

Abstract
Fluorescence spectroscopy, coupled with microscopy, opens new frontiers for the study of dynamic processes with high spatio-temporal resolution. The application of phasor plots to FLIM and hyperspectral imaging demonstrate unprecedented capabilities to study complex photophysics at the subcellular level. Using these approaches we studied the effects of an H2O2 bolus on NIH-3T3 membranes dynamics monitored by LAURDAN fluorescence. Exposure of NIH-3T3 cells to a bolus of H2O2 modifies the cell membranes and, in particular, the plasma membrane in a complex manner. The LAURDAN results reveal that the peroxide treatment decreases membrane fluidity but surprisingly increases dipolar relaxation around the excited probe. Using the Multidimensional-phasor approach we elucidated the complex photophysics of LAURDAN incorporated into cell membrane after H2O2 exposure. The results indicate the occurrence of LAURDAN fast-diffusion from gel↔ld phases in membranes exposed to a H2O2 bolus. An ad hoc hypothesis is presented to interpret the results in the context of H2O2 oxidative distress/eustress.
Keywords: LAURDAN, Phasor plots, membrane dynamics, dipolar relaxation, Hydrogen peroxide, Oxidative stress
Graphical Abstract
Introduction
LAURDAN (6-dodecanoyl-2-dimethylaminonaphthalene) belongs to the family of dimethylaminonaphthalene fluorescent probes original synthetized by Gregorio Weber to study polarity and dipolar relaxation in macromolecular systems [1–3]. Upon excitation LAURDAN exhibits a significant increase of its dipole moment and thus can reorient surrounding dipoles in the environment. A requirement for this process is that the solvent molecules must be able to move within the excited state lifetime of LAURDAN [4]. The energy expended on this environment reorganization is reflected in the shift of the LAURDAN fluorescence towards the red [5]. In membranes, LAURDAN fluorescence depends strongly on the physical state of the phase (e.g. local and translational mobility) [6]. Thus, for gel (Lβ) and liquid order (lo) membranes LAURDAN’s emission is centered at 440 nm (unrelaxed, long lifetime), while for liquid disorder (ld - fluid) the emission is 50 nm red shifted (relaxed, short lifetime) [7]. The relaxation time of LAURDAN in a Lβ phase is around 4.0x10−7 s, but in ld membranes this time decreases to 2.5x10−9 s [8]. Therefore, these observations point out to an important property of LAURDAN, namely to sense confined (rotational restricted) water at the membrane interphase, since the relaxation time of bulk water is on the picosecond time scale [9]. Thus, LAURDAN is able to sense the dipolar relaxation of few water molecules (0–5) at the membrane interphase, that can fluctuate in number due to the membrane physical state [9]. Apart from the physical state of the membrane determined by the fatty acid composition and the thermodynamic condition, LAURDAN is also sensitive to cholesterol concentration [10,11]. Cholesterol modifies the membrane order parameters in a complex manner regarding the physical phase; introducing disorder in a gel membrane but increasing the order in a fluid membrane [11]. An important property of LAURDAN on regards of cholesterol concentration in cells is the lack of segregation in different phases (lo/ld), as well as for the pH or headgroup, which is fundamental to work on in vivo condition with cells [6]. Originally, LAURDAN fluorescence was analyzed using steady-state data by the generalized polarization function (GP), as a normalized ratio of the intensity in the blue and red emission (GP= (IB-IR)/(IB+IR)) [6]. Excitation and emission GP provide information on the polarity and dipolar relaxation for the ground and excited states, respectively [12,13]. Within the last 20 years, the revolution in fluorescence microscopy has changed the way we use fluorescence to study cell structure and dynamics with unprecedented spatial and temporal resolutions. In particular, lifetime and hyperspectral imaging in combination with phasor plots allow a detailed understanding of LAURDAN’s photophysics for in vivo cell experiments [14,15]. More recently, the multidimensional phasor approach (MultiD-phasor) was used to compare the information between lifetime and spectra within a pixel due to the reciprocity properties of the phasor plots and the image [5]. Phasor plots are a powerful approach to understand LAURDAN photophysics in both model membranes and cells [5,15,16].
In this work we use LAURDAN fluorescence to study the effects of an H2O2 bolus on mouse fibroblast cell membranes (NIH-3T3). H2O2 is a ubiquitous molecule at subcellular levels that can either produce damage or signaling (oxidative distress and eustress, respectively), depending on its effective concentration and the redox response of the cell [17–21]. Contrary to the common beliefs that H2O2 is exclusively produced by inflammatory cells, it is now well established that all cells produce and carefully control the concentration of H2O2 in order to produce “redox switches” at specific targets [21]. An H2O2 bolus is a model of oxidative distress depending on the effective concentration and the exposure time [20]. Depending on the cell type and the enzymatic subcellular machinery available to handle the excess H2O2, a gradient inside-out of the cell was described [22]. This gradient is usually conceived as a 100-fold concentration difference between extra and intracellular compartments [20]. This difference could be 600–1000 times larger for cells with high active peroxiredoxins [20,21,23,24]. An important concept about the H2O2 role in cell signaling arises from the fact that some subcellular regions can be considered as zero-H2O2 concentration [20]. This concept is based on the distribution of a detoxifying enzyme at different subcellular levels, e.g. peroxisome (catalase) or cytosol/nucleus (peroxiredoxins/glutathione peroxidase) [20,21,25]. One interpretation for the modulation of key redox switches is known as the “oxidability”. The oxidability is related to the final subcellular concentration of H2O2, as well as their reactivity on the thiolate groups for specific enzymes towards the H2O2 (enzyme kD) [21]. The reactivity can change from 20 M−1s−1 for protein tyrosine phosphatases (PTPs) to 107 M−1s−1 for peroxiredoxins 2 (Prx-2) [21]. The “floodgate” model consider that high reactivity molecular switches (peroxiredoxins, Prx) would first sense for low H2O2 concentrations, while after Prx inactivation (over-oxidation) for higher H2O2 concentrations the less-reactive redox switches (PTPs) can get oxidized. Examples of these redox molecular switches can be peroxiredoxins (Prx), glutathione peroxidase (GPx), catalase, protein kinase C (PKC), Src tyrosine kinase or PTPs [21]. On the other hand, another interpretation based on protein signaling by H2O2 throughout a "Prx sensor" activity is gaining experimental support [19,26]. The mechanism proposes that peroxiredoxins will acts as a sensors for H2O2 based on their high reactivity and cell abundance and then transfer the oxidizing equivalents to less reactive redox switches (like PTPs) [19]. The signal transmission involves the oxidation of Prx to sulfenic acid or disulfide, follow by the formation of mixed disulfide (Prx-sensor/target protein) to finally end up in the oxidation of the regulatory protein and the regenerated Prx-sensor [19]. Transient mixed disulfide between Prx-2 and the transcription factor STAT3 (Signal transducer and activator of transcription 3) was found by Sabotta et al. [26]. Using a depletion of Prx-2 an increase in the activity of STAT3 was reported, implying a complex oxidation and transcriptional activity [26]. Peroxiredoxins are not the only actors in the sensor mechanism, thioredoxin (Trx) can recycle the Prx-sensor, thereby a competition between Trx and redox-target could help to control the spatial and temporal regulation of the signaling[27]. Hydrogen peroxide was considered to freely diffuse across the membrane, however, the fluidity of the membrane and the presence of specific aquaporin’s (peroxiporins) shows that this scenario is in doubt [28–32]. The fluidity of the yeast plasma membrane led to changes in the supramolecular structure driven by the phospholipid profile and modification on the sterol profile [30]. This diffusion control also compromises the consumption rate of the extracellular H2O2 gradient, keeping the gradient time longer [21]. Besides, the intracellular diffusion is controlled by the H2O2 consumption associate to the high reactive enzymes, like Prx′s, reducing the radii of activity and increasing the specificity [27].
Here, we evaluate the effect of an H2O2 bolus on the NIH-3T3 cell membrane organization and dynamics by using LAURDAN fluorescence with lifetime and hyperspectral 2-photon imaging. Phasor analysis of the lifetime and hyperspectral data allows us to infer that the H2O2 bolus modifies the cell membrane in a complex way. While the lifetime-phasor analysis for the blue wavelength channel indicates that the cell membranes are more ordered (less fluid) after H2O2 exposure, the green wavelength channel shows an increase in the dipolar relaxation of the LAURDAN emission (indicating higher water content in the LAURDAN environment). These results are also statistically stronger when masking is applied to select the plasma membrane. The use of MultiD-phasor elucidates LAURDAN’s photophysics and reveals the occurrence of a fast-diffusion process (LAURDAN diffusion during excited-state between gel/Lo ⇔ Ld environments).
Materials and Methods
Material
LAURDAN (6-dodecanoyl-2-dimethylaminonaphthalene), hydrogen peroxide (H2O2) and cell culture reagents were acquired from Thermo-Fisher Scientific Inc. (Huntington Beach, CA-USA). Coumarin 6 (3-(2-Benzothiazolyl)-N,N-diethylumbelliferylamine, 3-(2-Benzothiazolyl)-7-(diethylamino)coumarin) was acquired from Sigma-Aldrich Co. (Milwaukee, WI-USA)
Cell culture and hydrogen peroxide treatment
NIH-3T3 fibroblasts derived from mouse (ATCC® CRL-1658 ) were grown at 37°C with 5% CO2 in Dulbecco’s modified Eagle’s medium (Thermo Fisher Scientific Inc. Huntington Beach, CA-USA) supplemented with 10% fetal bovine serum, and 5 mL of Pen-Strep. The cells (2x104 cells/dish) were plated 24hs before the experiments onto a fibronectin-coated 35mm glass-bottom dishes (MatTek Corporation, MA-USA). On the day of the experiments the cells were incubated for 30-60 min with a final solution of 3 μM LAURDAN. Before imaging, LAURDAN was removed by fresh medium replacement. The concentration of LAURDAN was determined using a molar extinction coefficient of 20,000 M−1cm−1 (at 364 nm in methanol). For the hydrogen peroxide experiments, cells were incubated with a bolus of 1mM or 100μM final concentration in 1 mL of medium for 1h and before imaging the medium was replaced by a fresh one. A fresh opened bottle of 3% solution of hydrogen peroxide was used and the concentration was estimated by the absorbance at 240 nm, using 43.6 M−1cm−1 as the absorption extinction coefficient [33]. During LAURDAN and hydrogen peroxide incubations cells were kept at 37°C with 5% CO2. During the imaging on the Zeiss LSM710 microscope the cells were kept at 37°C with 5% CO2 using the Zeiss incubator.
Lifetime and hyperspectral fluorescence microscopy
For FLIM and hyperspectral fluorescence a Zeiss LSM710 META microscope (Carl Zeiss, Dublin, CA-USA) was used. The microscope was coupled to a femtosecond Ti:Sapphire laser (Spectra-Physics Mai Tai, Newport Beach CA-USA), the laser produce 80 fs pulses with a repetition rate of 80 MHz. For FLIM acquisition an A320 Fast-FLIMbox (ISS, Champaign, IL-USA) was used to collect the lifetime decay data. A 40× water immersion objective 1.2 N.A. (Carl Zeiss, Dublin, CA-USA) was used for all experiments. LAURDAN was excited using a wavelength of 780 nm. To avoid LAURDAN photoselection effects by the polarized light, a lambda/4 plate from Semrock (Rochester, NY-USA) was introduced into the excitation path. The average laser power at the sample was maintained at the mW level. For detection, two photomultiplier detectors (H7422P-40, Hamamatsu, Hamamatsu City, Japan) were used and the LAURDAN emission was split into two bands by the following filter set: a bandpass filter from Semrock (Rochester, NY-USA) was placed in front of each detector, 440/60nm (Channel-1, blue) and 500/60nm (Channel-2, green), the two bands were split by a 470nm longpass dichroic filter. The Lambda mode configuration of Zeiss LSM710 META was used for the hyperspectral image. A 32-channels configuration was used and each channel had a 9.7 nm bandwidth, the total range acquired was between 416 nm and 728 nm. Image acquisition in both FLIM and spectral mode was performed with a frame size of 256×256 and the pixel dwell time used for FLIM and spectral images were 25.61μs/pixel or 5.09μs/pixel, respectively. FLIM and spectral data were processed using the lifetime, spectral or MultiD-phasor routines in the SimFCS software developed at the Laboratory for Fluorescence Dynamics (www.lfd.uci.edu). When indicated, denoising of the phasor plots was performed by applying a median filter (3x3) using the SimFCS software as previous described [34]. For calibration of the lifetime measurements, a solution of 1 μM of coumarin 6 (Sigma-Aldrich Co., Milwaukee, WI-USA) was used with a reference lifetime of 2.5 ns (ISS webpage at www.iss.com).
FLIM phasor plot analysis and interpretation
The phasor plot is a geometrical representation introduced by Jameson et al. [35] and then applied to fluorescence lifetime imaging microscopy (FLIM) data by Digman et al. [36]. The phasor plot is a vector space; for each pixel in a FLIM image a Fourier transformation is applied. As exemplified in Figure 1A the fluorescence decay I(t) acquired pixel by pixel in an image using a FLIMbox is then transformed in the G(τ) and S(τ) coordinates (real and imaginary component of the Fourier transformation) following the math in equations 1 and 2. The phasor position for each pixel was plotted in a polar plot (that we call Phasor plot) using these two coordinates (G(τ) and S(τ)):
| (Eq. 1) |
| (Eq. 2) |
where ω is the angular modulation frequency, equal to 2πf, where f=1/T is the laser repetition frequency and T is the period of the laser frequency. The universal circle is defined as a semicircle centered at (0.5, 0) with a radius 0.5 in the phasor space. An important point to highlight is the model-free characteristics of the phasor transformation, which means that no previous assumption about number or types of decays is needed. For any single exponential decay at any given frequency, all the points should fall on the universal circle (see Figure S1). Having said that, we can notice for infinite unmodulated fluorescence the position on the phasor plot should fall at (0,0), which in other words means τ ≈ ∞. On the other hand, the position (1,0) represent τ = 0, which corresponds to no modulation in the fluorescence from the excitation light. The same reasoning can be applied for the phase shift; θ = 0 means τ = 0 and θ = 90 means τ ≈ ∞. The phasor plot has a tangent scale and this property is exemplified in Figure S1 (left panel). Phasors follow rules of vector addition and orthogonality, i.e., pixels that contain a linear combination of two independent fluorescent species will appear on the line joining the two independent emissions in the phasor plot (middle panel Figure 1S). Thereby, it is easy to identify single and multiple lifetime species in the phasor plot because the first should fall on the universal circle, however, multi lifetime species should be inside the universal circle (see middle panel in Figure S1). The rule of addition allows the phasor plot to decompose the contribution of two or more species as exemplified in Figure S1. A position outside the universal circle can be found in a situation wherein an exited-state reaction is occurring (Figure S1 left panel). An example of this situation is the dipolar relaxation around the fluorophore upon its excitation; this effect is experienced by fluorophores like PRODAN (6-Propionyl-2-(dimethylamino)naphthalene) or LAURDAN when molecules containing dipoles are near the excited fluorophore dipole [1,7]. Finally, an important property of the phasor transformation is the reciprocity maintained between the raw data and the phasor space. This property allows us to select a region of interest (ROI) in the phasor plot distribution and recognize the location of those pixels in the original image (see Figure S2); of course the opposite transposition is also true.
Figure 1. Phasor properties and graphical interpretation for lifetime and hyperspectral images.

A) Chart for the lifetime phasor plot representation, and its properties and mathematics. Note that the green arrow in the lifetime decay plot (left) indicates the period (T, at the integral for the Gτ and Sτ calculation). The blue and red curves represent a short and a long lifetime decay, respectively. Then these two decays were phasor transformed using the math at the right of panel A and the blue and red dot in the phasor plot (middle plot) represent the positions on the universal circle. Notice that these two points in the phasor plot are on the universal circle indicating a single exponential decay for each point. MB, MR, θB and θR represents the modulation and phase for the blue and red decays, respectively. Using the phasor representation it is easy to see that as the lifetime gets longer the modulation decrease and the phase increases. B) Chart for the spectral phasor representation, and its properties and mathematics. Note that the green arrow in the spectra plot (left) indicate the range (λmax, at the integral for the Gλ; and Sλcalculation). The blue and red curves represent two spectra with different maxima and full with at the half maximum (FWHM). Maximum for the red curve is more shifted to the red compared to the blue curve and the FWHM is broader in the red spectrum. The spectra were phasor transformed using the math at the right of the panel B and the blue and red dot in the spectral phasor plot (middle plot) represent the positions for blue and red spectra at the polar plots. Notice that the universal circle does not exist here. MB, MR, θB and θR represents the modulation and phase for the blue and red spectra, respectively. By spectral phasor representation we can notice that the reddest spectrum has the larger phase as it should, and as the spectra become broader the point moves closer to the coordinate (0,0) (modulation decrease). C) Chart for the three cursor analysis in the phasor representation. The gray cursor represents a point for the phasor distribution (light-blue contour plot behind). The cross-section between the line that joins a–b represents the fraction of a that is in the gray cursor, and the same is true for the line that join c-a, giving the fraction of c. The fraction at each position between a–b and c-a can be found in the histogram plot at the left, where red and green lines represent fractions of a and c, respectively. See Ref. [5, 15, 38] for a in-depth description of phasor plots.
Spectral phasor plot analysis and interpretation
The fluorescence spectra at each pixel was transformed into phasor coordinates (G(λ)) and (S(λ)) as described in equation 3 and 4, respectively [15,37]. G(λ) and S(λ) coordinates are the real and imaginary components of the Fourier transformation for a spectrum exemplified in Figure 1B. I(λ) represents the intensity at every wavelength (channel in our case), n is the number of the harmonic and λi the initial wavelength. The x and y coordinates were plotted in the spectral phasor plot as shown in Figure 1B (middle panel).
| (Eq. 3) |
| (Eq. 4) |
The position for every pixel in the spectral phasor plot can be defined by the phase angle and the modulus (M) given the coordinates G and S (both for spectral or lifetime)
| (Eq. 5) |
| (Eq. 6) |
The angular position in the spectral phasor plot relates to the center of mass of the emission spectrum and the modulus depends on the spectrum’s full width at the half maximum (FWHM). For instance, as exemplified in Figure 1B, if the spectrum is broadening the phasor cluster should be moving closer to the center (modulation is decreasing). On the other hand, for red shifted spectra, the position of the phasor will move counterclockwise towards increasing angles from position (1,0), i.e., the phase will increase. Spectral phasors share all vector properties with lifetime phasors. A in-depth description of the properties of spectral phasor plots is given in references [15,16].
Three cursor analysis of phasor contributions
The three-cursor analysis in the SimFCS software is useful to quantify the fraction of three components using the addition rules of the phasor plots [38]. In our case, we position the cursors a and c in Figure 1C to calculate the histogram for the pixel distribution along the line (ac) of the linear combination that refers to membrane fluidity (judged by the LAURDAN lifetime change). The term membrane fluidity refers to the membrane order parameters for a phospholipid. We consider that any process that can increase rotational or translational rates of lipids is increasing the membrane fluidity. Following the same reasoning, the third cursor (b cursor in Figure 1C) was positioned outside the universal circle in order to quantify the change in the dipolar relaxation (line ab). By moving a cursor of a given radius along the lines ab/c and ac/b it is possible to obtain the histograms for the fraction of dipolar relaxation and fluidity in each sample (see left plot in Figure 1C).
| (Eq. 7) |
| (Eq. 8) |
The histograms are presented as the number of pixels at each step along the line ab and ac, normalized by the total fraction. Then, we can plot the average value for each histogram ± standard deviation, as well the center of mass of the histogram for a quantitative analysis with descriptive statistics. The center of mass was calculated following the math below.
| (Eq. 9) |
where Fi is the fraction for each variable (namely fluidity or dipolar relaxation).
MultiD phasor plot analysis of LAURDAN fluorescence
The MultiD approach is based on establishing a relation between each point of different phasor plots and the pixels of the image [5]. As previously shown, it is possible to select an ROI in the lifetime phasor plot and identify those pixels in the image selected by this choice. One example could be selecting pixels corresponding to the Lo phase in the image based on the value of the lifetime of Laurdan in the Lo phase. Then in each of the other phasor plots we can identify the spectrum of the Lo phase or of any of the other properties used to construct the phasor plots. This approach is exemplified in Figure S2. In the present work, we acquire lifetime images in two channels and the hyperspectral image at the same focal plane followed by the phasor transformation. Using the channel_2 of lifetime as the master plot, it is possible to obtain the location the pixels selected by the cursor in the master plot in the secondary (channel_1 of lifetime and spectral) phasor plots (see Figure7).
Figure 7. Multidimensional phasor analysis of the effect of a 1mM bolus of H2O2 for 1h on the NIH-3T3 cell membranes by LAURDAN fluorescence.

A) MultiD phasor analysis for the LAURDAN labels NIH-3T3 cells (control group). From left to right we have the channel_2 lifetime phasor plot (notice that it was defined as a master phasor plot), common pseudocolor image generated by the cursor selection in the master phasor plot, channel_1 lifetime phasor plot (the insets are showing the distribution of pixels selected under each cursor at the master phasor plot) and the spectral phasor plot (the insets are showing the distribution of pixels selected under each cursor at the master phasor plot, notice that the theoretic triangle for the lo, ld and background (BG) is the same used for the spectral phasor analysis in Figure 6). The cursor included at the master phasor plot were positioned with the purpose to show the pixel migration in lo,/ld trajectory between the inside/outside the universal circle. The dashed lines exemplify the lo,/ld trajectory. B) MultiD phasor analysis for the LAURDAN label NIH-3T3 cells treated with a 1mM bolus of H2O2 (1mM H2O2 x1h group). From left to right the description is the same than for the A section but for the 1mM H2O2 x1h group.
Data reproducibility and statistics
Three independent biological replicates were used to perform the statistical analysis. Data are shown as means ± standard deviation (SD) and were analyzed using a two-tails unpaired student t-test. When more than 2 groups were compared, ANOVA with Tukey post-test for multiple comparisons was used. All the samples showed normal distributions as judged by the Kolmogorov-Smirnov test. A p<0.05 was considered statistically significant. Statistical analysis was realized using GraphPad Software, Inc.
Results
Effects of a hydrogen peroxide bolus on cell membranes organization evaluated by LAURDAN fluorescence
Figure 2 shows the FLIM-phasor results for the LAURDAN fluorescence in the blue channel when NIH-3T3 fibroblasts were either exposed or not exposed to a 1mM bolus of H2O2 for 1h. The phasor distribution was analyzed using the three-cursor approach (Figure 2A). Two dimensions were defined: blue-red cursors associated with membrane fluidity (fluidity-axis) and red-green cursors associated with dipolar relaxation (DR-axis). Using a color scale map as shown in each axes, it is possible to color-code the LAURDAN lifetime changes (Figure 2D). By producing a histogram of the fluidity and DR fraction we analyzed the effects of the H2O2 (see material and methods for a detailed explanation of the three cursor approach, Figure 1C). H2O2 does not significantly affect the fluidity of the cell membranes judged by the CM in the fluidity-axis for the blue channel (Figure 2B, bottom panel). However, a closer view of the fluidity histogram shows that the profile of the histograms looks different (Figure 2B, top panel). The DR-axis is increased 17% by the H2O2 incubation and the result is statistically significant (p<0.0001, Figure 2C, bottom). Also the histogram profiles show a shift towards increasing DR fractions (Figure 2C, top). An important result of the FLIM approach is that it provides the possibility to spatially discriminate the effects of the H2O2. Thereby, we apply masks to select the plasma membrane to discriminate between the effects of H2O2 exposure to different regions of the cell (Figure 3). Following the same reasoning as before, we applied the same logic in the analysis of the FLIM-phasor data. Figure 3B and C (bottom panels) shows that the H2O2 bolus decreased membrane fluidity and increased dipolar relaxation judged by the CM in the fluidity-axis and DR-axis histograms, respectively. Both effects were statistical significant with p values of 0.0270 and 0.0001, respectively. In the images (Figure 2D and 3D) it is possible to see the spatial effects of the H2O2 bolus on the LAURDAN lifetime changes. Even when the main effect, either in fluidity or dipolar relaxation, was observed at the plasma membrane, it is possible to see changes in the fluidity as well as in the DR in the interior of the cell (Figure 2D). In regard to fluidity, we can discriminate two main colors in the image, red-yellow for the PM related areas and blue-cyan for the intracellular regions. The DR images show an opposite color pattern (this was done deliberately to avoid confusion on the scales used). However, in both cases it is possible to see that the pseudo-coloring used is not homogeneous but shows spots of different size.
Figure 2. Effect of 1mM bolus of H2O2 for 1h on the NIH-3T3 cell membranes followed by LAURDAN fluorescence and lifetime phasor analysis of channel-1.

A) Phasor plot distribution for the control (Ctr) and H2O2 exposed cells (1mM H2O2 x1h). A three components analysis was performed and a dipolar relaxation (DR-axis) and fluidity axes (Fluidity-axis) were defined. The cells were colored using the scale indicated in each dimension (DR and fluidity). A histogram for fraction of DR and fluidity was generated for each cell and then averaged and standard deviations were calculated (top plots in B and C). B) Top, Fluidity fraction histogram for the LAURDAN lifetime distribution in channel-1. Data are represented as the mean plus the standard deviation (mean+SD, notice negative standard deviation was avoided to simplify the plots). The histogram is presented as the normalized number of pixels by area. Bottom, for each sample the center of mass of the histogram was determined as Eq. 9 and then plotted as mean±SD. C) Top, dipolar relaxation (DR) fraction histogram for the LAURDAN lifetime distribution in channel-1. Data are represented as the mean plus the standard deviation (mean+SD, notice negative standard deviation was avoided to simplify the plots). The histogram is presented as the normalized number of pixels by area. Bottom, for each sample histogram the center of mass was determined as Eq. 9 and then plotted as mean±SD. D) Representative images of LAURDAN fluorescence for control and 1mM H2O2 x1h treated cells. Intensity, fluidity-axis, DR-axis was color-coded as indicated at the right of the images from top to bottom, respectively. *** indicated p<0.001 vs Ctr, judged by the student t-test.
Figure 3. Effect of bolus of H2O2 (100 μM and 1mM) for 1h on the NIH-3T3 cell plasma membranes by LAURDAN fluorescence and lifetime phasor analysis of channel-1.

A) Phasor plot distribution for the control (Ctr) and H2O2 exposed cells (100 μM and 1mM H2O2 x1h). The data was represented and analyzed following same protocols explained in Figure 2A.. B) Top, Fluidity fraction histogram for the LAURDAN lifetime distribution in channel-1. Data are represented as mean+SD, notice negative standard deviation was avoided to simplify the plots. Bottom, center of mass for the histograms as mean±SD. C) Top, dipolar relaxation (DR) fraction histogram for the LAURDAN lifetime distribution in channel-1. Data are represented as in section B. Bottom, center of mass for the histograms as mean±SD. D) Representative images of the mask applied to the LAURDAN fluorescence for control and 1mM H2O2 x1h treated cells. Mask, fluidity-axis, DR-axis were color-coded as indicated at the right of the images from top to bottom, respectively. *** and * indicated p<0.001 or p<0.05 vs Ctr, respectively, judged by ANOVA and Tukey post-test. ●Indicated p<0.05 vs 100 μM H2O2 x 1h.
Furthermore, we applied the same analysis to the green channel of the FLIM data. Following the same line of reasoning we perform the analysis for the full image and plasma membrane by masking (Figure 4 and 5, respectively). The H2O2 bolus decreased the membrane fluidity by 7% and at the same time increases the dipolar relaxation by 38% (both with statistical significance, p equal to 0.001 and 0.0001 respectively). The fluidity result supports the difference obtained with the plasma membrane blue channel. Again the plasma membrane masking shows a clear significance in the fluidity and dipolar relaxation results (Figure 5B and C).
Figure 4. Effect of 1mM bolus of H2O2 for 1h on the NIH-3T3 cell membranes by LAURDAN fluorescence and lifetime phasor analysis of channel-2.

A) Phasor plot distribution for the control (Ctr) and H2O2 exposed cells (1mM H2O2 x1h). The data was represented and analyzed following same protocols explained in Figure 2A. B) Top, Fluidity fraction histogram for the LAURDAN lifetime distribution in channel-2. Data are represented as mean+SD, notice negative standard deviation was avoided to simplify the plots. Bottom, center of mass for the histograms as mean±SD. C) Top, dipolar relaxation (DR) fraction histogram for the LAURDAN lifetime distribution in channel-2. Data are represented as in section B. Bottom, center of mass for the histograms as mean±SD. D) Representative images of LAURDAN fluorescence for control and 1mM H2O2 x1h treated cells. Intensity, fluidity-axis, DR-axis were color-coded as indicated at the right of the images from top to bottom, respectively. *** and ** indicated p<0.0001 or p<0.001 vs Ctr, respectively, judged by the student t-test.
Figure 5. Effect of a bolus of H2O2 (100 μM and 1mM) for 1h on the NIH-3T3 cell plasma membranes by LAURDAN fluorescence and lifetime phasor analysis of channel-2.

A) Phasor plot distribution for the control (Ctr) and H2O2 exposed cells (100μM and 1mM H2O2 x1h). The data was represented and analyzed following same protocols explained in Figure 2A.. B) Top, Fluidity fraction histogram for the LAURDAN lifetime distribution in channel-1. Data are represented as mean+SD, notice negative standard deviation was avoided to simplify the plots. Bottom, center of mass for the histograms as mean±SD. C) Top, dipolar relaxation (DR) fraction histogram for the LAURDAN lifetime distribution in channel-1. Data are represented in section B. Bottom, center of mass for the histograms as mean±SD. D) Representative images of the mask applied to the LAURDAN fluorescence for control and 1mM H2O2 x1h treated cells. Mask, fluidity-axis, DR-axis were color-coded as indicated at the right of the images from top to bottom, respectively. *** indicated p<0.001 vs Ctr, judged by ANOVA and Tukey post-test. Indicated p<0.001 vs 100 μM H2O2 x 1h.. ¶Notice that the three-component positions are different from the channel-1.
To evaluate the effect of the addition of H2O2 we performed experiments with 100 μM final concentration of H2O2. When cells were incubated with 10 times less concentration than the 1mM data, similar results were obtained. Specifically, increased values for fluidity CM and DR were identified for the channel-1 (Figure 3B and C). In channel-2 (Figure 5B and C), the fluidity was higher than that obtained for 1 mM and the DR was in between the control and 1 mM H2O2 groups. The hyperspectral images were analyzed by the spectral phasor plot approach (Figure 6). With the spectral phasor plot it is possible to construct a triangle between the trajectory for lo-ld and the background (BG). Again using the three cursor analysis it is possible to get the fraction of lo/ld taking into account the pixels under the linear combination with the BG (Figure 6A). The color code used for images and the histogram for lo fraction shows that the H2O2 bolus strongly affects the plasma membrane fluidity (Figure 6B and C). By performing the CM analysis for the lo fraction histogram we quantified the increase in the lo fraction with strong significance (p=0.0001, see figure 6D).
Figure 6. Effect of 1mM bolus of H2O2 for 1h on the NIH-3T3 cell plasma membranes by LAURDAN fluorescence and spectral phasor analysis.

A) Spectral phasor plot distribution for the control (Ctr) and H2O2 exposed cells (1mM H2O2 x1h). A three components analysis was performed and the liquid order fraction (lo) was defined. The three positions were defined as lo, ld or background (BG). The cells were colored using the scale on top of the lo-fraction. B) Representative images of the transmission, intensity and lo-fraction for control (Ctr) and 1mM H2O2 x1h treated cells are presented, respectively. The masks used here were the same generated for the FLIM analysis. The scales at the right of the images indicated the range for each variable. C) A histogram for the lo-fraction was generated for each cell and then average and standard deviation were calculated. Data are represented as mean+SD, notice negative standard deviation was avoided to simplify the plots. D) Center of mass of the histograms was as mean±SD. *** indicated p<0.001 vs Ctr, judged by the student t-test.
In order to better understand the effects the H2O2 bolus on the cell membrane organization by LAURDAN fluorescence we used the MultiD analysis (Figure 7). The MultiD analysis was conceived to compare the data from different dimensions (namely two lifetime channels plus the hyperspectral data) using the reciprocity property of phasors and the common confocal plane for all the dimensions [5]. We used the green emission channel as a master phasor plot (Figure 7A and B), which means that we selected the ROI by cursors in this phasor plot and then these pixels selected in the images were propagated to the other dimensions (lifetime phasor for blue channel and spectral phasor). Two set of cursors were defined to identify the changes in the LAURDAN environment: red-green (inside the universal circle) and violet-pink (outside the universal circle, Figure 7). It is possible to see the changes in color between the control and cells treated by a H2O2 bolus. While control cells were more red-green, the cells treated with H2O2 bolus looked more violet-pink, which in other words means that the H2O2 bolus increased the DR fraction. Moreover, the connection with the other dimensions shows that for the controls cells the position of the pixels selected by the two cursors set are on top of the universal circle (Figure 7A, third panel from right). The pixels selected by the cursor in the green channel in the H2O2 bolus incubated cells are shifted towards the outside the universal circle (Figure 7B, third panel from right). Spectral phasors reveals the complexity in the LAURDAN photophysics (last panel in Figure 7A and B), while it is possible to connect short and long lifetimes on the blue channel with more fluid or lo membranes, respectively, the same is not true of the spectral shift. The spectral phasor shows that for long and short lifetimes the pixels recovered from the spectral phasor map to “pure” blue or red pixels, respectively, but also pixels with different ratios of linear combinations of lo/ld fractions.
The cell viability after 1h of 1mM H2O2 was measured using the combination of nuclear permeant dyes for healthy and non-healthy cells (see Table S1). The insult of 1mM H2O2 for 1 hour decreased the cell viability from 99 to 95%.
Discussion
LAURDAN fluorescence is a powerful tool to study the direct membrane perturbation and the lateral membrane organization changes induce by the H2O2 bolus. This unique characteristic is due to LAURDAN’s capability to sense its environment in terms of polarity (as an apparent dielectric constant) as well as by dipolar relaxation (DR) around its’ excited dipole (DR term refer to the capability of polar solvent molecules in the proximity of the LAURDAN to reorient) [5]. Gregorio Weber originally introduced the idea of independent analysis of these physical properties using different regions of the excitation/emission spectra of PRODAN [1]. LAURDAN fluorescence changes both in lifetime and spectra in response to the molecular environment [12]. In biological membranes this environment is located at the membrane interphase exactly behind the carbonyl group of the phospholipids [39]. After excitation, part of the energy of the LAURDAN excited state is expended on the reorientation of the water molecules (few molecules, ∼0–5), and the energy lost is reflected as a red-shift in the emission spectrum [9]. Membranes with different physical states can give different profiles of LAURDAN’s polarity and dipolar relaxation, and this information can be addressed by either lifetime or spectral shifts [14,15,40]. LAURDAN excitation spectrum also demonstrates sensitivity to the environment polarity and dipolar relaxation due to the ground state configuration. Two excitation bands (355 nm and 390 nm) are presented by LAURDAN in membranes (as well as in polar solvents) related to different ground state configurations. The red band (390 nm) is due to the stabilization of the Lα configuration (notice that this notation is related to the LAURDAN molecular orientation, not to the liquid crystalline phase) in the ground-state driven by the solvent molecules already relaxed around LAURDAN dipole [12]. In membranes, this band is particular intense in the gel phase and is also promoted in the gel/fluid coexistence by cholesterol addition [41]. This complex photophysics make LAURDAN one of the most useful dyes to study membrane dynamics in complex systems like the cell membranes. We defined an index, the “fluidity fraction”, as the decrease in the membrane order due to increasing penetration of water molecules at the interphase by the membrane physical state changes.
We acquired FLIM data using two bandpass filters with the ultimate purpose to discriminate the effects of H2O2 on the membrane polarity (fluidity, channel 1 or blue) and dipolar relaxation (hydration, channel 2 or green). Using the three-cursor approach [38] for the phasor distributions on the blue filter (Figures 2B and 3B, see materials & methods for the details of channel_1), we show that the H2O2 bolus increased the polarity of the cell membranes (decreased fluidity). However, using a similar analysis on the phasor distributions for the green filter (Figures 4C and 5C, see materials & methods for the details of channel_2), the H2O2 increased the dipolar relaxation. Using a mask that only selects the plasma membrane reveals that for this region of the cell (the plasma membrane), the results for the CM calculation of the fluidity-axis are significantly different after H2O2 treatment (Figure 3B, bottom panel). This difference in polarity was not significant for the whole cell (Figure 2B, bottom panel). These results are in accordance with the reported increased anisotropy for diphenylhexatriene in yeast plasma membrane (decreased fluidity) upon the adaptation to a H2O2 bolus [29]. The same group contributed to our understanding about the biochemical and genetically adaptation to H2O2 in yeast, concluding that cells increase the PC/PE ratio, sterols-rich micro domains change the sterol heterogeneity and some genes down-regulation related to fatty acid elongation and ceramide synthase were altered [30]. Also, H2O2 permeability through plasma membranes was compromised and this modification affected the rate of H2O2 detoxification by the antioxidant enzymes [30].
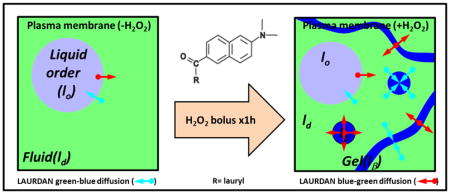
LAURDAN fluorescence in the channel 2 gives additional information about the physical state of the cell membranes. The increase in dipolar relaxation demonstrated by LAURDAN after the H2O2 bolus, both in the plasma membrane and throughout the cell, points out another important modification of the membranes related to increased water penetration. An interesting point is the lack of a significant difference in the membrane fluidity of the whole cell versus the decreased fluidity in the plasma membrane accompanied by the increased dipolar relaxation in the whole cell and plasma membranes. The apparent contradiction here is that less fluid membranes show increased dipolar relaxation. Before addressing this point, we want to focus on the increased dipolar relaxation found in channel 1 (Figure 2C). In principle, the blue channel should be more sensitive to changes in polarity than to dipolar relaxation. However, after the H2O2 bolus a shift of the pixels to the outside of the universal circle indicates an increased phase of the phasor (θ, which can only arise from a process occurring during the excited state). Since this process is not significant for gel/lo membranes the effect could be associated with some other form of relaxation. As discussed in the introduction, LAURDAN can sense both polarity and dipolar relaxation in the ground and in the excited-state. In the ground state, gel-like or lo domains will stabilize the LAURDAN with dipole molecules already relaxed around it, modifying its emission [10,12], (see Figure 8). Parasassi et al. describes a process experienced by LAURDAN during the excited-state called “fast interconversion between phases” due to translational diffusion [4]. Namely, LAURDAN could diffuse few angstroms during the excited state and the dye can be excited in one phase (gel) but emit while in the ld phase. Of course the translational distance involved needs to be very small (∼6–10 Å) but it is in the range of molecular diffusion measured in a membrane (∼1 μm2/s)[4]. The interconversion is mostly from ld→gel to lo, but the opposite is also possible in circumstances wherein the size of the gel domains are very small or a filament type of topology has developed. This hypothesis is supported by our results and the MultiD phasor analysis. By selecting pixels with pure gel/lo or ld on the channel 2 (green), the positions for those pixels can be identified in the lifetime channel 1 (blue) and spectral phasor (Figure 7). For the control situation, the pixels selected by green and red cursors fall on top of the universal circle in the blue channel (Figure 7A, third panel from left), while the spectral phasor mainly selects the pixels on the extreme of the triangle for lo/ld trajectory (Figure 7A, fourth panel from left). This situation indicates that the probe is emitting in the same phase where it was excited. When the cells were treated with a H2O2 bolus, a different situation is presented (Figure 7B). The pink/violet cursors were positioned with the intention to isolated the pixels from lo/ld trajectory on the green channel as previous proposed by Golfetto et al. [14]. We notice that those pixels fall outside the universal circle on the blue channel (Figure 7B, third panel from left). This effect is also seen in the spectral phasor where we can notice that an increased number of pixels with different degrees of linear combination between the lo/ld trajectory were highlighted (Figure 7B, fourth panel from left). This result supports the idea of fast interconversion of LAURDAN from gel→ld and ld→gel/lo. Why does this interconversion occur in that particular combination of gel, lo and ld? We believe that the interconversion from ld→gel/lo is not restricted by distance since the gel/lo domains are immersed in a ld sea. However, the distance to diffuse between gel/lo to ld needs to be few angstroms, thus either the domains must be very small or they should adopt a filament-like shapes to reduce the distance. Since lo/ld coexistence is a liquid-liquid phase segregation, the domain shape must be round [42], however for gel/ld coexistence the shape depends on the composition but there are many examples of supramolecular arrangements like filaments [43]. This idea is exemplified in Figure 8 which depicts a scheme of the plasma membrane organization before and after the H2O2 bolus, as well as LAURDAN’s photophysics and the fast interconversion concepts. In Figure 8 the left panel illustrates the plasma membrane in the control situation, where light blue circles represent liquid order domains immersed in a liquid disorder phase (light green). The small cyan and red arrows exemplify the fast interconversion for LAURDAN (dimensions are not at scale), the length of the arrow shows the process wherein LAURDAN is excited in one phase and emits in another (e.g. gel→ld). The color code for the arrows is illustrative of the spectral interconversion: cyan means excitation in the relaxed environment and emission in unrelaxed, whereas the red ones exemplify the opposite. LAURDAN photophysics is explained in detail and the two main variables; namely, polarity and dipolar relaxation are presented as blue-red arrows (central panel). In the right panel, the plasma membrane after the H2O2 bolus is represented, the occurrence of gel phase was drawn as small or filament-like domains in blue color (Figure 8, right panel). The illustration schematically shows the fast interconversion and the short distance of LAURDAN diffusion during the excited-state that support the results. In summary, we can say that while gel→ld justified the results of Figure 7B, on the other hand, the opposite interconversion ld→gel/lo observed in the emission explains the long lifetimes and blue spectra for LAURDAN in Figure 2 and 6.
Figure 8. Cartoon for the interpretation of LAURDAN results in cells after a bolus of H2O2.

At the center of the image the LAURDAN photophysics is shown. In the ground state it is possible to see the occurrence of different environments for LAURDAN which is sensitive to the presence of Cholesterol. Upon excitation, LAURDAN increases the transition dipole moment (represented as the enlargement of the blue double arrow) and depending on the environment, the probe can relaxed or not (represented by the blue and red double arrows). Cholesterol modifies the dipolar relaxation as shown in the figure represented by the black arrows (water molecules) oriented along the axis of the LAURDAN dipolar moment (red double arrow). On the left panel, the initial situation of the cell plasma membrane is represented as a coexistence of lo and ld phases. The cyan and red arrows represent the diffusion of LAURDAN from one phase to another during the excited state. The right panel is used to interpret the results. Basically, the occurrence of lβ phase (small or fiber-like domains) increases the chances for LAURDAN initially excited in a unrelaxed environment (lβ) to emit in a relaxed environment (ld). This process is known as “fast interconversion” of LAURDAN by translational diffusion between gel→ld and ld→gel/lo [4].
A second question is where the gel phase comes from? The hydrogen peroxide is currently recognized as a key molecule in many processes that involve oxidative eustress and distress [17,18,20,21,44]. Two interesting molecular targets of H2O2 in the membrane are protein kinase C (PKC) and phospholipase D (PLD). The relevance of these two molecular targets is significant to our discussion since PLD is responsible for hydrolysis of phosphatidylcholine (PC) to produce phosphatidic acid (PA) and choline [45]. The PA generated by hydrolysis of dimyristoylphosphocholine (DMPC) or diphosphatidylphosphocholine (DPPC) changes its melting temperature from 24°C to 51.3°C and from 41°C to 64.7°C, respectively, generating an abrupt phase transition in the DMPC from ld to gel at physiological temperatures [46]. The main function for PA in cells is signaling [45], but we cannot rule out that this phospholipid transformation can be responsible for the results that we discussed earlier. We discuss first the potential role of PKC/PLD on the plasma membrane remodeling by the H2O2 stimulus. PKC belong to a family of enzymes with crucial role on many of the physiological and pathophysiological processes due to the H2O2-induced signaling [47]. The PKC amino acid sequence contains a group of redox cysteines that can be modulated by H2O2. Depending on its redox status, the enzyme could be either activated or inactivated. The in vitro threshold for activation is 1mM and below, but for concentration higher than 1mM PKC loses its activity [47]. The regulatory region of PKC is responsible for the enzyme activation due to oxidation of cysteines related to zinc-binding domain. Upon oxidation, the autoinhibitory function of the regulator domain is released and the PKC is activated independent of any lipid cofactor. On the other hand, PLD can by directly activated by H2O2 presumably by a Ca+2-dependent protein tyrosine kinase(s), as well as by PKC [45,48–50]. Following this discussion, our results can be understood in terms of the definition of oxidative eustress or distress. We know there is a gradient through the plasma membrane caused by the H2O2 bolus, related to the permeability and detoxification by the intracellular enzymes [19–21,51]. It is relevant to discuss the validity of our ad hoc hypothesis regarding the H2O2 treatment and the PKC/PLD activation followed by a membrane supramolecular reorganization by the occurrence of gel phase associate to PA production. A 1mM bolus should be considering an oxidative distress based on the ruler presented by Sies [20]. However, newer estimates of this gradient pointed out that 100-fold difference between inside/outside can be increased up to 650–1000 times [21,24]. Considering this new estimate, one can imagine that 1–10 μM intracellular concentration is a reasonable concentration to modulate redox switches like the PKC/PLD. Evidently, the use of enzyme knock-out or PKC/PLD inhibitors would help to elucidate between the “Prx-sensor” or the direct “redox-switcher” oxidation mechanism. A concentration-response titration and temporal analysis will help to better comprehend the molecular basis of the H2O2 effect on plasma membrane. However, our data with a 10 times less concentration of H2O2 for the same amount of time showed a similar trend than 1 mM H2O2, even lower fluidity and increased DR. So far, our results point to a new interchange between H2O2-mediate signaling and plasma membrane remodeling after H2O2 challenge that would also be responsible for further effects in membrane signals.
Conclusions
Here we demonstrate the use of LAURDAN fluorescence in combination with phasor plots to detect membrane dynamic and structural organization. The treatment with an H2O2 bolus decreased the cell membrane fluidity in NIH-3T3 cells and at the same time increased water molecules penetration. This conclusion was made possible by the unique interpretation of LAURDAN’s complex photophysics afforded by the MultiD approach which has the capability to simultaneously use the lifetime and spectral information. This result was associated with the reorganization of gel domains and thus the protein-membrane signaling would be modified. We speculate that modulation of PKC/PLD activation by H2O2 redox-signaling could result from the plasma membrane changes, but further research is needed to confirm this ad hoc hypothesis.
Supplementary Material
Highlights.
LAURDAN fluorescence and phasor plot identify a complex effect of H2O2 on NIH-3T3 membranes.
A H2O2 bolus modifies the supramolecular organization of the plasma membrane in NIH-3T3.
Decreased membrane fluidity but increased dipolar relaxation was found after a H2O2 bolus.
MultiD-phasor revealed the occurrence LAURDAN fast-diffusion during the excited-state from gel→ld phases after a H2O2 bolus.
Acknowledgments
The authors would like to thanks to Milka Stakic for preparation of the cells and Prof. David M. Jameson and Prof. Ana Denicola for their valuable suggestions and discussions.
Funding
This work was supported by grants Grants NIH P41-GM103540 and NIH P50-GM076516. LM is supported for the Universidad de la República-Uruguay as a full time professor.
Abbreviations
- LAURDAN
6-dodecanoyl-2-dimethylaminonaphthalene
- FLIM
Fluorescence Lifetime Imaging Microscopy
- ROI
region of interest
- LSM
Laser scanning microscope
- PKC
protein Kinase C
- PLD
phospholipase D. CM, center of mass
- DR
dipolar relaxation
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Weber G, Farris FJ. Synthesis and spectral properties of a hydrophobic fluorescent probe: 6-propionyl-2-(dimethylamino)naphthalene. Biochemistry. 1979;18:3075–3078. doi: 10.1021/bi00581a025. [DOI] [PubMed] [Google Scholar]
- 2.Parasassi T, Conto F, Gratton E. Time-Resolved Fluorescence Emission Spectra of Laurdan in Phospholipid Vesicle by Multifrequency Phase and Modulation Fluorometry. Cell Mol Biol. 1986;32:103–108. [PubMed] [Google Scholar]
- 3.Macgregor RB, Weber G. Estimation of the polarity of the protein interior by optical spectroscopy. Nature. 1986;319:70. doi: 10.1038/319070a0. [DOI] [PubMed] [Google Scholar]
- 4.Parasassi T, Ravagnan G, Rusch RM, Gratton E. Modulation and dynamics of phase properties in phospholipid mixtures detected by Laurdan fluorescence. Photochem Photobiol. 1993;57:403–10. doi: 10.1111/j.1751-1097.1993.tb02309.x. [DOI] [PubMed] [Google Scholar]
- 5.Malacrida L, Jameson DM, Gratton E. A multidimensional phasor approach reveals LAURDAN photophysics in NIH-3T3 cell membranes. Sci Rep. 2017;7 doi: 10.1038/s41598-017-08564-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Parasassi T, De Stasio G, Ravagnan G, Rusch RM, Gratton E. Quantitation of lipid phases in phospholipid vesicles by the generalized polarization of Laurdan fluorescence. Biophys J. 1991;60:179–189. doi: 10.1016/S0006-3495(91)82041-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parasassi T, De Stasio G, d’Ubaldo A, Gratton E. Phase fluctuation in phospholipid membranes revealed by Laurdan fluorescence. Biophys J. 1990;57:1179–1186. doi: 10.1016/S0006-3495(90)82637-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parasassi T, Gratton E. Packing of phospholipid vesicles studied by oxygen quenching of Laurdan fluorescence. J Fluoresc. 1992;2:167–174. doi: 10.1007/BF00866931. [DOI] [PubMed] [Google Scholar]
- 9.Parasassi T, Gratton E, Yu WM, Wilson P, Levi M. Two-photon fluorescence microscopy of laurdan generalized polarization domains in model and natural membranes. Biophys J. 1997;72:2413–2429. doi: 10.1016/S0006-3495(97)78887-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parasassi T, Di Stefano M, Loiero M, Ravagnan G, Gratton E. Cholesterol modifies water concentration and dynamics in phospholipid bilayers: a fluorescence study using Laurdan probe. Biophys J. 1994;66:763–768. doi: 10.1016/S0006-3495(94)80852-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parasassi T, Giusti aM, Raimondi M, Gratton E. Abrupt modifications of phospholipid bilayer properties at critical cholesterol concentrations. Biophys J. 1995;68:1895–1902. doi: 10.1016/S0006-3495(95)80367-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parasassi T, Gratton E. Membrane lipid domains and dynamics as detected by Laurdan fluorescence. J Fluoresc. 1995;5:59–69. doi: 10.1007/BF00718783. [DOI] [PubMed] [Google Scholar]
- 13.Parasassi T, Krasnowska EK. Laurdan and Prodan as polarity-sensitive fluorescent membrane probes. J Fluoresc. 1998;8:365–373. doi: 10.1023/A:1020528716621. [DOI] [Google Scholar]
- 14.Golfetto O, Hinde E, Gratton E. Laurdan fluorescence lifetime discriminates cholesterol content from changes in fluidity in living cell membranes. Biophys J. 2013;104:1238–1247. doi: 10.1016/j.bpj.2012.12.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Malacrida L, Astrada S, Briva A, Bollati-Fogolín M, Gratton E, Bagatolli LA. Spectral phasor analysis of LAURDAN fluorescence in live A549 lung cells to study the hydration and time evolution of intracellular lamellar body-like structures. Biochim Biophys Acta - Biomembr. 2016;1858:2625–2635. doi: 10.1016/j.bbamem.2016.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Malacrida L, Gratton E, Jameson DM. Model-free methods to study membrane environmental probes: A comparison of the spectral phasor and generalized polarization approaches. Methods Appl Fluoresc. 2015;3 doi: 10.1088/2050-6120/3/4/047001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rhee SG. H2O2, a Necessary Evil for Cell Signaling. Science (80- ) 2006;312:1882–1883. doi: 10.1126/science.1130481. [DOI] [PubMed] [Google Scholar]
- 18.Veal EA, Day AM, Morgan BA. Hydrogen peroxide sensing and signaling. Mol Cell. 2007:1–14. doi: 10.1016/j.molcel.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 19.Winterbourn CC, Hampton MB. Redox biology: Signaling via a peroxiredoxin sensor. Nat Chem Biol. 2015;11:5–6. doi: 10.1038/nchembio.1722. [DOI] [PubMed] [Google Scholar]
- 20.Sies H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017;11:613–619. doi: 10.1016/j.redox.2016.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Antunes F, Brito PM. Quantitative biology of hydrogen peroxide signaling. Redox Biol. 2017;13:1–7. doi: 10.1016/j.redox.2017.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Antunes F, Cadenas E. Estimation of H2O2 gradients across biomembranes. FEBS Lett. 2000;475:121–126. doi: 10.1016/S0014-5793(00)01638-0. [DOI] [PubMed] [Google Scholar]
- 23.Adimora NJ, Jones DP, Kemp ML. A Model of Redox Kinetics Implicates the Thiol Proteome in Cellular Hydrogen Peroxide Responses. Antioxid Redox Signal. 2010;13:731–743. doi: 10.1089/ars.2009.2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang BK, Sikes HD. Quantifying intracellular hydrogen peroxide perturbations in terms of concentration. Redox Biol. 2014;2:955–962. doi: 10.1016/j.redox.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mbemba F, Houbion A, Raes M, Remacle J. Subcellular localization and modification with ageing of glutathione, glutathione peroxidase and glutathione reductase activities in human fibroblasts. Biochim Biophys Acta - Gen Subj. 1985;838:211–220. doi: 10.1016/0304-4165(85)90081-9. htt. [DOI] [PubMed] [Google Scholar]
- 26.Sobotta MC, Liou W, Stöcker S, Talwar D, Oehler M, Ruppert T, Scharf AND, Dick TP. Peroxiredoxin-2 and STAT3 form a redox relay for H2O2 signaling. Nat Chem Biol. 2014;11:64. doi: 10.1038/nchembio.1695. [DOI] [PubMed] [Google Scholar]
- 27.Randall LM, Ferrer-Sueta G, Denicola A. Peroxiredoxins as preferential targets in H2O 2-induced signaling. 1. Elsevier Inc; 2013. [DOI] [PubMed] [Google Scholar]
- 28.Branco MR, Marinho HS, Cyrne L, Antunes F. Decrease of H 2 O 2 Plasma Membrane Permeability during Adaptation to H 2 O 2 in Saccharomyces cerevisiae. J Biol Chem. 2004;279:6501–6506. doi: 10.1074/jbc.M311818200. [DOI] [PubMed] [Google Scholar]
- 29.Folmer V, Pedroso N, Matias AC, Lopes SCDN, Antunes F, Cyrne L, Marinho HS. H2O2induces rapid biophysical and permeability changes in the plasma membrane of Saccharomyces cerevisiae. Biochim Biophys Acta - Biomembr. 2008;1778:1141–1147. doi: 10.1016/j.bbamem.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 30.Pedroso N, Matias AC, Cyrne L, Antunes F, Borges C, Malhó R, de Almeida RFM, Herrero E, Marinho HS. Modulation of plasma membrane lipid profile and microdomains by H2O2in Saccharomyces cerevisiae. Free Radic Biol Med. 2009;46:289–298. doi: 10.1016/j.freeradbiomed.2008.10.039. [DOI] [PubMed] [Google Scholar]
- 31.Bienert GP, Schjoerring JK, Jahn TP. Membrane transport of hydrogen peroxide. Biochim Biophys Acta - Biomembr. 2006;1758:994–1003. doi: 10.1016/j.bbamem.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 32.Bienert GP, Chaumont F. Aquaporin-facilitated transmembrane diffusion of hydrogen peroxide. Biochim Biophys Acta - Gen Subj. 2014;1840:1596–1604. doi: 10.1016/j.bbagen.2013.09.017. [DOI] [PubMed] [Google Scholar]
- 33.Hadwan MH, Abed HN. Data supporting the spectrophotometric method for the estimation of catalase activity. Data Br. 2016;6:194–199. doi: 10.1016/j.dib.2015.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Digman MA, Gratton E. The phasor approach to fluorescence lifetime imaging: Exploiting phasor linear properties. In: Marcu L, French PMW, Elson DS, editors. Fluoresc Lifetime Spectrosc Imaging Princ Appl Biomed Diagnostics. 1. Chapter 10. CRC Press; 2014. pp. 235–248. [DOI] [Google Scholar]
- 35.Jameson DM, Gratton E, Hall RD. The Measurement and Analysis of Heterogeneous Emissions by Multifrequency Phase and Modulation Fluorometry. Appl Spectrosc Rev. 1984;20:55–106. doi: 10.1080/05704928408081716. [DOI] [PubMed] [Google Scholar]
- 36.Digman MA, Caiolfa VR, Zamai M, Gratton E. The Phasor Approach to Fluorescence Lifetime Imaging Analysis. Biophys J. 2008;94:L14–L16. doi: 10.1529/biophysj.107.120154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fereidouni F, Bader AN, Gerritsen HC. Spectral phasor analysis allows rapid and reliable unmixing of fluorescence microscopy spectral images. Opt Express. 2012;20:12729. doi: 10.1364/OE.20.012729. [DOI] [PubMed] [Google Scholar]
- 38.Ranjit SR, Dvornikov A, Dobrinskikh E, Wang X, Luo Y, Levi M, Gratton E. Measuring the effect of a Western diet on liver tissue architecture by FLIM autofluorescence and harmonic generation microscopy. Biomed Opt Express. 2017;8:371–378. doi: 10.1364/BOE.8.003143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bagatolli LA, Parasassi T, Fidelio GD, Gratton E. A Model for the Interaction of 6-Lauroyl-2-(N,N-dimethylamino)naphthalene with Lipid Environments: Implications for Spectral Properties. Photochem Photobiol. 1999;70:557–564. doi: 10.1111/j.1751-1097.1999.tb08251.x. [DOI] [PubMed] [Google Scholar]
- 40.Malacrida L, Gratton E, Jameson DM. Model-free methods to study membrane environmental probes: A comparison of the spectral phasor and generalized polarization approaches. Methods Appl Fluoresc. 2015;3:047001. doi: 10.1088/2050-6120/3/4/047001. [DOI] [PubMed] [Google Scholar]
- 41.Parasassi T, Di Stefano M, Loiero M, Ravagnan G, Gratton E. Influence of cholesterol on phospholipid bilayers phase domains as detected by Laurdan fluorescence. Biophys J. 1994;66:120–132. doi: 10.1016/S0006-3495(94)80763-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bernardino J, Serna D, Perez-gil J, Simonsen AC, Bagatolli LA. Cholesterol Rules. 2004;279:40715–40722. doi: 10.1074/jbc.M404648200. [DOI] [PubMed] [Google Scholar]
- 43.Fidorra M, Garcia a, Ipsen JH, Härtel S, Bagatolli La. Lipid domains in giant unilamellar vesicles and their correspondence with equilibrium thermodynamic phases: a quantitative fluorescence microscopy imaging approach. Biochim Biophys Acta. 2009;1788:2142–9. doi: 10.1016/j.bbamem.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 44.Sikes HD. Redox regulation: Scaffolding H2O2 signaling. Nat Chem Biol. 2017;13:818–819. doi: 10.1038/nchembio.2432. [DOI] [PubMed] [Google Scholar]
- 45.Ito Y, Nakashima S, Nozawa Y. Hydrogen Peroxide-Induced Phospholipase D Activation in Rat Pheochromocytoma PC12 Cells: Possible Involvement of Ca2+-Dependent Protein Tyrosine Kinase. J Neurochem. 2002;69:729–736. doi: 10.1046/j.1471-4159.1997.69020729.x. [DOI] [PubMed] [Google Scholar]
- 46.Marsh D. Handbook of Lipid Bilayers. 2. CRC Press; 2013. http://www.crcpress.com/product/isbn/9781420088328. [Google Scholar]
- 47.Gopalakrishna R, McNeill TH, Elhiani AA, Gundimeda U. Methods for studying oxidative regulation of protein kinase C. 1. Elsevier Inc; 2013. [DOI] [PubMed] [Google Scholar]
- 48.Natarajan V, Taher MM, Roehm B, Parinandi NL, Schmid HH, Kiss Z, Garcia JG. Activation of endothelial cell phospholipase D by hydrogen peroxide and fatty acid hydroperoxide. J Biol Chem. 1993;268:930–937. [PubMed] [Google Scholar]
- 49.Min DS, Kim E-G, Exton JH. Involvement of tyrosine phosphorylation and protein kinase C in the activation of phospholipase D by H2O2 in Swiss 3T3 fibroblasts. J Biol Chem. 1998;273:29986–29994. doi: 10.1074/jbc.273.45.29986. [DOI] [PubMed] [Google Scholar]
- 50.Usatyuk PV, Kotha SR, Parinandi NL, Natarajan V. Phospholipase D signaling mediates reactive oxygen species-induced lung endothelial barrier dysfunction. Pulm Circ. 2013;3:108–15. doi: 10.4103/2045-8932.109925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Winterbourn CC. The biological chemistry of hydrogen peroxide. 1. Elsevier Inc; 2013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.