

INTRODUCTION
Rare genetically defined neurodevelopmental disorders (GND) with increased risk of autism have recently become an entry point for autism-related drug discovery. Through exploration of downstream effects of the pathological mutations, specific mechanistic pathways have been identified as dysregulated. The identification of shared mechanisms across forms of autism opens the door for the development of novel “mechanism-based therapeutics”. However, confidence in the therapeutic mechanism does not diminish the need for well-designed clinical trials.
Progress has been made in the last ten years towards the development of pharmacotherapeutics for autism spectrum disorders (ASD) through a focus on rare genetically defined neurodevelopmental disorders (GND) with increased risk of autism. The disorders of greatest focus have been Tuberous Sclerosis Complex (TSC), Rett Syndrome (RTT) and Fragile X Syndrome (FXS), all of which have a defined genetic etiology and 30–60% co-morbidity with ASD1. Through an exploration of downstream effects of pathological mutations, specific mechanistic pathways have been identified as dysregulated. Strikingly, there is some convergence on disrupted pathways across these and many other GNDs associated with ASD and genetic risk variants in idiopathic ASD. The identification of shared mechanisms across forms of autism associated with distinct genetic causes opens the door for the development of novel treatment strategies. ASD, once a seemingly intractable target due to heterogeneity, now has a viable entry point for drug discovery through associated GNDs. The development of therapies targeting pathological mechanisms identified in these disorders has been termed “mechanism-based therapeutics”2. Mechanism-based therapies differ from previously explored treatment approaches in idiopathic ASD that were typically based on measurable endophenotypes, like hyperserotonemia or oxytocin insufficiency. The efficacy of mechanism-based therapies has been explored in several early stage clinical trials but the results have been tempered. Here we outline current mechanism-based therapies being pursued for genetically defined autism-related disorders, their clinical success and propose recommendations for enhancing efficacy in future trials.
Analysis of mutations associated with ASD has yielded convergence on several distinct mechanistic pathways, with disruption of transcriptional regulation and protein synthesis centrally implicated in several GNDs2. TSC is the result of loss-of-function mutation of TSC1/TSC2, which negatively regulate the protein kinase mTOR (mechanistic Target of Rapamycin), a pathway central to the regulation of protein synthesis3. RTT is caused by mutation of MeCP2 encoding a DNA binding protein, which amongst many functions, regulates factors involved in transcriptional repression4. FXS, due to silencing of FMR1, reduces expression of fragile X mental retardation protein (FMRP), a protein that affects multiple aspects of RNA metabolism and translation5. Disruption of these processes within the brain results in synaptic impairment and alterations in neuronal connectivity that are thought to underlie neurocognitive manifestations, including autism, intellectual disability and mood disorders, often comorbid with these disorders. The identification of common mechanisms, preclinically linked to behavioral phenotypes, empowers directed target identification for treatment of this symptomatic domain.
Fragile X Syndrome
Loss of FMRP is hypothesized to cause the behavioral symptoms of FXS through enhancement of mGluR1/5-dependent protein synthesis. Inhibition of mGluR5, either through genetic or pharmacological intervention, rescues both cellular and behavioral alterations in animal models of Fmr1 mutation, and consequently mGluR5 inhibitors have been developed for clinical testing5. Several clinical trials have tested efficacy of two different mGluR5 antagonists, mavoglurant and basimglurant, at ameliorating the symptoms associated with FXS. An early-phase trial demonstrated efficacy measured using Aberrant Behavior Checklist (ABC) in a post-hoc analysis of patients with complete silencing of the FMR1 gene (NCT00718341). However, three larger Phase IIb/III randomized clinical trials failed to demonstrate neurocognitive effects in any of the primary outcome measures (ABC or Anxiety, Depression and Mood Scale; ADAMS) with either compound (NCT01253629, NCT01357239, NCT01517698). Currently, though, a trial is underway testing the effect of basimglurant in young children with FXS based on the hypothesis that previous trials failed to demonstrate efficacy due to inclusion of a patient population outside the critical window for influencing cognitive development (NCT02920892).
Tuberous Sclerosis Complex
Dysregulation of the mTOR pathway, such as occurs in TSC, is associated with a common phenotype of intellectual disability, epilepsy and ASD. As there are several FDA approved inhibitors of mTOR, clinical testing of mechanism-based treatment strategies for “mTORopathy” related GNDs has been broadly undertaken3. mTOR inhibitors are approved for the treatment of several somatic manifestations in TSC and have recently been shown an effective treatment of seizures in TSC (NCT01070316). Evidence from preclinical research indicates that mTOR inhibitors are also able to rescue cognitive deficits and autistic-like features, suggesting they may also have efficacy for neurocognitive deficits associated with TSC3. An early-phase open label trial testing the efficacy of the mTOR inhibitor, sirolimus, for the treatment of renal angiomyolipomas in TSC assessed cognitive endpoints as a secondary outcome and found pro-cognitive effects (NCT00490789). On this basis, a randomized clinical trial was initiated to look at the cognitive effects of the mTOR inhibitor, everolimus, in TSC. However, the trial failed to show a positive effect in any of the neurocognitive assessment measures (NCT01289912).
Rett Syndrome
Mutation of the MeCP2 gene reduces the expression of growth factors, BDNF (brain derived neurotropic factor) and IGF1 (insulin-like growth factor), and negatively regulates the mTOR pathway. Consequently, of the numerous therapeutic strategies explored, some of the mechanism-based approaches have focused on upregulation of these growth factors. IGF1 treatment in preclinical models rescues a broad range of RTT-like phenotypes from heartrate and respiration to social and anxiety behaviors4. An open label investigation of IGF1 (mecasermin) revealed improvements in autonomic and cognitive measures, but a similarly designed study failed to show an effect on clinical impressions (Clinical Global Impressions; CGI) and reported worsened seizures (NCT01253317;6). A Phase II double-blind placebo-controlled study of the active tripeptide of IGF1 (trofinetide) reported improvement of clinical impressions and cognitive assessments (Vineland Adaptive Behavior Scales; VABS; NCT02715115). However, a recent randomized, placebo-controlled clinical trial of IGF1 failed to detect improvement of mood (ADAMS), clinical rating (CGI), or biomarkers of cardio-respiratory and EEG activity (NCT01777542). Two open-label trials testing the effects of glatiramer acetate, hypothesized to increase BDNF levels, demonstrated improved respiratory, motor function and memory but have severe adverse events associated with administration (NCT02153723, NCT02023424).
The Way Forward
Most mechanism-based therapies had promising results in early trials; however, none of the treatment strategies have gained FDA approval for neurocognitive endpoints nor have become commonly prescribed off-label treatment strategies3,5. The failure of clinical trials for neurocognitive endpoints in GNDs does not necessarily reflect a failure in mechanistic understanding, but rather highlights the parameters to be optimized beyond target identification for this symptomatic domain. The ability of many of these treatments to rescue non-cognitive domains supports proof-of-mechanism for the target. The strength of the preclinical evidence demonstrating the rescue of both physiological and behavioral phenotypes using mechanism-based treatment strategies supports the tractability of neurocognitive symptoms.
To enhance the likelihood of future trial success, there are several factors that must considered. First, placebo effect size, around 0.5 from pre to post treatment in GNDs, strongly influences the signal-to-noise ratio, and limits the value of open label trials7. Second, it is unclear which are the appropriate endpoints to measure cognitive and psychiatric features in GNDs, which are both sensitive enough to capture early indicators of efficacy but also robust enough to have strong test-retest reliability. The scales that have been used in the early trials have failed to yield reproducible results in later trials, which may be due to separation of these indices from the etiological mechanisms against which the therapies were designed. Objective measures, endophenotypes or biomarkers that reflect both symptomatic features and the underlying biological underpinnings may be more likely to reveal consistent results and earlier readouts of efficacy. For example, white matter fractional anisotropy in TSC both reflects ASD diagnosis and responds to everolimus treatment (Figure 1) and could be used as a measure of target engagement or proof of mechanism8,9. The incorporation of biomarkers as secondary endpoints for the aforementioned purposes will help validate (or refute) the strength of their association with measures of efficacy. Third, there is considerable phenotypic heterogeneity in GNDs and a developmental progression of phenotypic presentation that contributes to variability within and between subjects. The developmental progression of behavioral impairments suggests critical windows in which intervention will have the maximal effect. Finally, the dosage and duration of treatment necessary to induce disease modifying effects is poorly defined. The degree of target engagement within the brain necessary to evoke cognitive effects is unclear and dose-finding trials are seldom undertaken.
Figure 1:
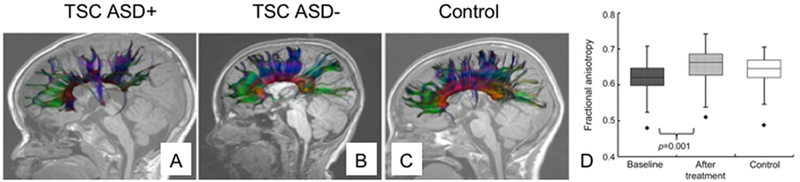
MRI based biomarker of ASD diagnosis in children with TSC.
Representative illustrations of the projections of the corpus callosum in a child with TSC and ASD (A. TSC ASD+), only TSC (B. TSC ASD-) and a typically developing child (C. Control) demonstrating a sparsity of connections in children with TSC and ASD relative to the other groups9. Treatment with everolimus increases fractional anisotropy, a marker of white matter connectivity, in patients with TSC(D)8.
Figure 1, panels a, b, c are adapted with permission from Peters, J. M. et al. Loss of white matter microstructural integrity is associated with adverse neurological outcome in tuberous sclerosis complex. Acad Radiol 19, 17–25, doi:10.1016/j.acra.2011.08.016 (2012).
Figure 1, panel d is adapted with permission from Tillema, J. M., Leach, J. L., Krueger, D. A. & Franz, D. N. Everolimus alters white matter diffusion in tuberous sclerosis complex. Neurology 78, 526–531, doi:10.1212/WNL.0b013e318247ca8d (2012).
There are several strategies that can be undertaken to improve the next generation of clinical trials for mechanism-based therapies. First, clinical trials in GNDs should be designed with sufficient power for the selected primary endpoints and secondary biomarkers (likely requiring a multi-site design) and randomized after the homogenetic selection with accommodations made to offset the high degree of placebo effect4. Second, patient stratification, for example by methylation state in FXS or ASD co-diagnosis, should be incorporated to reduce variability in the population to maximize the window to detect efficacy. Third, preclinical evidence suggests different neurocognitive domains have different critical windows of plasticity, thus the study population for efficacy trials should be of an age that is within the vulnerable window for the primary endpoint. Natural history studies, currently being conducted for the disorders discussed here, provide perspective on the course of the disease and variability in potential endpoints. Many of these studies include genetic assessment, cognitive behavioral measures, early assessments for associated neuropsychiatric disorders and neural biomarkers including EEG and MRI. Characterization of variability in these measures, association of specific phenotypes with specific biomarkers and change in measures over early development will provide a framework against which the most appropriate endpoints for clinical trials can be chosen. Biomarkers, identified and validated prior to the trial, should also be collected alongside primary endpoints to validate mechanistic intent of the treatment. Finally, careful attention must be paid to the fundamentals of clinical trial design, with particular attention to the neuropharmacokinetics and dynamics and the inclusion of markers of target engagement or early efficacy as necessary components of all Phase II/III studies.
Mechanism-based therapies coupled with mechanistic readouts of efficacy and careful trial design should increase the ability to reach definitive conclusions from clinical trials in GNDs. Trials that are undertaken only after “proof of mechanism” have achieved a higher level of successful advancement relative to those for which it has not been demonstrated10. Caution in early trial design, to minimize ambiguous results, will prevent the concept of mechanism-based therapies from being the baby thrown out with the bathwater and encourage continued investment. Only with these improvements will we be able to realize the potential of mechanism-based therapeutics for both GNDs and the idiopathic populations of neurocognitive impairments for which they serve as a model.
Acknowledgements
We would like to thank Denise McGinnis her invaluable comments on the manuscript. The Sahin lab has received grant funding from the US National Institutes of Health (NIH) (U01-NS082320, U01-NS092595, U54-HD090255 and U54-NS092090), US Department of Defense W81XWH-15–1–0189, Nancy Lurie Marks Family Foundation, Autism Speaks, TS Alliance, Roche, Novartis, Pfizer and LAM Therapeutics.
Footnotes
CONFLICT OF INTEREST
MEM has no conflicts to declare. MS has served on the Scientific Advisory Board of Sage Therapeutics and PTEN Research Foundation and PTEN Hamartoma Tumor Syndrome Foundation. He also serves on the professional advisory board of the Tuberous Sclerosis Alliance. He has received research funding from Roche, Novartis, Pfizer, and LAM Therapeutics.
Bibliography
- 1.Richards C, Jones C, Groves L, Moss J & Oliver C Prevalence of autism spectrum disorder phenomenology in genetic disorders: a systematic review and meta-analysis. Lancet Psychiatry 2, 909–916, doi: 10.1016/S2215-0366(15)00376-4 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Sahin M & Sur M Genes, circuits, and precision therapies for autism and related neurodevelopmental disorders. Science 350, doi: 10.1126/science.aab3897 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davis PE, Peters JM, Krueger DA & Sahin M Tuberous Sclerosis: A New Frontier in Targeted Treatment of Autism. Neurotherapeutics 12, 572–583, doi: 10.1007/s13311-015-0359-5 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Katz DM et al. Rett Syndrome: Crossing the Threshold to Clinical Translation. Trends Neurosci 39, 100–113, doi: 10.1016/j.tins.2015.12.008 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berry-Kravis EM et al. Drug development for neurodevelopmental disorders: lessons learned from fragile X syndrome. Nat Rev Drug Discov 17, 280–299, doi: 10.1038/nrd.2017.221 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaufmann WE, Stallworth JL, Everman DB & Skinner SA Neurobiologically-based treatments in Rett syndrome: opportunities and challenges. Expert Opin Orphan Drugs 4, 1043–1055, doi: 10.1080/21678707.2016.1229181 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Curie A et al. Placebo Responses in Genetically Determined Intellectual Disability: A Meta-Analysis. PLoS One 10, e0133316, doi: 10.1371/journal.pone.0133316 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tillema JM, Leach JL, Krueger DA & Franz DN Everolimus alters white matter diffusion in tuberous sclerosis complex. Neurology 78, 526–531, doi: 10.1212/WNL.0b013e318247ca8d (2012). [DOI] [PubMed] [Google Scholar]
- 9.Peters JM et al. Loss of white matter microstructural integrity is associated with adverse neurological outcome in tuberous sclerosis complex. Acad Radiol 19, 17–25, doi: 10.1016/j.acra.2011.08.016 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morgan P et al. Impact of a five-dimensional framework on R&D productivity at AstraZeneca. Nat Rev Drug Discov 17, 167–181, doi: 10.1038/nrd.2017.244 (2018). [DOI] [PubMed] [Google Scholar]