between‐subject standard deviation of log(AUC0–24h,Day10) used in the sample size calculation was 0.60. Twenty‐two subjects with evaluable PK profiles in the normal hepatic function group and 11 in the moderate hepatic impairment group provided at least 80% probability of achieving a 2‐sided 90% confidence interval (CI) for the ratio R of AUC0–24h,Day10 between these 2 groups contained within the interval [0.66*R; 1.51*R]. Because of the large inherent variability in semaglutide exposure following oral administration, the sample size was not powered to demonstrate equivalence, but was solely based on the desired precision of the CI; demonstrating equivalence would have required more patients than was considered ethically acceptable given the absence of any significant clinical benefit in this population.
The full analysis set consisted of all subjects who were exposed to at least 1 dose of trial product. The primary end point was AUC0–24h,Day10, which was compared between subjects with normal hepatic function and subjects in the 3 hepatic impairment groups. AUC0–24h,Day10 was log‐transformed and analyzed using a linear normal model, with age and logarithmic‐transformed weight as continuous covariates and sex and hepatic function group (4 levels) as categorical fixed effects; the model allowed for different variations in each of these 4 groups. Estimated differences in log‐transformed values between the group with normal hepatic function and each of the 3 groups with hepatic impairment were back‐transformed to original scale and presented as ratios together with the corresponding 2‐sided 90%CIs. Maximum semaglutide concentration after the 10th dose (Cmax,Day10) was measured as a secondary end point and compared between groups in a similar manner to AUC0–24h,Day10. Other secondary end points included time to maximum semaglutide concentration (tmax,Day10), half‐life of oral semaglutide (t1/2,Day10), and CLR after the 10th dose, which were analyzed descriptively, and the fraction of unbound semaglutide. End points derived for SNAC included AUC0–24h,Day10, Cmax,Day10, tmax,Day10, total apparent clearance (CL/F), and CLR, and the fraction of unbound SNAC. All statistical analyses of end points were performed using SAS 9.3 or 9.4.
Safety was assessed from day 1 to the follow‐up visit (0–14 days after the last PK sample) in all subjects who were exposed to at least 1 dose of trial product (safety analysis set). Safety assessments included the number of adverse events (AEs), hypoglycemic episodes (defined as confirmed if the episode was severe according to the American Diabetes Association [ADA], ie, requiring third‐party assistance,19 or verified by a plasma glucose level <56 mg/dL [3.1 mmol/L]), and changes in laboratory safety variables, physical examination, vital signs, and electrocardiogram.
Results
In total, 56 subjects were exposed to oral semaglutide (full analysis set and safety analysis set) and 52 subjects completed the trial. One subject in the severe hepatic impairment group was withdrawn prematurely due to a serious AE with fatal outcome (bacterial peritonitis in a subject with severe hepatic impairment and a previous history of peritonitis) and 3 subjects (2 with normal hepatic function and 1 with mild hepatic impairment) withdrew prematurely for other reasons not related to AEs. Of those subjects included in the trial, 24 had normal hepatic function and 32 had hepatic impairment (mild, n = 12; moderate, n = 12; and severe, n = 8). Baseline characteristics were generally well balanced between the hepatic function groups, although the proportion of males was higher in the severe hepatic impairment group compared with the normal hepatic function group (Table 1). Of the subjects with hepatic impairment, 6 had type 2 diabetes at baseline.
Table 1.
Demographic and Baseline Characteristics
| Hepatic Function Group (Full Analysis Set) | ||||
|---|---|---|---|---|
| Parameters | Normal (n = 24) | Mild (n = 12) | Moderate (n = 12) | Severe (n = 8) |
| Mean (SD) age, years | 49 (11) | 52 (10) | 54 (10) | 52 (8) |
| Sex, male, n (%) | 11 (45.8) | 7 (58.3) | 6 (50.0) | 5 (62.5) |
| Mean (SD) weight, kg | 79.2 (14.8) | 82.3 (16.9) | 82.9 (14.4) | 77.6 (20.2) |
| Mean (SD) body mass index, kg/m2 | 27.4 (5.7) | 28.9 (4.9) | 29.4 (5.3) | 26.6 (4.5) |
| Mean (SD) HbA1c, % | 5.6 (0.4) | 5.7 (0.5) | 5.7 (1.1) | 6.2 (1.9) |
| Mean (min–max) Child‐Pugh score | NA | 6 (5–6) | 8 (7–9) | 11 (10–12) |
HbA1c, glycosylated hemoglobin; NA, not applicable; SD, standard deviation.
Pharmacokinetics
Geometric mean concentration–time profiles of plasma semaglutide for one dosing interval (24 hours) after the 10th dosing appeared to be similar across the 4 hepatic function groups (Figure 2). In line with the concentration–time profiles, total semaglutide exposure (AUC0–24h,Day10) and maximum concentration (Cmax,Day10) were similar across the 4 hepatic function groups (Table 2 and Figure 2). The estimated ratio of the mean AUC0–24h,Day10 of semaglutide in each of the hepatic impairment groups to that in the normal hepatic function group ranged from 0.87 (90%CI, 0.57–1.31) in the moderate impairment group to 0.91 (90%CI, 0.60–1.40) in the mild impairment group (Figure 3). The estimated ratio of mean Cmax,Day10 of semaglutide ranged from 0.85 (90%CI, 0.55–1.30) in the moderate hepatic impairment group to 0.92 (90%CI, 0.60–1.40) in the mild hepatic impairment group (Figure 3).
Figure 2.
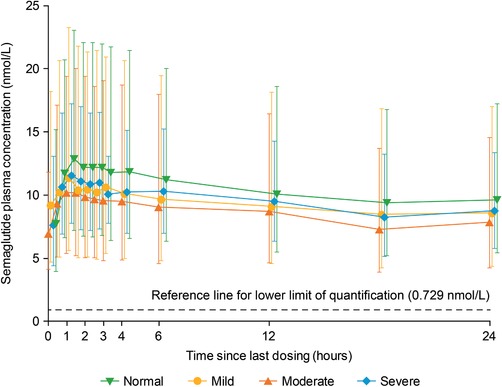
Geometric mean concentration–time profiles of semaglutide after the 10th dose, by hepatic function group. Full analysis set.
Table 2.
Pharmacokinetic End Points for Semaglutide and SNAC After the 10th Dosing
| Hepatic Function Group (Full Analysis Set) | ||||
|---|---|---|---|---|
| Parameters | Normal (n = 24) | Mild (n = 12) | Moderate (n = 12) | Severe (n = 8) |
| Semaglutide | ||||
| AUC0–24h,Day10, nmol·h/L | 250.3 (64.0) | 221.9 (78.3) | 204.2 (71.4) | 227.8 (41.6) |
| Cmax,Day10, nmol/L | 13.3 (62.3) | 11.8 (82.4) | 10.5 (73.5) | 12.0 (41.4) |
| tmax,Day10, h | 1.0 (0.5, 4.0) | 1.0 (0.5, 3.0) | 1.0 (1.0, 3.0) | 1.5 (1.0, 3.0) |
| t½,Day10, h | 156.4 (12.1) | 142.1 (7.6) | 146.7 (13.9) | 153.7 (12.5) |
| SNAC | ||||
| AUC0–24h,Day10, ng·h/mL | 1088 (28) | 1534 (24) | 2854 (63) | 3823 (36) |
| Cmax,Day10, ng/mL | 1309 (53) | 1261 (50) | 2358 (71) | 2358 (108) |
| tmax,Day10, h | 0.49 (0.30, 1.50) | 0.50 (0.17, 1.00) | 0.48 (0.17, 1.02) | 0.52 (0.47, 1.50) |
| CL/F (L/h) | 275.8 (27.9) | 195.6 (24.2) | 105.1 (63.1) | 78.5 (36.4) |
| CLR (L/h) | 0.076 (109.0) | 0.075 (125.3) | 0.054 (73.98) | 0.111 (88.52) |
AUC, area under the plasma concentration–time curve; Cmax, maximum concentration; SNAC, sodium N‐(8‐[2‐hydroxybenzoyl] amino) caprylate; tmax, time to reach maximum concentration; t½, terminal half‐life; CL/F, total apparent clearance; CLR, renal clearance.
Data are geometric means (coefficient of variation) except for tmax, for which median (minimum, maximum) values are presented.
Figure 3.
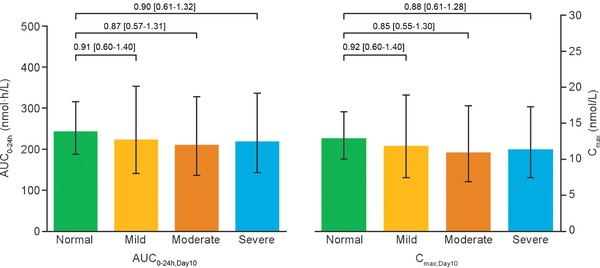
AUC0–24h,Day10 and Cmax,Day10 for semaglutide after the 10th dose, by hepatic function group. Full analysis set. Bars are estimated means and 95%CI. Treatment comparisons show estimated treatment ratio and 90%CI. AUC, area under the semaglutide plasma concentration–time curve; CI, confidence interval; Cmax, maximum semaglutide concentration.
There was no apparent effect of hepatic impairment on tmax,Day10 (range of medians, 1.0–1.5 hours) or t1/2,Day10 (range of geometric means, 142.1–156.4 hours) (Table 2). Semaglutide was not detected in urine samples of subjects with normal hepatic function or in samples of any of the subjects in the groups with hepatic impairment. The median fraction of unbound semaglutide was less than 1% across the groups, corresponding to a plasma protein binding of more than 99% in all subjects.
Exposure of SNAC (AUC0–24h,Day10 and Cmax,Day10) in plasma increased with a decrease in hepatic function (Table 2 and Figure 4). The ratios of the estimated mean of AUC0–24h of SNAC in each of the 3 hepatic impairment groups to that in the group with normal hepatic function ranged from 1.50 (90%CI, 1.28–1.74) in the mild impairment group to 3.64 (90%CI, 2.84–4.68) in the severe impairment group (Figure 5). The ratios of the estimated mean of Cmax of SNAC ranged from 1.10 (90%CI, 0.83–1.45) in the mild impairment group, to 2.00 (90%CI, 1.10–3.66) in the severe impairment group. No accumulation of SNAC was observed in any of the hepatic function groups, with exposure after administration of a single dose (day 1) similar to that observed after the 10th dose (day 10). Median tmax of SNAC was similar across the 4 hepatic function groups at approximately 0.5 hours. A small amount of SNAC was found to be excreted via urine (estimated mean CLR of SNAC: 0.052 [95%CI, 0.036–0.076]–0.096 [95%CI, 0.059–0.157] L/h), but no obvious pattern in the renal clearance of SNAC was observed in relation to the changes in hepatic function. Geometric mean CL/F decreased with decreasing hepatic function (Table 2). The median fraction of unbound SNAC was less than 1% across the groups, corresponding to a plasma protein binding of more than 99% in all subjects.
Figure 4.
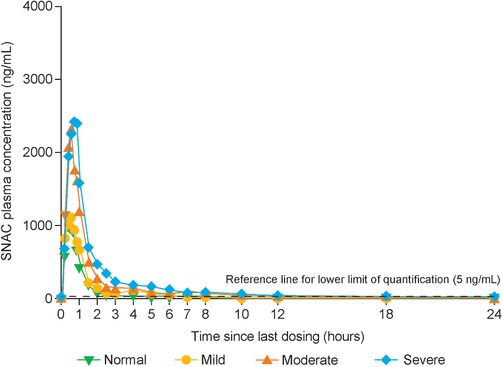
Mean concentration–time profiles of SNAC after the 10th dose, by hepatic function group. Full analysis set. SNAC, sodium N‐(8‐[2‐hydroxybenzoyl] amino) caprylate.
Figure 5.
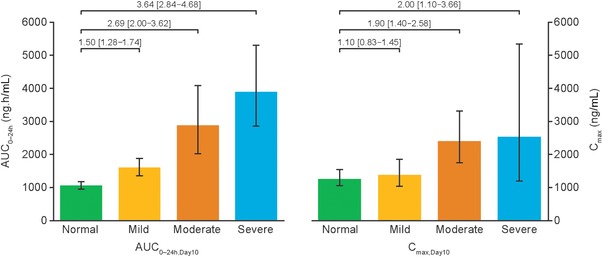
AUC0–24h,Day10 and Cmax,Day10 for SNAC after the 10th dose, by hepatic function group. Full analysis set. Bars are estimated means and 95%CI. Treatment comparisons show estimated treatment ratio and 90%CI. AUC, area under the plasma concentration–time curve; CI, confidence interval; Cmax, maximum concentration; SNAC, sodium N‐(8‐[2‐hydroxybenzoyl] amino) caprylate.
Safety and Tolerability
The safety analysis set comprised all 56 patients who received oral semaglutide. A total of 27 subjects (48.2%) reported 58 AEs (Table 3). No apparent pattern was observed in the incidence or severity of AEs between subjects with normal or impaired hepatic function or with increasing hepatic impairment. Almost all AEs reported were either moderate (17 subjects [30.4%], 30 AEs) or mild (15 subjects [26.8%], 27 AEs) in severity. Headache was the most frequently reported AE (14.3%), followed by dyspepsia (8.9%), vomiting (7.1%), decreased appetite (7.1%), and diarrhea (5.4%) (Table 3). GI disorders were the most common events considered at least possibly related to oral semaglutide administration. One subject with severe hepatic impairment and a history of peritonitis died due to bacterial peritonitis during the trial (serious AE); the investigator assessed this event as unlikely to be related to the trial product.
Table 3.
Adverse Events
| Hepatic Function Group [N (%), E] (Safety Analysis Set) | |||||
|---|---|---|---|---|---|
| System Organ Class and Preferred Term | Normal (n = 24) | Mild (n = 12) | Moderate (n = 12) | Severe (n = 8) | Total (N = 56) |
| Overall AEs | 16 (66.7), 31 | 5 (41.7), 18 | 1 (8.3), 1 | 5 (62.5), 8 | 27 (48.2), 58 |
| AEs occurring in >3% of subjects overall: | |||||
| Headache | 7 (29.2), 8 | 1 (8.3), 1 | 0 | 0 | 8 (14.3), 9 |
| Dyspepsia | 2 (8.3), 2 | 2 (16.7), 4 | 0 | 1 (12.5), 1 | 5 (8.9), 7 |
| Vomiting | 3 (12.5), 4 | 1 (8.3), 2 | 0 | 0 | 4 (7.1), 6 |
| Decreased appetite | 2 (8.3), 2 | 2 (16.7), 2 | 0 | 0 | 4 (7.1), 4 |
| Diarrhea | 1 (4.2), 1 | 1 (8.3), 2 | 0 | 1 (12.5), 1 | 3 (5.4), 4 |
| Abdominal pain | 1 (4.2), 3 | 0 | 0 | 1 (12.5), 1 | 2 (3.6), 4 |
AE, adverse event; E, number of AEs; N/n, number of subjects with AE; %, proportion of subjects with AE.
There were no episodes of severe hypoglycemic or symptomatic hypoglycemia with plasma glucose <56 mg/dL (3.1 mmol/L) reported during the trial. Twelve asymptomatic hypoglycemic episodes with plasma glucose level ≤70 mg/dL (3.9 mmol/L) (according to the ADA classification19) were reported in 7 patients (4 with normal hepatic function, 2 with mild hepatic impairment, and 1 with severe liver impairment).
Mean pulse rate increased from baseline to end of treatment in all 4 hepatic function groups. No clinically relevant changes were observed in other vital signs, electrocardiogram, physical examination, or clinical laboratory assessments.
Discussion
The results of this trial suggest that hepatic impairment has no apparent effect on the PK of oral semaglutide, regardless of the severity of hepatic dysfunction. In addition, there were no safety or tolerability concerns regarding the use of oral semaglutide in subjects with hepatic impairment. These findings indicate that dose adjustment of oral semaglutide is not warranted in subjects with impaired hepatic function.
The primary concern with hepatic impairment is a potential change in exposure to a drug that is extensively cleared by hepatic metabolism or biliary excretion via the liver. This could potentially exaggerate the pharmacodynamic action and/or side effects. However, GLP‐1 receptor agonists are thought to be primarily metabolized throughout the body, with multiple organ/tissue clearance. Thus, hepatic impairment is not expected to lead to increased exposure. In a previous study of the subcutaneous formulation of semaglutide, hepatic impairment did not affect exposure after a single 0.5‐mg injection, with the primary PK end point of AUC from time zero to infinity meeting a “no effect” criterion for each of the mild, moderate, and severe hepatic impairment groups compared with the normal hepatic function group.14 In addition, Cmax, tmax, and t1/2 were similar across hepatic function groups. These results are consistent with the absence of any apparent effect on the PK of oral semaglutide in subjects with hepatic impairment seen in the current trial.
Similarly, in a previous PK study with subcutaneous once‐daily liraglutide, drug exposure was not increased by impaired liver function.20 Indeed, the primary end point of AUC from time zero to infinity reduced with increasing hepatic impairment, although data were not sufficiently conclusive to suggest a dose increase was required for subjects with reduced hepatic function. No dose adjustment is required with liraglutide in patients with mild or moderate hepatic impairment and it is not currently recommended in patients with severe hepatic impairment,21 due to limited therapeutic experience in this group.
With a potential decrease in albumin due to hepatic impairment, it is possible that an increase in unbound semaglutide might decrease semaglutide exposure, because albumin binding slows the degradation of semaglutide in plasma and decreases renal clearance. In this trial, the fraction of protein‐bound semaglutide was >99% in all subjects across the hepatic function groups and no apparent difference between the impairment groups were observed. However, as has been previously noted,14 the observation that <1% of semaglutide was unbound should be interpreted with caution given that protein binding was assessed in vitro using blood samples collected before dosing. Additionally, due to the nature of the assay, the concentration of semaglutide relative to albumin was much higher than is seen in dosed subjects, in whom semaglutide concentrations are more than 10,000‐fold lower compared with physiological albumin concentrations.
Oral semaglutide is coformulated with SNAC, the absorption of which can be measured systemically. As with other fatty acid derivatives, SNAC is metabolized via β‐oxidation and glucoronidation and eliminated mainly via urine. However, there are no previous published studies on SNAC exposure levels in subjects with hepatic impairment and, consequently, no data available on the long‐term safety of SNAC in subjects with liver dysfunction. In the current trial, exposure to SNAC (AUC and Cmax) increased with a decrease in hepatic function. The maximal increase was 3.64 times and was observed in the group with severe hepatic impairment, which may reflect changes in β‐oxidation abilities during first‐pass metabolism. However, increased exposure was not considered clinically relevant given that SNAC is an absorption enhancer with no anticipated systemic pharmacodynamic effects. Increased exposure did not seem to translate into any accumulation of SNAC and was not associated with any increase in AEs. A small amount was excreted via urine but there was no obvious pattern in renal clearance of SNAC in relation to hepatic function. The fraction of protein‐bound SNAC was >99% in all subjects across the hepatic function groups.
The safety profile was as expected for the GLP‐1 receptor agonist class and there were no safety concerns with increasing severity of hepatic impairment. The most frequently reported AEs in all groups were GI disorders, which are considered a class effect of GLP‐1 receptor agonists. GI AEs were mild or moderate in severity and did not lead to premature withdrawal. The trial design involved a dose‐escalation aspect, with subjects receiving 5 mg for 5 days followed by 10 mg for 5 days, which was intended to improve GI tolerability. No severe hypoglycemic events were reported. One serious AE was reported, bacterial peritonitis with fatal outcome, which was considered unlikely to be related to oral semaglutide by the investigator. No significant changes in liver enzymes were observed with oral semaglutide, which is consistent with observations in clinical trials of other GLP‐1 receptor agonists alone or in combination with various other glucose‐lowering agents.12 Indeed, there is evidence that incretin‐based therapies may be beneficial in patients with chronic liver disease, as indicated by the histological resolution of nonalcoholic steatohepatitis seen with liraglutide.22 Further long‐term safety data on oral semaglutide will be provided by the ongoing PIONEER phase 3a clinical trial program in patients with type 2 diabetes.
Exposure‐response was not investigated in the current trial due to the planned population (ie, diagnosis of type 2 diabetes was not an inclusion criterion), the low number of subjects in each group, and the short treatment duration. However, a phase 2 study of oral semaglutide (including doses from 2.5–40 mg) observed a clear dose‐dependent response in HbA1c and a safety profile in line with other GLP‐1 receptor agonists.23 The doses investigated in the present trial (5 and 10 mg) are within the range of doses being investigated in oral semaglutide phase 3a trials. As such, the exposure levels observed are expected to be within therapeutic levels and the findings are therefore considered clinically relevant. Importantly, results are consistent with those seen with a single 0.5‐mg dose of subcutaneous semaglutide,14 indicating that semaglutide exposure does not seem to be affected by hepatic impairment.
Limitations of the trial include its open‐label design, short duration, and 10‐day dosing. This dosing period was used to ensure measurable semaglutide exposure while keeping the treatment period to a minimum in this vulnerable population. The parallel‐group design allowed for comparisons between groups after 10 days, although exposure had not reached steady state. The number of subjects in each group was low and unequal, with the fewest in the severely impaired group. The low number enrolled was for ethical reasons, ie, to minimize exposure of subjects, and because of the difficulty in recruiting subjects with hepatic impairment, particularly for the severe group. Subject numbers are, however, in accordance with the regulatory guidelines for these types of trials.15, 16 The trial population included subjects with and without type 2 diabetes to allow for the target population to be enrolled, while acknowledging that it would be difficult to only recruit subjects with both hepatic impairment and type 2 diabetes. This is considered acceptable, as the PK of semaglutide is comparable between healthy individuals and individuals with type 2 diabetes.24 Oral semaglutide at 10 mg falls within the range of doses tested in further development (3–14 mg).
Conclusions
To conclude, there was no apparent effect of hepatic impairment, regardless of severity, on the PK of semaglutide when administered via the oral route, suggesting that dose adjustment may not be necessary. Moreover, oral semaglutide was well tolerated in subjects with normal or impaired hepatic function, with a safety profile consistent with the GLP‐1 receptor agonist class.
Disclosures
This trial was funded by Novo Nordisk A/S. TAB, MT, and TWA are employees of Novo Nordisk A/S and own stock in the company; CWH is an employee of Novo Nordisk A/S; VK has no disclosures. Medical writing and editorial assistance was provided by Andy Bond and Emma Marshman of Spirit Medical Communications Ltd, and was supported by Novo Nordisk A/S.
Author Contributions
All authors participated in the trial design. VK was involved in the trial conduct and acquisition of data. All authors were involved in data analysis and interpretation and participated in writing the manuscript together with medical writing services provided by the sponsor. All authors have approved the submitted manuscript.
Acknowledgments
The authors would like to thank all subjects who participated in this trial. We also acknowledge the involvement of Dr Blanka Cieslarová (Pharmaceutical Research Associates CZ, s.r.o., Prague, Czech Republic) in the conduct of the trial, Michael Pilgaard Andersen (Novo Nordisk A/S) for support with the semaglutide and SNAC bioanalyses, and Morten Donsmark and Christin Løth Hertz (Novo Nordisk A/S) for their review of the manuscript.
Data Accessibility
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
- 1. Carls G, Huynh J, Tuttle E, Yee J, Edelman SV. Achievement of glycated hemoglobin goals in the US remains unchanged through 2014. Diabetes Ther. 2017;8:863–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Drucker DJ, Nauck MA. The incretin system: glucagon‐like peptide‐1 receptor agonists and dipeptidyl peptidase‐4 inhibitors in type 2 diabetes. Lancet. 2006;368:1696–1705. [DOI] [PubMed] [Google Scholar]
- 3. Verspohl EJ. Novel therapeutics for type 2 diabetes: incretin hormone mimetics (glucagon‐like peptide‐1 receptor agonists) and dipeptidyl peptidase‐4 inhibitors. Pharmacol Ther. 2009;124:113–138. [DOI] [PubMed] [Google Scholar]
- 4. Blundell J, Finlayson G, Axelsen M, et al. Effects of once‐weekly semaglutide on appetite, energy intake, control of eating, food preference and body weight in subjects with obesity. Diabetes Obes Metab. 2017;19:1242–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of hyperglycemia in type 2 diabetes, 2015: a patient‐centered approach: update to a position statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care. 2015;38:140–149. [DOI] [PubMed] [Google Scholar]
- 6. American Diabetes Association . 8. Pharmacologic approaches to glycemic treatment. Diabetes Care. 2017;40(suppl 1): S64–S74. [DOI] [PubMed] [Google Scholar]
- 7. Ismail R, Csóka I. Novel strategies in the oral delivery of antidiabetic peptide drugs ‐ insulin, GLP 1 and its analogs. Eur J Pharm Biopharm. 2017;115:257–267. [DOI] [PubMed] [Google Scholar]
- 8. Lau J, Bloch P, Schäffer L, et al. Discovery of the once‐weekly glucagon‐like peptide‐1 (GLP‐1) analogue semaglutide. J Med Chem. 2015;58:7370–7380. [DOI] [PubMed] [Google Scholar]
- 9. Kapitza C, Nosek L, Jensen L, Hartvig H, Jensen CB, Flint A. Semaglutide, a once‐weekly human GLP‐1 analog, does not reduce the bioavailability of the combined oral contraceptive, ethinylestradiol/levonorgestrel. J Clin Pharmacol. 2015;55:497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Buckley ST, Schéele SG, Kirk RK, Knudsen LB. Mechanism of absorption mediated by SNAC in an oral formulation of semaglutide. Diabetes. 2017;66(suppl 1):1206‐P. [Google Scholar]
- 11. Connor A, Borregaard J, Buckley ST, et al. Site of absorption of an oral formulation of semaglutide. Diabetes. 2017;66(suppl 1):1180‐P. [Google Scholar]
- 12. Scheen AJ. Pharmacokinetics in patients with chronic liver disease and hepatic safety of incretin‐based therapies for the management of type 2 diabetes mellitus. Clin Pharmacokinet. 2014;53:773–785. [DOI] [PubMed] [Google Scholar]
- 13. Jensen L, Helleberg H, Roffel A, et al. Absorption, metabolism and excretion of the GLP‐1 analogue semaglutide in humans and nonclinical species. Eur J Pharm Sci. 2017;104:31–41. [DOI] [PubMed] [Google Scholar]
- 14. Jensen L, Kupcova V, Arold G, et al. Pharmacokinetics and tolerability of semaglutide in people with hepatic impairment. Diabetes Obes Metab. 2018;20:998–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. US Department of Health and Human Services Food and Drug Administration . Center for Drug Evaluation and Research. Guidance for industry. Pharmacokinetics in patients with impaired hepatic function – study design, data analysis and impact on dosing and labeling. https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM072123.pdf. Published May 2003. Accessed August 2017.
- 16. European Medicines Agency. Committee for Medicinal Products for Human Use . Guideline on the evaluation of the pharmacokinetics of medicinal products in patients with impaired hepatic function. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003122.pdf. Published February 2005. Accessed August 2017.
- 17. Bækdal TA, Blicher TM, Donsmark M, Søndergaard FL. Safety, tolerability, and pharmacokinetics of single escalating doses of oral semaglutide in healthy male subjects. Diabetes. 2017;66(suppl 1):1191‐P. [Google Scholar]
- 18. Marbury TC, Flint A, Jacobsen JB, Derving Karsbøl J, Lasseter K. Pharmacokinetics and tolerability of a single dose of semaglutide, a human glucagon‐like peptide‐1 analog, in subjects with and without renal impairment. Clin Pharmacokinet. 2017;56:1381–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Seaquist ER, Anderson J, Childs B, et al. Hypoglycemia and diabetes: a report of a workgroup of the American Diabetes Association and the Endocrine Society. Diabetes Care. 2013;36:1384–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Flint A, Nazzal K, Jagielski P, Hindsberger C, Zdravkovic M. Influence of hepatic impairment on pharmacokinetics of the human GLP‐1 analogue, liraglutide. Br J Clin Pharmacol. 2010;70:807–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Victoza Summary of Product Characteristics. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/001026/WC500050017.pdf. Accessed November 2017.
- 22. Armstrong MJ, Gaunt P, Aithal GP, et al. Liraglutide safety and efficacy in patients with non‐alcoholic steatohepatitis (LEAN): a multicentre, double‐blind, randomised, placebo‐controlled phase 2 study. Lancet. 2016;387:679–690. [DOI] [PubMed] [Google Scholar]
- 23. Davies M, Pieber TR, Hartoft‐Nielsen ML, Hansen OKH, Jabbour S, Rosenstock J. Effect of oral semaglutide compared with placebo and subcutaneous semaglutide on glycemic control in patients with type 2 diabetes: a randomized clinical trial. JAMA. 2017;318:1460–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Granhall C, Donsmark M, Golor G, Søndergaard FL, Thomsen M. Safety, tolerability, and pharmacokinetics of multiple once‐daily dosing of oral semaglutide in healthy males and in males with T2D. Diabetes. 2017;66(suppl 1): 1192–P. [Google Scholar]