

Abstract
Mucopolysaccharidosis IVA (MPS IVA, Morquio A syndrome) is an autosomal recessive disorder caused by the deficiency of N-acetylgalactosamine-6-sulfate sulfatase. Deficiency of this enzyme leads to the accumulation of specific glycosaminoglycans (GAGs), chondroitin-6-sulfate (C6S) and keratan sulfate (KS), which are mainly synthesized in the cartilage. Therefore, the substrates are stored primarily in the cartilage and its extracellular matrix (ECM), leading to a direct impact on bone development and successive systemic skeletal spondylepiphyseal dysplasia. The skeletal-related symptoms for MPS IVA include short stature with short neck and trunk, odontoid hypoplasia, spinal cord compression, tracheal obstruction, obstructive airway, pectus carinatum, restrictive lung, kyphoscoliosis, platyspondyly, coxa valga, genu valgum, waddling gait, and laxity of joints. The degree of imbalance of growth in bone and other organs and tissues largely contributes to unique skeletal dysplasia and clinical severity. Diagnosis of MPS IVA needs clinical, radiographic, and laboratory testing to make a complete conclusion. To diagnose MPS IVA, total urinary GAG analysis which has been used is problematic since the values overlap with those in age-matched controls. Currently, urinary and blood KS and C6S, the enzyme activity of GALNS, and GALNS molecular analysis are used for diagnosis and prognosis of clinical phenotype in MPS IVA. MPS IVA can be diagnosed with unique characters although this disorder relates closely to other disorders in some characteristics.
In this review article, we comprehensively describe clinical, radiographic, biochemical, and molecular diagnosis and clinical assessment tests for MPS IVA. We also compare MPS IVA to other closely related disorders to differentiate MPS IVA. Overall, imbalance of growth in MPS IVA patients underlies unique skeletal manifestations leading to a critical indicator for diagnosis.
Keywords: mucopolysaccharidosis IVA, N-acetylgalactosamine-6-sulfate, keratan sulfate, chondroitin-6-sulfate, spondyloepiphyseal dysplasia
1. Introduction: Historical Aspect for MPS IVA
Mucopolysaccharidosis IVA (MPS IVA) also known as Morquio A syndrome is an autosomal recessive disorder caused by the deficiency of N-acetylgalactosamine-6-sulfate sulfatase (GALNS). This enzyme deficiency leads to a progressive accumulation of excessive glycosaminoglycans (GAGs): chondroitin-6-sulfate (C6S) and keratan sulfate (KS).
These excessive GAGs are stored primarily in the cartilage and its extracellular matrix (ECM) because C6S and KS are mainly produced in the cartilage [1]. The storage of GAGs within the lysosomes disrupts cell function and metabolism in cartilage [2], resulting in a direct impact on cartilage and bone development, failure of ossification, imbalance of growth, and successive unique systemic skeletal dysplasia [1,3,14]. Excessive C6S and KS are secreted in blood and excreted in the urine of patients with MPS IVA. Staprans et al. have described that GAGs in plasma are not covalently linked to plasma proteins and are isolated as complexed with plasma proteins in noncovalent linkages [4].
In 1929, Dr. Luis Morquio, a pediatrician in Uruguay, first reported 4 Swedish siblings with unique skeletal dysplasia, later classified as Morquio A syndrome (MPS IVA). Those patients reported had disproportionate short-trunk, hypermobile joints, prominent forehead, abnormal face with a large mandible, short neck, pectus carinatum, kyphoscoliosis, flaring of the rib cage, genu valgum, and pes planus [5]. At that same year, Dr. Brailsford, a radiologist in the UK, also reported a patient with similar skeletal manifestations, except that aortic valve disease and corneal clouding were not described in the original publication [6]. In 1962, Pedrini et al. reported urinary excretion of KS in three patients with Morquio syndrome, showing that this metabolic disorder differs from other mucopolysaccharidoses [6]. In 1965, McKusick et al. classified Morquio as MPS IVA [7]. Dr. Orii et al. reported patients with milder forms of MPS IVA [8,9].
These reports have demonstrated that patients with MPS IVA have a wide range of clinical heterogeneity and that the diagnosis of MPS IVA, especially with the attenuated form, can be difficult and delayed. Clinical phenotype and consequent prognosis are primarily determined by the extent of imbalance of growth between bones (flat bones - skull, hip, sternum, ribs; spine; short and long bones; sesamoid bones), trachea, vessels, and organs, leading to serious morbidity and mortality with spinal cord compression and restrictive lung and obstructive airway [9].
2. Diagnosis
2.1. Clinical Diagnosis
Diagnosis of MPS IVA begins with the development of unique clinical manifestations, which can occur from either clinical findings and/or radiographic findings [10]. Some symptoms that can be noticed at birth include prominent forehead, pectus carinatum, kyphosis (gibbus), and abnormal findings in the lumbar vertebral bodies in X-rays [12–15]. Gibbus, in particular, is often the first sign noticed in MPS IVA [11]. Diagnosis of MPS IVA patients is often not made until a few years of age since the majority of patients seem normal at birth, and normal levels of total urine GAG are observed in some cases [12].
GAG accumulation along the upper airway induces enlarged tongue, adenoidal, tonsillar, and vocal cord hypertrophy, and large mandible. GAG accumulation in the lower airway and cervicothoracic part results in an imbalance of growth between spine, rib, and manubrium vs. trachea and vessels in thoracic inlet causing consequent bulging and twisted narrowing trachea [13]. Respiratory involvement is seen with recurrent respiratory infections, upper and lower airway obstruction, tracheomalacia, restrictive lung, shortness of breath, loud snoring, look up to the sky position (head and neck maintained in extension), and sleep apnea [14]. Unlike other types of MPS, hypermobile joints (fingers, wrists, neck, and knees) are commonly observed; however, decreased joint mobility can be observed in the large joints including shoulder, elbows, and hips [15].
There is a broad phenotypic spectrum in MPS IVA ranging from a rapid progressive early-onset form (severe) to a slowly progressive late-onset form (attenuated) [16].In patients with the severe phenotype of MPS IVA, spinal cord compression, airway compromise, and later valvular heart disease are the leading causes of high morbidity and mortality, contributing to the shortened lifespan of individuals to the first few decades of life if untreated [13,17]. The severe form is characterized by hypermobile wrist joints, kyphosis, scoliosis, pectus carinatum, deformity of vertebral bodies, disproportionate stature with short trunk, knock knee (genu valgum), and waddling gait [16]. Due to the progressive and often life-threatening nature of the disease [16], early and accurate diagnosis is critical for optimal patient management [10], which provides a better quality of life and prolonged lifespan [17]. Respiratory failure has been shown as the primary cause of death in patients (63%), followed by cardiac failure (11%), post-traumatic organ failure (11%), complications of surgery (11%), and myocardial infarction (4%) [17].
Patients with the attenuated form of the disease can survive for over 70 years if well-managed [18] and might have late-disease onset during late childhood or adolescence, and often first manifests hip problems (pain, stiffness) as the primary symptom [9] (Fig. 1). Clinical features are limited to minor skeletal abnormalities, hip pain, and moderate short stature associated with the early-onset osteoporotic phenotype [19].
Figure 1.
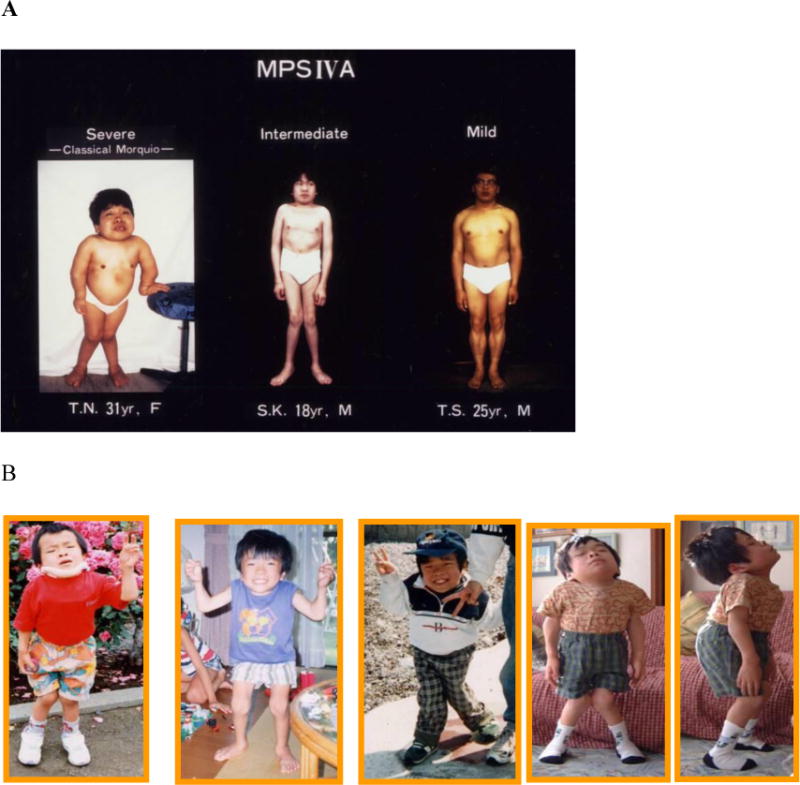
Patients with mild to severe form of MPS IVA. A. Images of patients with distinct differences in clinical features from mild form, immediate form, and severe form of MPS IVA (adapted from Educational CD for Morquio and permitted by Carol Ann Foundation). B. Images of a patient with severe form of MPS IVA. From left to right: patient age is 7, 8, 11, 16, and 16 years (adapted from Educational CD for Morquio and permitted by Carol Ann Foundation) [33].
According to the International Morquio A Registry, the most common initial signs are short stature (49.9%), genu valgum (45.1%), kyphosis (44.4%), pectus carinatum (43.6%), and abnormal gait (37.8%). The most common current symptoms and the percentage of the present signs and symptoms are short stature (84.7%), genu valgum (78.7%), pectus carinatum (71.4%), kyphosis (70.4%), abnormal gait (64.4%), and laxity of wrist joints (63.2%) [20] (Fig. 2).
Figure 2.
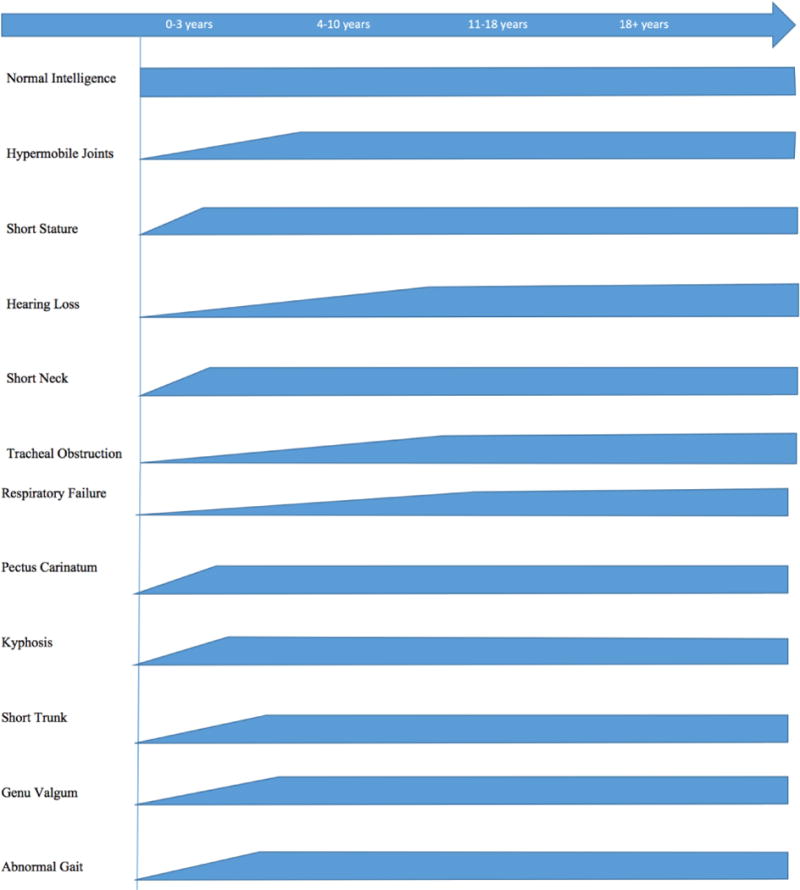
Time course of clinical symptoms in MPS IVA
As the disease progresses regardless of the phenotype of the disease, more signs and symptoms begin to appear. With increasing age, joint laxity and skeletal deformities become more evident, and patients typically require support by orthopedic interventions [18]. The combination of bone and joint impairment leads to pain and arthritis causing the subsequent inability of walking, restriction of range of motion, and consequently decreased activity of daily living [9].
2.2. Biochemical Diagnosis
2.2.1. Urinary and Blood GAG
Urine-based testing for total GAG is considered only a screen and provides substantial false negatives; it is recommended that KS measurement, as well as enzymatic testing, are used to confirm the diagnosis of MPS IVA [20]. Measurement of urinary GAGs is performed both quantitatively and qualitatively [20]. While a gross elevation of urinary GAGs is normally specific for patients with other types of MPS, borderline or slightly evaluated values are common in those with MPS IVA [21–24].
False negative results are found for almost 15% of all MPS patients [23] and even more for MPS IV patients [21–23]. The urinary excretion of GAGs is high in infants and young children, decreases with age, plateaus by the second decade of life, and remains constant through adulthood [22,23,25–27]. The qualitative analysis of total urinary GAGs is usually performed by spectrophotometric analysis using dimethylmethylene blue (DMMB) [28]. For qualitative urine-based testing methods, the GAGs are first isolated from the urine and then separated by thin layer chromatography or electrophoresis [29–32].
2.2.2. Keratan Sulfate (KS)
KS is mostly synthesized and accumulated in the cartilage and cornea in MPS IVA patients, and excessive accumulation of KS in lysosome leads to disruption of chondrocytes and subsequent increase of KS levels in blood and urine [33]. We had two autopsied cases. The first autopsied case died of respiratory complication with tracheal obstruction after cervical fusion at 20 years old [35]. The second case died of respiratory failure with tracheal obstruction as well. In both casess, the chondrocytes in various bones had accumulation of storage materials [34]. We have analyzed over 20 cases of surgical remnants with various bones. All chondrocytes were vacuolated and were severely affected [13,34–36]. Primary synthesis of KS in chondrocytes and successive storage of KS in chondrocytes because of the deficiency of GALNS enzyme proves the cartilage as the main source of KS in blood circulation.
The detection of KS in biological specimens with tandem mass spectrometry (MS/MS) has been developed first by Oguma et al. [37] and applied to specimens in MPS IVA patients and distinguished between control subjects and MPS IVA patients without the false negatives [38–40]. Blood and urine KS levels are higher in MPS IVA patients, compared with age-matched controls [12, 37–39]. Urine KS levels remain higher or subnormal in MPS IVA patients than those in controls after 20 years of age; however, blood KS levels tend to be normalized by the age of 20 years [38–40].
Using sandwich ELISA [21,41] and LC/MS/MS [37,39] assay, the study was conducted for KS levels in blood and urine from MPS IVA patients and healthy controls to evaluate the comparability of results. Blood, urine, and other biological samples were purified by filtration and GAGs are digested with chrondroitnase ABC and keratanase II to yield disaccharides of C6S and KS, respectively. After digestion, the samples were injected into the LC-MS/MS [38,40,42–45].
The levels of blood and urine KS correlate with clinical severity during the early and progressive stage of the disease, and therefore, it is a good prognostic biomarker at this stage [12]. Blood KS directly displays growth, turnover disruption, and/or repair of cartilage where it is mainly synthesized [12]. The advantage of measurement of KS in dried blood spot (DBS) testing is its convenience for transport of samples and screening purposes [12]. In contrast, urine KS has a broader range of value and may not reflect cartilage condition directly. KS is synthesized mainly in cartilage. Depending upon the extent of destruction of cartilage in MPS (not only IVA but other types), KS level was increased in blood [12,21,46]. The levels of blood and urine KS also correlated with clinical severity during the early and progressive stage of the disease [21] and therefore, it is a good prognostic biomarker at this stage. Urine KS comes via kidney, and urine KS is filtered in kidney. Therefore, only selected smaller molecules are excreted in urine [12,47]. When MPS IVA patients are treated with ERT, it is likely that urine KS levels are reduced rapidly since the enzyme is delivered to kidney and digests the KS stored in the kidney [48]. Thus, urinary KS is useful to differentiate MPS IVA from other forms of MPS and to demonstrate pharmacodynamic effects of therapy while it does not provide a valuable predictor (biomarker) of skeletal or clinical improvement during these therapies for MPS IVA [49]. It is noteworthy that the origin and character of urinary GAGs are different from those of blood GAGs as seen is KS [50,51].
The ratio of di-sulfated KS to total KS in urine is much higher than that in blood [52]. There is a negative correlation between blood and urine for levels of mono- and di-sulfated KS in ERT-treated MPS IVA patients, suggesting reduction of urine KS level does not reflect blood KS level [52].
GALNS plays a role as galactose-6-sulfatase (G6S) because the enzyme hydrolyzes the sulfated galactose of KS and converts di-sulfated KS to mono-sulfated KS [49,52]. Therefore, deficiency of GALNS activity leads to the accumulation of more di-sulfated KS, and therefore, the ratio of di-sulfated KS to total KS of patients with MPS IVA increases, compared with normal controls especially at an early stage [49]. Levels of both mono-sulfated and di-sulfated KS in blood were measured by LC/MS/MS, and patients with MPS IVA had higher KS with both forms, compared with age-matched controls [53]. The elevation of di-sulfated KS in MPS IVA patients was more significant than that of mono-sulfated KS [49]. The proportion of di-sulfated KS vs. total KS in blood rose with age in control subjects while it was age-independent in patients with MPS IVA. The proportion of di-sulfated KS is better at distinguishing younger MPS IVA patients than older patients from age-matched controls. Levels of mono- and di-sulfated KS in the urine of MPS IVA patients were also higher when compared to those in age-matched controls for all studied ages. A significant difference in sulfation levels of KS between control subjects and patients with MPS IVA indicates that di-sulfated KS is another potential biomarker for MPS IVA [49]. Overall, determination of urine KS concentration provides a potential biomarker to screen for a high risk of MPS IVA patients and to measure pharmacodynamics effects [54]. This is important for assessing the clinical status at the initial progressive stage. Blood KS could be used for 1st tier newborn screening followed by the enzyme essay or vice versa [12] and is a potential biomarker to provide the clinical severity and therapeutic effect for the bone at the initial progressive stage [47].
2.2.3. Chondroitin-6-sulfate (C6S)
The GALNS enzyme plays an important role in converting C6S to CS, by hydrolyzing the sulfate group in C6S. Measurement of C6S has been developed by LC/MS/MS [55]. It is important that the data of C6S and KS are reported together, as the information provided enhances discrimination of patients from controls compared to C6S or KS levels alone [3,55]. Thus, C6S could be another useful biomarker for MPS IVA [55]. Overall, blood and urine KS and its sulfation level should be measured for diagnosis, the prognosis of the phenotype, and assessment of pharmacokinetic efficacy. Blood KS and/or C6S could be a biomarker for therapeutic effect only at presymptomatic to the early progressive stage but not beyond the progressive stage.
2.2.4. Enzyme assay
In 1976, the enzyme deficiency in MPS IVA (GALNS deficiency) was identified [56]. GALNS acts on 2 substrates: N-acetylgalactosamine-6-sulfate (GalNAc-6S) [57] and galactose-6-sulfate (Gal-6S) [58,59].
GalNAc-6S is a component of C6S, and Gal-6S is a component of KS [57–59]. These substrates are both used currently to demonstrate deficient GALNS activity [10]. The GalNAc-6S based assay uses radio-labeled natural substrate [60], and the Gal-6S based assay uses a fluorogenic artificial substrate [61]. GALNS activity is determined based on the amount of radioactivity released from this substrate with a lack of GALNS activity, resulting in the low generation of the signal [61]. The fluorogenic assay uses the 4-methylumbelliferyl-β-D-galactopyranoside-6-sulfate (4MU-Gal6S) substrate [61]. GALNS present in the sample first removes the 6-sulfate, and then exogenous β-galactosidase removes the galactoside, freeing the 4-methylumbelliferone adduct, which will fluorescent under high pH [61]. The addition of exogenous β-galactosidase to the reaction mixture is critical because conditions in which β-galactosidase is deficient would result in significant GALNS activity underestimation and possibly misdiagnoses [61].
To facilitate prenatal diagnosis, prenatal samples, such as dissected chorionic villi, cultured chorionic villus cells, and amniocytes can also be used [62,63]. Fibroblasts and leukocytes are recommended for diagnosis of the deficiency in GALNS activity [64]. Protocols for evaluating GALNS activity in DBS samples have recently been proposed as screening methods [65,66]. Leukocytes that are isolated from whole blood are better for more rapid analysis as cell culture is not required [67].
Fibroblast samples are recommended for enzyme activity analysis due to the impact of environmental and logistical factors during shipment can be minimized and corrected through culturing of the cells [10]. The measurement of GALNS activity in DBS is useful for screening, but it is not as robust as it is in fibroblasts or leukocytes due to the low number of cells present in the sample [10]. More data is needed to evaluate GALNS stability in DBS because DBS samples are more likely to be unprotected to environmental extremes during shipping in comparison to leukocytes [10]. The activity of a reference enzyme with similar stability in the same sample should be measured to confirm that the low GALNS activity is not the cause of sample degradation [10]. If the DBS sample is used, measuring a reference enzyme in the same sample to confirm integrity is recommended, but it may not be enough to rule out an effect of handling on GALNS activity due to the stability of GALNS as compared to other enzymes in a DBS which remains unknown [10].
The MS/MS-based method using a novel substrate has recently been developed [68]. Incorporation of this new assay into a multiplexed lysosomal storage disease panel for use in newborn screening programs is being considered [69].
MPS IVA can also be diagnosed with DNA sequencing methods, where the coding regions of the GALNS gene and small segments of immediately adjacent intronic regions are evaluated. Multiple MPS IVA mutations are believed to be found in only one individual or even one family [70]. DNA sequencing-based methods identify missense and nonsense mutations, small insertions and deletions in the coding regions; however, splicing alterations or changes in copy number can be missed [71,72]. A variety of methods are available including PCR, comparative genomic hybridization and multiplex ligation-dependent probe amplification [70].
No example of pseudodeficiency allele or complex allelic interaction is currently described for MPS IVA; however, Morrone et al. believes that the possibility warrants caution and the analysis of mutations in the context of complete and accurate patient genotypes [73].
2.3. Radiographic diagnosis
2.3.1. Skeletal Manifestations
When MPS IVA is suspected, radiographic imagining should be performed as a component of the diagnostic process [10]. A patient should obtain a skeletal survey to allow evaluation of the skull, complete spine (including flexion-extension lateral views), chest, hips, and limbs (particularly, hands, wrists, and knees) because of the wide variation and subtleties of radiographic findings in MPS IVA [10].
2.3.2. Skull
In 8 out of 14 individuals with MPS IVA, subtle abnormal brain MRI findings such as prominent perivascular space, enlarged lateral ventricles, and prominent frontal cerebrospinal fluid (CSF) were reported [74].
3.3.3. Spine
Children with MPS IVA have instability of the neck, odontoid hypoplasia, ligamentous laxity, incomplete ossification of the anterior and posterior rings of the atlas, and spinal cord compression [12,16,75,76] (Fig. 3). Spinal cord compression may occur in any spinal segment; however, cervical spinal compression is the most common site [9]. Spinal cord compression may be due to several factors such as cervical instability, ossified fibrocartilage associated with an abnormal odontoid process, ligamentous laxity, cartilaginous and ligamentous hypertrophy at the atlantoaxial joint, GAG deposition in the extradural space, disc protrusion, kyphoscoliosis, and acquired central canal stenosis [18,77–80].
Figure 3.
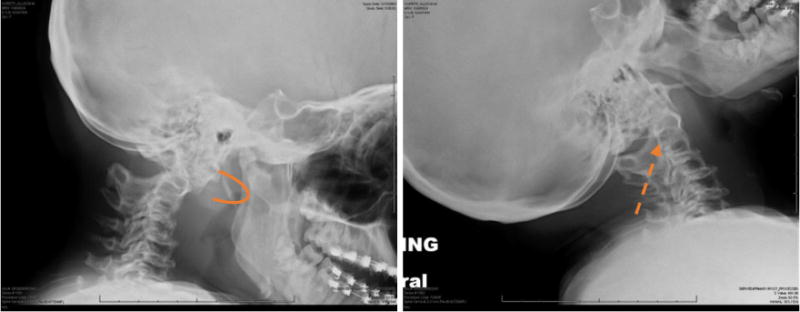
Lateral radiographs of the cervical spine in flexion (a) and extension (B) show atlantoaxial instablity in an 8-year-old female patient with MPS IVA. The anterior arch of C1 moves anteriorly in flexion (arrow) in relation to C2. There is hypoplasia of the dens (dashed arrow) and generalized platyspondyly.
If MPS IVA is suspected or diagnosed in a patient, frontal and lateral radiographs of the spine should be evaluated [80]. Spinal involvement in MPS IVA occurs at 2 distinct sites; cervical and thoracic [80]. Cervical spinal involvement, particularly instability and compression at the C1-C2 level, is a nearly universal finding and predisposes patients to myelopathy, paralysis, and sudden death [18]. Knowledge of the unique anatomy and pathology of the atlantoaxial (C1-C2) system facilitates radiological detection of upper cervical spine anomalies [80]. Spinal cord compression at the cervicothoracic or the thoracolumbar level due to kyphotic deformity can lead to paraplegia with gradual onset and all of its devastating consequences, although it is not common [81,82]. A lateral cervical spine x-ray of a 6-year-old female patient showed hypoplastic odontoid, platyspondyly, and anterior subluxation on C7 on T1 [9]. If cervical spine instability is suspected on plain film or with inconclusive radiographic findings, then flexion-extension cervical spine MRI can be used to evaluate cord compression [9]. Flexion-extension CT with sagittal reformation and soft-tissue filtration showed cone-shaped dens, a thickened cruciate ligament, and small thick cartilaginous posterior arch of C1. With flexion, there is a narrowing of the canal between the body of C2 and the cartilaginous posterior arch of C1.
2.3.4. Upper Extremities
Characteristic radiographic findings in hand and distal forearms of individuals with MPS IVA include a short ulna, ulnar deviation of the radial epiphysis, and delayed maturation of the carpal bones; the scaphoid, however, may not be radiographically present [20,83]. Metacarpals may be short and the proximal ends of the second through fifth metacarpals are typically rounded or pointed [20,83]. The upper extremities involvement is progressive and affects the wrist strength in some activities of daily living [9].
2.3.5. Lower Extremities
The lower extremity involvement is universal and progressive if untreated [84,85]. The most common lower extremity deformities are knee and ankle valgus. Genu valgum results from distal femoral and proximal tibial involvement and joint laxity [9]. Children with MPS IVA have unique waddling gait, a slower walking speed, reduced cadence, and reduced stride length [86]. The hips are either normal or slightly subluxated to start and may progressively dislocate over time [12]. The capital femoral epiphyses are smaller and progressively flattened, and many become fragmented over time [12]. The articular cartilage is abnormal and will degenerate promptly and build up early arthrosis, especially in the lower extremities [12].
2.3.6. Hips
Early in the disease course, the capital femoral epiphyses are small, and the acetabuli are shallow [9]. Affected individuals can become wheel-chair bound due to the following gradual and destructive changes in the femoral head and acetabuli resulting in hip dislocation, arthritis, and severe joint restriction [18] (Fig. 4). A hip x-ray of an 8-year-old female showed a bilateral irregular flattening of the capital femoral epiphyses and irregular dysplastic acetabuli with lateral joint subluxation [9].
Figure 4.
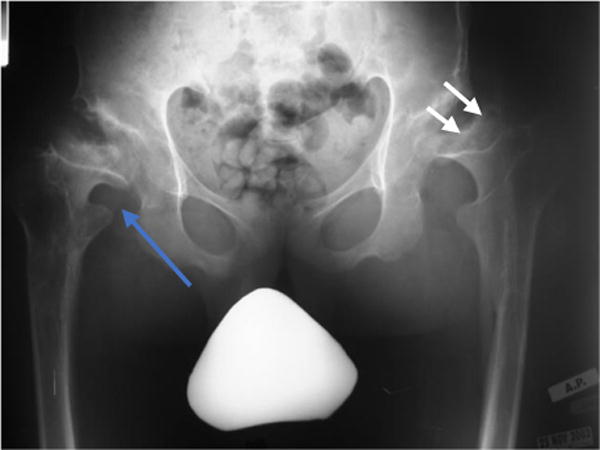
Erect frontal radiograph of the hips from a patient with MPS IVA. Shown here is progressive proximal and lateral migration (blue arrow) as well as the flattening and eventual disappearance of the proximal femoral epiphysis (white arrows).
2.3.7. Knee
The knee is the most commonly affected lower extremity joint in MPS IVA. Laxity of the collateral ligaments aggravates the deformity [87]. The proximal tibia contributes to more knee deformity than the distal femur, and there may also be procurvatum of the distal femur with recurvatum of the proximal tibia [87]. Arthrograms or magnetic resonance imaging (MRI) of the knees in MPS IVA patients shows that the proximal lateral portion of the tibia is unossified and the fibula is short; these results offer clarity of the bony and cartilaginous nature of the deformity [18]. Knee valgus causes the ground force to be shifted laterally, which forces the knee to go into further valgus [87] (Fig. 5). Correction of knee valgus using guided growth, such as the 8-plate hemiepiphysiodesis, is proven to be effective as the child is growing [84]. Multiplanar and more severe deformities may be corrected with osteotomies around the knee [88].
Figure 5.
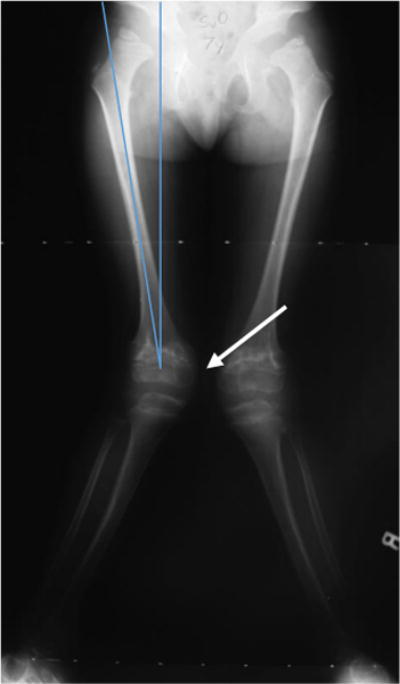
Erect frontal radiographs of the lower extremities of a patient with genu valgum.
2.3.8. Ankle
The ankles of MPS IVA patients are typically in valgus with wedging of the distal tibial epiphysis and shortening of the fibula [87]. The patients might have hindfoot valgus, some degree of equinus in the hindfoot, and adductus in the forefoot [87] (Fig. 6). Ankle valgus can be managed by orthotics but sometimes may require surgical correction such as guided growth or an osteotomy [84]. On lateral images, tarsal bones are irregular, and the talus appears to be plantarflexed [87]. Recurrence of the knee and/or ankle valgus is not uncommon in MPS IVA patients [89].
Figure 6.
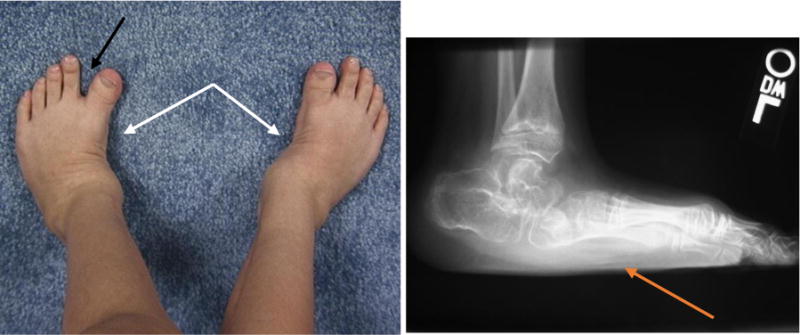
Feet of a patient with MPS IVA. Photograph of the feet (a) and lateral radiograph of the left foot (b) of this MPS IVA patient show skew foot posture in both feet (white arrows), pronated pes planus (orange arrow), and increased sandal gap between the first and second toes (black arrow).
Imaging of x-rays, computed tomography (CT), and MRI is important for the diagnosis of MPS IVA because of the evaluation of cervical spine instability, stenosis, and cord compression [80]. Radiographs are the mainstay for assessing and monitoring lower extremity bone involvement; two dimensional CT provides detailed information on the bone structure and also allows for precise determination of the bony dimensions [87,90]. However, progressive fragmentation and collapse of the osseous structures can lead to false assumptions regarding the anatomy around joints [87]. To get a more accurate picture of the non-ossified structures and the articular surfaces, arthrography or MRI will be needed [87]. The cartilaginous part of the epiphysis is often not as deformed as could be predicted by the plain radiographs [87]. CT does not offer the ability to assess the health of the spinal cord itself [80]. The detection of canal stenosis, cord compression, and myelomalacia is critical to check for the disease progression [80]. MRI is the most useful method for evaluating the neural axis in MPS IVA and is also currently considered the imaging method of choice for evaluating the spinal cord [80]. No other imaging method provides superior information about the soft tissues, including the cartilage, ligaments, dura, spinal cord, and nerve roots [80].
2.3.9. Imbalance of Growth
MPS IVA is characterized by accumulation of GAGs in chondrocytes, the incomplete ossification, and the successive imbalance of growth, which cause unique clinical features of disproportionate short trunk, hypermobile joints in hands and fingers, a prominent forehead, an abnormal face with a large mandible, short neck, cervical spinal cord compression, tracheal obstruction with crowd thoracic inlet, pectus carinatum, flaring of the rib cage, coxa valga, genu valgum, and pes planus [5,8,18,20,57,91–93].
The frontal bone, parietal bone, occipital bone, and the mandible keep growing, causing MPS IVA patients to have an enlarged head, a prominent forehead, and a large mandible. The cervical spine in the region C1-C7 halts growing earlier in patients with MPS IVA, causing the patients to have a short neck. Cervical spine shows platyspondyly and typical beaking of the anterior margin of the vertebral bodies, causing kyphosis as well as instability of the atlantoaxial joint and spinal cord compression (Fig. 7). Tracheal abnormalities which include stenosis, tortuosity ultimately resulting in obstruction is due to several factors, including the imbalance of growth in patients with MPS IVA. The thoracic spine from T1 to T12, as well as the lumbar region of the spine from L1 to L5, stop growing early causing patients with MPS IVA to have a short trunk. Pectus carinatum is marked deformity of the anterior chest wall because the ribs (costal cartilage) overgrow compared with other parts of the body, causing restrictive lung [94]. The wrist joints show marked hyperlaxity because the ulna stops growing earlier and the radius continues to grow, resulting in an ulnar deviation of the wrist. The metacarpals and carpals grow slowly in patients affected by MPS IVA. The growth plates are wide with mild generalized brachydactyly and hypoplastic carpal centers [95] (Fig. 9). Hip deformities are caused by a combination of the bilateral flattening and fragmentation of the capital femoral epiphyses and acetabular dysplasia with lateral joint subluxation [9]. The pelvis shows wide flared iliac bones laterally [96]. Genu valgum is caused by the mechanical axis shift laterally, where pathologic stress is placed on the lateral femur and tibia which inhibits growth [97]. The lower limbs malalignment is evaluated with the measurements of the mechanical axis deviation (MAD) [98]. The patellofemoral joint may become shallow, incongruous, or unstable, causing activity-related knee pain in patients [97]. Patients will not only have knee pain and laxity, but they can develop a circumduction gait, in which each leg is swung outward to avoid knocking their knees together due to genu valgum [97].
Figure 7.
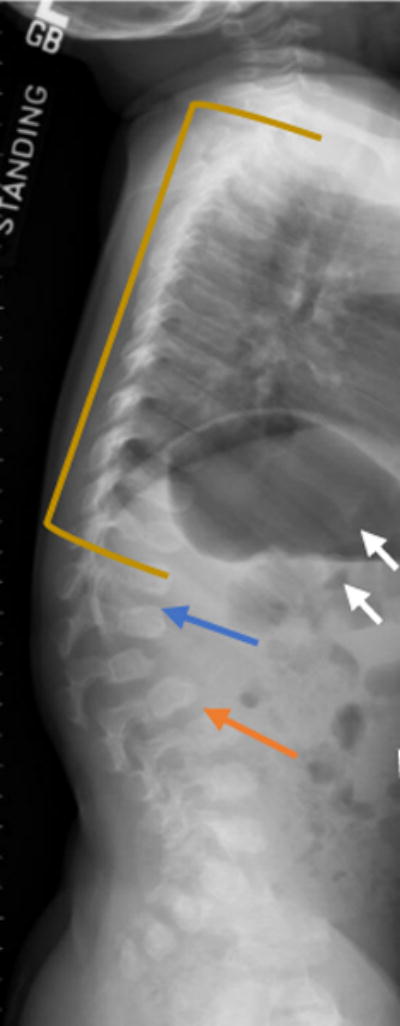
Lateral radiograph of the spine of a two-year-old MPS IVA patient with vertebral wedging (blue arrow). Platyspondyly (yellow line), anterior inferior beaking (orange arrow), rib flaring (white arrows) are seen in this X-ray radiograph of a patient with MPS IVA.
Figure 9.
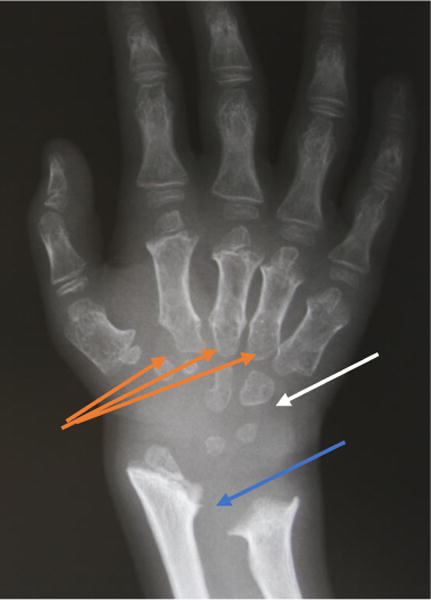
X-ray radiograph of an MPS IVA patient hand. Clearly seen are the tapering of the proximal portion for the metacarpals (orange arrows), small irregular carpal bones (white arrow), and the distal portion of the radius being tilted toward the ulna (blue arrow).
2.4. Non-Skeletal Manifestations
2.4.1. Ophthalmology
The most common ophthalmologic findings are related with slowly progressive corneal clouding (Fig. 10). Generally, in patients with MPS, corneal opacification of varying severity is frequently seen, as well as retinopathy, optic nerve swelling and atrophy, ocular hypertension and glaucoma [99]. Other ophthalmologic findings that are less common are astigmatism, cataracts, punctate lens opacities, open-angle glaucoma, optic disc swelling, optic atrophy and retinopathy [89,100]. Ocular manifestations are common in MPS and may result in significant visual impairment [99]. Corneal opacification often causes reduced vision in the early childhood of MPS IVA patients, necessitating penetrating keratoplasty for which the outcome can vary [9]. In a study with 20 patients aged 1-65 years, with MPS IV, in which the subtype A or B was not specified, 10 eyes had no corneal clouding, 17 eyes had mild corneal clouding, 4 eyes had moderate corneal clouding, and 4 eyes had severe corneal clouding (corneal clouding was not graded in 5 eyes) [101]. The severity of corneal clouding was related to the increasing age of the patient and results in the reduction in visual acuity [101]. The corneal deposits can also interfere with examination of the other ocular structures, such as the trabecular meshwork, the retina, and the optic nerve [100].
Figure 10.
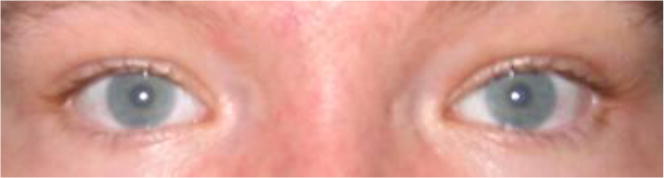
Eyes of a patient with MPS IVA. Image of a 24-year-old patient with MPS IVA shows fine stromal corneal clouding (adapted from Educational CD for Morquio and permitted by Carol Ann Foundation).
While visual acuity is better in MPS IVA patients compared to other MPS patients, the corneal clouding, refractive errors, glaucoma, and cataracts can affect the visual acuity as the patient ages [100]. Glaucoma or ocular hypertension seems to be unusual in MPS IVA [102]. Electron microscopy shows distended trabecular endothelial cells with a thickened basement membrane in MPS IVA [100]. The inclusions in the trabecular meshwork are primarily multi membranous, whereas other types of MPS inclusions are more fibrillogranular [103,104]. This most probably explains the reduced prevalence of ocular hypertension and glaucoma in individuals with MPS IVA [103,104].
Annual eye examinations should be performed in MPS IVA patients and should include the following: slit-lamp biomicroscopy of the cornea, measurement of intraocular pressure, assessment of refractive error, and examination of the posterior segment [100]. If the vision reduces, evaluation with low-vision aids should be considered [100].
2.4.2. Auditory system
Hearing impairment is common among patients with MPS IVA [9,11,16,100,105–107]. Conductive hearing loss caused by serous otitis media due to frequent upper respiratory tract infections may be present anytime from birth and onwards [9, 94]. Many patients with MPS IVA have one or more sets of ear pressure equalization tubes to treat conductive hearing loss due to recurrent ear infections [16,105]. Some undergo an ear tube insertion after childhood. Hearing loss in MPS IVA is progressive [108–110] and bilateral [9, 93] in general, and its severity ranges from mild to moderate [9,18,94] but can be severe [105,109,110]. Permanent hearing loss in MPS IVA is not usually identified until adolescence [100] but has been found in children younger than eight years old [108]. Younger patients exhibited conductive hearing loss and the sensorineural hearing loss element is added to the conductive component as the disease progressed [100,105,108]. A recent study reported that some MPS IVA patients who had normal audiometric results exhibited abnormal auditory neurophysiological responses based on otoacoustic emissions and/or abnormal auditory brainstem responses [105]. Their study suggests that the sensorineural hearing impairment may be in progress before patients notice [93].
Conductive hearing loss can be secondary to recurrent serous otitis media [100] or deformity of the ossicles due to the GAG accumulation in the middle ear [94]. The cause of sensorineural hearing loss is unknown, but it is likely due to the GAG accumulation in the cochlea and retrocochlear auditory nerves [100,111,112].
A recent study [105] on hearing function in patients with MPS IVA showed a strong correlation between height and hearing sensitivity and a strong correlation between height and the cochlear outer hair cell function. The strong relationship between short height and hearing loss suggests that patients with severe skeletal dysplasia may be at higher risk for developing severe hearing loss [105].
In addition to annual behavioral audiometric testing appropriate for the age of patients [100,107,113], regardless of age or skeletal severity, neurophysiological hearing testing such as otoacoustic emissions and ABR should be tested annually to monitor hearing disorders due to the progressive nature of the hearing impairment and the increased risk of developing a sensorineural hearing loss with age [88, 93].
2.4.3. Dental
Deciduous teeth erupt normally and are widely spaced and discolored with thin irregular (stippled) enamel and small pointed cusps which flatten over time with normal wear [9] (Fig. 11). Permanent teeth also have hypoplastic enamel [114]. Some common features have been reported with thin tooth enamel as well as multiple cavities.
Figure 11.
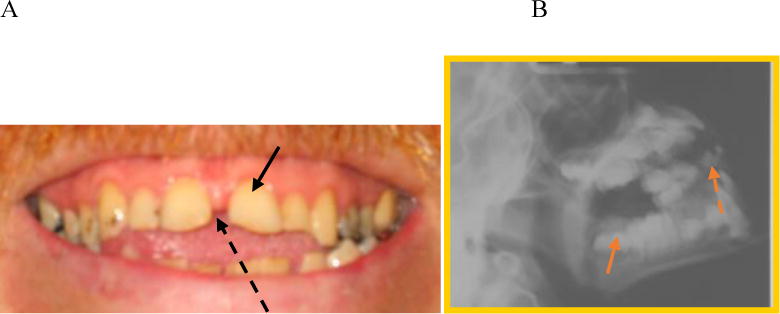
Teeth of a patient with MPS IVA. A. Image of a 24-year-old patient with widely spaced teeth (black arrow) and spade shaped incisors (black dotted arrow) (adapted from Educational CD for Morquio and permitted by Carol Ann Foundation). B. X-ray of a MPS IVA patient with concave occlulsal sufraces (orange arrow) and spade-shaped incisors (orange dotted arrow).
Levin et al. described the classic oral abnormalities found among 12 patients with MPS IVA [115]. Tooth morphology is highly specific for MPS IVA [116]. The maxillary anterior teeth are widely spaced and flared, and the posterior teeth are tapered with pointed cusp tips. The enamel can be of normal hardness, but some patients have pitted enamel with decreased thickness [115]. In roentgenograms, the enamel was less than one-fourth of normal thickness but was of normal radio density [115]. The dental abnormalities in the MPS IVA are of a type that is unique among the group of genetic MPS [115]. The teeth of patients with MPS IVA are typically small, and the enamel is thin with a greyish color. The cusps of the permanent teeth are sharpened [18]. Due to both dental and upper extremity abnormalities, maintaining oral health can be particularly challenging [110].
3.4.4. Cardiology
Cardiac complications include ventricular hypertrophy and early onset, severe valvular involvement as well as coronary intimal sclerosis [100]. Cases of cardiac valve thickening, regurgitation, and/or stenosis have been reported in patients [117–121]. In a case study in 1990, 10 patients with MPS IVA underwent echocardiographic assessment, and abnormalities were detected in 6 cases with mitral valve involvement in 5 patients and aortic valve disease in 4 [118]. One patient had severe mitral leaflet thickening to the point of mitral stenosis [118]. Two patients had evidence of myocardial involvement by way of echocardiographic ventricular hypertrophy [118]. Overall, there is a high prevalence of silent cardiac abnormalities in patients with MPS IVA with predominantly left-sided valve involvement [118]. Valve thickening that can be seen on cardiac ultrasound has been found to be nearly as common in patients who do not accumulate dermatan sulfate as in those who do [122]. All MPS IVA cardiac valves may show GAG deposition, although the left-sided cardiac valves are more severely affected [100].
The single cardiac ultrasound study that specifically addresses MPS IVA lacks the color flow Doppler technology which improves the detection of valve stenosis and insufficiency [118]. Despite the absence of color Doppler, valvular heart disease was found to be quite common in the single study devoted to MPS IVA [118]. 5 of the 10 (50%) patients studied had mitral valve regurgitation (1 of whom had mitral valve stenosis as well), 30% had aortic valve regurgitation, and 20% had both mitral and aortic regurgitation in conjunction with left ventricular hypertrophy [100]. Aortic and/or mitral valve thickening was present in 40% of patients tested [100].
Electrocardiography (ECG) examinations were carried out in 20 out of 37 patients [123], and measurements were compared with normal data [124]. All 20 patients had low R wave voltage in V6 [123]. Among the 37 patients who had echocardiography, cardiovascular abnormalities progressed with age although most had mild clinical signs and symptoms [123].
2.4.5. Respiratory
Respiratory complications are a major cause of morbidity and mortality in MPS IVA. This includes airway obstruction, sleep-disordered breathing, and restrictive lung [9]. The GAG accumulation in the following locations: adenoids, tonsils, pharynx, larynx, trachea, and bronchial tree leads to adenotonsillar hypertrophy, tracheal distortion, trachea- and bronchomalacia and obstructive sleep apnea [125,126]. Restrictive lung results from a small thorax, chest wall abnormalities, spine deformities, and neuromuscular compromise from cervical myelopathy and hepatomegaly causes an upward displacement of the diaphragm [100,126].
Patients with MPS IVA often prefer to sleep prone on a flat surface without a pillow to keep the neck extended and to minimize the tortuosity of the airway [9]. Due to the short stature and skeletal dysplasia, alteration in growth and development provides an additional mechanism for respiratory compromise which is frequently noted in patients [18]. Patients with MPS IVA are at increased risk for complications that include recurrent infections, progressive loss of respiratory function, sleep-disordered breathing, and ultimately respiratory failure [16,18,127].
The cause of restrictive lung in patients with MPS IVA is likely multifactorial; however, thoracic deformity (pectus carinatum, rib deformity) appears to be a primary cause [20]. Pulmonary function tests (PFT) such as forced vital capacity (FVC) and forced expiratory volume in 1 second (FEV1) should be performed regularly to assess changes in lung volume and obstruction [20]. Pulmonary hypertension, which may be caused by obstructive sleep apnea, is observed in patients with MPS IVA as commonly as observed in other MPS [20]. Patients may complain of nocturnal dyspnea, in which case polysomnography can be used to assess sleep disturbances [20]. 22 MPS IV patients (7 male, 15 females) ranged from 3 to 40 years of age, underwent noninvasive PFTs with vital signs [128].
For lung function studies, the age, size, and fitness are all important considerations [100]. As patients with MPS IVA exhibit a restrictive and obstructive element of respiratory compromise, both aspects need to be assessed [100]. To assess airflow limitation, the most common test utilized is the spirometry [100]. It is also a readily available technique utilized to measure the vital capacity (VC; the maximum amount of air that can be inhaled/ exhaled from full inflation to maximum deflation) which indicates the size of the lungs [100]. Airway resistance is assessed through a measurement known as impulse oscillation (IOS), where it utilizes a speaker to impose external oscillating pressure and airflow impulses over these subject’s tidal breathing [100]. The “gold standard” assessment of lung restriction is by measurement of total lung capacity (TLC) [100]. This can be assessed by a variety of techniques such as physiological, which includes whole body plethysmography, gas (typically helium) dilution, and nitrogen washout, or by the radiographic means [conventional chest radiographs or (CT)] [100].
2.4.6. Tracheal Obstruction
Unlike other MPS patients, MPS IVA patients develop severe tracheal stenosis with a characteristic appearance on imaging studies and on direct bronchoscopic examination. Thus it is important to remember that MPS IVA patients have obstruction of large airways in addition to the upper airway obstruction reported in the earlier literature. Characteristics of the tracheal abnormality include right ward deviation of the trachea, twisting and buckling of the trachea which may at a younger age appear simply tortuous. Factors that contribute to this unusual pathological features are both external and internal to the trachea itself. External factors consist of the compression of the trachea due to the crossing of the brachiocephalic artery across and anterior to the trachea, proximity of the cervicothoracic spine which moves forward, the sternum (the manubrium) with the clavicular heads, and the pectus carinatum all competing for space in a crowded thoracic inlet, each contributing varying degrees of compression and narrowing of the trachea [8]. Internal factors contributing to the tracheal pathology include deposits of GAG increasing the thickness of the tracheal walls; unbalanced longitudinal growth of the trachea (in comparison to the growth of the thoracic cavity) resulting in trachea having to ‘fold upon itself’ which is now known as ‘buckling’ of the trachea [20] [13]. Patients with MPS IVA can develop respiratory failure secondary to reduced chest wall compliance and airway collapse due to the irregularly shaped vocal cords and trachea [127]. Tracheal obstruction also leads to life-threating complications during anesthesia because of the difficulty in managing both upper and the lower airways in MPS IVA primarily manifested by the difficulty in intubating the trachea [13] (Fig. 12). Tracheal narrowing increases with age as does anesthetic risk from both upper airway and tracheal pathology[13].
Figure 12.
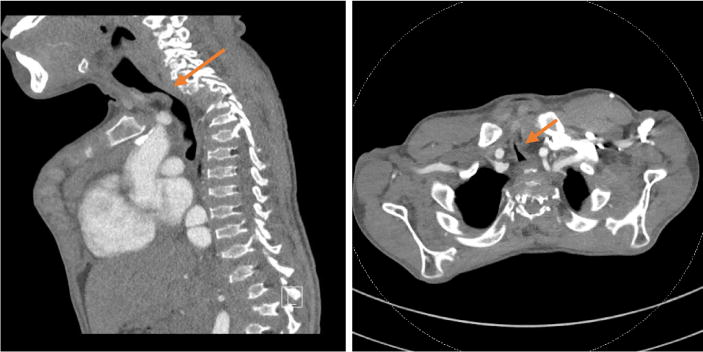
Sagittal (a) and axial (b) CT images of a 16-year-old boy with MPS IVA focal severe tracheal narrowing at the thoracic inlet (arrows). Tracheal cross-sectional area measures 10 mm2 at the thoracic inlet and 110 mm2 intrathoracic below the area of narrowing.
Sagittal MRI images of the cervical spine of 28 MPS IVA patients (12 ± 8.14 years) showed that 19 out of the 28 (67.9%) patients had at least 25% tracheal narrowing. 8 out of 28 (28.6%) patients were categorized as severe (>75%) tracheal narrowing when images were evaluated in neutral head and neck position. The tortuous brachiocephalic artery was the most common cause in the 19 patients with tracheal narrowing (n = 15). Evidence of such tracheal narrowing was evident as early as 2 years of age. Tracheal narrowing increased with age, with all 8 patients over 15 years of age having greater than 50% narrowing. The severity score of the subjects over 15 years of age was nearly twice higher than the score of the 10–15 years old [13]. Greater attention to the trachea is needed when evaluating cervical spine MRIs as well as other imaging and clinical investigations, with the main goal of establishing a timely treatment protocol to reduce the mortality rate in the MPS IVA population.
3. Clinical tests
Since MPS is a progressive disorder that affects many organ systems simultaneously, it is very challenging to identify signs and symptoms that can be applied as suitable parameters for clinical trials. Some clinical endpoint tests are performed by measuring the general endurance since the impairment of stamina is common in all forms of MPS. The longitudinal, prospective MPS IVA clinical assessment program reviewed a decline of endurance tests, 6-minute walk test (6-MWT) and 3-minute stair climb test (3-MSCT) suggesting decreased functional ability over time [129]. Other tests can be used as clinical endpoints in studies that are aimed to demonstrate the efficacy of a new drug for MPS patients such as the stair climb test, lung function, joint range of motion, pain, and quality of life.
3.1. 6 Minute Walk Test (6-MWT)
The 6-minute walk test (6-MWT) is a supervised test that measures the distance a patient can walk on a hard flat surface over a 6-minute period [130]. According to the American Thoracic Society (ATS) guidelines, the 6-MWT is a simple test that requires a 30 meters hallway with no exercise equipment of advanced training for technicians. This test evaluates the global and integrated responses of all body system such as the pulmonary and cardiovascular systems, systemic circulation, peripheral circulation, blood, neuromuscular units and muscle metabolism. It is important to note that most patients do not achieve maximal exercise capacity during the 6-MWT, as the patients choose their own intensity of exercise and are allowed to stop and rest during their test. The strongest indication for the 6-MWT is for measuring the response to medical interventions in patients with moderate to severe heart or lung disease [131].
The advantage of 6-MWT is that it is the easiest to perform compared with other endurance tests and has a good correlation with the functions of the heart and lungs. In contrast, the 6-MWT has several limitations as this test has been studied mostly in adult patients and rarely in children. Nixon et al. used the 6-MWT in severely ill children before they received a lung or a heart transplantation. According to their study, there was a strong correlation of oxyhemoglobin saturation on the bike test compared to the walk test. In general, the results of the various studies of the 6-MWT including MPS patients have to be interpreted with caution as several factors have to be taken in consideration such as the length of the corridor used, the learning effects, and/or encouragement [132]. It is impossible to apply the 6-MWT in young MPS patients. Therefore, the 6-MWT may not be reliable due to its lack of effectiveness in MPS IVA patients.
Treatment with 2 mg/kg elosulfase alfa weekly resulted in a modest improvement in the 6-MWT in the first clinical trial of 6 months; however, there was no improvement in the extension clinical trial (weekly infusion group) at week 72 [3]. After 120 weeks, results showed mild improvement in 6-MWT [133]. For patients treated with elosulfase alfa at 2.0 mg/kg/week throughout the study, mean 6-MWT improvements from baseline were 32.0 and 39.9 m from the intent to treat and modified per protocol populations, respectively, in relation to the 22.5 m improvement reported in the pivotal study [134]. The effectiveness of 6-MWT as a clinical endpoint remains ambiguous especially in a short term.
3.2. 3 Minute Stair Climb Test (3-MSCT)
The 6-MWT is a submaximal exercise test widely used to measure endurance and flexibility a range of patients [49,135,136], whereas the 3-minute stair climb test evaluates the effort required by the cardiovascular, pulmonary and/or musculoskeletal systems to perform an activity [136,137]. The risk with 3-MSCT is that patients with MPS IVA may get injured with falling down, as well as that patients who are handicapped can not perform this test. There was no significant improvement for patients treated in neither the 2 mg/kg WT nor 2 mg/kg QOW treatment group, compared to the placebo group at week 24. The two treatments groups and the placebo group mean difference in the stair climb rate was only 1.1 stairs per minute [134]. On week 72, both groups maintained the change in 3 MSCT without worsening below baseline. After 120 weeks, the results were higher than those at 24 weeks [139].
3.3. Pulmonary Function Test
Respiratory function tests are difficult to perform in MPS IVA patients due to their characteristic skeletal dysplasia, small body size and lack of cooperation of young patients, causing conventional spirometry for pulmonary function to be very challenging in some cases [128]. The non-invasive pulmonary tests: impulse oscillometry system (IOS), pneumotachography (PNT), and respiratory inductance plethysmography (RIP) in conjunction with conventional spirometry were evaluated in MPS IVA patients, all subjects had normal vital signs at rest including and age-appropriate heart rate [128]. All patients also preserved normal values in IOS, PNT, RIP, and forced expiratory volume in 1 second/forced expiratory volume total which were normal and not significantly impacted by age; however, the predicted forced expiratory total decreased with age and was below normal [128]. The proposed non-invasive pulmonary function tests can cover a greater number of patients (young patients and/or wheel-chair bound), which provides a new diagnostic approach for the assessment of lung function in MPS IVA that in many cases may be difficult to evaluate [128]. In a natural history study, changes in the FVC and maximum voluntary ventilation (MVV) were observed. For patients less than 14 years, the values increased, as would be expected for growth; however, the values decreased in older patients, which raised concerns about disease progression [129]. The compromised respiratory function and decreased FVC and MVV are related to multiple factors, including the progression of bone abnormalities and airway obstruction [138].
3.4. Range of Motion
Patients typically show significant differences in the active and passive range of motion (ROM) at the wrist joint, which shows the loss of stability at the joint [139]. Patients typically show a pronounced decrease in grip and pinch strength and have difficulties performing day-to-day tasks that require strength [139]. This regular assessment of ROM and strength provides inside into the degree and progression of functional impairment of the hands [87]. A goniometer can show both active and passive ROM of the wrists and digits [87].
3.5. Sleep Study
Sleep-disordered breathing (SDB) is common in all MPS diseases and may precede the development of overt respiratory failure during wakefulness [100,125]. Ventilatory abnormalities during sleep include obstructive sleep apnea due to several factors such as GAG accumulation in the upper airway, sustained hypoventilation due to the chest wall deformity and/or respiratory muscle weakness [100]. Sleep studies should also be performed in addition to lung function assessments [100]. Symptoms suggestive of SDB, such as apneas, gasping respirations, snoring, difficult wakening, daytime somnolence, and restless sleep should be assessed annually as part of the clinical evaluation of all patients [100]. Overnight sleep studies can be used to both diagnose the type and severity of SDB and also to evaluate nocturnal ventilatory treatments for the underlying respiratory disorder [100].
3.6. Bone Mineral Density
In the last few years, MPS IVA has been reported to be related with an early-onset osteoporotic phenotype, which can affect the clinical course of the condition [19]. The accumulation of KS disturbing bone mass acquisition and perturbing the regular microarchitecture of bone tissue has been reported to reflect on the bone mass density (BMD) [140]. Bone growth and mineralization have been reported to be affected by GAGs accumulation in animal models of MPS I, II, III, IVA, VI, and VII; there are, however, limited publications on the assessment of bone mineral density in patients with MPS [46,141–144].
In a study with 18 patients (16 unrelated) with MPS IVA with an average age of 21.4 years (3.3 to 40.8 years). In this prospective cross-sectional study, BMD of the whole body (WB), lumbar spine (LS), and lateral distal femur (LDF) was acquired by dual-energy X-ray absorptiometry (DXA) on patients with MPS IVA (Fig. 13). WB DXA could be obtained on only 6 patients (5 full-time ambulators) because of the respiratory compromise caused by the position, presence of hardware, or positioning difficulties. Mean WB Z-score was −2.0 (range − 0.3 to −4.1). Technical issues invalidating LS DXA in 8 patients included kyphosis at the thoracolumbar junction results in overlap of vertebrae in the posterior-anterior view. Mean LS BMD Z-score in full-time ambulators was −3.4 (range − 1.6 to −5.0) and in the non-/partial ambulator was −4.0 (−3.7 to −4.2). Lateral distal femur BMD was acquired on every patient, and average Z-scores were −2 or less at all the sites; fulltime ambulators exhibited higher BMD. LDF proved to be the most feasible site to measure in patients with MPS IV A [145]. Low BMD of the lower extremities as measured by the LDF, DXA is directly associated with lack of ambulation or weight bearing [146–150].
Figure 13.
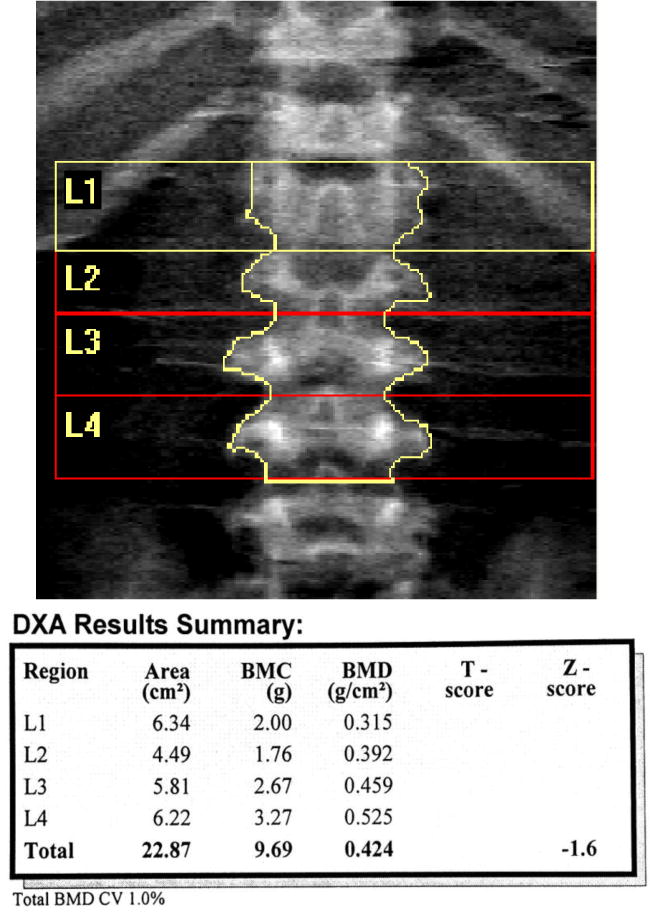
DXA image of the spine of a MPS IVA patient. Lumbar spine DXA requires careful and precise measurement of vertebral body levels and margins (Adapted from Kecskemethy et al. Mol Genet Metab; 2017, 144-149) [145].
3.7. Dexterity Test
The Functional Dexterity Test (FDT) is validated and timed pegboard test that is not only easy to administer, but also it assesses the ability of the patients to perform functional daily tasks that require a 3-jaw chuck pretension pattern (also referred to as the lamar pinch, pencil pinch or tripod grip) such as writing and buttoning [151,152]. Each patient with MPS IVA syndrome requires regular assessment of upper limb function (fine motor skills) [89]. To obtain consistent measurements, it is important to record the position of the wrist and also to record whether the wrist was supported or not during testing [89]. Passive and active assessments of the range of motion of elbows and shoulders can be useful but do not have therapeutic consequences [89].
The FDT was developed as a measure of dexterity that will take a minimum amount of time to administer, yet still provides information regarding the patient’s ability to use the hand for daily tasks requiring a 3-jaw pretension between the fingers and the thumb [152]. The overall, recommended pediatric modifications to the FDT are to use speed (pegs per second) instead of time (seconds) to report the results, and also to not assess penalties [151].
3.8. Gait pattern
Tests on gait and mobility can easily be done by a simple physical exam and an interrogation of the patients [89]. Instrumented gait analysis proves information about the dynamic alignment of the lower extremities and is also helpful in decision making in various conditions [153]. In a study of 9 MPS IV children (who had no previous lower extremity surgery), underwent a 3D gait analysis to describe the gait kinetics and kinematics in children with MPS IV. The mean age at gait analysis for the study was 10.6±4 years, mean height was 105.2±15.6 cm (z = −4.5), and mean weight was 22.3±7.2 kg (z = −1.434). The measurements obtained from gait analysis in the patients were compared with data from 10 healthy young individuals with age ranging from 9.5 to 11.5 years; the mean height was of 138±6.6 cm, and the mean weight was of 32.5±7.1 kg [86]. There were significant differences in the temporal-spatial characteristics, kinetics, and kinematics in children with MPS IV in comparison with the normal population on instrumented gait analysis. There was a decreased forward velocity, cadence, and stride length that was compared with the normal population (p<0.05). The height-adjusted, normalized forward velocity and stride length were also reduced in comparison with the normal population (p<0.01). The forward tilt of the trunk and pelvis, hip flexion, hip adduction, and external hip rotation were increased in comparison with the normal population (p<0.05). There was increased knee flexion, genu valgus (abduction), and external tibial torsion compared with normal during stance (p<0.05). Dynamic knee varus–valgus joint laxity showed a mean difference of 9.5° between the minimum and maximum genu valgus (abduction) that was recorded during stance, which is compared with a difference of 0.9° in the normal population (p<0.05). While there was a strong correlation between genu valgus measured on gait analysis and standing radiographs (r = 0.89), there was a moderate correlation between genu valgus measured on gait analysis and clinical examination (r = 0.69) [86].
3.9. Activity of daily living
The current options for treating MPS IVA in a multisystem manner to address overall quality of life are limited [100]. Currently, due to the absence of an effective and safe systemic treatment option, patients require extensive management and regular intervention to maximize their quality of life [100]. To track and manage the quality of life, patients with MPS IVA should see both physiotherapist and experts on MPS annually to evaluate the degree of impairment, including determination of the range of motion, muscle strength, daily activity, and participation limitations [100]. As MPS IVA disorder process progresses, the quality of life for the patient declines. Patients increasingly become more dependent on caregivers as deteriorating vision, hearing, oral health, respiratory and cardiac function, muscular strength, and endurance make routine daily activities increasingly difficult to complete tasks [100].
The activity of daily living (ADL) questionnaire is made up of three sections: “Movement,” “Movement with Cognition,” and “Cognition.” Each section has four subcategories rated on a 5-point scale based on the level of assistance. The questionnaire was then collected from 145 healthy controls and 82 patients with MPS IVA. Of the 82 patients questioned, 63 patients were severe and 17 patients had attenuated phenotypes (2 were undefined); 4 patients treated with hematopoietic stem cell transplantation (HSCT), 33 patients with enzyme replacement therapy (ERT) for more than one year and 45 untreated patients. MPS IVA patients showed a decline in ADL scored after 10 years of age. Patients with a severe phenotype had a lower ADL score than healthy control subjects, and lower scores than patients with an attenuated phenotype in domains of “Movement” and “Movement with cognition.” Patients who underwent HSCT were followed up for over 10 years, had higher ADL scores and fewer surgical interventions than untreated patients [154]. Further investigation of large study on HSCT is required since it is limited number.
3.10. Growth
Growth impairment is commonly observed in patients with MPS [155]. Patients with MPS IVA are most severely affected in growth and final height, which are used as an indicator of disease severity [16,156]. Short stature is the main evident symptom, and the male and female patients have the growth impairment to approximately an equal degree [16,156,157]. Growth in patients with MPS IVA stops around 7 to 8 years age [156,158].
In 2007, Montano et al. described that the phenotypic classification for severe MPS IVA by height was below 120 cm while the classification for the attenuated type was above 120 cm [158]. Some patients with attenuated MPS IVA continue growing even into their teens and reach over 140 cm [16]. In 2008, Montano et al. proposed that a standard growth chart for each gender of MPS IVA patients should be made for better results of phenotypic classification [158]. Height and weight measurements from MPS IVA patients were collected in the International MPS IVA Registry, and the growth charts for male and female patients were established, compared to reference growth curves of healthy children provided by the CDC [16,20]. In 2012, more height and weight measurements from 193 girls and 195 boys with MPS IVA were collected to revise the growth charts [147]. The mean birth length of boys was 52.4 ± 3.9 cm, and the mean height for males at 18 years of age was 119.3 ± 22.6. The mean birth length of girls was 52.1 ± 2.9 cm, and the mean height for girls at 18 years of age was 113.5 ± 23.1 cm. These values correspond to −8.0 SD and −7.7 SD of the mean height for normal males and females. The mean birth weights for boys were 3.56 ± 0.5 and for girls were 3.5 ± 0.7 kg. The growth patterns in MPS IVA patients were characterized by impaired growth velocity after 1 and 2 years of age [156]. According to the isopleth upon which the patient falls, patients above the 90th centile on the growth chart for each gender of MPS IVA are more likely to be defined as mild attenuated while patients between the 75th centile and 90th centile on the growth chart are more likely to be defined as intermediate attenuated. Patients less than the 75th centile are classified as classic severe.
In another study with MPS IVA from Taiwan, 92% of the patients had short stature [159]. Patients treated with ERT showed no statistical significance in height/growth rate [134,160]. Overall, the height and growth velocity are the simplest and objective tests to define the clinical severity and to monitor therapeutic efficacy.
4. Molecular Diagnosis
Molecular analysis is used to confirm the diagnosis and to provide genetic counseling for the family and prenatal analysis [17]. The GALNS gene is located on chromosome 16q24.3, contains 14 exons, and generates a 1566 nucleotide mRNA [161–164]. Multiple MPS IVA mutations are believed to be exclusive and found in only one individual or family [70].
As of February of 2018, 334 mutations in the GALNS gene have been reported missense/nonsense mutations account for a total of 203, 35 deletions, 22 splicing site mutations, 7 insertions and 3 complex rearrangements [165]. Several mutations are common; the most prevalent recurrent mutations in the GALNS gene are c.1156C>T (p.R386C), c.901G>T (p.G301C), c.337A>T (p.I113F), c.1A>G (p.M1V), c.757C>T (p.R253W), c.871G>A (p.A291 T), c.935C>G (p.T312S), and c.1171A>G (p.M391V), accounting for 8.9%, 6.8%, 5.7%, 2.3%, 2.1%, 1.8%, 1.8%, 1.8%, and 1.8%, respectively [166–172].
In a study with 37 MPS IVA patients that were based on clinical data, biochemical assays, molecular analyses, and in silico structural analyses of associated mutations, the results displayed that standard sequencing procedures, albeit identifying novel GALNS genetic lesions, and failed to characterize the 2nd disease-causing mutation in the 16% of the patients’ cohort [173]. To address this drawback and uncover potential gross GALNS rearrangements, the study developed molecular procedures assays, quantitative fluorescent-PCRs (QF-PCRs), endorsed by comparative genome hybridization (CGH) arrays [173]. Using this approach, the study was able to characterize two new large deletions and their corresponding breakpoints [173].
5. Challenges of Diagnosing MPS IVA
The signs and symptoms of MPS IVA overlap with those of other MPS, all of which have a broad spectrum of clinical manifestations [9] (Table 1). In comparison to the other MPS, patients with MPS IVA have normal intellectual abilities as well as less coarsening of the facial features [9]. Laxity of joints (atlanto-axial, fingers, hands, knee) is a major condition for MPS IVA. Atlanto-axial instability, weakness of wrist, and knock-knee are more common in patients with MPS IVA in comparison to other MPS types [9]. Patients with a severe phenotype accounted for nearly 75% of the 399 MPS IVA patients registered in International MPS IV Registry [20]. With the skeletal involvement, significant morbidity of patients can also result from obstructive sleep apnea, corneal clouding, hearing impairment, respiratory compromise, valvular heart disease and spinal cord compression.
Table 1.
Differential diagnosis between MPS IVA and others diseases
| MPSIVA | MPSIVB | MP S I | MP S II | MP S VI | MPVVII | GM1 | SED | LCPD | DMC | SMC | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Abnormal Behavior | − | − | − | ++ | − | + | − | − | − | − | − |
| Anterior Breaking: vertebral bodies | +++ | ++ | +++ | +++ | ++ | ++ | +++ | + | − | ++ | ++ |
| Cardiac Valve Abnormalities | ++ | ++ | +++ | ++ | +++ | +++ | +++ | − | − | − | − |
| Cervical Instability | +++ | ++ | +++ | ++ | +++ | +++ | − | − | − | − | − |
| Chest Abnormalities: rib flaring | +++ | +++ | − | ++ | ++ | +++ | − | + | − | ++ | ++ |
| Claw Hands (rigid fingers and hands) | − | − | ++ | ++ | ++ | ++ | − | − | − | + | + |
| CNS Impairment | − | − | ++ | ++ | − | ++ | ++ | − | − | ++ | − |
| Coarse Face | + | + | ++ | ++ | ++ | ++ | ++ | − | − | ++ | ++ |
| Coarse Hair | + | + | ++ | ++ | + | ++ | − | − | − | − | − |
| Corneal Clouding | ++ | ++ | ++ | + | ++ | ++ | ++ | − | − | − | − |
| Cranofacial Dysmorphia | − | − | ++ | ++ | ++ | − | ++ | − | − | − | − |
| Dental Abnormalities: widely spaced teeth | ++ | ++ | ++ | ++ | ++ | ++ | − | − | − | − | − |
| Developmental Delay | − | − | ++ | ++ | − | ++ | ++ | − | − | ++ | − |
| Disproportionate Dwarfism | ++ | ++ | ++ | ++ | ++ | ++ | − | + | + | + | + |
| Fetal Hydrops | − | − | − | − | − | ++ | + | − | − | − | − |
| Hearing Loss | ++ | ++ | ++ | ++ | ++ | ++ | ++ | ++ | − | − | − |
| Hepatosplenomegaly | + | + | ++ | ++ | ++ | ++ | ++ | − | − | − | − |
| Hip Deformity | ++ | ++ | ++ | ++ | ++ | ++ | − | ++ | ++ | − | − |
| Hydrocephalus | − | − | ++ | + | ++ | ++ | − | − | − | − | − |
| Hyperactivity | − | − | − | ++ | − | ++ | − | − | − | − | − |
| Hypermobile joint (weak wrist and double fingers) | ++ | ++ | − | − | − | − | − | − | − | − | − |
| Hypertrophic adenoid and tonsil | ++ | ++ | − | ++ | ++ | + | − | − | − | − | − |
| Inguinal Hernia | + | + | ++ | ++ | ++ | ++ | − | − | − | − | − |
| Joint Pain | ++ | ++ | ++ | ++ | ++ | ++ | ++ + | + | − | − | − |
| Joint Stiffness | − | − | ++ | ++ | ++ | ++ | − | + | − | ++ | ++ |
| Knock Knee (genu valgum) | ++ | ++ | ++ | − | ++ | ++ | ++ | ++ | ++ | ++ | |
| Kyphosis/Gibbus | ++ | ++ | ++ | + | ++ | ++ | ++ | ++ | ++ | ++ | |
| Large Head | ++ | ++ | ++ | ++ | ++ | ++ | − | − | − | − | − |
| Large Mandible | ++ | ++ | + | − | + | − | − | − | − | − | − |
| Large Tongue | ++ | + | ++ | ++ | ++ | ++ | − | − | − | − | − |
| Loud breathing, shortness of breath | ++ | ++ | ++ | − | − | ++ | − | − | − | − | − |
| Muscular Weakness | ++ | ++ | ++ | ++ | + | + | ++ | − | ++ | ++ | ++ |
| Normal Intelligence | ++ | ++ | + | + | ++ | ++ | ++ | ++ | ++ | + | ++ |
| Odontoid Hypoplasia | ++ | ++ | − | + | ++ | ++ | − | − | − | ++ | ++ |
| Pectus Carinatum | ++ | ++ | − | − | ++ | ++ | − | − | − | ++ | ++ |
| Platyspondyly: vertebral bodies | ++ | ++ | ++ | − | − | ++ | − | ++ | − | ++ | ++ |
| Recurrent otitis media | ++ | ++ | + | ++ | ++ | ++ | − | − | − | − | − |
| Recurrent respitatory infections | ++ | ++ | ++ | ++ | ++ | ++ | − | − | − | − | − |
| Rigidity of Joints (shoulder, elbow) | + | + | ++ | ++ | ++ | ++ | − | − | − | − | − |
| Scoliosis | ++ | ++ | ++ | − | ++ | ++ | ++ + | ++ | − | ++ | ++ |
| Short Neck | ++ | ++ | ++ | − | ++ | ++ | − | − | − | − | ++ |
| Short Stature | ++ | ++ | ++ | ++ | ++ | ++ | ++ + | ++ | ++ + | ++ | ++ |
| Short Trunk | ++ | ++ | + | + | − | ++ | ++ | − | ++ | ++ | |
| Sleep Apnea | ++ | ++ | ++ | ++ | ++ | ++ | − | − | − | − | − |
| Sleep Distrubance | − | − | ++ | ++ | ++ | + | − | − | − | − | − |
| Snoring | ++ | ++ | − | ++ | + | ++ | − | − | − | − | − |
| Speech Delay | − | − | + | ++ | − | ++ | ++ | − | − | ++ | − |
| Umbilical Hernia | + | + | ++ | ++ | ++ | ++ | − | − | − | − | − |
| Visual Impariment | ++ | ++ | ++ | + | ++ | + | ++ | ++ | − | − | − |
| Waddling Gait | ++ | ++ | − | − | ++ | + | ++ | − | ++ | − | − |
MPS IVA can be diagnosed with the above unique characters although this disorder relates closely to other disorders in some characteristics. The disorder that most commonly resembles MPS IVA is MPS IVB. MPS IVB differs from MPS IVA in that there is an accumulation of only KS due to the deficiency in the enzyme β-galactosidase, and several disease characteristics overlap although MPS IVA is usually more severe than MPS IVB.
Mucopolysaccharidosis type VI known as Maroteaux-Lamy syndrome is an autosomal recessive disorder, caused by mutations in the arylsulfatase B (ARSB) gene and leads to a deficiency in the N-acetylgalactosamine 4-sulfatase enzyme [168]. The clinical presentation of MPS VI like MPS IVA varies depending on the age of onset and rate of disease progression [175]. Unlike MPS VI, MPS IVA patients have hypermobile joints, platyspondyly, loud breathing and short trunk [95,176,177].
Mucopolysaccharidosis type VII known as Sly syndrome is an extremely rare recessive lysosomal storage disease caused by the mutations in the GUSB gene leading to a deficiency in the β-glucuronidase enzyme causing accumulation in the DS, HS, and C6S. Unlike MPS VI, MPS IVA patients have hypermobile joints and a large mandible [178–181].
GM1 gangliosidosis is an autosomal recessive storage disorder and is caused by the deficiency of β-galactosidase which is the same as MPS IVB [182]. GM1 gangliosidosis like MPS IVA, have anterior breaking of the vertebral bodies, joint pain, muscle weakness, short stature, coarse face, corneal clouding, visual impairment, hearing loss, cardiac valve abnormalities, hepatosplenomegaly, kyphosis/gibbus, scoliosis, and waddling gait. Unlike MPS IVA, GM1 gangliosidosis patients have CNS impairment, developmental delay, craniofacial dysmorphia and fetal hydrops. GM1 gangliosidosis patients however lack sleep disturbance, disproportionate short stature, hypermobile joints, large head, coarse hair, hypertrophic adenoid and tonsil, large tongue, dental abnormalities, recurrent otitis media, recurrent respiratory infections, large mandibular, short neck, cervical instability, odontoid hypoplasia, chest abnormalities, pectus carinatum, hip deformity, short trunk, platyspondyly, genu valgum, and inguinal hernia.[183,184].
Spondyloepiphyseal dysplasia congenita (SEDc) has very similar radiographic findings to MPS IVA. SEDc is caused by the autosomal dominant mutations in the TRPV4 gene. SEDc like MPS IVA, have anterior breaking of the vertebral bodies, short stature, normal intelligence, joint pain, hearing loss, chest abnormalities, kyphosis/gibbus, scoliosis, hip deformity, short trunk and genu valgum. While MPS IVA patients have hypermobile joints, SEDc patients have joint stiffness [185–188].
Legg-Calve-Perthes disease (LCPD) is an autosomal dominant disorder of the COL2A1 gene mutations, in which mild MPS IVA with only hip pain at the early stages can lead to an initial misdiagnosis of Legg-Calve-Perthes disease. Perthes disease, unlike MPS IVA, is a rare childhood condition that only affects the hip Signs and symptoms for LCPD correlate to MPS IVA because symptoms include muscular weakness, normal intelligence, short stature, hip deformity, genu valgum, and waddling gait. Usually, LCPD only involves one hip. However, both hips can be affected in some children [189–191].
Dyggve-Melchior-Clausen syndrome (DMC), originally reported as Morquio-Ullrich’s disease, is caused by mutations in the DYM gene and is autosomal recessive. Mutations in the same DYM gene can also cause Smith-McCourt syndrome (SMC), which is a condition that is radiographically identical to DMC in which intelligence and psychomotor developments are normal in the patients [192]. DMC can be clinically differentiated from MPS IVA because DMC patients are intellectually disabled, whereas MPS IVA patients are not [10,193,194]. A patient with DMC or SMC will have a lace-like appearance of iliac crests, concavity of the end of the scapula because of defective ossification, a horizontal acetabular roof and a double-humped end-plates of vertebrae, which lacks in MPS IVA patients [195–197].
Although clinical findings and radiographs can provide great insight, laboratory testing is required to reach a diagnosis of MPS IVA [10]. When alternative skeletal dysplasia is suspected, and MPS IVA is considered enough of a possibility that the clinician chooses to perform a laboratory test to rule out the MPS disorders, then urinary screening is not an acceptable method to use by itself because of the high false negative rates for MPS IVA [10].
6. Conclusion
Variable clinical presentation and laboratory testing caveats make MPS IVA particularly challenging to diagnose [10]. Both skeletal and non-skeletal symptoms need to be tested and assessed for clinical suspicion. Especially, imbalance of growth causes major clinical signs and symptoms leading to serious morbidity and mortality. Radiographic, biochemical, and molecular tests are critical for precise diagnosis and prognosis for MPS IVA. The age of diagnosis, the natural progression of the disease, and prognosis of the disease play a role in determining the precise management of MPS IVA.
Figure 8.
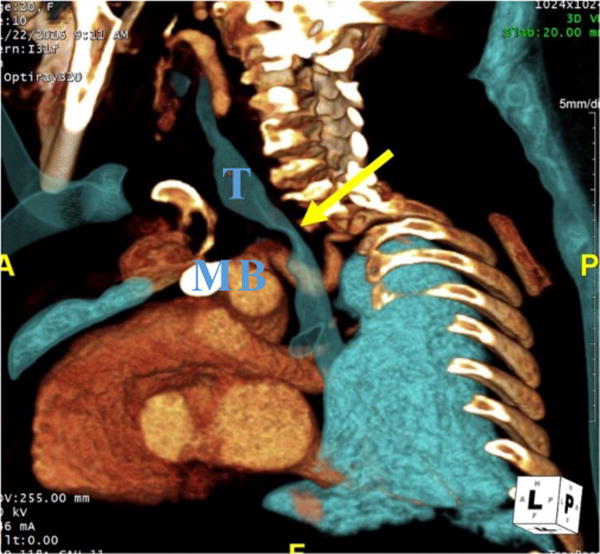
CT angiography (CTA) of a 20-year-old woman with MPS IVA. 3D oblique reconstruction of CTA demonstrates marked narrowing of the trachea at the thoracic inlet (yellow arrow). T– Trachea, B- Brachiocephalic artery, M- Manubrium.
Highlights.
Mucopolysaccharidosis IVA is an autosomal recessive disorder caused by the deficiency of GALNS.
This enzyme deficiency leads to a progressive accumulation of excessive C6S and KS.
Spinal cord compression, airway compromise, and valvular heart disease are the leading causes of high morbidity and mortality.
The attenuated form may not become evident until late childhood or adolescence, and often first manifests hip problems as the primary symptom.
Imbalance of growth in bones, organs, and tissues causes the hallmark of clinical manifestations.
Acknowledgments
This work was supported by grants from National MPS Society, the Austrian MPS Society, and The Carol Ann Foundation, Angelo R. Cali & Mary V. Cali Family Foundation, Inc., The Vain and Harry Fish Foundation, Inc., The Bennett Foundation, and Jacob Randall Foundation. This research was supported by the project for baby and infant in research of health and development to Adolescent and young adult from Japan Agency for Medical Research and development, AMED, under grant number JP18gk0110017. R.W.M., S.T., and K.N. were supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of National Institutes of Health (NIH) under grant number P30GM114736. The content of the article has not been influenced by the sponsors.
Abbreviations
- 3-MSCT
3-Minute Stair Climb Test
- 4MU-Gal6S
4-methylumbelliferyl-β-D-galactopyranoside-6-sulfate
- 6-MWT
6-Minute Walk Test
- ABR
Auditory Brainstem Response
- ADL
Activity of Daily Living
- BMD
Bone Mineral Density
- C6S
Chondroitin-6-sulfate
- CDC
Centers for Disease Control and Prevention
- CSF
Cerebrospinal Fluid
- CT
Computed Tomography
- DBS
Dried Blood Spot
- DMMB
Dimethylmethylene Blue
- DMC
Dyggve-Melchior-Clausen syndrome
- DS
Dermatan Sulfate
- DXA
Dual Energy X-Ray Absorptiometry
- ECG
Electrocardiography
- ECM
Extracellular Matrix
- ELISA
Enzyme-linked Immunosorbent Assay
- ERT
Enzyme Replacement Therapy
- FDT
Functional Dexterity Test
- FET
Forced Expiratory Time
- FEV1
Forced Expiratory Volume in 1 Second
- FIVC
Forced Inspiratory Vital Capacity
- FVC
Forced Vital Capacity
- G6S
Galactose-6-sulfatase
- GAG
Glycosaminoglycans
- Gal-6S
Galactose-6-Sulfate
- GalNAc-6S
N-acetylgalactosamine-6-sulfate
- GALNS
N-acetylgalactosamine-6-sulfate sulfatase
- GM1
GM1-Gangliosidosis
- HS
Heparan Sulfate
- HSCT
Hematopoietic Stem Cell Transplantation
- IOC
Impulse Oscillation
- ITT
Intent-to-Treat
- KS
Keratan Sulfate
- LCPD
Legg-Calve Perthes Disease
- LDF
Lateral Distal Femur
- LS
Lumbar Spine
- LSD
Lysosomal Storage Disorders
- MPS
Mucopolysaccharidoses
- MPS IVA
Morquio A Syndrome
- MRI
Magnetic Resonance Imaging
- MVV
Maximum Voluntary Ventilation
- PFT
Pulmonary Function Tests
- QF-PCRs
Quantitative Fluorescent-PCRs
- ROM
Range of Motion
- SDB
Sleep Disordered Breathing
- SEDc
Spondoephisyseal Dysplasia Congenita
- SMC
Smith-McCort Syndrome
- TLC
Total Lung Capacity
- VC
Vital Capacity
- VRA
Visual Reinforcement Audiometry
- WB
Whole Body
- WHO
World Health Organization
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest:
All the authors contributed to the Review Article and had no conflict of interest with any other party. Hira Peracha, Kazuki Sawamoto, Lauren Averill, Heidi Kecskemethy, Mary Theroux, Mihir Thacker, Kyoko Nagao, Christian Pizarro, William Mackenzie, Hironori Kobayashi, Seiji Yamaguchi, Yasuyuki Suzuki, Kenji Orii, Tadao Orii, Toshiyuki Fukao, Shunji Tomatsu declare that they have no conflict of interests.
Contributions to the project:
Hira Peracha has contributed to the concept and planning of the project, collection of data from the publications, the draft of the manuscript, and reporting of the work described as the primary author.
Kazuki Sawamoto has contributed to the concept and planning of the project, collection of data, data analysis, the draft of the manuscript, and reporting of the work described.
Lauren Averill has contributed to the concept and planning of the project, analysis of images on x-rays, MRI, and CT, and reporting of the work described.
Heidi Kecskemethy has contributed to the concept and planning of the project, analysis of images on DXA, and reporting of the work described.
Mary Theroux has contributed to the concept and planning of the project, data and interpretation on airway, and reporting of the work described.
Mihir Thacker has contributed to the concept and planning of the project, interpretation of x-rays, clinical manifestations of spine and lower extremities, and reporting of the work described. Kyoko Nagao has contributed to the concept and planning of the project, hearing test, and reporting of the work described.
Christian Pizarro has contributed to the concept and planning of the project, tracheal surgery, and reporting of the work described.
William Mackenzie has contributed to the concept and planning of the project, spine and knee surgeries, and reporting of the work described.
Hironori Kobayashi has contributed to the concept and planning of the project, interpretation of GAG data, and reporting of the work described.
Seiji Yamaguchi has contributed to the concept and planning of the project, interpretation of GAG data, and reporting of the work described.
Yasuyuki Suzuki has contributed to the concept and planning of the project, interpretation of clinical data, and reporting of the work described.
Kenji Orii has contributed to the concept and planning of the project, interpretation of clinical data, and reporting of the work described. Tadao Orii has contributed to the concept and planning of the project, interpretation of clinical pictures, images, and GAG data, interpretation of published data and reporting of the work described.
Toshiyuki Fukao has contributed to the concept and planning of the project, interpretation of clinical data, and reporting of the work described.
Shunji Tomatsu is a Principal Investigator for this project and has contributed to the concept and planning of the project, interpretation of published data, and reporting of the work described.
References
- 1.Gentili C, Cancedda R. Cartilage and bone extracellular matrix. Curr Pharm Des. 2009;15:1334–48. doi: 10.2174/138161209787846739. http://www.ncbi.nlm.nih.gov/pubmed/19355972 (accessed October 28, 2017) [DOI] [PubMed] [Google Scholar]
- 2.Alliston T. Chondroitin sulfate and growth factor signaling in the skeleton: Possible links to MPS VI. J Pediatr Rehabil Med. 2010;3:129–38. doi: 10.3233/PRM-2010-0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khan S, Alméciga-Díaz CJ, Sawamoto K, Mackenzie WG, Theroux MC, Pizarro C, Mason RW, Orii T, Tomatsu S. Mucopolysaccharidosis IVA and glycosaminoglycans. Mol Genet Metab. 2017;120:78–95. doi: 10.1016/j.ymgme.2016.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Staprans I. Isolation and characterization of glycosaminoglycans in human plasma. J Clin Invest. 1985;76:1984–91. doi: 10.1172/JCI112198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morquio L. Sur une forme de dystrophie osseuse familiale. Bull Soc Pediatr Paris. 1929;27:145–52. [Google Scholar]
- 6.Brailsford J. Chondro-osteo-dystrophy. Roentgenographic and clinical features of a child with dislocation of vertebrae. Am J Surg. 1929;7:404–410. [PubMed] [Google Scholar]
- 7.McKusick VA, Kaplan D, Wise D, Hanley WB, Suddarth SB, Sevick ME, Maumanee AE. The genetic mucopolysaccharidoses. Medicine (Baltimore) 1965;44:445–83. doi: 10.1097/00005792-196511000-00001. http://www.ncbi.nlm.nih.gov/pubmed/4221470 (accessed August 30, 2017) [DOI] [PubMed] [Google Scholar]
- 8.Orii T, Minami R, Chiba T, Yamaguchi M, Tsugawa S, Nakao T, Horino K, Sakuma K. Study on Morquio syndrome bone metabolism (in Japanese) 1971;5:72–78. [Google Scholar]
- 9.Regier D, Oetgen M, Tanpaiboon P. Mucopolysaccharidosis Type IVA. In: Adam M, Ardinger H, Pagon R, Wallace S, Bean L, Stephens K, Amemiya A, editors. GeneReviews. 2013. pp. 1993–2018. doi:NBK148668 [bookaccession] [Google Scholar]
- 10.Wood TC, Harvey K, Beck M, Burin MG, Chien YH, Church HJ, D’Almeida V, Van Diggelen OP, Fietz M, Giugliani R, Harmatz P, Hawley SM, Hwu WL, Ketteridge D, Lukacs Z, Miller N, Pasquali M, Schenone A, Thompson JN, Tylee K, Yu C, Hendriksz CJ. Diagnosing mucopolysaccharidosis IVA. J Inherit Metab Dis. 2013;36:293–307. doi: 10.1007/s10545-013-9587-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Northover H, Cowie RA, Wraith JE. Mucopolysaccharidosis type IVA (Morquio syndrome): a clinical review. J Inherit Metab Dis. 1996;19:357–65. doi: 10.1007/BF01799267. http://www.ncbi.nlm.nih.gov/pubmed/8803780 (accessed August 21, 2017) [DOI] [PubMed] [Google Scholar]
- 12.Tomatsu S, Yasuda E, Patel P, Ruhnke K, Shimada T, Mackenzie W, Mason R, Thacker M, Theroux M, Motano A, C A-D, Luis A. Morquio A Syndrome: Diagnosis and Current and Future Therapies The pathogenesis of systemic skeletal dysplasia in Morquio A syndrome remains an enigmatic. Pediatr Endocrinol Rev. 2014;12:141–151. doi: 10.1016/j.jsbmb.2011.07.002.Identification. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tomatsu S, Averill LW, Sawamoto K, Mackenzie WG, Bober MB, Pizarro C, Goff CJ, Xie L, Orii T, Theroux M. Obstructive airway in Morquio A syndrome, the past, the present and the future. Mol Genet Metab. 2016;117:150–156. doi: 10.1016/j.ymgme.2015.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Muhlebach MS MJ, Wooten W. Respiratory Manifestations in Mucopolysaccharidoses. Pediatr Respir. 2011;12:133–138. doi: 10.1016/j.prrv.2010.10.005. [DOI] [PubMed] [Google Scholar]
- 15.Neufeld E, Muenzer J. The Mucopolysaccharidoses. In: Scriver, Beaudet, Valle, Sly, Childs, Kinzler, Vogelstein, editors. Metab Mol Basis Inherit Dis. 8th. 2001. pp. 3421–3452. [Google Scholar]
- 16.Montaño AM, Tomatsu S, Gottesman GS, Smith M, Orii T. International Morquio A Registry: Clinical manifestation and natural course of Morquio A disease. J Inherit Metab Dis. 2007;30:165–174. doi: 10.1007/s10545-007-0529-7. [DOI] [PubMed] [Google Scholar]
- 17.Lavery C, Hendriksz C. Mortality in patients with morquio syndrome a. JIMD Rep. 2015;15:59–66. doi: 10.1007/8904_2014_298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tomatsu S, Montaño AM, Oikawa H, Smith M, Barrera L, Chinen Y, Thacker MM, Mackenzie WG, Suzuki Y, Orii T. Mucopolysaccharidosis type IVA (Morquio A disease): clinical review and current treatment. Curr Pharm Biotechnol. 2011;12:931–945. doi: 10.2174/138920111795542615. http://www.ncbi.nlm.nih.gov/pubmed/21506915 (accessed October 30, 2017) [DOI] [PubMed] [Google Scholar]
- 19.Tummolo A, Gabrielli O, Gaeta A, Masciopinto M, Zampini L, Pavone LM, Di Natale P, Papadia F. Bisphosphonate Treatment in a Patient Affected by MPS IVA with Osteoporotic Phenotype. Case Rep Med. 2013;2013:891596. doi: 10.1155/2013/891596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hendriksz CJ, Harmatz P, Beck M, Jones S, Wood T, Lachman R, Gravance CG, Orii T, Tomatsu S. Review of clinical presentation and diagnosis of mucopolysaccharidosis IVA. Mol Genet Metab. 2013;110:54–64. doi: 10.1016/j.ymgme.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tomatsu S, Okamura K, Taketani T, Orii KO, Nishioka T, Gutierrez MA, Velez-Castrillon S, Fachel AA, Grubb JH, Cooper A, Thornley M, Wraith E, Barrera LA, Giugliani R, Schwartz IV, Frenking GS, Beck M, Kircher SG, Paschke E, Yamaguchi S, Ullrich K, Isogai K, Suzuki Y, Orii T, Kondo N, Creer M, Noguchi A. Development and testing of new screening method for keratan sulfate in mucopolysaccharidosis IVA. Pediatr Res. 2004;55:592–7. doi: 10.1203/01.PDR.0000113767.60140.E9. [DOI] [PubMed] [Google Scholar]
- 22.Whitley CB, Draper KA, Dutton CM, Brown PA, Severson SL, France LA. Diagnostic test for mucopolysaccharidosis. II. Rapid quantification of glycosaminoglycan in urine samples collected on a paper matrix. Clin Chem. 1989;35:2074–81. http://www.ncbi.nlm.nih.gov/pubmed/2507197 (accessed August 21, 2017) [PubMed] [Google Scholar]
- 23.Gray G, Claridge P, Jenkinson L, Green A. Quantitation of urinary glycosaminoglycans using dimethylene blue as a screening technique for the diagnosis of mucopolysaccharidoses – an evaluation. Ann Clin Biochem. 2007;44:360–363. doi: 10.1258/000456307780945688. [DOI] [PubMed] [Google Scholar]
- 24.Chih-Kuang C, Shuan-Pei L, Shyue-Jye L, Tuen-Jen W. MPS screening methods, the berry spot and acid turbidity tests, cause a high incidence of false-negative results in sanfilippo and morquio syndromes. J Clin Lab Anal. 2002;16:253–258. doi: 10.1002/jcla.10051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Jong JG, Wevers RA, Laarakkers C, Poorthuis BJ. Dimethylmethylene blue-based spectrophotometry of glycosaminoglycans in untreated urine: a rapid screening procedure for mucopolysaccharidoses. Clin Chem. 1989;35:1472–7. http://www.ncbi.nlm.nih.gov/pubmed/2503262 (accessed August 21, 2017) [PubMed] [Google Scholar]
- 26.Piraud M, Boyer S, Mathieu M, Maire I. Diagnosis of mucopolysaccharidoses in a clinically selected population by urinary glycosaminoglycan analysis: a study of 2,000 urine samples. Clin Chim Acta. 1993;221:171–81. doi: 10.1016/0009-8981(93)90031-x. http://www.ncbi.nlm.nih.gov/pubmed/8149634 (accessed August 21, 2017) [DOI] [PubMed] [Google Scholar]
- 27.Wood T, Bodamer OA, Burin MG, D’Almeida V, Fietz M, Giugliani R, Hawley SM, Hendriksz CJ, Hwu WL, Ketteridge D, Lukacs Z, Mendelsohn NJ, Miller N, Pasquali M, Schenone A, Schoonderwoerd K, Winchester B, Harmatz P. Expert recommendations for the laboratory diagnosis of MPS VI. Mol Genet Metab. 2012;106:73–82. doi: 10.1016/j.ymgme.2012.02.005. [DOI] [PubMed] [Google Scholar]
- 28.Whitley CB, Ridnour MD, Draper KA, Dutton CM, Neglia JP. Diagnostic test for mucopolysaccharidosis. I. Direct method for quantifying excessive urinary glycosaminoglycan excretion. Clin Chem. 1989;35:374–9. http://www.ncbi.nlm.nih.gov/pubmed/2493341 (accessed August 21, 2017) [PubMed] [Google Scholar]
- 29.Hata R, Nagai Y. A rapid and micro method for separation of acidic glycosaminoglycans by two-dimensional electrophoresis. Anal Biochem. 1972;45:462–8. doi: 10.1016/0003-2697(72)90208-4. http://www.ncbi.nlm.nih.gov/pubmed/4258509 (accessed August 29, 2017) [DOI] [PubMed] [Google Scholar]
- 30.Hopwood JJ, Harrison JR. High-resolution electrophoresis of urinary glycosaminoglycans: an improved screening test for the mucopolysaccharidoses. Anal Biochem. 1982;119:120–7. doi: 10.1016/0003-2697(82)90674-1. http://www.ncbi.nlm.nih.gov/pubmed/6803608 (accessed August 29, 2017) [DOI] [PubMed] [Google Scholar]
- 31.Humbel R, Chamoles NA. Sequential thin layer chromatography of urinary acidic glycosaminglycans. Clin Chim Acta. 1972;40:290–3. doi: 10.1016/0009-8981(72)90287-2. http://www.ncbi.nlm.nih.gov/pubmed/4262516 (accessed August 29, 2017) [DOI] [PubMed] [Google Scholar]
- 32.Wessler E. Analytical and preparative separation of acidic glycosaminoglycans by electrophoresis in barium acetate. Anal Biochem. 1968;26:439–44. doi: 10.1016/0003-2697(68)90205-4. http://www.ncbi.nlm.nih.gov/pubmed/5716194 (accessed August 29, 2017) [DOI] [PubMed] [Google Scholar]
- 33.Yasuda E, Fushimi K, Suzuki Y, Shimizu K, Takami T, Zustin J, Patel P, Ruhnke K, Shimada T, Boyce B, Kokas T, Barone C, Theroux M, Mackenzie W, Nagel B, Ryerse J, Orii K, Iida H, Orii T, Tomatsu S. Pathogenesis of Morquio A syndrome: an autopsied case reveals systemic storage disorder. Mol Genet Metab. 2013;109:301–311. doi: 10.1016/j.ymgme.2013.04.009. [DOI] [PubMed] [Google Scholar]
- 34.Doherty C, Averill LW, Theroux M, Mackenzie WG, Pizarro C, Mason RW, Tomatsu S. Natural history of Morquio A patient with tracheal obstruction from birth to death. Mol Genet Metab Reports. 2018;14:59–67. doi: 10.1016/j.ymgmr.2017.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tomatsu S, Sawamoto K, Alméciga-Díaz C. Impact of enzyme replacement therapy and hematopoietic stem cell transplantation in patients with Morquio A syndrome. Drug Des Dev Ther. 2015;9:1937–1953. doi: 10.2147/DDDT.S68562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pizarro C, Davies RR, Theroux M, Spurrier EA, Averill LW, Tomatsu S. Surgical Reconstruction for Severe Tracheal Obstruction in Morquio A Syndrome. Ann Thorac Surg. 2016;102:e329–e331. doi: 10.1016/j.athoracsur.2016.02.113. [DOI] [PubMed] [Google Scholar]
- 37.Oguma T, Tomatsu S, Okazaki O. Analytical method for determination of disaccharides derived from keratan sulfates in human serum and plasma by high-performance liquid chromatography/turbo-ionspray ionization tandem mass spectrometry. Biomed Chromatogr. 2007;21:356–362. doi: 10.1002/bmc.760. [DOI] [PubMed] [Google Scholar]
- 38.Hintze J, Tomatsu S, Fujii T, Montaño A, Yamaguchi S, Suzuki Y, Orii T. Comparison of Liquid Chromatography–Tandem Mass Spectrometry and Sandwich ELISA for Determination of Keratan Sulfate in Plasma and Urine. Biomark Insights. 2011;6:69–78. doi: 10.4137/BMI.S7451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tomatsu S, Montaño AM, Oguma T, Dung VC, Oikawa H, de Carvalho TG, Gutiérrez ML, Yamaguchi S, Suzuki Y, Fukushi M, Kida K, Kubota M, Barrera L, Orii T. Validation of keratan sulfate level in mucopolysaccharidosis type IVA by liquid chromatography-tandem mass spectrometry. J Inherit Metab Dis. 2010;33(Suppl 3):S35–42. doi: 10.1007/s10545-009-9013-x. [DOI] [PubMed] [Google Scholar]
- 40.Martell LA, Cunico RL, Ohh J, Fulkerson W, Furneaux R, Foehr ED. Validation of an LC-MS/MS assay for detecting relevant disaccharides from keratan sulfate as a biomarker for Morquio A syndrome. Bioanalysis. 2011;3:1855–66. doi: 10.4155/bio.11.172. [DOI] [PubMed] [Google Scholar]
- 41.Sweet MB, Coelho A, Schnitzler CM, Schnitzer TJ, Lenz ME, Jakim I, Kuettner KE, Thonar EJ. Serum keratan sulfate levels in osteoarthritis patients. Arthritis Rheum. 1988;31:648–52. doi: 10.1002/art.1780310510. http://www.ncbi.nlm.nih.gov/pubmed/2967706 (accessed March 9, 2018) [DOI] [PubMed] [Google Scholar]
- 42.Tomatsu S, Montano AM, Ohashi A, Gutierrez MA, Oikawa H, Oguma T, Dung VC, Nishioka T, Orii T, Sly WS. Enzyme replacement therapy in a murine model of Morquio A syndrome. Hum Mol Genet. 2007;17:815–824. doi: 10.1093/hmg/ddm353. [DOI] [PubMed] [Google Scholar]
- 43.Montaño AM, Oikawa H, Tomatsu S, Nishioka T, Vogler C, Gutierrez MA, Oguma T, Tan Y, Grubb JH, Dung VC, Ohashi A, Miyamoto K, Orii T, Yoneda Y, Sly WS. Acidic amino acid tag enhances response to enzyme replacement in mucopolysaccharidosis type VII mice. Mol Genet Metab. 2008;94:178–189. doi: 10.1016/j.ymgme.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 44.Tomatsu S, Montaño AM, Dung VC, Ohashi A, Oikawa H, Oguma T, Orii T, Barrera L, Sly WS. Enhancement of drug delivery: enzyme-replacement therapy for murine Morquio A syndrome. Mol Ther. 2010;18:1094–102. doi: 10.1038/mt.2010.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ru M, Tol L, Vlies N, Bigger B, Hollak C, IJlst L, Kulik W, van Lenthe H, Saif M, Wagemans T, van der Wal W, Wanders R, Wijburg F. Plasma and urinary levels of dermatan sulfate and heparan sulfate derived disaccharides after long-term enzyme replacement therapy (ERT) in MPS I: correlation with the timing of ERT and with total urinary excretion of glycosaminoglycans. J Inherit Metab Dis. 2013;36:247–255. doi: 10.1007/s10545-012-9538-2. [DOI] [PubMed] [Google Scholar]
- 46.Rowan DJ, Tomatsu S, Grubb JH, Montaño AM, Sly WS. Assessment of bone dysplasia by micro-CT and glycosaminoglycan levels in mouse models for mucopolysaccharidosis type I, IIIA, IVA, and VII. J Inherit Metab Dis. 2013;36:235–246. doi: 10.1007/s10545-012-9522-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu Y, Yi F, Kumar AB, Kumar Chennamaneni N, Hong X, Scott CR, Gelb MH, Turecek F. Multiplex Tandem Mass Spectrometry Enzymatic Activity Assay for Newborn Screening of the Mucopolysaccharidoses and Type 2 Neuronal Ceroid Lipofuscinosis. Clin Chem. 2017;63:1118–1126. doi: 10.1373/clinchem.2016.269167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tomatsu S, Yasuda E, Patel P, Ruhnke K, Shimada T, Mackenzie W. Morquio A syndrome: diagnosis and current and future therapies. Pediatr Endocrinol Rev. (Suppl 1):141–151. (n.d.) [PMC free article] [PubMed] [Google Scholar]
- 49.Tomatsu S, Sawamoto K, Shimada T, Bober MB, Kubaski F, Yasuda E, Mason RW, Khan S, Alméciga-Díaz CJ, Barrera LA, Mackenzie WG, Orii T. Enzyme replacement therapy for treating mucopolysaccharidosis type IVA (Morquio A syndrome): effect and limitations. Expert Opin Orphan Drugs. 2015;3:1279–1290. doi: 10.1517/21678707.2015.1086640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Erickson RP, Sandman R, Epstein CJ. Lack of relationship between blood and urine levels of glycosaminoglycans and lysomal enzymes. Biochem Med. 1975;12:331–9. doi: 10.1016/0006-2944(75)90064-2. http://www.ncbi.nlm.nih.gov/pubmed/126060 (accessed February 26, 2018) [DOI] [PubMed] [Google Scholar]
- 51.Saville JT, McDermott BK, Fuller M. Glycosaminoglycan fragments as a measure of disease burden in the mucopolysaccharidosis type I mouse. Mol Genet Metab. 2018;123:112–117. doi: 10.1016/j.ymgme.2017.12.007. [DOI] [PubMed] [Google Scholar]
- 52.Khan S, Mason RW, Giugliani R, Orii K, Fukao T, Suzuki Y, Yamaguchi S, Kobayashi H, Orii T, Tomatsu S. Glycosaminoglycans analysis in blood and urine of patients with mucopolysaccharidosis. Mol Genet Metab (in Press) doi: 10.1016/j.ymgme.2018.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shimada T, Tomatsu S, Mason R, Yasuda E, Mackenzie W, Hossain J, Shibata Y, Montaño A, Kubask IF, Giugliani R, Yamaguchi S, Suzuki Y, Orii K, Fukao T, Orii T. Di-sulfated keratan sulfate as a novel biomarker for mucopolysaccharidosis IVA. J Inherit Metab Dis Reports. 2015;21:1–13. doi: 10.1007/8904_2014_330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tomatsu S, Fujii T, Fukushi M, Oguma T, Shimada T, Maeda M, Kida K, Shibata Y, Futatsumori H, Montaño AM, Mason RW, Yamaguchi S, Suzuki Y, Orii T. Newborn screening and diagnosis of mucopolysaccharidoses. Mol Genet Metab. 2013;110:42–53. doi: 10.1016/j.ymgme.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shimada T, Tomatsu S, Yasuda E, Mason RW, Mackenzie WG, Shibata Y, Kubaski F, Giugliani R, Yamaguchi S, Suzuki Y, Orii K, Orii T. Chondroitin 6-Sulfate as a Novel Biomarker for Mucopolysaccharidosis IVA and VII. JIMD Rep. 2014;16:15–24. doi: 10.1007/8904_2014_311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Singh J, Di Ferrante N, Niebes P, Tavella D. N-acetylgalactosamine-6-sulfate sulfatase in man. Absence of the enzyme in Morquio disease. J Clin Invest. 1976;57:1036–1040. doi: 10.1172/JCI108345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Matalon R, Arbogast B, Justice P, Brandt IK, Dorfman A. Morquio’s syndrome: deficiency of a chondroitin sulfate N-acetylhexosamine sulfate sulfatase. Biochem Biophys Res Commun. 1974;61:759–65. doi: 10.1016/0006-291x(74)91022-5. http://www.ncbi.nlm.nih.gov/pubmed/4218100 (accessed September 10, 2017) [DOI] [PubMed] [Google Scholar]
- 58.Yutaka T, Okada S, Kato T, Inui K, Yabuuhi H. Galactose 6-sulfate sulfatase activity in Morquio syndrome. Clin Chim Acta. 1982;122:169–80. doi: 10.1016/0009-8981(82)90276-5. http://www.ncbi.nlm.nih.gov/pubmed/6809361 (accessed September 10, 2017) [DOI] [PubMed] [Google Scholar]
- 59.Glössl J, Kresse H. Impaired degradation of keratan sulphate by Morquio A fibroblasts. Biochem J. 1982;203:335–8. doi: 10.1042/bj2030335. http://www.ncbi.nlm.nih.gov/pubmed/6213226 (accessed September 10, 2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Glössl J, Kresse H. A sensitive procedure for the diagnosis of N-acetyl-galactosamine-6-sulfate sulfatase deficiency in classical Morquio’s disease. Clin Chim Acta. 1978;88:111–9. doi: 10.1016/0009-8981(78)90157-2. http://www.ncbi.nlm.nih.gov/pubmed/98244 (accessed September 5, 2017) [DOI] [PubMed] [Google Scholar]
- 61.van Diggelen OP, Zhao H, Kleijer WJ, Janse HC, Poorthuis BJ, van Pelt J, Kamerling JP, Galjaard H. A fluorimetric enzyme assay for the diagnosis of Morquio disease type A (MPS IV A) Clin Chim Acta. 1990;187:131–9. doi: 10.1016/0009-8981(90)90339-t. http://www.ncbi.nlm.nih.gov/pubmed/2107987 (accessed September 10, 2017) [DOI] [PubMed] [Google Scholar]
- 62.Zhao H, Van Diggelen OP, Thoomes R, Huijmans J, Young E, Mazurczak T, Kleijer WJ. Prenatal diagnosis of Morquio disease type A using a simple fluorometric enzyme assay. Prenat Diagn. 1990;10:85–91. doi: 10.1002/pd.1970100204. http://www.ncbi.nlm.nih.gov/pubmed/2111546 (accessed August 21, 2017) [DOI] [PubMed] [Google Scholar]
- 63.Kleijer WJ, Geilen GC, Garritsen V, Huijmans JG, Los FJ, Voznyi YV, van Diggelen OP. First-trimester diagnosis of Morquio disease type A. Prenat Diagn. 2000;20:183–5. http://www.ncbi.nlm.nih.gov/pubmed/10719317 (accessed August 21, 2017) [PubMed] [Google Scholar]
- 64.Tomatsu S, Okamura K, Maeda H, Taketani T, Castrillon SV, Gutierrez MA, Nishioka T, Fachel AA, Orii KO, Grubb JH, Cooper A, Thornley M, Wraith E, Barrera LA, Laybauer LS, Giugliani R, Schwartz IV, Frenking GS, Beck M, Kircher SG, Paschke E, Yamaguchi S, Ullrich K, Haskins M, Isogai K, Suzuki Y, Orii T, Kondo N, Creer M, Okuyama T, Tanaka A, Noguchi A. Keratan sulphate levels in mucopolysaccharidoses and mucolipidoses. J Inherit Metab Dis. 2005;28:187–202. doi: 10.1007/s10545-005-5673-3. [DOI] [PubMed] [Google Scholar]
- 65.Camelier MV, Burin MG, De Mari J, Vieira TA, Marasca G, Giugliani R. Practical and reliable enzyme test for the detection of Mucopolysaccharidosis IVA (Morquio Syndrome type A) in dried blood samples. Clin Chim Acta. 2011;412:1805–1808. doi: 10.1016/j.cca.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 66.Duffey TA, Khaliq T, Scott CR, Turecek F, Gelb MH. Design and synthesis of substrates for newborn screening of Maroteaux-Lamy and Morquio A syndromes. Bioorg Med Chem Lett. 2010;20:5994–6. doi: 10.1016/j.bmcl.2010.08.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Burin M, Dutra-Filho C, Brum J, Mauricio T, Amorim M, Giugliani R. Effect of collection, transport, processing and storage of blood specimens on the activity of lysosomal enzymes in plasma and leukocytes. Brazilian J Med Biol Res. 2000;33:1003–13. doi: 10.1590/s0100-879x2000000900003. http://www.ncbi.nlm.nih.gov/pubmed/10973130 (accessed October 24, 2017) [DOI] [PubMed] [Google Scholar]
- 68.Duffey TA, Khaliq T, Scott CR, Turecek F, Gelb MH. Design and synthesis of substrates for newborn screening of Maroteaux–Lamy and Morquio A syndromes. Bioorg Med Chem Lett. 2010;20:5994–5996. doi: 10.1016/j.bmcl.2010.08.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Khaliq T, Sadilek M, Scott CR, Turecek F, Gelb MH. Tandem Mass Spectrometry for the Direct Assay of Lysosomal Enzymes in Dried Blood Spots:Application to Screening Newborns for Mucopolysaccharidosis IVA. Clin Chem. 2011;57:128–131. doi: 10.1373/clinchem.2010.149880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tomatsu S, Montaño AM, Nishioka T, Gutierrez MA, Peña OM, Trandafirescu GG, Lopez P, Yamaguchi S, Noguchi A, Orii T. Mutation and polymorphism spectrum of the GALNS gene in mucopolysaccharidosis IVA (Morquio A) Hum Mutat. 2005;26:500–512. doi: 10.1002/humu.20257. [DOI] [PubMed] [Google Scholar]
- 71.Fukuda S, Tomatsu S, Masuno M, Ogawa T, Yamagishi A, Rezvi GM, Sukegawa K, Shimozawa N, Suzuki Y, Kondo N, Imaizumi K, Kuroki Y, Okabe T, Orii T. Mucopolysaccharidosis IVA: submicroscopic deletion of 16q24.3 and a novel R386C mutation of N-acetylgalactosamine-6-sulfate sulfatase gene in a classical Morquio disease. Hum Mutat. 1996;7:123–34. doi: 10.1002/(SICI)1098-1004(1996)7:2<123::AID-HUMU6>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 72.Pajares S, Alcalde C, Couce M, Del Toro M, González-Meneses A, Guillén E, Pineda M, Pintos G, Gort L, Coll M. Molecular analysis of mucopolysaccharidosis IVA (Morquio A) in Spain. Mol Genet Metab. 2012;106:196–201. doi: 10.1016/j.ymgme.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 73.Morrone A, Tylee KL, Al-Sayed M, Brusius-Facchin AC, Caciotti A, Church HJ, Coll MJ, Davidson K, Fietz MJ, Gort L, Hegde M, Kubaski F, Lacerda L, Laranjeira F, Leistner-Segal S, Mooney S, Pajares S, Pollard L, Ribeiro I, Wang RY, Miller N. Molecular testing of 163 patients with Morquio A (Mucopolysaccharidosis IVA) identifies 39 novel GALNS mutations. Mol Genet Metab. 2014;112:160–70. doi: 10.1016/j.ymgme.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Davison JE, Kearney S, Horton J, Foster K, Peet AC, Hendriksz CJ. Intellectual and neurological functioning in Morquio syndrome (MPS IVa) J Inherit Metab Dis. 2013;36:323–8. doi: 10.1007/s10545-011-9430-5. [DOI] [PubMed] [Google Scholar]
- 75.Tomatsu S, Theroux WG, Sly MC, Almeciga-Diaz C, Yasuda MM, Suzuki TH, Ruhnke AM, Chinen D, Mackenzie W, Thacker CJ, Mason LA, Montano Y, Rowan E, Barrera K, Orii Y, Shaffer T. Current and emerging treatments and surgical interventions for Morquio A syndrome: a review. Res Reports Endocr Disord. 2012;2012:65–77. doi: 10.2147/RRED.S37278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hughes DG, Chadderton RD, Cowie RA, Wraith JE, Jenkins JP. MRI of the brain and craniocervical junction in Morquio’s disease. Neuroradiology. 1997;39:381–5. doi: 10.1007/s002340050429. http://www.ncbi.nlm.nih.gov/pubmed/9189888 (accessed August 21, 2017) [DOI] [PubMed] [Google Scholar]
- 77.Lipson SJ. Dysplasia of the odontoid process in Morquio’s syndrome causing quadriparesis. J Bone Joint Surg Am. 1977;59:340–4. http://www.ncbi.nlm.nih.gov/pubmed/403192 (accessed August 21, 2017) [PubMed] [Google Scholar]
- 78.Ransford AO, Crockard HA, Stevens JM, Modaghegh S. Occipito-atlanto-axial fusion in Morquio-Brailsford syndrome. A ten-year experience. J Bone Joint Surg Br. 1996;78:307–13. http://www.ncbi.nlm.nih.gov/pubmed/8666648 (accessed August 21, 2017) [PubMed] [Google Scholar]
- 79.McKay SD, Al-Omari A, Tomlinson LA, Dormans JP. Review of cervical spine anomalies in genetic syndromes. Spine (Phila Pa 1976) 2012;37:E269–77. doi: 10.1097/BRS.0b013e31823b3ded. [DOI] [PubMed] [Google Scholar]
- 80.Solanki GA, Martin KW, Theroux MC, Lampe CCG, White KK, Shediac R, Lampe CCG, Beck M, Mackenzie WG, Hendriksz CJ, Harmatz PR. Spinal involvement in mucopolysaccharidosis IVA (Morquio-Brailsford or Morquio A syndrome): presentation, diagnosis and management. J Inherit Metab Dis. 2013;36:339–55. doi: 10.1007/s10545-013-9586-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Stevens JM, Kendall BE, Crockard HA, Ransford A. The odontoid process in Morquio-Brailsford’s disease. The effects of occipitocervical fusion. J Bone Joint Surg Br. 1991;73:851–8. doi: 10.1302/0301-620X.73B5.1910048. http://www.ncbi.nlm.nih.gov/pubmed/1910048 (accessed August 21, 2017) [DOI] [PubMed] [Google Scholar]
- 82.Baratela WAR, Bober MB, Thacker MM, Belthur MV, Oto M, Rogers KJ, Mackenzie WG. Cervicothoracic Myelopathy in Children With Morquio Syndrome A. J Pediatr Orthop. 2014;34:223–228. doi: 10.1097/BPO.0000000000000074. [DOI] [PubMed] [Google Scholar]
- 83.Eggli KD, Dorst JP. The mucopolysaccharidoses and related conditions. Semin Roentgenol. 1986;21:275–94. doi: 10.1016/0037-198x(86)90039-8. http://www.ncbi.nlm.nih.gov/pubmed/3097828 (accessed September 7, 2017) [DOI] [PubMed] [Google Scholar]
- 84.Dhawale AA, Thacker MM, Belthur MV, Rogers K, Bober MB, Mackenzie WG. The Lower Extremity in Morquio Syndrome. J Pediatr Orthop. 2012;32:534–540. doi: 10.1097/BPO.0b013e318259fe57. [DOI] [PubMed] [Google Scholar]
- 85.Holzgreve W, Gröbe H, von Figura K, Kresse H, Beck H, Mattei JF. Morquio syndrome: clinical findings in 11 patients with MPS IVA and 2 patients with MPS IVB. Hum Genet. 1981;57:360–5. doi: 10.1007/BF00281685. http://www.ncbi.nlm.nih.gov/pubmed/6793501 (accessed August 21, 2017) [DOI] [PubMed] [Google Scholar]
- 86.Dhawale AA, Church C, Henley J, Holmes L, Thacker MM, Mackenzie WG, Miller F. Gait pattern and lower extremity alignment in children with Morquio syndrome. J Pediatr Orthop B. 2013;22:59–62. doi: 10.1097/BPB.0b013e32835a0e6d. [DOI] [PubMed] [Google Scholar]
- 87.White KK, Jester A, Bache CE, Harmatz PR, Shediac R, Thacker MM, Mackenzie WG. Orthopedic management of the extremities in patients with Morquio A syndrome. J Child Orthop. 2014;8:295–304. doi: 10.1007/s11832-014-0601-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Atinga M, Hamer AJ. Total knee replacements in a patient with the Morquio syndrome. J Bone Jt Surg - Br Vol. 2008;90-B:1631–1633. doi: 10.1302/0301-620X.90B12.20641. [DOI] [PubMed] [Google Scholar]
- 89.Hendriksz CJ, Berger KI, Giugliani R, Harmatz P, Kampmann C, Mackenzie WG, Raiman J, Villarreal MS, Savarirayan R. International guidelines for the management and treatment of Morquio A syndrome. Am J Med Genet Part A. 2015;167:11–25. doi: 10.1002/ajmg.a.36833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Borowski A, Thacker MM, Mackenzie WG, Littleton AG, Grissom L. The Use of Computed Tomography to Assess Acetabular Morphology in Morquio-Brailsford Syndrome. J Pediatr Orthop. 2007;27:893–897. doi: 10.1097/bpo.0b013e31815a6007. [DOI] [PubMed] [Google Scholar]
- 91.Brailsford JF. The classics: Chondro-osteo-dystrophy. roentgenographic and clinical features of a child with dislocation of vertebrae. Clin Orthop Relat Res. 1929:404–410. http://www.ncbi.nlm.nih.gov/pubmed/770041 (accessed August 21, 2017) [PubMed]
- 92.Pedrini V, Lennzi L, Zambotti V. Isolation and identification of keratosulphate in urine of patients affected by Morquio-Ullrich disease. Proc Soc Exp Biol Med. 1962;110:847–849. doi: 10.3181/00379727-110-27668. http://www.ncbi.nlm.nih.gov/pubmed/14484871 (accessed October 31, 2017) [DOI] [PubMed] [Google Scholar]
- 93.Hecht JT, Scott CI, Smith TK, Williams JC, Opitz JM. Mild manifestations of the Morquio syndrome. Am J Med Genet. 1984;18:369–371. doi: 10.1002/ajmg.1320180222. [DOI] [PubMed] [Google Scholar]
- 94.Check T. Pectus Carinatum: Pigeon Chest. Surg Technolgists. 2010:495–504. http://www.ast.org/pdf/323.pdf (accessed November 19, 2017)
- 95.Lachman RS, Burton BK, Clarke LA, Hoffinger S, Ikegawa S, Jin DK, Kano H, Kim OH, Lampe C, Mendelsohn NJ, Shediac R, Tanpaiboon P, White KK. Mucopolysaccharidosis IVA (Morquio A syndrome) and VI (Maroteaux-Lamy syndrome): under-recognized and challenging to diagnose. Skeletal Radiol. 2014;43:359–69. doi: 10.1007/s00256-013-1797-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Parashari UC, Khanduri S, Bhadury S, Rawat S. Roentgenographic diagnosis of mucopolysaccharidosis with particular reference to Morquio syndrome. South African J Radiol. 2012;16:32–34. doi: 10.4102/sajr.v16i1.233. [DOI] [Google Scholar]
- 97.Stevens PM. Pediatric Genu Valgum: Background, Anatomy, Pathophysiology, Medscape. 2016 https://emedicine.medscape.com/article/1259772-overview#a9 (accessed January 22, 2018)
- 98.Al Kaissi A, Kenis V, Melchenko E, Ben Ghachem M, Csepan R, Grill F, Ganger R. Corrections of diverse forms of lower limb deformities in patients with mucopolysaccharidosis type IVA (Morquio syndrome) Afr J Paediatr Surg. 2016;13:88–94. doi: 10.4103/0189-6725.182563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ashworth JL, Biswas S, Wraith E, Lloyd IC. Mucopolysaccharidoses and the Eye. Surv Ophthalmol. 2006;51:1–17. doi: 10.1016/j.survophthal.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 100.Hendriksz CJ, Al-Jawad M, Berger KI, Hawley SM, Lawrence R, Mc Ardle C, Summers CG, Wright E, Braunlin E. Clinical overview and treatment options for non-skeletal manifestations of mucopolysaccharidosis type IVA. J Inherit Metab Dis. 2013;36:309–22. doi: 10.1007/s10545-012-9459-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Couprie J, Denis P, Guffon N, Reynes N, Masset H, Beby F. Manifestations ophtalmologiques de la maladie de Morquio. J Fr Ophtalmol. 2010;33:617–622. doi: 10.1016/j.jfo.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 102.Davis DB, Currier FP. Morquio’s disease. J Am Med Assoc. 1934;102:2173. doi: 10.1001/jama.1934.02750260019007. [DOI] [Google Scholar]
- 103.Iwamoto M, Nawa Y, Maumenee IH, Young-Ramsaran J, Matalon R, Green WR. Ocular histopathology and ultrastructure of Morquio syndrome (systemic mucopolysaccharidosis IV A) Graefes Arch Clin Exp Ophthalmol. 1990;228:342–9. doi: 10.1007/BF00920060. http://www.ncbi.nlm.nih.gov/pubmed/2119328 (accessed September 9, 2017) [DOI] [PubMed] [Google Scholar]
- 104.Leslie T, Siddiqui MAR, Aitken DA, Kirkness CM, Lee WR, Fern AI. Morquio syndrome: electron microscopic findings. Br J Ophthalmol. 2005;89:925–926. doi: 10.1136/bjo.2004.055400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Nagao K, Morlet T, Haley E, Padilla J, Nemith J, Mason RW, Tomatsu S. Neurophysiology of hearing in patients with mucopolysaccharidosis type IV. Mol Genet Metab. 2018 doi: 10.1016/J.YMGME.2018.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gökdoğan Ç, Altinyay Ş, Gökdoğan O, Tutar H, Gündüz B, Okur İ, Tümer L, Kemaloğlu YK. Audiologic evaluations of children with mucopolysaccharidosis. Braz J Otorhinolaryngol. 2016;82:281–284. doi: 10.1016/j.bjorl.2015.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Giugliani R, Harmatz P, Wraith JE. Management guidelines for mucopolysaccharidosis VI. Pediatrics. 2007;120:405–18. doi: 10.1542/peds.2006-2184. [DOI] [PubMed] [Google Scholar]
- 108.Riedner ED, Levin LS. Hearing patterns in Morquio’s syndrome (mucopolysaccharidosis IV) Arch Otolaryngol. 1977;103:518–20. doi: 10.1001/archotol.1977.00780260048003. http://www.ncbi.nlm.nih.gov/pubmed/409383 (accessed March 9, 2018) [DOI] [PubMed] [Google Scholar]
- 109.Käsmann-Kellner B, Weindler J, Pfau B, Ruprecht KW. Ocular changes in mucopolysaccharidosis IV A (Morquio A syndrome) and long-term results of perforating keratoplasty. Ophthalmologica. 1999;213:200–5. doi: 10.1159/000027420. [DOI] [PubMed] [Google Scholar]
- 110.James A, Hendriksz CJ, Addison O. The Oral Health Needs of Children. Adolescents and Young Adults Affected by a Mucopolysaccharide Disorder. JIMD Rep. 2011;2:51–58. doi: 10.1007/8904_2011_46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ruckenstein MJ, Macdonald RE, Clarke JT, Forte V. The management of otolaryngological problems in the mucopolysaccharidoses: a retrospective review. J Otolaryngol. 1991;20:177–83. http://www.ncbi.nlm.nih.gov/pubmed/1908026 (accessed March 26, 2018) [PubMed] [Google Scholar]
- 112.Friedmann I, Spellacy E, Crow J, Watts RW. Histopathological studies of the temporal bones in Hurler’s disease [mucopolysaccharidosis (MPS) IH] J Laryngol Otol. 1985;99:29–41. doi: 10.1017/s0022215100096250. http://www.ncbi.nlm.nih.gov/pubmed/3918131 (accessed March 26, 2018) [DOI] [PubMed] [Google Scholar]
- 113.Lin H, Shih S, Chuang C, Lee K, Chen M, Lin H, Chiu P, Niu D, Lin S. Assessment of hearing loss by pure-tone audiometry in patients with mucopolysaccharidoses. Mol Genet Metab. 2014;111:533–538. doi: 10.1016/j.ymgme.2014.02.003. [DOI] [PubMed] [Google Scholar]
- 114.Onçağ G, Ertan Erdinç AM, Cal E. Multidisciplinary treatment approach of Morquio syndrome (Mucopolysaccharidosis Type IVA) Angle Orthod. 2006;76:335–40. doi: 10.1043/0003-3219(2006)076[0335:MTAOMS]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 115.Levin LS, Jorgenson RJ, Salinas CF. Oral findings in the Morquio syndrome (mucopolysaccharidosis IV) Oral Surg Oral Med Oral Pathol. 1975;39:390–5. doi: 10.1016/0030-4220(75)90082-1. http://www.ncbi.nlm.nih.gov/pubmed/803669 (accessed September 4, 2017) [DOI] [PubMed] [Google Scholar]
- 116.Nelson J, Kinirons M. Clinical findings in 12 patients with MPS IV A (Morquio’s disease). Further evidence for heterogeneity. Part II: Dental findings. Clin Genet. 1988;33:121–5. doi: 10.1111/j.1399-0004.1988.tb03422.x. http://www.ncbi.nlm.nih.gov/pubmed/3129222 (accessed September 4, 2017) [DOI] [PubMed] [Google Scholar]
- 117.Ireland MA, Rowlands DB. Mucopolysaccharidosis type IV as a cause of mitral stenosis in an adult. Br Heart J. 1981;46:113–5. doi: 10.1136/hrt.46.1.113. http://www.ncbi.nlm.nih.gov/pubmed/6791672 (accessed August 21, 2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.John RM, Hunter D, Swanton RH. Echocardiographic abnormalities in type IV mucopolysaccharidosis. Arch Dis Child. 1990;65:746–749. doi: 10.1136/adc.65.7.746. http://www.ncbi.nlm.nih.gov/pubmed/2117422 (accessed August 21, 2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Barry MO BA, Beadslee MA. Morquio’s syndrome: severe aortic regurgitation and late pulmonary autograft failure. J Hear Valve Dis. 2006;15:839–842. [PubMed] [Google Scholar]
- 120.Nicolini F, Corradi D, Bosio S, Gherli T. Aortic Valve Replacement in a Patient with Morquio Syndrome. Heart Surg Forum. 2008;11:E96–E98. doi: 10.1532/HSF98.20071197. [DOI] [PubMed] [Google Scholar]
- 121.Pagel PS, Almassi GH. Perioperative implications of Morquio syndrome in a 31-year-old woman undergoing aortic valve replacement. J Cardiothorac Vasc Anesth. 2009;23:855–7. doi: 10.1053/j.jvca.2008.12.009. [DOI] [PubMed] [Google Scholar]
- 122.Leal GN, de Paula AC, Leone C, Kim CA. Echocardiographic study of paediatric patients with mucopolysaccharidosis. Cardiol Young. 2010;20:254–261. doi: 10.1017/S104795110999062X. [DOI] [PubMed] [Google Scholar]
- 123.Chen M, Lin S, Hwang H, Yu C. Cardiovascular changes in mucopolysaccharidoses in Taiwan. Acta Cardiol. 2005;60:51–53. doi: 10.2143/AC.60.1.2005049. [DOI] [PubMed] [Google Scholar]
- 124.Henry WL, Gardin JM, Ware JH. Echocardiographic measurements in normal subjects from infancy to old age. Circulation. 1980;62:1054–61. doi: 10.1161/01.cir.62.5.1054. http://www.ncbi.nlm.nih.gov/pubmed/7418156 (accessed September 2, 2017) [DOI] [PubMed] [Google Scholar]
- 125.Semenza GL, Pyeritz RE. Respiratory complications of mucopolysaccharide storage disorders. Medicine (Baltimore) 1988;67:209–19. doi: 10.1097/00005792-198807000-00002. http://www.ncbi.nlm.nih.gov/pubmed/3134589 (accessed August 21, 2017) [DOI] [PubMed] [Google Scholar]
- 126.Walker PP, Rose E, Williams JG. Upper airways abnormalities and tracheal problems in Morquio’s disease. Thorax. 2003;58:458–9. doi: 10.1136/thorax.58.5.458. http://www.ncbi.nlm.nih.gov/pubmed/12728175 (accessed August 21, 2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Pelley CJ, Kwo J, Hess DR. Tracheomalacia in an adult with respiratory failure and Morquio syndrome. Respir Care. 2007;52:278–82. http://www.ncbi.nlm.nih.gov/pubmed/17328826 (accessed September 9, 2017) [PubMed] [Google Scholar]
- 128.Kubaski F, Tomatsu S, Patel P, Shimada T, Xie L, Yasuda E, Mason R, Mackenzie WG, Theroux M, Bober MB, Oldham HM, Orii T, Shaffer TH. Non-invasive pulmonary function test on Morquio patients. Mol Genet Metab. 2015;115:186–192. doi: 10.1016/j.ymgme.2015.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Harmatz PR, Mengel KE, Giugliani R, Valayannopoulos V, Lin SP, Parini R, Guffon N, Burton BK, Hendriksz CJ, Mitchell JJ, Martins AM, Jones SA, Guelbert N, Vellodi A, Wijburg FA, Yang K, Slasor P, Decker C. Longitudinal analysis of endurance and respiratory function from a natural history study of Morquio A syndrome. Mol Genet Metab. 2015;114:186–194. doi: 10.1016/j.ymgme.2014.10.015. [DOI] [PubMed] [Google Scholar]
- 130.Pellegrino R, Viegi G, Brusasco V, Crapo RO, Burgos F, Casaburi R, Coates A, van der Grinten CPM, Gustafsson P, Hankinson J, Jensen R, Johnson DC, MacIntyre N, McKay R, Miller MR, Navajas D, Pedersen OF, Wanger J. Interpretative strategies for lung function tests. Eur Respir J. 2005;26:948–968. doi: 10.1183/09031936.05.00035205. [DOI] [PubMed] [Google Scholar]
- 131.ATS. Statement: Guidelines for the Six-Minute Walk Test. Am Thorac Soc Am J Respir Crit Care Med. 2002;166:111–117. doi: 10.1164/rccm.166/1/111. [DOI] [PubMed] [Google Scholar]
- 132.Nixon PA, Joswiak ML, Fricker FJ. A six-minute walk test for assessing exercise tolerance in severely ill children. J Pediatr. 1996;129:362–6. doi: 10.1016/s0022-3476(96)70067-7. http://www.ncbi.nlm.nih.gov/pubmed/8804324 (accessed May 7, 2018) [DOI] [PubMed] [Google Scholar]
- 133.Hendriksz CJ, Parini R, AlSayed MD, Raiman J, Giugliani R, Solano Villarreal ML, Mitchell JJ, Burton BK, Guelbert N, Stewart F, Hughes DA, Berger KI, Slasor P, Matousek R, Jurecki E, Shaywitz AJ, Harmatz PR. Long-term endurance and safety of elosulfase alfa enzyme replacement therapy in patients with Morquio A syndrome. Mol Genet Metab. 2016;119:131–143. doi: 10.1016/j.ymgme.2016.05.018. [DOI] [PubMed] [Google Scholar]
- 134.Hendriksz CJ, Burton B, Fleming TR, Harmatz P, Hughes D, Jones SA, Lin SP, Mengel E, Scarpa M, Valayannopoulos V, Giugliani R, Slasor P, Lounsbury D, Dummer W, Dummer W. Efficacy and safety of enzyme replacement therapy with BMN 110 (elosulfase alfa) for Morquio A syndrome (mucopolysaccharidosis IVA): a phase 3 randomised placebo-controlled study. J Inherit Metab Dis. 2014;37:979–990. doi: 10.1007/s10545-014-9715-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Butland RJ, Pang JA, Geddes DM. Carbimazole and exercise tolerance in chronic airflow obstruction. Thorax. 1982;37:64–7. doi: 10.1136/thx.37.1.64. http://www.ncbi.nlm.nih.gov/pubmed/7041324 (accessed September 4, 2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.McDonald A, Steiner R, Kuehl K, Turbeville S. Clinical utility of endurance measures for evaluation of treatment in patients with mucopolysaccharidosis VI (Maroteaux-Lamy syndrome) J Pediatr Rehabil Med. 2010;3:119–27. doi: 10.3233/PRM-2010-0114. [DOI] [PubMed] [Google Scholar]
- 137.Mocellin R. Exercise testing in children with congenital heart disease. Pediatrician. 1986;13:18–25. http://www.ncbi.nlm.nih.gov/pubmed/3534827 (accessed September 4, 2017) [PubMed] [Google Scholar]
- 138.Regier DS, Tanpaiboon P. Role of elosulfase alfa in mucopolysaccharidosis IVA. Appl Clin Genet. 2016;9:67–74. doi: 10.2147/TACG.S69080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Aslam R, van Bommel ACM, Hendriksz CJ, Jester A. Subjective and Objective Assessment of Hand Function in Mucopolysaccharidosis IVa Patients, in. JIMD Rep. 2012:59–65. doi: 10.1007/8904_2012_179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.KK W. Orthopaedic surgery for mucopolysaccharidosis. Curr Orthop Pr. 2012;23:394–399. [Google Scholar]
- 141.Fung EB, Johnson JA, Madden J, Kim T, Harmatz P. Bone density assessment in patients with mucopolysaccharidosis: A preliminary report from patients with MPS II and VI. J Pediatr Rehabil Med. 2010;3:13–23. http://www.ncbi.nlm.nih.gov/pubmed/20617160 (accessed September 17, 2017) [PMC free article] [PubMed] [Google Scholar]
- 142.Rigante D, Buonuomo PS, Caradonna P. Early-onset osteoporosis with high bone turnover in children with Morquio-Brailsford syndrome. Rheumatol Int. 2006;26:1163–1164. doi: 10.1007/s00296-006-0150-3. [DOI] [PubMed] [Google Scholar]
- 143.Rigante D, Caradonna P. Secondary skeletal involvement in Sanfilippo syndrome. QJM. 2004;97:205–9. doi: 10.1093/qjmed/hch041. http://www.ncbi.nlm.nih.gov/pubmed/15028850 (accessed September 17, 2017) [DOI] [PubMed] [Google Scholar]
- 144.Turner AS, Norrdin RW, Gaarde S, Connally HE, Thrall MA. Bone mineral density in feline mucopolysaccharidosis VI measured using dual-energy X-ray absorptiometry. Calcif Tissue Int. 1995;57:191–5. doi: 10.1007/BF00310257. http://www.ncbi.nlm.nih.gov/pubmed/8574935 (accessed September 17, 2017) [DOI] [PubMed] [Google Scholar]
- 145.Kecskemethy HH, Kubaski F, Harcke HT, Tomatsu S. Bone mineral density in MPS IV A (Morquio syndrome type A) Mol Genet Metab. 2016;117:144–149. doi: 10.1016/j.ymgme.2015.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Szalay EA, Cheema A. Children with Spina Bifida are at Risk for Low Bone Density. Clin Orthop Relat Res. 2011;469:1253–1257. doi: 10.1007/s11999-010-1634-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Henderson RC, Berglund LM, May R, Zemel BS, Grossberg RI, Johnson J, Plotkin H, Stevenson RD, Szalay E, Wong B, Kecskemethy HH, Harcke HT. The relationship between fractures and DXA measures of BMD in the distal femur of children and adolescents with cerebral palsy or muscular dystrophy. J Bone Miner Res. 2010;25:520–526. doi: 10.1359/jbmr.091007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Henderson RC, Lark RK, Gurka MJ, Worley G, Fung EB, Conaway M, Stallings VA, Stevenson RD. Bone density and metabolism in children and adolescents with moderate to severe cerebral palsy. Pediatrics. 2002;110:e5. doi: 10.1542/peds.110.1.e5. http://www.ncbi.nlm.nih.gov/pubmed/12093986 (accessed October 12, 2017) [DOI] [PubMed] [Google Scholar]
- 149.Harcke HT, Kecskemethy HH, Conklin D, Scavina M, Mackenzie WG, McKay CP. Assessment of Bone Mineral Density in Duchenne Muscular Dystrophy Using the Lateral Distal Femur. J Clin Neuromuscul Dis. 2006;8:1–6. doi: 10.1097/01.cnd.0000244623.06318.23. [DOI] [Google Scholar]
- 150.Haas RE, Kecskemethy HH, Lopiccolo MA, Hossain J, Dy RT, Bachrach SJ. Lower extremity bone mineral density in children with congenital spinal dysfunction. Dev Med Child Neurol. 2012;54:1133–1137. doi: 10.1111/j.1469-8749.2012.04420.x. [DOI] [PubMed] [Google Scholar]
- 151.Gogola GR, Velleman PF, Xu S, Morse AM, Lacy B, Aaron D. Hand Dexterity in Children: Administration and Normative Values of the Functional Dexterity Test. J Hand Surg Am. 2013;38:2426–2431. doi: 10.1016/j.jhsa.2013.08.123. [DOI] [PubMed] [Google Scholar]
- 152.Aaron DH, Jansen CWS. Development of the Functional Dexterity Test (FDT): construction, validity, reliability, and normative data. J Hand Ther. 16:12–21. doi: 10.1016/s0894-1130(03)80019-4. n.d. http://www.ncbi.nlm.nih.gov/pubmed/12611441 (accessed September 7, 2017) [DOI] [PubMed] [Google Scholar]
- 153.Wren TAL, Gorton GE, Õunpuu S, Tucker CA. Efficacy of clinical gait analysis: A systematic review. Gait Posture. 2011;34:149–153. doi: 10.1016/j.gaitpost.2011.03.027. [DOI] [PubMed] [Google Scholar]
- 154.Yasuda E, Suzuki Y, Shimada T, Sawamoto K, Mackenzie WG, Theroux MC, Pizarro C, Xie L, Miller F, Rahman T, Kecskemethy HH, Nagao K, Morlet T, Shaffer TH, Chinen Y, Yabe H, Tanaka A, Shintaku H, Orii KE, Orii KO, Mason RW, Montaño AM, Fukao T, Orii T, Tomatsu S. Activity of daily living for Morquio A syndrome. Mol Genet Metab. 2016;118:111–122. doi: 10.1016/j.ymgme.2016.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Melbouci M, Mason RW, Suzuki Y, Fukao T, Orii T, Tomatsu S. Growth impairment in mucopolysaccharidoses. Mol Genet Metab. 2018 doi: 10.1016/j.ymgme.2018.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Tomatsu S, Montaño AM, Oikawa H, Giugliani R, Harmatz P, Smith M, Suzuki Y, Orii T. Impairment of Body Growth in Mucopolysaccharidoses. Handb Growth Growth Monit Heal Dis. 2012;1:2091–2117. doi: 10.1007/978-1-4419-1795-9_126. [DOI] [Google Scholar]
- 157.Harmatz P, Mengel KE, Giugliani R, Valayannopoulos V, Lin SP, Parini R, Guffon N, Burton BK, Hendriksz CJ, Mitchell J, Martins A, Jones S, Guelbert N, Vellodi A, Hollak C, Slasor P, Decker C. The Morquio A Clinical Assessment Program: Baseline results illustrating progressive, multisystemic clinical impairments in Morquio A subjects. Mol Genet Metab. 2013;109:54–61. doi: 10.1016/j.ymgme.2013.01.021. [DOI] [PubMed] [Google Scholar]
- 158.Montaño AM, Tomatsu S, Brusius A, Smith M, Orii T. Growth charts for patients affected with Morquio A disease. Am J Med Genet Part A. 2008;146A:1286–1295. doi: 10.1002/ajmg.a.32281. [DOI] [PubMed] [Google Scholar]
- 159.Schleier E, Streubel H, Chen M, Chiu P, Ke Y, Niu D, Tsai F, Hwu W, Lin J, Lin S. Phoniatrische Aspekte bei Kindern mit Mukopolysaccharidose. Folia Phoniatr Logop. 1976;28:65–72. doi: 10.1159/000264033. [DOI] [PubMed] [Google Scholar]
- 160.Hendriksz CJ, Giugliani R, Harmatz P, Mengel E, Guffon N, Valayannopoulos V, Parini R, Hughes D, Pastores GM, Lau HA, Al-Sayed MD, Raiman J, Yang K, Mealiffe M, Haller C, Haller C. Multi-domain impact of elosulfase alfa in Morquio A syndrome in the pivotal phase III trial. Mol Genet Metab. 2015;114:178–185. doi: 10.1016/j.ymgme.2014.08.012. [DOI] [PubMed] [Google Scholar]
- 161.Baker E, Guo XH, Orsborn AM, Sutherland GR, Callen DF, Hopwood JJ, Morris CP. The morquio A syndrome (mucopolysaccharidosis IVA) gene maps to 16q24.3. Am J Hum Genet. 1993;52:96–8. http://www.ncbi.nlm.nih.gov/pubmed/8434612 (accessed September 11, 2017) [PMC free article] [PubMed] [Google Scholar]
- 162.Masuno M, Tomatsu S, Nakashima Y, Hori T, Fukuda S, Masue M, Sukegawa K, Orii T. Mucopolysaccharidosis IV A: Assignment of the Human N-Acetylgalactosamine-6-Sulfate Sulfatase (GALNS) Gene to Chromosome 16q24. Genomics. 1993;16:777–778. doi: 10.1006/geno.1993.1266. [DOI] [PubMed] [Google Scholar]
- 163.Tomatsu S, Fukuda S, Masue M, Sukegawa K, Fukao T, Yamagishi A, Hori T, Iwata H, Ogawa T, Nakashima Y. Morquio disease: isolation, characterization and expression of full-length cDNA for human N-acetylgalactosamine-6-sulfate sulfatase. Biochem Biophys Res Commun. 1991;181:677–83. doi: 10.1016/0006-291x(91)91244-7. http://www.ncbi.nlm.nih.gov/pubmed/1755850 (accessed September 11, 2017) [DOI] [PubMed] [Google Scholar]
- 164.Nakashima Y, Tomatsu S, Hori T, Fukuda S, Sukegawa K, Kondo N, Suzuki Y, Shimozawa N, Orii T. Mucopolysaccharidosis IV A: Molecular Cloning of the Human N-Acetylgalactosamine-6-sulfatase Gene (GALNS) and Analysis of the 5′-Flanking Region. Genomics. 1994;20:99–104. doi: 10.1006/geno.1994.1132. [DOI] [PubMed] [Google Scholar]
- 165.Cooper DN, Ball EV, Stenson PD, Phillips AD, Evans K, Heywood S, Hayden MJ, Azevedo L, Mort ME, Hussain M. The Human Gene Mutation Database. 2018 n.d. http://www.hgmd.cf.ac.uk/ac/gene.php?gene=GALNS (accessed February 7, 2018)
- 166.Kato Z, Fukuda S, Tomatsu S, Vega H, Yasunaga T, Yamagishi A, Yamada N, Valencia A, Barrera LA, Sukegawa K, Orii T, Kondo N. A novel common missense mutation G301C in the N-acetylgalactosamine-6-sulfate sulfatase gene in mucopolysaccharidosis IVA. Hum Genet. 1997;101:97–101. doi: 10.1007/s004390050594. http://www.ncbi.nlm.nih.gov/pubmed/9385378 (accessed October 12, 2017) [DOI] [PubMed] [Google Scholar]
- 167.Montaño AM, Kaitila I, Sukegawa K, Tomatsu S, Kato Z, Nakamura H, Fukuda S, Orii T, Kondo N. Mucopolysaccharidosis IVA: characterization of a common mutation found in Finnish patients with attenuated phenotype. Hum Genet. 2003;113:162–9. doi: 10.1007/s00439-003-0959-8. [DOI] [PubMed] [Google Scholar]
- 168.Tomatsu S, Dieter T, Schwartz IV, Sarmient P, Giugliani R, Barrera LA, Guelbert N, Kremer R, Repetto GM, Gutierrez MA, Nishioka T, Serrato OP, Montaño AM, Yamaguchi S, Noguchi A. Identification of a common mutation in mucopolysaccharidosis IVA: correlation among genotype, phenotype, and keratan sulfate. J Hum Genet. 2004;49:490–4. doi: 10.1007/s10038-004-0178-8. [DOI] [PubMed] [Google Scholar]
- 169.Tomatsu S, Filocamo M, Orii KO, Sly WS, Gutierrez MA, Nishioka T, Serrato OP, Di Natale P, Montaño AM, Yamaguchi S, Kondo N, Orii T, Noguchi A. Mucopolysaccharidosis IVA (Morquio A): identification of novel common mutations in the N-acetylgalactosamine-6-sulfate sulfatase (GALNS) gene in Italian patients. Hum Mutat. 2004;24:187–8. doi: 10.1002/humu.9265. [DOI] [PubMed] [Google Scholar]
- 170.Tomatsu S, Fukuda S, Cooper A, Wraith JE, Rezvi GM, Yamagishi A, Yamada N, Kato Z, Isogai K, Sukegawa K. Mucopolysaccharidosis type IVA: identification of six novel mutations among non-Japanese patients. Hum Mol Genet. 1995;4:741–3. doi: 10.1093/hmg/4.4.741. http://www.ncbi.nlm.nih.gov/pubmed/7633425 (accessed October 12, 2017) [DOI] [PubMed] [Google Scholar]
- 171.Yamada N, Fukuda S, Tomatsu S, Muller V, Hopwood JJ, Nelson J, Kato Z, Yamagishi A, Sukegawa K, Kondo N, Orii T. Molecular heterogeneity in mucopolysaccharidosis IVA in Australia and Northern Ireland: nine novel mutations including T312S, a common allele that confers a mild phenotype. Hum Mutat. 1998;11:202–8. doi: 10.1002/(SICI)1098-1004(1998)11:3<202::AID-HUMU4>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 172.Tomatsu S, Nishioka T, Montaño AM, Gutierrez MA, Pena OS, Orii KO, Sly WS, Yamaguchi S, Orii T, Paschke E, Kircher SG, Noguchi A. Mucopolysaccharidosis IVA: identification of mutations and methylation study in GALNS gene. J Med Genet. 2004;41:e98. doi: 10.1136/jmg.2003.018010. http://www.ncbi.nlm.nih.gov/pubmed/15235041 (accessed October 12, 2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Caciotti A, Tonin R, Rigoldi M, Ferri L, Catarzi S, Cavicchi C, Procopio E, Donati MA, Ficcadenti A, Fiumara A, Barone R, Garavelli L, Di Rocco M, Filocamo M, Antuzzi D, Scarpa M, Mooney SD, Li B, Skouma A, Bianca S, Concolino D, Casalone R, Monti E, Pantaleo M, Giglio S, Guerrini R, Parini R, Morrone A. Optimizing the molecular diagnosis of GALNS: novel methods to define and characterize Morquio-A syndrome-associated mutations. Hum Mutat. 2015;36:357–68. doi: 10.1002/humu.22751. [DOI] [PubMed] [Google Scholar]
- 174.Maroteaux P, Leveque B, Marie J, Lamy M. A new dysostosis with urinary elimination of chonroitin sulfate B. Presse Med. 1963;71:1849–52. http://www.ncbi.nlm.nih.gov/pubmed/14091597 (accessed October 29, 2017) [PubMed] [Google Scholar]
- 175.Valayannopoulos V, Nicely H, Harmatz P, Turbeville S. Mucopolysaccharidosis VI. Orphanet J Rare Dis. 2010;5(5):1–20. doi: 10.1186/1750-1172-5-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Tessitore A, Pirozzi M, Auricchio A. Abnormal autophagy, ubiquitination, inflammation and apoptosis are dependent upon lysosomal storage and are useful biomarkers of mucopolysaccharidosis VI. Pathogenetics. 2009;2(4):1–12. doi: 10.1186/1755-8417-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Vairo F, Federhen A, Baldo G, Riegel M, Burin M, Leistner-Segal S, Giugliani R. Diagnostic and treatment strategies in mucopolysaccharidosis VI. Appl Clin Genet. 2015;8:245–55. doi: 10.2147/TACG.S68650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Montaño AM, Lock-Hock N, Steiner RD, Graham BH, Szlago M, Greenstein R, Pineda M, Gonzalez-Meneses A, Çoker M, Bartholomew D, Sands MS, Wang R, Giugliani R, Macaya A, Pastores G, Ketko AK, Ezgü F, Tanaka A, Arash L, Beck M, Falk RE, Bhattacharya K, Franco J, White KK, Mitchell GA, Cimbalistiene L, Holtz M, Sly WS. Clinical course of sly syndrome (mucopolysaccharidosis type VII) J Med Genet. 2016;53:403–18. doi: 10.1136/jmedgenet-2015-103322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Delbecque K, Gaillez S, Schaaps JP. Histopathological diagnosis of a type VII mucopolysaccharidosis after pregnancy termination. Fetal Pediatr Pathol. 2009;28:1–8. doi: 10.1080/15513810802547943. [DOI] [PubMed] [Google Scholar]
- 180.Lew V, Pena L, Edwards R, Wang RY. Cardiovascular Histopathology of a 11-Year Old with Mucopolysaccharidosis VII Demonstrates Fibrosis, Macrophage Infiltration, and Arterial Luminal Stenosis. JIMD Rep. 2017:1–7. doi: 10.1007/8904_2017_43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Metcalf JA, Zhang Y, Hilton MJ, Long F, Ponder KP. Mechanism of shortened bones in mucopolysaccharidosis VII. Mol Genet Metab. 2009;97:202–211. doi: 10.1016/j.ymgme.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Caciotti A, Garman SC, Rivera-Colón Y, Procopio E, Catarzi S, Ferri L, Guido C, Martelli P, Parini R, Antuzzi D, Battini R, Sibilio M, Simonati A, Fontana E, Salviati A, Akinci G, Cereda C, Dionisi-Vici C, Deodato F, d’Amico A, d’Azzo A, Bertini E, Filocamo M, Scarpa M, di Rocco M, Tifft CJ, Ciani F, Gasperini S, Pasquini E, Guerrini R, Donati MA, Morrone A. GM1 gangliosidosis and Morquio B disease: an update on genetic alterations and clinical findings. Biochim Biophys Acta. 2011;1812:782–90. doi: 10.1016/j.bbadis.2011.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Regier DS, Tifft CJ. GLB1-Related Disorders. University of Washington; Seattle: 1993. http://www.ncbi.nlm.nih.gov/pubmed/24156116 (accessed August 21, 2017) [PubMed] [Google Scholar]
- 184.Morrone A, Bardelli T, Donati MA, Giorgi M, Di Rocco M, Gatti R, Parini R, Ricci R, Taddeucci G, D’Azzo A, Zammarchi E. ?-galactosidase gene mutations affecting the lysosomal enzyme and the elastin-binding protein in GM1-gangliosidosis patients with cardiac involvement. Hum Mutat. 2000;15:354–366. doi: 10.1002/(SICI)1098-1004(200004)15:4<354::AID-HUMU8>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 185.Dahiya R, Cleveland S, Megerian CA. Spondyloepiphyseal dysplasia congenita associated with conductive hearing loss. Ear Nose Throat J. 2000;79:178–82. http://www.ncbi.nlm.nih.gov/pubmed/10743764 (accessed November 28, 2017) [PubMed] [Google Scholar]
- 186.Soultanis KC, Payatakes AH, Chouliaras VT, Mandellos GC, Pyrovolou NE, Pliarchopoulou FM, Soucacos PN. Rare causes of scoliosis and spine deformity: experience and particular features. Scoliosis. 2007;2:15. doi: 10.1186/1748-7161-2-15. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 187.Zhang Z, JW H, WZ F, Zhang C, Zhang Z. Identification of three novel mutations in the COL2A1 gene in four unrelated Chinese families with spondyloepiphyseal dysplasia congenita. Biochem Biophys Res Commun. 2011;413:504–508. doi: 10.1016/J.BBRC.2011.08.090. [DOI] [PubMed] [Google Scholar]
- 188.Ehtesham N, Cantor RM, King LM, Reinker K, Powell BR, Shanske A, Unger S, Rimoin DL, Cohn DH. Evidence that Smith-McCort dysplasia and Dyggve-Melchior-Clausen dysplasia are allelic disorders that result from mutations in a gene on chromosome 18q12. Am J Hum Genet. 2002;71:947–51. doi: 10.1086/342669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Tercier S, Shah H, Siddesh ND, Joseph B. Does proximal femoral varus osteotomy in Legg-Calvé-Perthes disease predispose to angular mal-alignment of the knee? A clinical and radiographic study at skeletal maturity. J Child Orthop. 2013;7:205–11. doi: 10.1007/s11832-013-0487-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Miyamoto Y, Matsuda T, Kitoh H, Haga N, Ohashi H, Nishimura G, Ikegawa S. A recurrent mutation in type II collagen gene causes Legg-Calvé-Perthes disease in a Japanese family. Hum Genet. 2007;121:625–629. doi: 10.1007/s00439-007-0354-y. [DOI] [PubMed] [Google Scholar]
- 191.Loder RT, Skopelja EN. The Epidemiology and Demographics of Legg-Calvé-Perthes’ Disease. ISRN Orthop. 2011;2011:1–14. doi: 10.5402/2011/504393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Cohn DH, Ehtesham N, Krakow D, Unger S, Shanske A, Reinker K, Powell BR, Rimoin DL. Mental Retardation and Abnormal Skeletal Development (Dyggve-Melchior-Clausen Dysplasia) Due to Mutations in a Novel, Evolutionarily Conserved Gene. Am J Hum Genet. 2003;72:419–428. doi: 10.1086/346176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.El Ghouzzi V, Dagoneau N, Kinning E, Thauvin-Robinet C, Chemaitilly W, Prost-Squarcioni C, Al-Gazali LI, Verloes A, Le Merrer M, Munnich A, Trembath RC, Cormier-Daire V. Mutations in a novel gene Dymeclin (FLJ20071) are responsible for Dyggve-Melchior-Clausen syndrome. Hum Mol Genet. 2003;12:357–64. doi: 10.1093/hmg/ddg029. http://www.ncbi.nlm.nih.gov/pubmed/12554689 (accessed September 9, 2017) [DOI] [PubMed] [Google Scholar]
- 194.Dyggve HV, Melchior JC, Clausen J. Morquio-Ullrich’s Disease: An Inborn Error of Metabolism? Arch Dis Child. 1962;37:525–34. doi: 10.1136/adc.37.195.525. http://www.ncbi.nlm.nih.gov/pubmed/21032395 (accessed September 9, 2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.İlkay Koray Bayrak HBD, Selim Nural Mehmet. Dyggve-Melchior-Clausen syndrome without mental retardation (Smith-McCort dysplasia) Turkish Soc Radiol. 2005;11:163–165. http://www.dirjournal.org/sayilar/6/buyuk/pdf_DIR_37.pdf (accessed November 28, 2017) [PubMed] [Google Scholar]
- 196.Seven M, Koparir E, Gezdirici A, Aydin H, Skladny H, Fenercioğlu E, Güven G, Karataş ÖF, Koparir A, Özen M, Ulucan H. A novel frameshift mutation and infrequent clinical findings in two cases with Dyggve–Melchior–Clausen syndrome. Clin Dysmorphol. 2014;23:1–7. doi: 10.1097/MCD.0000000000000020. [DOI] [PubMed] [Google Scholar]
- 197.Aglan MS, Temtamy SA, Fateen E, Ashour AM, Eldeeb K, Hosny GA. Dyggve-Melchior-Clausen syndrome: clinical, genetic, and radiological study of 15 Egyptian patients from nine unrelated families. J Child Orthop. 2009;3:451–8. doi: 10.1007/s11832-009-0211-8. [DOI] [PMC free article] [PubMed] [Google Scholar]