

ABSTRACT
The adult subventricular zone (SVZ) is a highly organized microenvironment established during the first postnatal days when radial glia cells begin to transform into type B‐cells and ependymal cells, all of which will form regenerative units, pinwheels, along the lateral wall of the lateral ventricle. Here, we identify p73, a p53 homologue, as a critical factor controlling both cell‐type specification and structural organization of the developing mouse SVZ. We describe that p73 deficiency halts the transition of the radial glia into ependymal cells, leading to the emergence of immature cells with abnormal identities in the ventricle and resulting in loss of the ventricular integrity. p73‐deficient ependymal cells have noticeably impaired ciliogenesis and they fail to organize into pinwheels, disrupting SVZ niche structure and function. Therefore, p73 is essential for appropriate ependymal cell maturation and the establishment of the neurogenic niche architecture. Accordingly, lack of p73 results in impaired neurogenesis. Moreover, p73 is required for translational planar cell polarity establishment, since p73 deficiency results in profound defects in cilia organization in individual cells and in intercellular patch orientation. Thus, our data reveal a completely new function of p73, independent of p53, in the neurogenic architecture of the SVZ of rodent brain and in the establishment of ependymal planar cell polarity with important implications in neurogenesis. © 2015 Wiley Periodicals, Inc. Develop Neurobiol 76: 730–747, 2016
Keywords: p73, ciliogenesis, ependymal cells, neurogenic pinwheel, planar cell polarity
INTRODUCTION
Maintenance of neural stem cell (NSC) capacity to self‐renew is dependent upon intrinsic regulatory networks and extrinsic cues within the NSC niches (Miller and Gauthier‐Fisher, 2009). The members of the p53 family of tumor suppressor genes, TP53, TP63, and TP73, are important players in NSC biology (Killick et al., 2011). In particular, TP73 is deeply involved in NSC maintenance and organization of the sub‐ventricular zone (SVZ) germinal center (Agostini et al., 2010; Fujitani et al., 2010; Gonzalez‐Cano et al., 2010; Talos et al., 2010) and regulates the balance between symmetric and asymmetric cell divisions of NSC (Gonzalez‐Cano et al., 2013). The hydrocephalus phenotype of Trp73−/− mice (p73KO from now on) (Yang et al., 2000) suggested a possible p73 role in ependymal ciliary function that might be linked to its regulation of the neurogenic environment. However, this has never been addressed.
The architecture of the SVZ is established during the first postnatal days when a select group of radial glia cells (RGCs) begins to transform into NSC (Merkle et al., 2004), while other subpopulation gives rise to ependymal cells (ECs). ECs will form pinwheel structures with B‐cells (NSC) intercalated among them. B‐cells have a small apical surface with a single primary cilium contacting the ventricle and large basal process contacting blood vessels, and exhibit ultrastructural characteristics and markers of astroglial cells, including GFAP and GLAST expression (Ihrie and Alvarez‐Buylla, 2011). The apical processes of the B‐cells form the core of the pinwheel, which itself is formed by multi and bi‐ciliated ECs (Mirzadeh et al., 2008). This highly organized microenvironment is necessary for maintaining NSC self‐renewal and differentiation capacity as well as the neurogenic niche homeostasis (Lim et al., 2000; Chmielnicki et al., 2004; Ramirez‐Castillejo et al., 2006; Andreu‐Agullo et al., 2009).
Multiciliated ECs have a prominent role in the maintenance of the neurogenic niche, since they induce neurogenesis and suppress gliogenesis by secreting the bone morphogenetic protein (BMP) inhibitor, Noggin (Lim et al., 2000; Chmielnicki et al., 2004). ECs are defined as large‐apical surface multiciliated cells that express S100ß and Vimentin (Spassky et al., 2005; Raponi et al., 2007; Mirzadeh et al., 2008; Pastrana et al., 2009). ECs are generated from RG in a multistep process orchestrated by the primary cilium and its basal body apparatus (Spassky et al., 2005). RG planar cell polarity (PCP) is first established during perinatal development when the primary cilium migrates toward the rostral end. Later on, from P5 until P20, cilia clusters in maturing ECs become densely packed, with basal bodies aligned and positioned as a patch on the downstream side of the EC apical surface with respect to the direction of cerebrospinal fluid (CSF) flow (Bayly and Axelrod, 2011). Thus, EC cilia display two types of PCP: rotational PCP (rPCP) which refers to the parallel alignment of the basal bodies within each multi‐ciliated cell and translational PCP (tPCP), defined by the basal body cluster anterior position on the cell apical surface (Mirzadeh et al., 2010). Both forms of polarity correlate with the onset of coordinated cilia beating in a uniform direction (Hirota et al., 2010).
Defects in cilia are associated with a range of human diseases, such as primary ciliary dyskinesia or hydrocephaly (Badano et al., 2006; Kishimoto and Sawamoto, 2012). In the brain, EC cilia are required for CSF circulation and neurogenesis (Boutin et al., 2014). Disruption in PCP establishment results in dysfunctions of ependymal cilia and their directional beating. Thus, the identification of the main players in EC maturation and PCP establishment might have important therapeutic implications.
Here we describe that p73 deficiency impairs ependymal cell maturation and ciliogenesis, as well as their organization in neurogenic pinwheel structures. Moreover, lack of p73 dramatically alters the establishment of PCP and affects the neurogenic capacity of the germinal center, independently of p53.
MATERIAL AND METHODS
Mice Husbandry, Genotyping, and BrdU Treatment
Housing and animal experiments were conducted in agreement with European and Spanish regulations on the protection of animals used for scientific purposes (Council Directive 2010/63/UE and RD 53/2013, respectively) with the appropriate institutional committee approval. Mice heterozygous for Trp73 on a mixed background C57BL/6 x 129/svJae (Yang et al., 2000) were backcrossed to C57BL/6, at least five times, to enrich for C57BL/6 background. Heterozygous animals were crossed to obtain the p73KO mice. C57BL/6 p53 knock‐out mice (p53KO) were previously described (Donehower et al., 1992). To generate the double p73;p53 knock‐out mice (DKO), heterozygous Trp73+/− animals were crossed with p53KO mice, obtaining the double heterozygous mice, Trp73+/−; Tp53+/−. Then, double heterozygous were inter‐crossed to originate the DKO animals. Genotyping of adult animals was performed by PCR analysis as described before (Yang et al., 2000; Flores et al., 2005). The thymidine analogue BrdU (Sigma, St. Louis, MO) was administered intraperitoneal (90 mg kg−1) in pulse injections every 2 h, during 8 h. Male and female mice were used in the experiments.
Immunohistochemistry
Animals were fully anesthetized using an analgesic/anesthetic mixture of medetomidine hydrochloride (Domtor®, Orion Corporation, Espoo, Finlandia) and ketamine (Imalgene® 500, Merial, Duluth, GA). Medetomidine (1 mg kg−1)/ketamine (75 mg kg−1) euthanasic mixture was prepared in phosphate buffer saline (PBS) and injected intraperitoneally. Animals were euthanized in agreement with European and Spanish regulations by transcardial perfusion with 0.1M PBS supplemented with 1% heparin until the drained blood became clear, and then with ∼20–40 mL of fresh 4% paraformaldehyde (PFA) in 0.1M PBS, depending of the age of the animal.
After perfusion, brains were dissected and post‐fixed overnight (o/n) in fresh 4% PFA in PBS solution at 4°C. For cryopreservation, samples were incubated for 24 h in 30% sucrose/PBS solution. Samples were then frozen in dry ice and stored at −80°C. A 30‐µm‐thick coronal sections were prepared using a Microm HM450 sliding microtome (Thermo Scientific, Walthman, MA) and stored in PBS supplemented with 0.05% sodium azide at 4°C. BrdU and Ki67 staining required antigen retrieval for which sections were incubated in 2N HCl for 15 min at 37°C. Then, sections were washed and stained according to the published protocol (Gonzalez‐Cano et al., 2010). Sections were incubated with the indicated antibodies (Table 1). Confocal microscopy images were obtained with Nikon Eclipse TE2000 confocal microscope (Nikon, Chiyoda‐Tokyo, Japan) and Olympus FluoView FV10i Confocal Laser Scanning Microscope (Olympus, Shinjuku‐Tokyo, Japan). Figures show single level representative pictures.
Table 1.
Primary Antibodies Used for Immunohistochemistry
| Antibody | Host | Dilution | Manufacturer, Catalog Number |
|---|---|---|---|
| BrdU | Mouse | 1:200 | Dako,·M0744 |
| BrdU | Mouse | 1:500 | BD Biosciences,·347583 |
| ß‐Catenin | Rabbit | 1:100 | Cell signaling,·9587 |
| ß‐Catenin | Mouse | 1:100 | BD Biosciences, 610153 |
| Dcx | Goat | 1:150 | Santa Cruz Biotech,·SC8066 |
| GFAP | Chicken | 1:500 | Chemicon,·Ab5541 |
| GLAST | Rabbit | 1:100 | Tocris, 2064 |
| IB4 | Mouse | 1:20 | Sigma,·L2140 |
| Ki67 | Rabbit | 1:100 | Abcam,·ab15580 |
| s100ß | Rabbit | 1:100 | Dako,·Z0311 |
| Ac‐Tubulin | Mouse | 1:1000 | Sigma,·T6793 |
| γ‐Tubulin | Goat | 1:200 | Santa Cruz Biotech,·SC7396 |
| Vimentin | Guinea Pig | 1:500 | Fitzgerald,·20R‐VP004 |
Isolation and Immunostaining of SVZ Wholemounts
Wholemounts of the lateral wall of the ventricles were dissected from 7, 15, and 160 postnatal day mice. Animals were anesthetized and euthanized in agreement with European and Spanish regulations and the brain was dissected and placed in cold 0.1M PBS. Dissection of the WMs was made under the stereomicroscope as described (Mirzadeh et al., 2010). WMs were transferred ventricle side up to a 24‐well plate with 4%PFA/0.1% Triton‐X100 (Tx100), fixed o/n at 4°C and stored in 0.1M PBS at 4°C.
WMs were incubated for 2 h at room temperature in blocking buffer (0.1M PBS/10% NDS/0.5% Tx100). Then, sections were incubated for 48 h at 4°C with the appropriate primary antibodies (Table 1) diluted in blocking buffer. Immunofluorescent detections were carried out with Alexa Fluor (Invitrogen, Carlsbad, CA) or DyLight (Pierce, Whaltman, MA) and Cy2, Cy3, and Cy5 (Jackson ImmunoResearch Laboratories, West Grove, PA) conjugated secondary antibodies. DAPI (1 µg mL−1) was used for nuclear counterstaining. Samples were analysed in an Olympus FluoView FV10i confocal laser scanning microscope (Olympus, Shinjuku‐Tokyo, Japan). Images were acquired and processed using FV10‐ASW 2.1 viewer software.
Scanning Electron Microscopy (SEM)
Lateral wall isolated from P7 and P15 mice were fixed with 0.1M PBS/2% glutaraldehyde, pH 7.2–7.4 at 4°C. Then, samples were dehydrated through a series of aqueous ethanol solutions (30, 70, 90, 96, and 100% ethanol) and finally dried with liquid CO2 (critical point at 8°C). To obtain SEM images, samples were covered with a cycle of evaporation of gold atoms induced by argon in vacuum conditions (0.05–0.07 mbar). SEM images were obtained using a JEOL Scanning Electron Microscope (JEOL, Akishima‐Tokyo, Japan). Images of the wholemounts were acquired every 0.5 µm and maximum projection of three to five consecutive Z levels are shown.
Image Data Quantification and Statistical Analyses
For all the analyses, at least three independent mice from each genotype were considered. Analysis of SVZ coronal sections was performed considering anterior, medial and posterior sections. For each section, Z‐stacks (20 µm) from at least six dorso‐ventral regions along the wall of the ventricle were acquired. To determine the numbers of positive cells, images were counted approximately every 7 µm ensuring that different cells were considered. The percentage of positive or double positive cells was calculated relative to the total number of nuclei (DAPI staining) at each Z level. When comparing the number of B cells versus GFAP positive cells, the number of GFAP and positive cells was first determined independently. Then, the percentage of GFAP/Ki67 cells out of the GFAP positive was calculated. Quantification of GLAST cells refers to the number of positive cells touching the ventricle relative to the analyzed lateral wall length (linear measurement = 130 μm).
Wholemount image analysis was performed counting four non overlapping fields (130 μm × 130 μm) of every specified region and genotype. Images of the WM were acquired every 0.5 µm and maximum projections of three to five consecutive Z levels are shown. In some cases, due to the cell heterogeneity depending on the region and the genotype, data are presented relative to the total surface analysed.
A customized software developed by Wimasis (Wimasis GmbH, Munich, Germany) was used to determine cell areas, percentage of cell polarization and angle displacement. Ten independent representative images per genotype were analyzed. For cell polarization analysis, the contours of ependymal cells and BB patches were defined and the relative distance between the geometric center of the apical cell surface and the BB cluster centroid was determined and divided by the cell radio. Angle displacement was determined relative to a reference angle for each microscopy field by drawing a vector from the cell center to the cluster centroid (Boutin et al., 2014).
Comparison of the cellular populations that constitute the SVZ was carried out using a Kruskal–Wallis test together with Dunn's multiple comparisons test. For all the other analyses, Mann–Whitney non‐parametric tests were performed. Angle displacements were compared by circular statistical analysis. Data were graphically represented using Oriana software (Kovach Computing Services, Anglesey, Wales) and Watson's U 2 test was performed. Differences were considered significant when P < 0.05 (*P < 0.05, **P < 0.01, ***P < 0.001) and all data are reported as mean ± standard deviation of the mean (SD).
RESULTS
p73 Deficiency Alters RG Cells Transition into ECs, Generating Immature Cells with Abnormal Identities and Resulting in Loss of Ventricular Integrity
ECs are generated from RGCs during perinatal development. This ependymal layer is crucial for the SVZ germinal center organization and function. Thus, to address whether p73 deficiency affected the maturation and organization of the EC layer, we analyzed P7 and P15 coronal sections using EC markers (Vimentin and S100β) together with GFAP. In P7‐wild type (WT) mice [Fig. 1(A)], most of the ependymal‐Vimentin+/S100β+ cells already formed a mono‐stratified epithelium lining the ventricle, although there were areas that remained pseudo‐stratified at this stage (white arrows). In the absence of p73, formation of the monostratified epithelium looked halted and chains of S100β+/Vimentin+ cells appeared to progress inwards with remarkable separations between the cells [Fig. 1(A), white arrows]. In WT ECs Vimentin and S100β proteins did not have the same cellular localization, with S100β being expressed mostly at the apical surface (white arrowheads). On the contrary, in p73KO ECs these proteins co‐localized (white arrowhead) also coinciding with GFAP expression around the cell soma and radial extensions [Fig. 1(B), yellow arrowheads].
Figure 1.
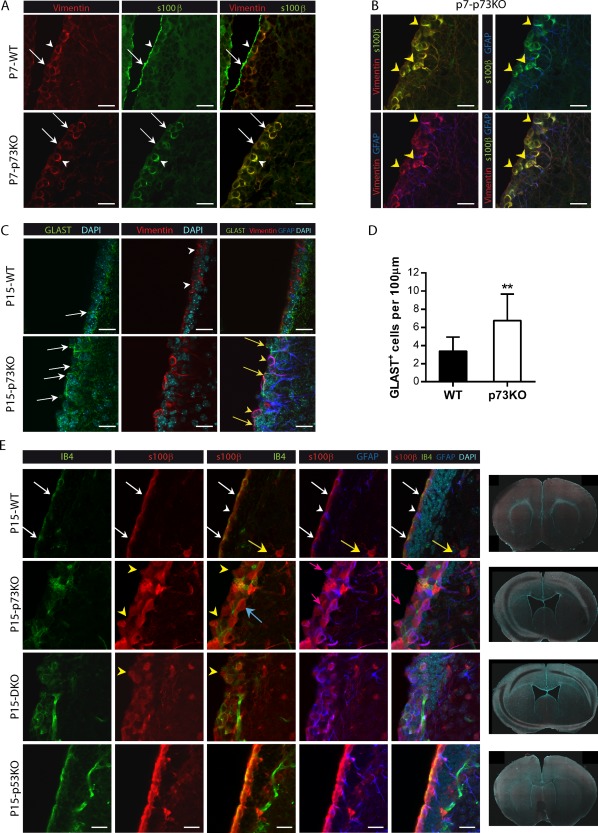
Lack of p73 results in a defective ependymal cell layer and loss of ventricular integrity. (A,B) Expression of Vimentin (A,B, red), S100β (A, B, green), and GFAP (B, blue) in confocal images of brain coronal sections from P7 WT and p73KO mice. Mono‐ or pseudo‐stratified ependymal layers are indicated by white arrows. Colocalization of S100β+/Vimentin+ is indicated by white arrowheads (A) and S100ß+/Vimentin+/GAFP+ by yellow arrowheads (B). (C) Analysis of GLAST (green), Vimentin (red), and GFAP (blue) expression in P15 mice after RGC transition into ependymal cells. Vimentin+ cells are indicated by white arrowheads. Different stages of RGCs transition were detected: immature GLAST+‐RGCs (white arrows), RGCs in transition GLAST+/Vimentin+ (yellow arrows) and aberrant GLAST+/Vimentin+/GFAP+ cells (yellow arrowheads). (D) Quantification of GLAST+ cells touching the lateral wall of the ventricule of SVZ. (E) Coronal sections of P15 WT, p73KO, p53KO, and DKO mice were stained with the indicated antibodies and the LW of the lateral ventricle was analyzed by confocal microscopy. WT LW showed a mono‐stratified epithelium of ependymal S100β+/IB4+ cells (red/green, white arrows), with some S100ß+/GFAP+ positive mature astrocytes in the striatum (yellow arrows) and a few GFAP+/S100β− B‐cells (white arrowheads). TP73 defective cells formed a pseudo‐stratified and discontinuous layer (yellow arrowheads) with S100β+/GFAP+ cells (pink arrows) with periventricular soma and long RG‐like striatal processes. Some S100β+ ependymal cells were IB4− (blue arrows). Tile scan of coronal sections images (×10) from WT, p73KO, p5KO, and DKO mice. Scale bars: 20 μm. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Many of these triple positive cells (S100β/Vimentin/GFAP) were contacting the ventricle and could correspond with immature cells in a transitional state from RGCs to ECs [Fig. 1(B)]. To address this, we analyzed expression and localization of the RGC marker GLAST, together with Vimentin, at a stage were RG transition into ECs has just been completed (P15) (Lavado and Oliver, 2011). As previously described (Mirzadeh et al., 2008), GLAST+‐RGCs were mostly absent from the ventricle wall in P15 WT brains and Vimentin+‐ECs lacked GLAST expression [Fig. 1(C), white arrowheads]. However, p73KO brains had significantly more GLAST+‐RGCs that remained in contact with the ventricle [Fig. 1(C,D), white arrows]. Moreover, many of these cells expressed Vimentin in their processes (Vimentin+/GLAST+ cells, yellow arrows). This pattern of marker expression strongly suggests an immature phenotype in the p73KO brains. We also detected an aberrant triple positive population (Vimentin+/GLAST+/GFAP+) in P15‐p73KO mice [Fig. 1(C), yellow arrowheads]. This population most likely represent an intermediate cellular state during RG transformation into GFAP+ astrocytes and B cells (Spassky et al., 2005), indicating that p73 might be required for RGCs cell fate determination.
To further study the effect of p73 deficiency on ependymal cell maturation and morphology, P15 coronal sections were stained with antibodies against S100ß (also expressed by mature GFAP‐astrocytes) and isolectin B4 (IB4), which also labels endothelial cells. At this stage, WT mice already exhibited an organized and continuous mono‐stratified epithelium of mature ECs (S100β+/IB4+) [Fig. 1(E), white arrows] with few B‐cells (GFAP+/S100β−) contacting the ventricle (white arrowheads) and some mature GFAP+/S100β+/IB4− astrocytes only in the striatum (yellow arrows). In contrast, lack of p73, resulted in a pseudo‐stratified and discontinuous ependyma with scattered S100β+‐ECs [Fig. 1(E), yellow arrowheads]. It is worth noticing that not all S100β+‐ECs were positive for IB4 (blue arrow) but, in turn, p73KO brains had abundant GFAP+/S100β+ cells, some of which were in contact with the ventricle (pink arrow), suggesting either an incomplete maturation or an astrocytic nature. p73‐deficient ependymal layer had gaps, invaginations, and “bumps” [Fig. 1(E), yellow arrowheads and Supporting Information Fig. 1], indicating that lack of p73 compromises ependymal barrier integrity. Defects in the ependymal barrier have been associated to hydrocephalus (Jimenez et al, 2014), a condition observed in the p73KO mice (Yang et al., 2000). However, it has been proposed that hydrocephalus in p73KO could be the result of induced apoptosis associated to an exacerbated p53 activity, due to the lack of the prosurvival DNp73 isoform (Pozniak et al., 2002; Lee et al., 2004). To address this possibility, even though the number of S100β+ cells was not lower in p73KO SVZ than in WT brains (data not shown), we analyzed coronal sections from p53 knock‐out (p53KO) and double mutant p73KO/p53KO (DKO) brains. DKO, like p73KO, showed severe hydrocephalus and enlarged ventricles [Fig. 1(E)], suggesting that p73‐loss has a dominant effect. Consistently, DKO also had numerous S100β+ ECs, some of which were IB4+, but these ECs did not form a monostratified epithelium [Fig. 1(E)] demonstrating that p53 abolishment did not restore the phenotype. Moreover, DKO mice, but not the single p53KO, displayed defective ependymal layers [Fig. 1(E), yellow arrowheads].
It has been reported that disruption of the mature ependyma disturbs neurogenic activity (Jimenez et al., 2014). Mature ECs create a permissive neurogenic environment by secreting Noggin (Lim et al., 2000). Noggin and S100ß expression co‐localized within the ependymal layer in WT mice (Supporting Information Fig. 1). However, in the p73KO, Noggin was detected throughout the SVZ and did not completely match the S100β+ expressing cells, highlighting the lack of appropriate organization and maturation of ECs (Supporting Information Fig. 1). This finding not only was consistent with the idea that p73 is required for the transition of RG into the different cell types that comprise the SVZ but, more importantly, it also suggested possible defects in the neurogenic environment of this region in p73‐defective mice. To address this, we analyzed the different cellular populations that constitute the SVZ. P15 brain coronal sections were first stained against Doublecortin (Dcx), combined with the proliferation markers Ki67 or BrdU to quantify the proliferating neuroblasts [Fig. 2(A–C)]. It has been published that p53KO mice had more proliferating neuroblasts than WT (Ferrón et al., 2009). However, we observed that, lack of p73, alone or in the context of p53KO (DKO), resulted in a significant reduction of the proliferating neuroblasts compared to WT or p53KO, respectively [Fig. 2(C)], indicating that p73 deficiency leads to a reduction in neurogenesis in the SVZ, despite p53 absence. However, p53 ablation in the context of p73 deficiency (DKO) did not elevate the number of proliferating neuroblasts, indicating that p73 effect was not due to enhanced neuroblast cell death resultant from p53 compensatory activation.
Figure 2.
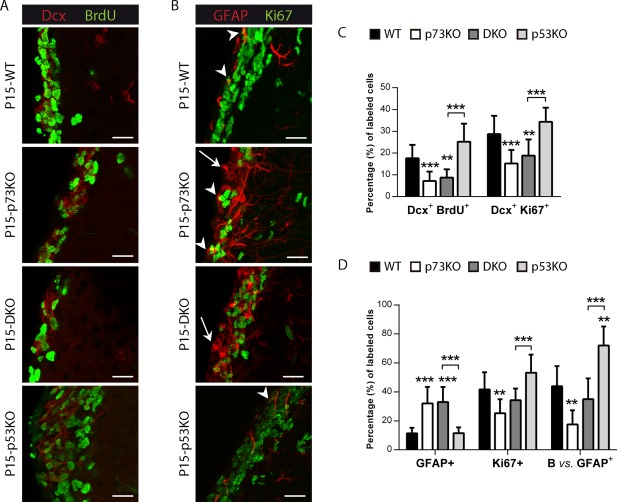
p73 deficiency affects proliferating neuroblasts and neural stem cells, even in the absence of p53. Coronal sections of P15 WT, p73KO, p53KO, and DKO mice were stained with the indicated antibodies and the LW of the lateral ventricle was analyzed by confocal microscopy (A, B) and cellular populations were quantified (C, D). (A, C) p73 loss resulted in less proliferating neuroblasts [Doublecortin (Dcx+)/BrdU+ (red/green)], even in the absence of p53. (B, D) Mice lacking p73 had more non proliferating astrocytes indicated by white arrows (GFAP+, red), but a reduced proportion of B‐cells, indicated by white arrowheads (GFAP+/Ki67+, red/green). Scale bars: 20 μm. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
RGC will also give raise to neural stem cells (B‐cell) and astrocytes. Thus, we next analyzed these populations: astrocytes (non‐proliferative GFAP cells) and B‐cells, identified as the subpopulation of proliferating astrocytes (GFAP/Ki67 double positive) in the SVZ (Merkle et al., 2004). Strikingly, lack of p73 gave raise to GFAP+ cells that had a periventricular cell soma in contact with the ventricle, GFAP+ filaments that formed tangles surrounding the nucleus, and long GFAP+ processes that extended across the striatum towards the pial surface, altogether resembling the morphology of RG cells [Fig. 2(B), white arrows]. However, most of these GFAP+ cells were non‐proliferating cells [Fig. 2(B,D), white arrows], discarding their possible identity as B‐cells. While 43.86% of GFAP+ cells represented B‐cells in the WT, only 17.56% of GFAP+ cells were B‐cells in p73KO. Moreover, p73 deficiency, alone or in the context of p53 ablation (DKO), resulted in significant reduction in the proportion of B‐cells in the SVZ compared to WT and p53KO, respectively [Fig. 2(D)], suggesting a p73 requirement for adult neural stem cells establishment and maintenance.
Altogether the alterations in the p73KO SVZ indicate that p73 loss results in an aberrant maturation of the ECs, affecting the SVZ environment, which might cause, at least in part, the impaired neurogenic capacity of the germinal centers, independently of p53.
p73 is Essential for the Ependymal Cell Assembly into Neurogenic SVZ Pinwheels
EC generation involves the transformation of monociliated RGCs into multiciliated ECs during the first postnatal days (P0‐P15) (Spassky et al., 2005). Within the first postnatal week, RGCs gradually increase their apical surfaces and upon reaching certain size, multiply the number of basal bodies (BB) to develop clusters of motile cilia (Mirzadeh et al., 2010). To analyze this maturation, P7‐WT and p73KO mice wholemounts dissections were stained with γ‐tubulin to label the cilia basal bodies (BBs), and β‐catenin to delimit the cell membrane, and the apical cell surface size was quantified [Fig. 3(A)]. Most of the mono‐BB cells in WT mice were small cells with an apical surface ≤25 µm2 corresponding to B‐cells and RGCs according to Mirzadeh et al. (2010). WT mice also had mono‐BB cells with expanded apical surface, between 30 and 60 µm2, which are RGCs in transition (Mirzadeh et al., 2010), but we rarely detected mono‐BB cells with an apical surface bigger than 60 µm2. However, in p73KO mice the percentages of mono‐BB cells with intermediate size or with apical surfaces bigger than 60 µm2 were significantly higher [Fig. 3(A)]. The later cells correspond, most likely, to abnormal stages of RGC transitioning into ECs.
Figure 3.
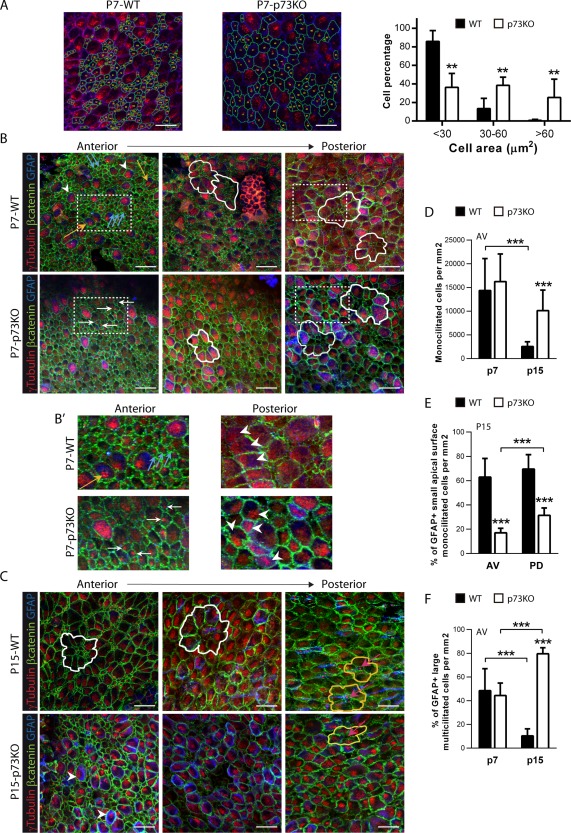
Lack of p73 delays the establishment of the lateral ventricle wall cytoarchitecture and the organization of ependymal cells into pinwheels. Confocal Z‐stacks images of lateral ventricle wall whole‐mounts from the indicated age and phenotype immunostained for β‐catenin (green), γ‐tubulin (red), and GFAP (blue). (A) Confocal images of P7 WT and p73KO SVZ with trace of the cell contour (green) and quantification of mono‐ basal body (BB) cell size. (B, B’) In P7 mice, RGCs are identified by the presence of a single BB and small apical surface (blue arrows). Immature multiciliated cells are shown by either yellow arrows (cells with deuterosomes) or by white arrowheads (cells with multiple BB covering the apical surface). The orientation of RGCs and multi‐BB cells is indicated by white arrows and white arrowheads, respectively. (B, C) In P7 and P15 mice white and yellow lines mark pre‐pinwheels and pinwheels respectively, and pink arrows indicate pinwheel core cells. (D–F) Quantification of the indicated populations at anterior‐ventral (AV) and posterior‐dorsal (PD) regions in P7 and P15 mice. The images represent Z‐stacks (20 µm) from at least 6 dorso‐ventral regions. Scale bars: 20 μm. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
The neurogenic capacity of the rodents SVZ is dependent upon ependymal cells integrity and their capacity to assemble into the unique structures that define this region: the pinwheels (Kuo et al., 2006; Paez‐Gonzalez 2011). Multi and bi‐ciliated ECs surround mono‐ciliated GFAP+‐B‐cells forming pinwheels in which the apical processes of the neural stem cells constitute the core of the structure (Mirzadeh et al., 2008). To address whether p73‐deficiency affected SVZ cytoarchitecture, P7 and P15 wholemounts were stained with γ‐tubulin, β‐catenin and GFAP antibodies to identify ventricle‐contacting monociliated GFAP+‐neural stem cells (B‐cells) and multiciliated GFAP‐ ECs [Fig. 3(B–D)].
P7‐WT AV region showed mono‐BB cells which do not express GFAP and correspond to RGCs [Fig. 3(B, B’)], blue arrows] and RGCs with extended apical surface and γ‐tubulin stainings marks like dense punctuated deposits known as deuterosomes (Spassky et al., 2005) [Fig. 3(B, B’), yellow arrows]. There were also cells with multiple BBs, which are sparse and randomly oriented in the apical surface, that correspond to immature multiciliated cells [Fig. 3(B), white arrowheads].
Toward posterior regions, WT‐ECs seemed to be organizing into pinwheel structures, with immature large GFAP+‐multi BB cells surrounding groups of small mono‐BB cells (pre‐pinwheels) [Fig. 3(B), white lines], confirming the gradual transformation of the lateral ventricle wall in WT mice. The maturing p73KO‐ECs were arranged into aberrant structures, with many intermediate‐size mono‐BB cells in the core, but without a pre‐pinwheel organization [Fig. 3(B), white lines]. Moreover, while in the WT brains neighbors cells have begun to coordinate their BB clusters [Fig. 3(B’) white arrowheads], in p73KO the cells do not show any coordinated orientation at this stage (white arrowheads). Nevertheless, p73 deficient mice did not show significant differences in the number of total mono‐BB cells [Fig. 3(D)], neither in multi‐BB cells (WT 32.83 ± 12.45 vs. p73KO 22.02 ± 8.086; p = 0.0734).
P15‐WT mice [Fig. 3(C)] showed a progressive maturation with pre‐pinwheel patterns (white lines) at AV zone and pinwheels (yellow lines) in PD regions, very similar to those described in the adult mice (Mirzadeh et al., 2008). The monociliated cells at the pinwheel core had a small apical surface with long GFAP+ processes oriented tangentially to the epithelial surface, what identified them as B‐cells [Fig. 3(C), pink arrows]. Surprisingly, P15‐p73KO SVZ lacked this cytoarchitecture [Fig. 3(C)]. First, the number of total monociliated cells was significantly higher in p73KO than in the WT [Fig. 3(D)]. However, 62.9% ± 15.4% of monociliated cells in WT were GFAP+ B‐cells, while in p73KO only 17.0% ± 3.8% (AV region) and 31.4% ± 6.1% (PD region) expressed this marker [Fig. 3(E)], indicating that p73 deficient monociliated cells were not bona‐fide B‐cells. As we progressed toward the posterior regions, a more organized distribution was observed, but structures resembling pinwheel patterns were seldom detected [yellow lines and pink arrows]. In the absence of p73, it is noteworthy that, a significant number of large‐surface multi‐BB cells touching the ventricle, which by definition are ECs (Spassky et al., 2005), were GFAP+, even at P15 [Fig. 3(C,F), white arrowheads]. These large multiciliated‐GFAP+ cells [Fig. 4(A), white arrowheads] seemed to correspond with the previously described cells contacting the ventricle that coexpressed S100β, Vimentin and GFAP, some of which were also GLAST+ [Fig. 1(B,C), yellow arrowheads]. These cells had an aberrant membrane morphology with waves and pleats [Fig. 4(A), yellow arrowheads], suggesting that the intercellular junctions at the apical surface of these cells might be affected.
Figure 4.
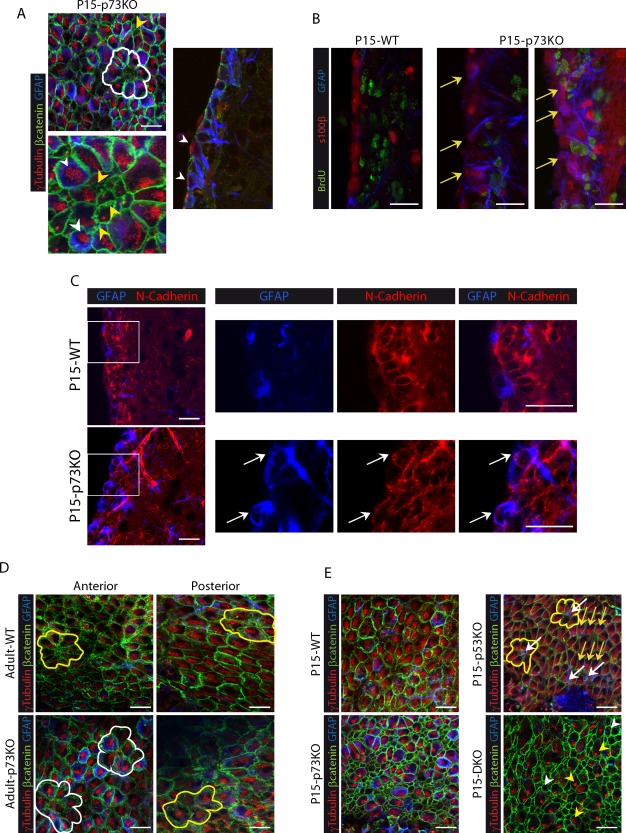
p73 deficiency results in immature and aberrant multiciliated ependymal cells that persist in adulthood, independently of p53. (A) Magnification of confocal WMs Z‐stacks micrographs (left) and coronal section (right) from P15 p73KO mice immunostained for β‐catenin (green), γ‐tubulin (red), and GFAP (blue). Yellow arrowheads indicate multiciliated cells with aberrant membrane morphology, while multiciliated cells with abnormal identities (GFAP+) are marked by white arrowheads. (B) Confocal images of brain coronal section from WT and p73KO mice. Inmunostaining for BrdU (green), S100β (red), and GFAP (blue) indicate that most of the S100β+/GFAP+ cells in the p73KO SVZ were non‐proliferating cells (BrdU−) (yellow arrows). (C) Confocal images and magnifications of P15 coronal sections immunostained for N‐Cadherin (red), and GFAP (blue). White arrows indicate N‐cadherin+/GFAP+ cell touching the ventricle. (D) Analysis of lateral ventricle wall whole‐mounts of the indicated phenotypes at P160 (D) and P15 (E) immunostained for β‐catenin (green), γ‐tubulin (red), and GFAP (blue). Pinwheel structures are marked by white lines and pre‐pinwheel structures by yellow lines. (E) Wholemount analysis of P15 mice from the four genotypes. While p53KO mice presented pinwheel structures (yellow lines) with B cells (white arrows), DKO mice showed aberrant expanded apical surface monociliated GFAP+ cells (white arrowheads) and cells with deuterosomes (yellow arrowheads), but not organized structures. Yellow arrows follow basal body polarization of ependymal cells. Scale bars: 20 μm. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
It has been reported that p73KO brains suffer ependymal denudation (Medina‐Bolivar et al., 2014). This process triggers neighboring astrocytes to proliferate and form a superficial cell layer covering the denuded ventricular surface (Roales‐Buján et al., 2012). The reactive astrocytes found at the denuded ventricular walls resemble the ependymal cells in several aspects, like the expression of vimentin, but maintain specific differences like lack of N‐cadherin expression and absence of cilia (Roales‐Buján et al., 2012). Thus, we sought to determine whether the GFAP‐expressing cells in p73KO VZ/SVZ were indeed, reactive astrocytes. We analyzed the proliferation capacity of S100β+/GFAP+ cells, since remodeling astrocytes have been shown to express proliferative markers (Kuo et al., 2006; Luo et al., 2008), and found that most of the S100β+/GFAP+ cells in the p73KO SVZ were nonproliferating BrdU−‐cells [Fig. 4(B), yellow arrows]. Furthermore, the cells lining the ventricles in p73 deficient mice, including the GFAP+ cells, expressed N‐cadherin at the lateral plasma membrane [Fig. 4(C), white arrows]. A similar phenotype was observed in DKO mice (Supporting Information Fig.2). Moreover, the presence of BB clusters in these GFAP+ cells revealed their fate as multicilliated ependymal cells [Fig. 4(A), white arrowheads]. Altogether, our data is consistent with a defect in RGC maturation and the presence of aberrant and immature multiciliated‐ECs in p73KO VZ/SVZ, rather than with the presence of remodeling astrocytes. It is meaningful that adult p73KO maintained the observed phenotype in adulthood with the presence of multiple large‐surface ependymal cells with BBs clusters and altered membrane junctions [Fig. 4(D)]. Adult p73KO multiciliated cells had strong GFAP expression and very condensed basal bodies clusters forming mainly pre‐pinwheel structures [Fig. 4(D) white lines] and only a small number of pinwheels (yellow line). Thus, in the absence of p73, EC maturation is halted, with the immature ECs remaining in the lateral ventricle. These data confirm that p73 deficiency results in a deregulation of the transition process from RGC to ependymal and B‐cells, leading to the emergence of aberrant cells not capable of assemble into neurogenic structures.
To further address whether p53 function played a relevant role on neurogenic pinwheel organization, we performed a comparative study of wholemounts from the four genotypes at P15, since the DKO mice do not survive any longer. p53 deficient brains displayed an organized VZ with mature multiciliated ECs and monociliated GFAP+‐B cells forming pinwheels [Fig. 4(E), yellow lines]. Thus, lack of p53 did not affect the cytoarchitecture of the lateral ventricle wall, despite the numerous B‐cells touching the ventricle [Fig. 4(E), white arrows]. Strikingly, DKO mice were very similar to p73KO, with numerous monociliated cells with expanded apical surface, some of them GFAP+ [Fig. 4(E), white arrowhead]. Moreover, many multiciliated cells still had deuterosomes, with no detectable polarization, characteristic of immature ECs [yellow arrowheads]. These results demonstrate that while p73 function is required for the establishment of SVZ architecture while p53 is not essential for these processes.
Lack of p73 Impairs Ciliogenesis and Planar Cell Polarity Establishment
To address whether lack of p73 affected the genesis and organization of ependymal cilia, we performed a comparative study of P7 and P15 WT and p73KO mice using SEM. It has been described that cilia tufts appear around P5 and reach their mature density by P10 (Tissir et al., 2010). Accordingly, P7‐WT AV regions displayed some monociliated cells [Supporting Information Fig. 3(A), black arrows; Fig. 5(A),] and abundant multiciliated cells [Supporting Information Fig. 3(A,B)] covering most of the ventricular surface. At the same stage, p73KO showed an immature phenotype with many monociliated cells [Supporting Information Fig. 3(A,B), black arrows] and less cilia tufts [Supporting Information Fig. 3(A,B)]. Moreover, it is noteworthy the presence of cells with cilia of different sizes distributed through the whole apical surface [Fig. 5(A), double line]. As reported (Spassky et al., 2005), we observed that by P15 mature ECs dominate the VZ displaying compacted cilia clusters on the apical surface which have begun to be coordinately oriented [Fig. 5(B)]. In contrast, cilia tufts in p73KO cells displayed an abnormal organization and, in some cases, the individual cilia within a given cell did not point to the same direction and some cilia seem to be angled or even truncated [Fig. 5(B), black triangles]. This data suggests that in the p73KO mice the cilia fail to form normally.
Figure 5.
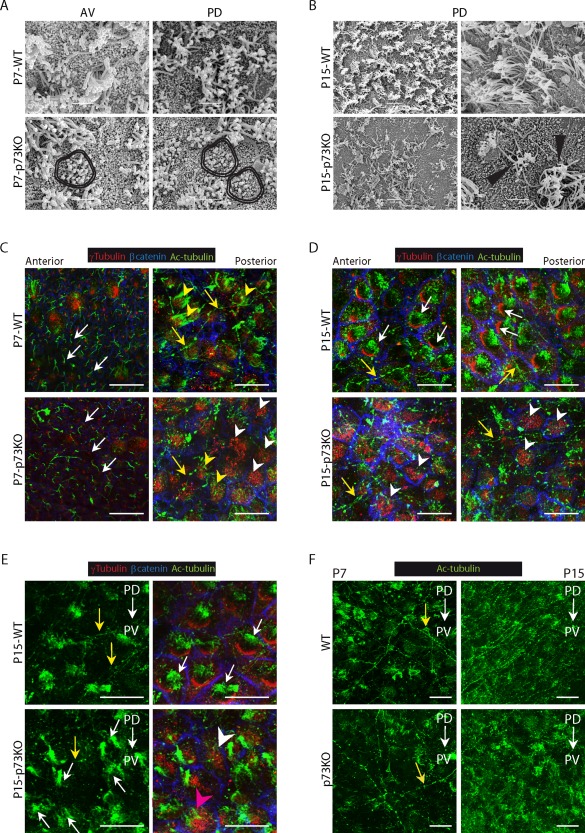
Lack of p73 alters motile cilia generation and planar cell polarity establishment during the transformation of monociliated RG into ependymal cells. SEM analysis of WT and p73KO lateral ventricle wall wholemounts at P7 (A) and P15 (B). (A) P7‐p73KO mice displayed aberrant groups of cilia of different length (double lines). Scale bar: 5 μm (B) P15 WT PD region showed organized multiciliated cells while in p73KO the cells had angled and truncated cilia (black triangles). Scale bar: 5 and 10 μm. (C–F) Confocal Z‐stacks images of WMs immunostained for γ‐tubulin (red), β‐catenin (blue) and Ac‐tubulin (green) from P7 (C, F) and P15 (D–F) mice. (C) At P7 RGCs had a primary cilium associated to a single basal body (white arrows) and immature ECs had long cilia emanating from basal bodies patches (yellow arrowheads). Most p73KO ECs had multiple basal bodies but lacked motile cilia (white arrowheads) and only few displayed cilia, but disorganized and of different lengths (yellow arrowheads). (D,E) Mature WT‐EC showed characteristic oriented basal bodies patches associated to cilia tufts (white arrows). ECs in p73KO had non oriented BB clusters either associated to disorganized cilia (pink arrowhead) or without cilia (white arrowheads). (F) Comparison of the supra‐ependymal axons (C‐F, yellow arrows) network between WT and p73KO mice. AV: anterior‐ventral; PD: posterior‐dorsal. PD→AD: anterior‐posterior axis. PD→PV: dorso‐ventral axis. Scale bar: 20 μm. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
We found intriguing that while the SEM analysis revealed less cilia covered area in p73KO lateral ventricle wall [Fig. 5(B)], our previous en‐face wholemount staining of the walls of the lateral ventricle (Fig. 3) demonstrated that there were no differences in the number of total multiciliated cells in the p73KO, as accounted by the presence of BB clusters. Thus, we hypothesized that p73‐deficient VZ ependymal cells had BB clusters, but lacked cilia due to a defect in the ciliogenesis process. To address this, we analyzed wholemounts of P7 and P15 SVZ stained for acetylated tubulin (Ac‐tubulin) which marks ependymal long motile cilia and RGCs short primary cilia. At P7 anterior regions, WT mice displayed RGCs with a primary cilium associated to a single basal body [Fig. 5(C), white arrows], while at posterior regions, ECs have long cilia tufts emanating from BBs patches (yellow arrowheads). The majority, although not all, p73‐deficient RGCs had a primary cilium associated to a basal body [Fig.5(C), white arrows]. However, many ECs had BB clusters that lacked cilia (white arrowheads). Few p73KO ECs had BB patches with associated cilia, but in these cases, the EC displayed disorganized cilia of different lengths [Fig.5(C), yellow arrowheads], similar to those observed by SEM [Fig. 5(A), double lines].
P15 WT ECs displayed densely packed BB clusters, all associated to cilia tufts [Fig. 5(D,E), white arrows]. The cilia dysfunction was patent in p73KO at this stage where many of the large surface cells had disorganized BB clusters that were not associated with cilia [Fig. 5(D,E), white arrowheads]. Ac‐tubulin antibodies also stained some long and slender processes with varicosities [Fig. 5(C–F), yellow arrows], corresponding with supra‐ependymal axons (Tong et al., 2014). Supra‐ependymal axons run parallel to the anterior–posterior axis (PD→AD) of the lateral ventricle on the dorsal V‐SVZ and along the dorso‐ventral axis (PD→PV) of the WT mice. Surprisingly, the lateral ventricular wall of the P7‐p73KO mice had a less dense network of supra‐ependymal axons which lack direction [Fig. 5(F) and Supporting Information Fig.4(A), yellow arrows]. This was also evident at P15 stage where a fine and organized network of supra‐ependymal axons running on a dorso‐ventral direction was observed in the WT mice [Fig. 5(F) and Supporting Information Fig. 4(B), yellow arrows]), whereas p73‐deficient mice showed a less dense and disorganized network, presumably contributing to their neurogenic deficiency.
Our results confirmed that p73 deficiency results in defective ciliogenesis during the transition from monociliated RGCs into multiciliated ECs. A correlation between defective ciliogenesis and alterations in ependymal planar polarization has been previously reported (Mirzadeh et al., 2010). In agreement, throughout this work we had detected, in p73KO cells, irregularities in the position of BBs on the apical surface of the cells, i.e., defects in translational cell polarity (tPCP). For example, while neighboring RGCs in P7‐WT mice had their cilia BB displaced to the same side of the apical surface [Fig. 3(B’), blue arrows], neighboring p73KO‐RGCs had their cilia positioned on opposite sides [Fig. 3(B’), white arrows], suggesting that p73 deficiency affected RGCs primary cilium polarization. Most multiciliated P15 WT ECs were polarized at tissue level (tPCP established), since they had their BB patches compacted and clustered on the “downstream” side of the apical cell surface, with respect to the direction of CSF flow, and cilia tufts following the same orientation [Fig. 5(D,E), white arrows]. However, in p73‐deficient cells most BB clusters were not polarized [Fig. 5(D), white arrowheads), independently of the presence of cilia tufts [Fig. 5(E), white arrows, pink arrowhead] or its absence [white arrowheads], suggesting that p73 effect on tPCP was upstream of ciliogenesis.
Moreover, WT ECs that conform a pinwheel structure had their BB clusters polarized with a similar orientation [Fig. 6(A), yellow lines and white arrows,]. That was not the case in p73KO ECs, which had their BBs either distributed throughout the apical surface, or oriented following different directions [Fig.6 (A,B), white arrows). Moreover, they display different arrays of cilia cluster distribution: some cells had very tightly compacted basal bodies [Fig. 6(B), yellow arrows], others formed more than one cluster located on one side of the apical surface, or even in two opposing poles [white lines]. Altogether, suggesting that p73 is essential to organize BB in individual cells and also for intercellular BB patch orientation. To further address p73 requirement for the establishment of tPCP we quantify the degree of cluster polarization relative to the apical surface. For this purpose we use a program generated by Wimasis® to calculate the polarization ratio [Fig. 6(C,D)]. We measured the relative distance between the geometric center of the apical surface and the BB cluster centroid (Boutin et al., 2014). We observed significant differences between WT ECs, with a mean polarization ratio of 77.72 ± 19.77, and p73KO cells with a ratio of 25.99 ± 12.13 [Fig. 6(D)], indicating that p73 is essential to organize translational polarity at single cell level. Furthermore, we asked whether p73 deficiency affected the coordination of BB patches displacement at tissue level by drawing a vector from the cell center to the cluster centroid (Boutin et al., 2014). Vectors of neighbor WT ECs had similar direction, but in p73KO ECs they were more divergent [Fig. 6(C), white lines]. Calculation of the angle of displacement [Fig. 6(E)] indicated that the angular deviation of individual vectors from the mean was ± 35.76° in the WT while the p73 deficient cells displayed a significantly higher deviation, with a mean of ± 57.83° (Watson's U2 test p < 0.005). Altogether, our data strongly support the hypothesis that p73 is essential to translational PCP.
Figure 6.
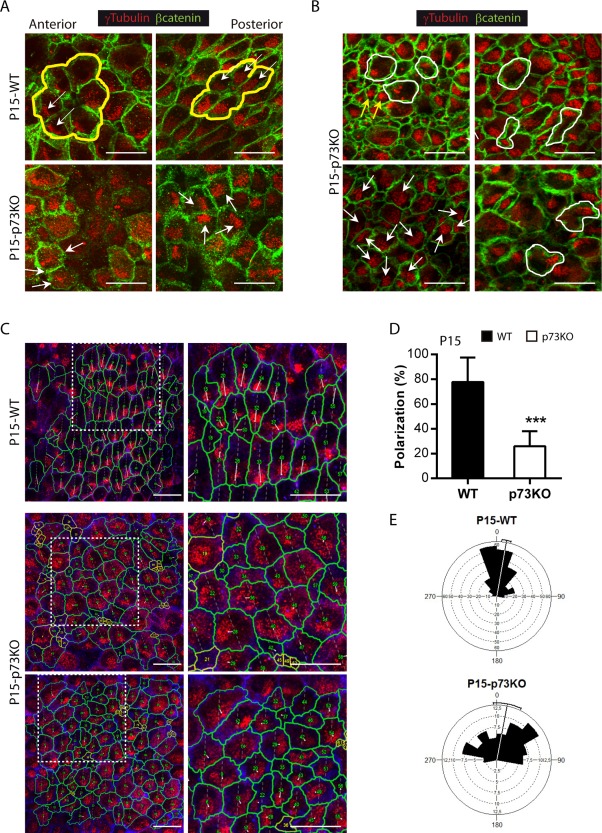
Lack of p73 impairs translational planar cell polarity in ependymal cells. Confocal Z‐stacks images of lateral ventricle wall wholemounts from the indicated age and phenotype immunostained for β‐catenin (green, A, B or blue, C) and γ‐tubulin (red). (A) P15‐WT ECs, but not the p73KO, were organized in pinwheel structures (yellow lines) and displayed translational PCP with cilia patches clustered on the “downstream” side of the apical cell surfaces, and oriented with respect to the direction of CSF flow (white arrows). (B) p73KO multiciliated ECs had the basal bodies distributed throughout the apical surface (white arrows) and showed different cilia clusters distribution: e.g. BB patches compacted very tightly (yellow arrows) or multiple clusters (white lines). (C) Trace of the cell contour (green) and magnifications of the white line inserts (right panels). Lines from the cell center to the BB centroid show BB cluster's angle displacement. Scale bar: 20 μm. (D) Quantification of BB polarization (the relative distance between the geometric center of the apical surface and the BB cluster centroid) (E) Angular distribution of vectors of BB displacement around the mean. In WT, the majority of vectors were closely distributed showing the coordination of BB displacement among neighbor cells. In the absence of p73, this coordination is lost, leading to a broader distribution. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
DISCUSSION
The appropriate functioning of ependymal cells is fundamental to create and maintain the neurogenic milieu of the germinal niche in the SVZ of rodent brain (Lledo et al., 2008). In this regard, since p73 is expressed in the ependymal cells (Hernandez‐Acosta et al., 2011; Medina‐Bolivar et al., 2014), we hypothesized that p73 had a possible role in EC biology. In agreement with our hypothesis, ependymal cell denudation in p73 deficient mice has recently been described (Medina‐Bolivar et al 2014), although the nature of the ECs defects have not been described.
Our results indicated that p73 ablation did not result in less S100β+ cells, but rather in ECs immaturity with the consequent cellular disorganization in the VZ and loss of brain ventricular integrity. One of the hallmarks of the p73 deficient brain is the presence of immature ECs that express GFAP and had the basal bodies clustered in deuterosomes. These immature ECs fail to organize into neurogenic pinwheels, disrupting p73KO‐SVZ cytoarchitecture and thus, niche structure and function (Kokovay et al., 2012). These features are maintained in the adult p73KO VZ, suggesting that the EC maturation was halted and confirming that p73 deficiency results in a deregulation of the transition process from RGC to ependymal and B‐cells, leading to the emergence of aberrant cells with abnormal intermediate identities within the lateral wall of the ventricle. p73 deficient ependymal cells had an aberrant membrane morphology with pleats and invaginations that could be the cause of their failure in establishing appropriate cell to cell adhesions. This p73KO phenotype was also detected in the absence of p53 (DKO mice), suggesting that p73‐deficient effect is not due to an enhanced cell death resultant from p53 compensatory activation in the absence of the anti‐apoptotic DNp73. Indeed, the ependymal layer in the absence of p73, including in DKO, is pseudostratified and discontinuous indicating p73 requirement for brain ventricular integrity. In this regard, studies have demonstrated that ependymal cell–cell adhesion regulates NSC activation under physiological and regenerative conditions (Porlan et al., 2014). Thus, lack of cellular contacts in the p73KO SVZ could lead to activation and subsequent depletion of the normally quiescent NSCs.
In P15 p73KO mice we detect monociliated cells (accounted by BB staining) that cannot be identified as bona fide B1‐cells (neural stem cells). These cells resemble the monociliated‐small apical surface radial glial cells observed in WT brains at early immature stages (P7) that did not yet display GFAP expression. It is possible that in the absence of p73, only few RGC are capable to progress into functional neural stem cells (GFAP expressing B1‐cells), leading to a diminished neural stem cell population in the SVZ. This lack of an appropriate pool of neural stem cells could be the underlying mechanism of the impaired neurogenic capacity in p73KO mice brain. This suggests that p73 is not only essential for neural stem cell maintenance (Agostini et al., 2010; Fujitani et al., 2010; Gonzalez‐Cano et al., 2010; Talos et al., 2010), but also for the de novo generation of neural stem cells during the postnatal generation of the neurogenic niche in mice SVZ. In agreement with impaired neurogenic function, p73KO lateral wall had a less dense network of supra‐ependymal axons, known to directly interact with NSCs to regulate neurogenesis (Tong et al., 2014). These axons normally encircle the base of ependymal cilia tufts; therefore, their reduction could be secondary to abnormal cilia generation and growth. It has been proposed that glutamate provided by these axons may supplement metabolic pathways in multiciliated ECs in order to fuel the energy demand of the ciliary beating (Robinson et al., 1996). Consistent with p73 requirement for the maintenance of the neurogenic niche function, our analysis revealed that p73KO mice had significantly less proliferating neuroblasts and B cells than WT mice. This effect it not due to p53‐induced cell death since DKO mice also showed a significant reduction in these cellular populations in the SVZ. These results support an impairment of neurogenesis and probably a decrease in the number of new neurons that migrate to the OB. Thus, p73 deficiency has a profound effect on SVZ maintenance and neurogenesis.
The most striking alteration of the p73KO brain is the profound defects in ECs planar cell polarity. Our data strongly suggest that p73 is necessary for the primary cilia organization in RGCs, which orchestrates the planar polarized architecture of both radial glia and their progeny ependymal cells (Mirzadeh et al., 2010), and also for the establishment of ependymal tPCP. Moreover, in the absence of p73 many ECs had basal body clusters but lacked cilia, or the number of total cilia tufts was lower, indicating that p73 deficiency alters or delays ciliogenesis.
We demonstrate that p73 function is necessary to organize the basal bodies within individual cells and, even though it is not required for basal body cluster displacement from the center, it is essential for intercellular patch orientation, since the p73KO cells did not have a common direction of displacement. Lack of basal body cluster orientation in p73KO ECs was independent of the presence of cilia tufts, suggesting that p73 effect on PCP is upstream of ciliogenesis. This is in agreement with reported observations that support the notion that PCP signaling influences cilium, while definitive evidence for regulation of PCP signaling by cilium is still missing (Gray et al., 2011). Many PCP core regulator genes like Cadherin EGF LAG Seven‐Pass G‐Type 1‐3 (Celsr 1‐3), Frizzled Class Receptor 3 (Fzd3), Van Gogh like1‐2 (Vangl1‐2) and Disheveled1‐3 (Dvl1‐3) or Non‐Muscle Myosin II (NMII) have also been implicated in cilia development and function (Ibañez‐Tallon, et al. 2004; Lechtreck et al., 2008; Guirao et al., 2010; Hirota et al., 2010; Ohata et al., 2014). However, the mechanism of p73 regulation of PCP remains elusive.
Our results show that lack of p73 results in profound defects in the timing of ependymal cell maturation, and in the establishment of planar cell polarity, altogether affecting the correct assembly of the neurogenic cytoarchitecture of the SVZ germinal center. These alterations could be the subjacent mechanisms that lead to the hydrocephalus and defective neurogenic capacity of the p73 deficient mice, highlighting the important role of p73 in brain development and brain homeostasis.
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
The authors thank Dr. Luis Fernando de la Fuente Crespo (Departamento de Produccion Animal, Universidad de Leon) for the advice with the statistical analysis.
REFERENCES
- Agostini M, Tucci P, Chen H, Knight RA, Bano D, Nicotera P, McKeon F, et al. 2010. P73 regulates maintenance of neural stem cell. Biochem Biophys Res Commun 403:13–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreu‐Agullo C, Morante‐Redolat JM, Delgado AC, Farinas I. 2009. Vascular niche factor PEDF modulates notch‐dependent stemness in the adult subependymal zone. Nat Neurosci 12:1514–1523. [DOI] [PubMed] [Google Scholar]
- Badano JL, Mitsuma N, Beales PL, Katsanis N. 2006. The ciliopathies: an emerging class of human genetic disorders. Annu Rev Genomics Hum Genet 7:125–148. [DOI] [PubMed] [Google Scholar]
- Bayly R, Axelrod JD. 2011. Pointing in the right direction: New developments in the field of planar cell polarity. Nat Rev Genet 12:385–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutin C, Labedan P, Dimidschstein J, Richard F, Cremer H, Andre P, Yang Y, Montcouquiol M, Goffinet AM, Tissir F. 2014. A dual role for planar cell polarity genes in ciliated cells. Proc Natl Acad Sci U S A 111:E3129–E3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chmielnicki E, Benraiss A, Economides AN, Goldman SA. 2004. Adenovirally expressed noggin and brain‐derived neurotrophic factor cooperate to induce new medium spiny neurons from resident progenitor cells in the adult striatal ventricular zone. J Neurosci 24:2133–2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA Jr, Butel JS Bradley A. 1992. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 356:215–221. [DOI] [PubMed] [Google Scholar]
- Ferrón SR, Marqués‐Torrejón MA, Mira H, Flores I, Taylor K, Blasco MA, Fariñas I. 2009. Telomere shortening in neural stem cells disrupts neuronal differentiation and neuritogenesis. J Neurosci 29(46):14394–14407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores ER, Sengupta S, Miller JB, Newman JJ, Bronson R, Crowley D, Yang A, et al. 2005. Tumor predisposition in mice mutant for p63 and p73: Evidence for broader tumor suppressor functions for the p53 family. Cancer Cell 7:363–373. [DOI] [PubMed] [Google Scholar]
- Fujitani M, Cancino GI, Dugani CB, Weaver IC, Gauthier‐Fisher A, Paquin A, Mak TW, et al. 2010. TAp73 acts via the bHLH Hey2 to promote long‐term maintenance of neural precursors. Curr Biol 20:2058–2065. [DOI] [PubMed] [Google Scholar]
- Gonzalez‐Cano L, Herreros‐Villanueva M, Fernandez‐Alonso R, Ayuso‐Sacido A, Meyer G, Garcia‐Verdugo JM, Silva A, et al. 2010. P73 deficiency results in impaired self renewal and premature neuronal differentiation of mouse neural progenitors independently of P53. Cell Death Dis 1:e109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez‐Cano L, Hillje AL, Fuertes‐Alvarez S, Marques MM, Blanch A, Ian RW, Irwin MS, et al. 2013. Regulatory feedback loop between TP73 and TRIM32. Cell Death Dis 4:e704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray RS, Roszko I, Solnica‐Krezel L. 2011. Planar cell polarity: Coordinating morphogenetic cell behaviors with embryonic polarity. Dev Cell 21:120–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guirao B, Meunier A, Mortaud S, Aguilar A, Corsi JM, Strehl L, Hirota Y, et al. 2010. Coupling between hydrodynamic forces and planar cell polarity orients mammalian motile cilia. Nat Cell Biol 12:341–350. [DOI] [PubMed] [Google Scholar]
- Hernandez‐Acosta NC, Cabrera‐Socorro A, Morlans MP, Delgado FJ, Suarez‐Sola ML, Sottocornola R, Lu X, Gonzalez‐Gomez M, Meyer G. 2011. Dynamic expression of the p53 family members p63 and p73 in the mouse and human telencephalon during development and in adulthood. Brain Res 1372:29–40. [DOI] [PubMed] [Google Scholar]
- Hirota Y, Meunier A, Huang S, Shimozawa T, Yamada O, Kida YS, Inoue M, et al. 2010. Planar polarity of multiciliated ependymal cells involves the anterior migration of basal bodies regulated by non‐muscle myosin II. Development 137:3037–3046. [DOI] [PubMed] [Google Scholar]
- Ihrie RA, Alvarez‐Buylla A. 2011. Lake‐front property: a unique germinal niche by the lateral ventricles of the adult brain. Neuron 70:674–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez AJ, Domínguez‐Pinos MD, Guerra MM, Fernández‐Llebrez P, Pérez‐Fígares JM. 2014. Structure and function of the ependymal barrier and diseases associated with ependyma disruption. Tissue Barriers 2:e28426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Killick R, Niklison‐Chirou M, Tomasini R, Bano D, Rufini A, Grespi F, Velletri T, et al. 2011. P73: A multifunctional protein in neurobiology. Mol Neurobiol 43:139–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishimoto N, Sawamoto K. 2012. Planar polarity of ependymal cilia. Differentiation 83:S86–S90. [DOI] [PubMed] [Google Scholar]
- Kokovay E, Wang Y, Kusek G, Wurster R, Lederman P, Lowry N, Shen Q, et al. 2012. VCAM1 is essential to maintain the structure of the SVZ niche and acts as an environmental sensor to regulate SVZ lineage progression. Cell Stem Cell 11:220–230. [DOI] [PubMed] [Google Scholar]
- Kuo CT, Mirzadeh Z, Soriano‐Navarro M, Rasin M, Wang D, Shen J, Sestan N, et al. 2006. Postnatal deletion of Numb/Numblike reveals repair and remodeling capacity in the subventricular neurogenic niche. Cell 127:1253–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavado A, Oliver G. 2011. Six3 is required for ependymal cell maturation. Development 138:5291–5300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechtreck KF, Delmotte P, Robinson ML, Sanderson MJ, Witman GB. 2008. Mutations in hydin impair ciliary motility in mice. J Cell Biol 180:633–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AF, Ho DK, Zanassi P, Walsh GS, Kaplan DR, Miller FD. 2004. Evidence that DeltaNp73 promotes neuronal survival by p53‐dependent and p53‐independent mechanisms. J Neurosci 24:9174–9184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim DA, Tramontin AD, Trevejo JM, Herrera DG, Garcia‐Verdugo JM, Alvarez‐Buylla A. 2000. Noggin antagonizes BMP signaling to create a niche for adult neurogenesis. Neuron 28:713–726. [DOI] [PubMed] [Google Scholar]
- Lledo PM, Merkle FT, Alvarez‐Buylla A. 2008. Origin and function of olfactory bulb interneuron diversity. Trends Neurosci 31:392–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Shook BA, Daniels SB, Conover JC. 2008. Subventricular zone‐mediated ependyma repair in the adult mammalian brain. J Neurosci 28:3804–3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina‐Bolivar C, Gonzalez‐Arnay E, Talos F, Gonzalez‐Gomez M, Moll UM, Meyer G. 2014. Cortical hypoplasia and ventriculomegaly of p73‐deficient mice: Developmental and adult analysis. J Comp Neurol 522:2663–2679. [DOI] [PubMed] [Google Scholar]
- Merkle FT, Tramontin AD, Garcia‐Verdugo JM, Alvarez‐Buylla A. 2004. Radial glia give rise to adult neural stem cells in the subventricular zone. Proc Natl Acad Sci USA 101:17528–17532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller FD, Gauthier‐Fisher A. 2009. Home at last: Neural stem cell niches defined. Cell Stem Cell 4:507–510. [DOI] [PubMed] [Google Scholar]
- Mirzadeh Z, Han YG, Soriano‐Navarro M, Garcia‐Verdugo JM, Alvarez‐Buylla A. 2010. Cilia organize ependymal planar polarity. J Neurosci 30:2600–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzadeh Z, Merkle FT, Soriano‐Navarro M, Garcia‐Verdugo JM, Alvarez‐Buylla A. 2008. Neural stem cells confer unique pinwheel architecture to the ventricular surface in neurogenic regions of the adult brain. Cell Stem Cell 3:265–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohata S, Nakatani J, Herranz‐Perez V, Cheng J, Belinson H, Inubushi T, Snider WD, et al. 2014. Loss of dishevelleds disrupts planar polarity in ependymal motile cilia and results in hydrocephalus. Neuron 83:558–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paez‐Gonzalez P, Abdi K, Luciano D, Liu Y, Soriano‐Navarro M, Rawlins E, Bennett V, Garcia‐Verdugo JM, Kuo CT. 2011. Ank3‐dependent SVZ niche assembly is required for the continued production of new neurons. Neuron 71:61–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastrana E, Cheng LC, Doetsch F. 2009. Simultaneous prospective purification of adult subventricular zone neural stem cells and their progeny. Proc Natl Acad Sci U S A 106:6387–6392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porlan E, Marti‐Prado B, Morante‐Redolat JM, Consiglio A, Delgado AC, Kypta R, Lopez‐Otin C, et al. 2014. MT5‐MMP regulates adult neural stem cell functional quiescence through the cleavage of N‐cadherin. Nat Cell Biol 16:629–638. [DOI] [PubMed] [Google Scholar]
- Pozniak CD, Barnabe‐Heider F, Rymar VV, Lee AF, Sadikot AF, Miller FD. 2002. p73 is required for survival and maintenance of CNS neurons. J Neurosci 22:9800–9809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez‐Castillejo C, Sanchez‐Sanchez F, Andreu‐Agullo C, Ferron SR, Aroca‐Aguilar JD, Sanchez P, Mira H, et al. 2006. Pigment epithelium‐derived factor is a niche signal for neural stem cell renewal. Nat Neurosci 9:331–339. [DOI] [PubMed] [Google Scholar]
- Raponi E, Agenes F, Delphin C, Assard N, Baudier J, Legraverend C, Deloulme JC. 2007. S100B expression defines a state in which GFAP‐expressing cells lose their neural stem cell potential and acquire a more mature developmental stage. Glia 55:165–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roales‐Buján R, Páez P, Guerra M, Rodríguez S, Vío K, Ho‐Plagaro A, García‐Bonilla M, et al. 2012. Astrocytes acquire morphological and functional characteristics of ependymal cells following disruption of ependyma in hydrocephalus. Acta Neuropathol 124:531–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson SR, Noone DF, O'Dowd BS. 1996. Ependymocytes and supra‐ependymal axons in rat brain contain glutamate. Glia 17:345–348. [DOI] [PubMed] [Google Scholar]
- Spassky N, Merkle FT, Flames N, Tramontin AD, Garcia‐Verdugo JM, Alvarez‐Buylla A. 2005. Adult ependymal cells are postmitotic and are derived from radial glial cells during embryogenesis. J Neurosci 25:10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talos F, Abraham A, Vaseva AV, Holembowski L, Tsirka SE, Scheel A, Bode D, et al. 2010. p73 is an essential regulator of neural stem cell maintenance in embryonal and adult CNS neurogenesis. Cell Death Differ 17:1816–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tissir F, Qu Y, Montcouquiol M, Zhou L, Komatsu K, Shi D, Fujimori T, et al. 2010. Lack of cadherins Celsr2 and Celsr3 impairs epen6dymal ciliogenesis, leading to fatal hydrocephalus. Nat Neurosci 13:700–707. [DOI] [PubMed] [Google Scholar]
- Tong CK, Chen J, Cebrian‐Silla A, Mirzadeh Z, Obernier K, Guinto CD, Tecott LH, et al. 2014. Axonal control of the adult neural stem cell niche. Cell Stem Cell 14:500–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang A, Walker N, Bronson R, Kaghad M, Oosterwegel M, Bonnin J, Vagner C, et al. 2000. P73‐deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature 404:99–103. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information