

Abstract
Arginine is a semi-essential amino acid which serves as a substrate for nitric oxide (NO) production by nitric oxide synthase (NOS) and a precursor for various metabolites including ornithine, creatine, polyamines, and agmatine. Arginase competes with nitric oxide synthase for substrate arginine to produce orthinine and urea. There is contradictory evidence in the literature on the role of nitric oxide in the pathophysiology of traumatic brain injury (TBI). These contradictory perspectives are likely due to different NOS isoforms - endothelial (eNOS), inducible (iNOS) and neuronal (nNOS) which are expressed in the central nervous system. Of these, the role of nNOS in acute injury remains less clear. This study aimed to employ a genetic approach by overexpressing arginase isoforms specifically in neurons using a Thy-1 promoter to manipulate cell autonomous NO production in the context of TBI. The hypothesis was that increased arginase would divert arginine from pathological NO production. We generated 2 mouse lines that overexpress arginase I (a cytoplasmic enzyme) or arginase II (a mitochondrial enzyme) in neurons of FVB mice. We found that two-weeks after induction of controlled cortical injury, overexpressing arginase I but not arginase II in neurons significantly reduced contusion size and contusion index compared to wild-type (WT) mice. This study establishes enhanced neuronal arginase levels as a strategy to affect the course of TBI and provides support for the potential role of neuronal NO production in this condition.
Keywords: Traumatic Brain Injury, Arginine, Nitric Oxide, Arginase, Nitric oxide synthase
Introduction
Arginine is a semi-essential amino acid that serves as a precursor for several metabolites including ornithine, agmatine, creatine, polyamines and nitric oxide (NO). These metabolites have a wide range of physiological functions including modulation of cerebral blood flow, energy production, regeneration and extracellular matrix remodeling. These processes influence outcomes following traumatic brain injury by protecting or repairing the brain. Interestingly, severe traumatic brain injury patients have reduced levels of plasma arginine and alterations in several of its downstream metabolites.1 Administration of L-arginine post induction of controlled cortical injury in rats has been shown to increase cerebral blood flow and reduce contusion volume.2 Studies have shown that this effect of L-arginine administration is due to increased NO production and is absent in mice deficient in endothelial nitric oxide synthase (eNOS), an enzyme that utilizes arginine to produce NO.3, 4
In addition to endothelial NO, several reports in the literature have implicated neuronal or glial NO in traumatic brain injury (TBI). However, the literature reveals conflicting data on whether NO has neurotoxic or neuroprotective effect post-TBI. For instance, administration of L-NAME, an inhibitor of all forms of NOS, is associated with both positive and adverse outcomes in TBI.5,6 Inhalation of nitric oxide post-TBI has been shown to increase cerebral blood flow (CBF) and prevent ischemia in C57/BL6 mice.7 The same study showed that after prolonged exposure to inhaled NO, the mice had reduced lesion volume and improved neurological function. On the contrary, another study has shown that higher levels of NO metabolites in extracellular fluid adjacent to neurons, correlates with poorer survival in TBI patients.8 Moreover, a placebo controlled randomized phase II trial in humans for nitric oxide synthase inhibitor, VAS203, resulted in better Glasgow Scale Outcomes for patients 6 months after administration.9 These contradictory findings may be due in part to the spatial and temporal differences in NO production.10 NO can be generated from arginine by three isoforms of the enzyme nitric oxide synthase (NOS), which has cell-specific expression. Endothelial NOS (eNOS) is expressed in vascular endothelium and the choroid plexus, neuronal NOS (nNOS) is expressed in neurons while inducible NOS (iNOS) is expressed in macrophages and glial cells. While multiple studies have been performed to understand the role of eNOS on CBF, the role of NO derived from nNOS is underreported and still widely debated. The aim of this study was to understand the role of arginine, and consequently NO, in neurons after TBI.
In addition to lower plasma levels of L-arginine post-TBI, multiple studies have also reported an increase in plasma arginase levels and activity.11 Increased arginase levels and activity has also been noted in different tissues such as mesenteric arteries and mononuclear cells.12, 13 Arginase is an enzyme that utilizes arginine as a substrate to generate ornithine and urea. It competes with NOS for utilization of arginine. Genetic modulation of arginase serves as a useful tool to manipulate arginine levels in a cell-specific manner. Therefore, we generated genetic mouse models that overexpress arginase only in neuronal cells. The rationale is that overexpression of arginase in neurons would channel arginine to ornithine and urea production, away from nitric oxide synthesis, thereby lending insight into the role neuronal nitric oxide and its effect on TBI.
Materials and Methods
All animal experiments performed in this study followed the National Institute of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee at Baylor College of Medicine.
Animal Models
In order to generate the animal models needed in this study, we used two different constructs. For the neuronal overexpression lines, we generated the constructs by cloning either Arginase I or Arginase II cDNA under the promoter of the neuronal Thy-1 gene. Both constructs also contain the tyrosinase gene under the control of the K14 (keratinocyte specific promoter), to detect insertion of the construct using eye color change, as well as insulators. These constructs were then injected into embryos of FVB/N mice. Founders were bred with WT FVB females to establish and maintain the lines as heterozygous transgenic mice. To confirm presence of the transgene, animals were genotyped using DNA isolated from 1–2mm tail clippings. Polymerase chain reaction was performed for 30 cycles (1 min denaturing, 1 min annealing at 58° Celcius and 1 min elongation at 72° Celcius) annealing temperature for 1min, followed by running the product on a gel. For all experimental procedures described, both male and female mice were used
Western Blotting
Expression of Arginase I or II protein was confirmed by Western blotting. Whole brain tissues from neuronal arginase overexpressing mice were used to prepare protein lysates. Brain lysates from WT mice were used for comparison. Protein lysates from liver and kidney tissues of wild-type (WT) mice were used as positive controls for Arginase I and Arginase II expression respectively. Lysates from WT brain tissues were used as negative control. Antibodies for arginase I and II were purchased from Santa Cruz Biotechnology (catalog numbers sc-18354 and sc-20151 respectively).
Enzyme Activity Assay
Arginase enzyme activity assay was performed using the technique described by Zawada et.al.14 Mouse brains were homogenized for 10 seconds at speed 6 in Ultra-turrax homogenizer in a buffer composed of 50mM Tris-HCl pH7.5, 150mM NaCl, 1% Triton X and phenylmethylsufonyl fluoride protease inhibitor. Proteins were quantified using the microBCA Protein Assay Kit from Thermo Scientific Pierce (catalog number 23235). The following components were mixed for the reaction: 200ug extracted brain protein, 50mM arginine, in 10mM potassium phosphate buffer (pH 8.5) for a total of 100ul. The reaction was performed at 37° Celsius for 30 minutes and stopped by boiling. Precipitated proteins were then spun down and the supernatant was collected. Urea was quantified by adding 150ul of primaquine/o-phtalaldehyde solution and reading at 450 nm after 30 minutes, in parallel with a standard curve for urea.
Amino Acid Measurements
Brain arginine and ornithine levels were measured in transgenic and WT mice using a Biochrom 30 HPLC amino acid analyzer. In brief 30–50ul of tissue homogenate was mixed with equal volumes of Seraprep SP100 (Pickering Laboratories) and lithium dilution buffer. Protein was precipitated by centrifugation and 10ul supernatant was injected to the analyzer. Physiological amino acid standards (Sigma-Aldridge) were used to calibrate and determine analyte concentration. Results were analyzed using EZchrom Elite software.
Surgical Preparation
The mice were anesthetized with 5% isoflurane in 100% oxygen in a vented anesthesia chamber and maintained on 2% isoflurane for the duration of the experiment using a volume-controlled ventilator (Hugo Sachs Elektronik-Harvard Apparatus, March-Hugstetten - Germany) adjusted to obtain and end-tidal C02 level [EtC02] between 35–40 mmHg monitored with microcapnography (Columbus Instruments, Columbus, OH, USA). Rectal temperature was maintained between 36 to 37 °C using a heat pad and a rectal thermistor. Blood pressure was monitored via a femoral artery cannula.
Controlled Cortical Impact
Controlled cortical impact (CCI) injury was produced as previously described.15 Briefly, after craniectomy and dural exposure, the 3 mm diameter impactor tip was positioned perpendicular to the exposed surface of the brain at an angle of approximately 45 degrees to the vertical. CCI (3 m/sec, 1.5 mm deformation, 100 msec of duration) was performed using a voltage driven impactor (Benchmark Stereotaxic Impactor, myNeuroLab, St Louis, MO, USA).
Histology
Intact mouse brains were collected two weeks post-injury. First, the mice were anesthetized and then perfused with phosphate-buffered saline followed by 10% formalin. Brains were harvested from the mice cut in half and fixed in 10% formalin. The paraffin blocks were sectioned in slices of 6 microns and mounted on slides.
For H&E staining for contusion volume measurements, samples were de-paraffinized and rehydrated with reducing concentration of ethanol, rinsed with distilled water and stained with Mayer hematoxylin solution. Next, samples with treated with 1% sodium bicarbonate for 1 minute and then rinsed with distilled water followed by 80% ethanol before counterstaining with eosin-y solution for 1 minute. Last, samples were dehydrated with increasing concentrations of ethanol, and treated with xylene twice for two minutes each and mounted with a xylene based medium. For neuron morphology, Nissl staining on cortical sections and cerebellar sections was performed after deparaffinization and rehydration. A 0.1% cressyl violet solution was used for the Nissl staining.
Pathological Analysis: Contusion volume and contusion index
The whole slide H&E stained sections were scanned using a Pathscan Enabler. The contusion volume (CV) was determined by multiplying the cross-sectional area of injury in each coronal image and by the thickness of the tissue between the slices. This slab volume technique was implemented on the image processing program Optimas 5.2 (Optimas Corp., Seattle, WA). The contusion index (CI) is a measure of contusion severity and is determined by the degree of parenchymal penetration of a contusion through cortex and underlying white matter as follows: no contusion CI = 0, contusion present but not extending through the full thickness of the cortex CI = 1, contusion present and extends through the full thickness of the cortex CI = 2, contusion present extending through the full thickness of the cortex and into the interdigitating white matter CI = 3, and contusion extending through the full thickness of the cortex and into the deep white matter CI = 4.16 The contusion volume and contusion index provide two measures of contusion severity. It is possible to have a high volume but superficial contusion which would have a large CV and low CI, but it is also possible to have an intermediate CV with a high CI. All contusion volume and index measurements were performed by a trained neuropathologist who was blinded to the genotypes of the mice.
Statistical Analysis
Statistical analysis was performed using R software. For quantitative data like arginine levels, ornithine levels and contusion volumes, we used Welch’s t-test for unequal variances. For qualitative data such as contusion score, we used the Kruskal- Wallis rank sum test. All error bars shown in the graphs represent standard deviation.
Results
Generation and characterization of neuronal arginase overexpression mice
We generated transgenic constructs for two neuronal mouse lines overexpressing Arginase I and Arginase II, respectively (Thy1-ArgI and Thy1-ArgII, Figure 1). Western blotting was performed to measure expression of Arginase I or Arginase II in whole brain lysate. Protein lysate from a WT mouse liver was used as positive control for Arginase I expression and negative control for Arginase II expression. Conversely, protein lysate from a WT mouse kidney was used as positive control for Arginase II expression but negative control for Arginase I expression. Neuronal line Thy1-ArgI showed overexpression of Arginase I in brain lysates in comparison to WT brain lysates (Figure 2A), while neuronal line Thy1-ArgII showed overexpression of Arginase II in brain lysates in comparison to WT brain lysate (Figure 2B). Enzyme activity assay results confirmed higher brain arginase activity in Thy1-ArgI and Thy1-ArgII compared to WT mice (Figure 3). Arginase activity in WT liver was measured as a positive control.
Figure 1.
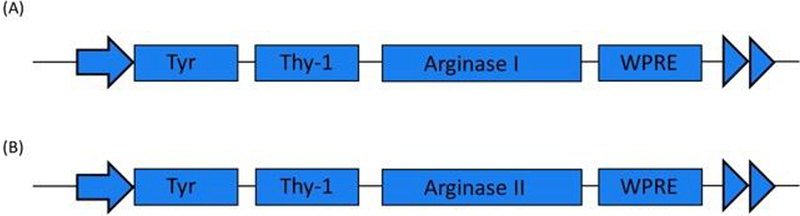
(A) and (B) show constructs used for generation of the neuronal arginase I or arginase II overexpression mouse lines respectively. The transgenes Arginase I or Arginase II are expressed under the neuron-specific Thy-1 promoter.
Figure 2.
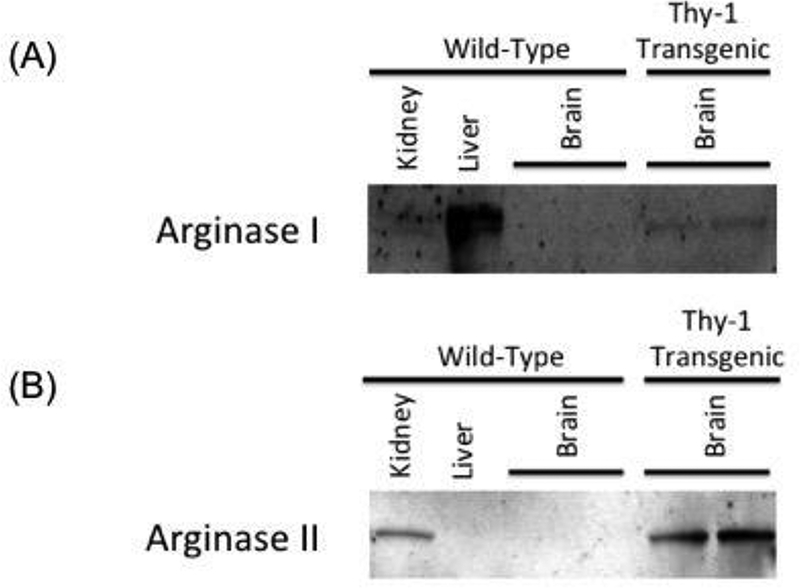
(A) and (B) Western blots showing overexpression of arginase I or arginase II expression in brain lysates from Thy1-ArgI and Thy1-ArgII mice respectively as compared to WT mouse brain lysate. Liver and kidney tissues were used as positive controls for Arginase I and Arginase II respectively.
Figure 3.
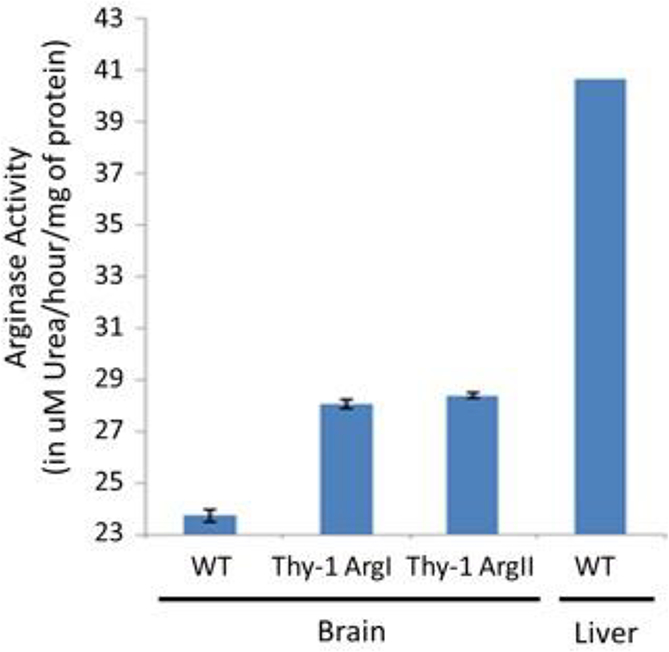
Arginase enzyme activity assay data showing brain lysates of transgenic mice have higher arginase activity compared to WT mice. Liver was used as a positive control.
Nissl staining was performed to evaluate for potential differences in gross morphology and neuronal numbers due to neuronal arginase overexpression. No differences were found in overall gross morphology between the overexpression mouse lines compared to WT mice (data not shown). The numbers of cortical neurons in Thy1- ArgI and Thy1-ArgII mice were comparable to WT mice (Figure 4A). Cerebellar morphology in Thy1-ArgI mice was normal. No differences were seen between Thy1-ArgII mice compared to WT mice in neuronal number or morphology. Similarly, H&E examination of coronal sections showed no microscopic abnormalities of cerebral cortex or hippocampus.
Figure 4.
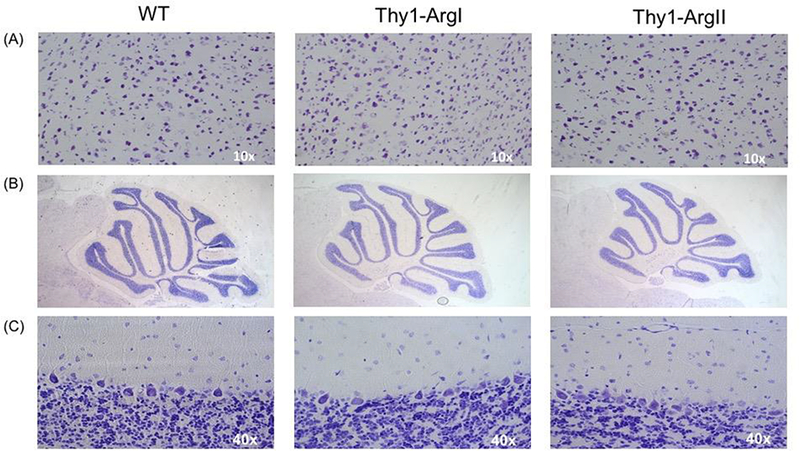
(A) Nissl staining of the cortex. (B) and (C) Nissl staining of cerebellum of Thy1-ArgI and Thy-1 ArgII transgenic mice respectively.
We also assessed levels of brain arginine and ornithine in the transgenic mice compared to WT mice. As expected, both Thy1-ArgI and Thy1-ArgII had significantly reduced levels of arginine compared to WT mice (p=0.0032 and p=0.0024 respectively) (Figure 5A). Brain ornithine levels were significantly higher in Thy1-ArgI mice compared to WT mice (p=0.0006) while brain ornithine levels in Thy1-ArgII mice trended towards being higher than those in WT mice (p=0.0987) (Figure 5B).
Figure 5.
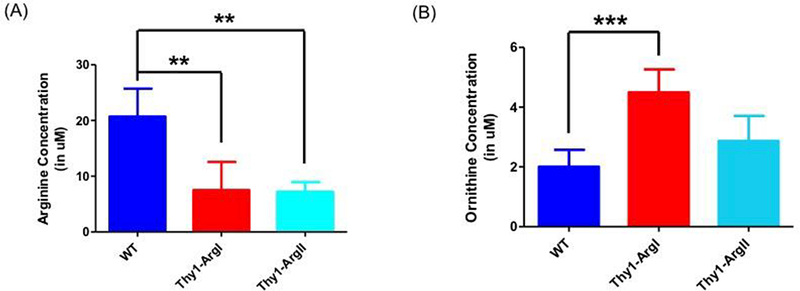
(A) Brain arginine levels were significantly lower in Thy-1 ArgI and Thy-1 ArgII transgenic mice compared to WT, n=5 per group (B) Brain ornithine levels were significantly higher in Thy-1 Arg I mice and trended towards being higher in Thy-1 Arg II transgenic mice compared to WT, n=5 per group. Error bars depict standard deviation. Double asterisk (**) denotes p≤0.01, triple asterisk (***) denotes p≤0.001.
Effect of arginase overexpression on contusion volume produced by controlled cortical injury
To understand the effect of arginase overexpression on traumatic brain injury, controlled cortical injury (CCI) was induced surgically on the right cortex in 3 different groups: wild-type, Thy1-ArgI, and Thy1-ArgII mice (n=7–15 per group). As a control, a second set of WT mice underwent a sham surgery without induction of cortical injury (n=3).
Injury size was determined by measuring contusion volume (CV) and injury severity was determined by assessing the contusion index (CI). As expected, WT mice had significantly higher CV (p=0.0002) and CI (p=0.0149) compared to the sham control group which did not receive the CCI (Figure 6A and 6B). Additionally, the Thy1-ArgI mice had significantly reduced CV (p=0.0055) and lower CI (p=0.01) compared to WT, thus implying a neuroprotective role of arginase 1 neuronal overexpression. On the other hand, Thy1-ArgII overexpression mice trended towards reduced CV as compared to WT mice (p=0.0518), CI was not significantly different between the two groups (p=0.1519).
Figure 6.
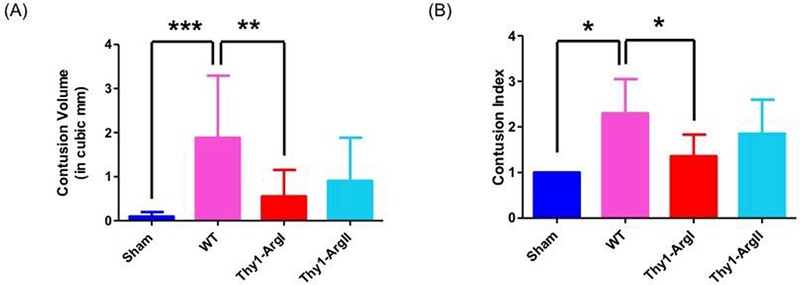
(A) Graph depicting average volume of contusion by experimental group and genotype. As expected, WT mice had significantly higher contusion volume as compared to the sham group. The contusion volume was significantly reduced in the Thy-1 ArgI mice compared to WT mice while the contusion volume in Thy1 Arg II mice trended towards being significantly reduced as compared to WT mice. (B) Graph depicting average contusion index by experimental group and genotype. As expected, WT mice had significantly higher contusion index as compared to the sham group. The contusion index was significantly reduced in the Thy-1 ArgI mice compared to WT mice while the contusion index in Thy1 Arg II mice was not significantly different than WT mice. For the contusion volume and contusion index measurements, the sham group had n=3. For the WT and transgenic mice, n=7–15 mice. Error bars depict standard deviation. Single asterisk (*) denotes p≤0.05, double asterisk (**) denotes p≤0.01, triple asterisk (***) denotes p≤0.001.
Discussion
Pharmacologic studies attempting to understand the role of nitric oxide in traumatic brain injury pathology have often taken a systemic approach where animals were either administered L-arginine which is a substrate of all nitric oxide synthases or administered an inhibitor of all nitric oxide synthases. However, this does not give a clear picture of the role of L-arginine on different cell types or NO from the three major cell specific NOS isoforms. The spatial and temporal differences in NOS expression supports potential cell-specific and isoform-specific differences in the role of NO in TBI. Here, we attempted a genetic approach which complements NOS deletion in a cell-specific fashion as the reduction of arginine substrate in cell autonomous fashion would overcome potential redundant up regulation of compensating NOS proteins. This may also provide a potential therapeutic approach in using activators of arginase expression in the context of TBI.
Arginase I is a cytosolic enzyme and is perceived to be responsible primarily for ureagenesis in the liver, while Arginase II is expressed in other tissue types and was traditionally thought to be responsible for regulating other functions of arginase besides ureagenesis including synthesis of polyamines, glutamate and nitric oxide. However, this theory has been questioned since Arginase II is localized in the mitochondria and therefore, unlikely to be involved in cytoplasmic activities such as inhibition of nitric oxide synthesis.17 Moreover, a study evaluating the expression of arginase isoforms in mouse brain showed that both isoforms were expressed in neurons thus, deflating the belief that only one isoform is expressed in a given cell type.18 Given that both isoforms are expressed in neurons but differ in subcellular localization, it is likely that they utilize different pools of arginine as substrate. It is unknown whether functional redundancy exists between the two isoforms. Hence, it was important to study both isoforms to understand how modulation of arginine levels can affect TBI.
Additionally, very few studies have been conducted on the neuronal NOS isoform in TBI. In some studies, increase in nNOS expression has been reported post-TBI and pharmacologic inhibitors of nNOS activity result in better neurologic outcomes - both morphologic and behavioral.19,20 One mechanism by which excess NO is proposed to be neurotoxic is that it reacts with superoxide to produce peroxynitrite which can lead to oxidative damage and cellular death.21,22 Moreover, excessive NO can lead to increased nitrosative stress and nitration of several enzymes responsible for decreasing oxidative stress thereby reducing their activity and contributing even further to cellular damage from oxidative stress.23, 24 On the other hand, arginase overexpression and arginine depletion can lead to eNOS uncoupling, which also results in superoxide production and oxidative stress.25, 26, 27 Thus, the role of nNOS-derived NO in TBI has remained elusive.
This study used a genetic approach - transgenic overexpression of arginase selectively in neurons, to better understand the cell-specific effects of reduced availability of arginine for NO production. Neuronal overexpression of arginase I and decreased availability of arginine in the brain led to reduced contusion volume and score; therefore, it appears that nNOS derived NO may be detrimental in TBI. This agrees with its proposed role of NO causing neurotoxicity reported in pharmacological studies mentioned earlier. A potential weakness of the study is that we cannot exclude contributing effect due to the altered flux of arginine to its other downstream metabolites, which has been implicated in axon growth, cytoskeletal stability and neural repair. Still, these data complement the modeling of nNOS function in TBI models and support potential arginase upregulation in the approach to treating TBI.
Conclusion
NO has many roles in TBI beyond being a mediator of CBF and the role of nNOS-derived NO in TBI has remained unclear. This study established arginase overexpression as a tool to modulate arginine levels in a cell-specific manner. Additionally, the study demonstrates that manipulating arginase levels can be used as a potential approach to affect the course of TBI and indirectly lends insight into the role of neuronal NO production in this condition. We believe that understanding these cell-specific differences will be key in designing better interventions for TBI.
Acknowledgements
This work was supported by the NIH R01 DK102641 (B. Lee), the Baylor College of Medicine (BCM) Intellectual and Developmental Disabilities Research Center (HD024064) from the Eunice Kennedy Shriver National Institute Of Child Health & Human Development, the BCM Advanced Technology Cores with funding from the NIH (AI036211, CA125123, and RR024574). Simran Madan was supported by Howard Hughes Med into Grad Scholarship awarded through the Translational Biology and Molecular Medicine Graduate Program at Baylor College of Medicine.
Footnotes
Author Disclosure Statement
No competing financial interests exist
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jeter CB, Hergenroeder GW, Ward NH, Moore AN, and Dash PK (2012). Human Traumatic Brain Injury Alters Circulating L-Arginine and Its Metabolite Levels: Possible Link to Cerebral Blood Flow, Extracellular Matrix Remodeling, and Energy Status. [DOI] [PubMed] [Google Scholar]
- 2.Cherian L, Chacko G, Goodman C, and Robertson CS (2003). Neuroprotective Effects of L-Arginine Administration after Cortical Impact Injury in Rats: Dose Response and Time Window. J. Pharmacol. Exp. Ther 304, 617–623. [DOI] [PubMed] [Google Scholar]
- 3.Cherian L, and Robertson CS (2003). L-Arginine and Free Radical Scavengers Increase Cerebral Blood Flow and Brain Tissue Nitric Oxide Concentrations after Controlled Cortical Impact Injury in Rats. J. Neurotrauma 20, 77–85. [DOI] [PubMed] [Google Scholar]
- 4.Lundblad C, Gra P, and Bentzer P (2009). Hemodynamic and Histological Effects of Traumatic Brain Injury in eNOS-Deficient Mice.1962, 1953–1962. [DOI] [PubMed] [Google Scholar]
- 5.Lu YC, Liu S, Gong QZ, Hamm RJ, and Lyeth BG (1997). Inhibition of nitric oxide synthase potentiates hypertension and increases mortality in traumatically brain-injured rats. Mol. Chem. Neuropathol 30, 125–37. [DOI] [PubMed] [Google Scholar]
- 6.Wada K, Chatzipanteli K, Busto R, and Dietrich WD (1999). Effects of L-NAME and 7-NI on NOS catalytic activity and behavioral outcome after traumatic brain injury in the rat. J. Neurotrauma 16, 203–12. [DOI] [PubMed] [Google Scholar]
- 7.Terpolilli N. a, Kim S-W, Thal SC, Kuebler WM, and Plesnila N (2013). Inhaled nitric oxide reduces secondary brain damage after traumatic brain injury in mice. J. Cereb. Blood Flow Metab 33, 311–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tisdall MM, Rejdak K, Kitchen ND, Smith M, and Petzold A (2013). The prognostic value of brain extracellular fluid nitric oxide metabolites after traumatic brain injury. Neurocrit. Care 19, 65–8. [DOI] [PubMed] [Google Scholar]
- 9.Terpolilli NA, Zweckberger K, Trabold R, Schilling L, Schinzel R, Tegtmeier F, and Plesnila N (2009). The novel nitric oxide synthase inhibitor 4-amino-tetrahydro-L-biopterine prevents brain edema formation and intracranial hypertension following traumatic brain injury in mice. J. Neurotrauma 26, 1963–75. [DOI] [PubMed] [Google Scholar]
- 10.Cherian L, Hlatky R, and Robertson CS (2004). Nitric oxide in traumatic brain injury. Brain Pathol. 14, 195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ochoa JB (2012). Clinical Relevancy Statement. J. Parenter. Enter. Nutr 36, 53–59. [Google Scholar]
- 12.Ochoa JB, Bernard AC, O ‘brien WE, Griffen MM, Maley ME, Rockich AK, Tsuei BJ, Boulanger BR, Kearney PA, and Morris SM (2001). Arginase I Expression and Activity in Human Mononuclear Cells After Injury. [DOI] [PMC free article] [PubMed]
- 13.Villalba N, Sackheim AM, Nunez IA, Hill-Eubanks DC, Nelson MT, Wellman GC, and Freeman K (2017). Traumatic Brain Injury Causes Endothelial Dysfunction in the Systemic Microcirculation through Arginase-1-Dependent Uncoupling of Endothelial Nitric Oxide Synthase. J. Neurotrauma 34, 192–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zawada RJX, Kwan P, Olszewski KL, Llinas M, and Huang S-G (2009). Quantitative determination of urea concentrations in cell culture medium. Biochem. Cell Biol 87, 541–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cherian L, Chacko G, Goodman JC, and Robertson CS (1999). Cerebral hemodynamic effects of phenylephrine and L-arginine after cortical impact injury. Crit. Care Med 27, 2512–7. [DOI] [PubMed] [Google Scholar]
- 16.Adams JH, Scott G, Parker LS, Graham DI, and Doyle D (1980). The contusion index: a quantitative approach to cerebral contusions in head injury. Neuropathol. Appl. Neurobiol 6, 319–24. [DOI] [PubMed] [Google Scholar]
- 17.Cederbaum SD, Yu H, Grody WW, Kern RM, Yoo P, and Iyer RK (2004). Arginases I and II: do their functions overlap? Mol. Genet. Metab 81 Suppl 1, S38–44. [DOI] [PubMed] [Google Scholar]
- 18.Yu H, Iyer RK, Kern RM, Rodriguez WI, Grody WW, and Cederbaum SD (2001). Expression of arginase isozymes in mouse brain. J. Neurosci. Res 66, 406–422. [DOI] [PubMed] [Google Scholar]
- 19.Sharma HS, Wiklund L, Badgaiyan RD, Mohanty S, and Alm P (2006). Intracerebral administration of neuronal nitric oxide synthase antiserum attenuates traumatic brain injury-induced blood-brain barrier permeability, brain edema formation, and sensory motor disturbances in the rat. Acta Neurochir Suppl 96, 288–94. [DOI] [PubMed] [Google Scholar]
- 20.Khan M, Dhammu TS, Matsuda F, Annamalai B, Dhindsa TS, Singh I, and Singh AK (2016). Targeting the nNOS/peroxynitrite/calpain system to confer neuroprotection and aid functional recovery in a mouse model of TBI. Brain Res. 1630, 159–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beckman JS, Beckman TW, Chen J, Marshall PA, and Freeman BA (1990). Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. U. S. A 87, 1620–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pacher P, Beckman JS, and Liaudet L (2007). Nitric oxide and peroxynitrite in health and disease. Physiol. Rev 87, 315–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bayir H, Kagan VE, Clark RSB, Janesko-Feldman K, Rafikov R, Huang Z, Zhang X, Vagni V, Billiar TR, and Kochanek PM (2007). Neuronal NOS-mediated nitration and inactivation of manganese superoxide dismutase in brain after experimental and human brain injury. J. Neurochem 101, 168–181. [DOI] [PubMed] [Google Scholar]
- 24.Beckman JS, and Koppenol WH (1996). Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am. J. Physiol 271, C1424–37. [DOI] [PubMed] [Google Scholar]
- 25.Luo S, Lei H, Qin H, and Xia Y (2014). Molecular mechanisms of endothelial NO synthase uncoupling. Curr. Pharm. Des 20, 3548–53. [DOI] [PubMed] [Google Scholar]
- 26.Chan J, and Wu K (2014). NOS uncoupling in oxidative stress-associated neurogenic hypertension induced by the high fructose diet (682.2). FASEB J 28, 682.2. [Google Scholar]
- 27.Sullivan JC, and Pollock JS (2006). Coupled and Uncoupled NOS: Separate But Equal? Circ. Res 98. [DOI] [PubMed] [Google Scholar]