

Abstract
Psychiatric disorders are thought to have a complex genetic pathology consisting of interplay of common and rare variation. Traditionally, pedigrees are used to shed light on the latter only, while here we discuss the application of polygenic risk scores to also highlight patterns of common genetic risk. We analyze polygenic risk scores for psychiatric disorders in a large pedigree (n ~ 260) in which 30% of family members suffer from major depressive disorder or bipolar disorder. Studying patterns of assortative mating and anticipation, it appears increased polygenic risk is contributed by affected individuals who married into the family, resulting in an increasing genetic risk over generations. This may explain the observation of anticipation in mood disorders, whereby onset is earlier and the severity increases over the generations of a family. Joint analyses of rare and common variation may be a powerful way to understand the familial genetics of psychiatric disorders.
Simone de Jong et al. examine the balance of common and rare risk for psychiatric disorders in a large family with high incidence of Bipolar Disorder and Major Depressive Disorder. They find that increased polygenic risk over generations could be partially due to assortative mating, which may explain the observation of anticipation in mood disorders.
Introduction
The development of polygenic risk scoring (PRS) has greatly advanced the field of psychiatric genetics. This approach allows for even sub-genome-wide significant threshold results from large genome-wide meta analyses to be leveraged to explore genetic risk in smaller studies1. The effect sizes at many individual single-nucleotide polymorphisms (SNPs), estimated by large genome-wide association studies (GWAS) on the disorder of interest, are used to calculate an individual level genome-wide PRS in individuals from an independent genetic dataset. The PRS based on the summary statistics of the schizophrenia (SCZ) GWAS by the Psychiatric Genomics Consortium (PGC)2,3 has proven to be most powerful in predicting not only SCZ1,4 but also other psychiatric disorders5–7. In addition, updated, more powerful, summary statistics from the Psychiatric Genomics Consortium from the latest GWAS for bipolar disorder (BPD) and major depressive disorder (MDD) are available via the PGC Data Access Portal (https://www.med.unc.edu/pgc/shared-methods).
Aside from increasing power in traditional case-control designs, PRS algorithms also open up new avenues for studying common variation. In this study, we consider the application of PRS within a family context. While pedigree studies have been traditionally used to explore rare genetic variation through linkage analyses, studying patterns of PRS throughout a pedigree would allow for assessment of phenomena like assortative mating and anticipation. Assortative (non-random) mating is a common phenomenon where mated pairs are more phenotypically similar for a given characteristic than would be expected by chance8. Results from a recent study by Nordsletten et al.9 show extensive assortative mating within and across psychiatric, but not physical disorders. This could explain some of the features of the genetic architecture of this category of disorders9–11. This includes anticipation, a phenomenon where later generations exhibit more severe symptoms at an earlier age, robustly reported (although not explained) in BPD12, and recently highlighted in genetic studies of MDD13,14.
In the current study, we aim to discuss the application of polygenic risk scoring for SCZ, MDD, and BPD to explore patterns of common risk variation within a family context. We illustrate our discussion by investigating the relationship between PRS and apparent assortative mating, and anticipation within a complex multigenerational pedigree affected with mood disorders.
Results
Study overview
We identified a large pedigree in Brazil, the Brazilian Bipolar Family (BBF), after examination of a 45-year-old female who presented with severe Bipolar Type 1 (BPI) disorder. She stated there were dozens of cases of mood disorders in the family, most of whom lived in a small village in a rural area of a large state north of São Paulo (see Methods for details). We conducted 308 interviews using the Portuguese version of the Structured Clinical Interview for DSM-IV Axis I Disorders (SCID-I)16 for family members over the age of 16 and the Portuguese version of Kiddie-SADS-Present and Lifetime Version (K-SADS-PL)17 for family members aged 6–16. Following diagnostic interviews, we conducted genotype analysis of all interviewees using the Illumina Infinium PsychArray-24. Polygenic risk scores (PRS) were assigned to each family member using PRS thresholds most predictive in discriminating affected from unaffected family members (see Methods).
Affection status
The PRS thresholds were selected to optimally discriminate between affected (n = 78) versus unaffected (n = 147) family members with a higher score in affecteds for SCZ:PRS (Beta = 0.069, SE = 0.032, Z-ratio = 2.117, p = 0.035, R2 = 0.021), and BPD:PRS (Beta = 0.094, SE = 0.030, Z-ratio = 3.123, p = 0.002, R2 = 0.039). None of the PRS significantly discriminated between individuals having experienced a psychotic episode at some point in their lives (n = 25) versus the unaffected group (n = 147). Visualization of PRS in different diagnostic categories is shown in Supplementary Figure 1.
Assortative mating
Married-in individuals were defined as individuals married to a BBF member, but having no parents in the family themselves. Of the 70 married-in individuals ascertained (irrespective of having genotype data) 19 (27%) were affected with a psychiatric disorder. This is significantly higher than the 17% population prevalence of the most common of the three disorders: MDD (Fisher’s exact p = 0.02)15. The unaffected married-in group does not differ from the general healthy population as evidenced by no significant differences in PRS as compared to the population control group (BRA; see Methods). The above led us to investigate whether we can observe assortative mating on a genetic level, using PRS. In spouse pairs, we were unable to predict the PRS of the husband, using that of his wife, even when selecting concordant (both affected or both unaffected) pairs only. We considered the possibility that the married-in individuals might confer a different genetic predisposition to mood disorders to their offspring than the original family members. The number of children contributed per spouse pair to each offspring category is shown in Supplementary Table 1. Demographics of the offspring in the different offspring categories (no affected parents (n = 54); one affected family member parent (n = 69); one affected married-in parent (n = 15) and two affected parents (n = 38)) are given in Supplementary Tables 2 and 3. Indeed, we find that offspring of an affected married-in parent show increased SCZ:PRS (Beta = 0.209, SE = 0.064, Z-ratio = 3.288, p = 0.002, R2 = 0.186, Fig. 1) and BPD:PRS (Beta = 0.172, SE = 0.066, Z-ratio = 2.613, p = 0.013, R2 = 0.126, Fig. 1) as compared to having no affected parents.
Fig. 1.

Violin plots of SCZ:PRS (dark blue plots) MDD:PRS (light blue plots) and BPD:PRS (green plots) for offspring of all spouse pair possibilities. The first category represents PRS in individuals with no affected parents, the next for individuals with an affected family member parent, followed by offspring of an affected married-in individual, and finally offspring of two affected parents. The last two sets of violin plots represent offspring of unknown spouse pairs and the BRA controls. The dot and error bars represent mean ± standard deviation of standardized PRSs
Anticipation
The BBF shows patterns of anticipation, with individuals having an earlier age at onset (AAO) in later generations. For 104 individuals (irrespective of having genotype data), the average age at onset significantly decreases over generations with G2 (n = 1, AAO = 8), G3 (n = 23, AAO = 30.2 yrs ± 21.1), G4 (n = 53, AAO = 31.2 yrs ± 12.3), G5 (n = 23, AAO = 19.7 yrs ± 9.5), and G6 (n = 4, AAO = 13 yrs ± 3.6) (Supplementary Figure 2) with older participants recalling their AAO directly and younger participants confirmed using clinical records or parental recall (Beta = −4.549, SE = 1.793, Z-ratio = −2.537, p = 0.013, R2 = 0.059). We hypothesized that this decrease in AAO would be reflected in a negative correlation with PRS, subsequently resulting in a pattern of increased PRS over generations. Because of a limited sample size of affected individuals per generation, a direct correlation of AAO and PRS does not reach significance, although the youngest generation (G5) does show trends towards negative correlations for SCZ:PRS and MDD:PRS (Supplementary Figure 3). The SCZ:PRS does show a significant increase over generations (Fig. 2) where n = 197 family members were included (46 married-in individuals were excluded from the analysis to capture inheritance patterns of SCZ:PRS) in a linear regression with generation as independent variable (Beta = 0.131, SE = 0.049, Z-ratio = 2.668, p = 0.008, R2 = 0.025). The presence of such an effect when comparing generations suggests ascertainment effects such as relying on the recall of older family member with very long duration of illness in previous generations may be masking an overall effect across the entire family.
Fig. 2.
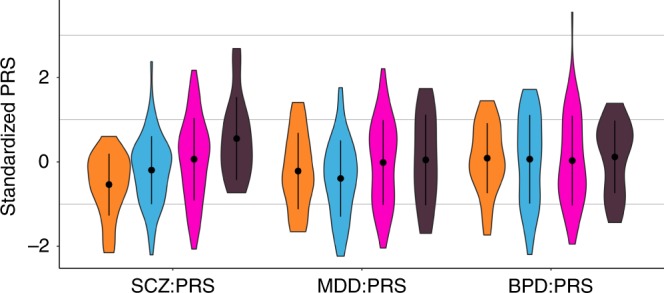
Violin plots of SCZ:PRS, MDD:PRS and BPD:PRS per generation for family members only, with results for the generations G3 (n = 25, orange plots), G4 (n = 72, light blue plots), G5 (n = 80, pink plots), and G6 (n = 16, dark purple plots) (excluding the oldest generation G2 and youngest generation G7 because of n = 2 sample size). The dot and error bars represent mean ± standard deviation of standardized PRSs
Balance of common and rare genetic risk
Transmission disequilibrium test analysis within the chr2p23 linkage region resulted in identification of rs1862975, a SNP originally typed on the Affymetrix linkage array (combined test p = 0.003). The homozygous T genotype was detected in 68% affected family members, 57% affected married-ins, 36% unaffected family members and 24% unaffected married-ins. Since this SNP was present only on the Affymetrix array, we identified rs12996218 as a proxy in CEU/TSI populations (D′ = 1.0, R2 = 0.92) via the LDproxy option in LDlink (Machiela et al.16, https://analysistools.nci.nih.gov/LDlink/). Of the 57 BRA controls, 9 individuals (15%) carried the GG genotype equivalent to the rs1862975 TT risk genotype. The distribution of the rs1862975 genotypes in affected and unaffected individuals over generations is given in Supplementary Figure 4. The number of individuals carrying the TT does not significantly change over generations in either group. None of the PRS showed a significant difference when comparing PRS for rs1862975 genotypes in affected and unaffected individuals (Supplementary Figure 5).
Discussion
The current study is one of the first the first to probe patterns of common genetic variation within a traditional pedigree design. While increased polygenic scores in patients as compared to unaffected family members have been demonstrated recently17, we aimed to illustrate the possibilities of this approach by investigating apparent assortative mating and anticipation in a large multigenerational pedigree affected with mood disorders through polygenic risk scores for SCZ2, MDD18, and BPD19, and thereby improve mechanistic understanding of common genetic risk for psychiatric disorders.
Highlighting the possibilities of PRS applications within a family context, we set out to utilize patterns of common variation to illuminate phenomena within the family that are out of reach from traditional case/control studies. Assortative mating is one of the features in this family, where many married-in individuals are more affected with a mood disorder than the general population. As opposed to the family members, the married-in individuals were more often affected with (r)MDD instead of BP. As diagnoses were determined after the couples were married, we cannot rule out that this could be a result from a causal effect of a spouse’s mental health on that of their partner. However, non-random mating patterns have been reported in the population regarding body type, socio-economic factors and psychiatric traits9,10. The BBF provides a unique opportunity to look at the genetic correlation between spouse pairs and the contribution of married-in individuals to overall psychiatric morbidity. A recent study has found genetic evidence for assortative mating when studying BMI and height in spouse pairs11. In the BBF; the affected married-in individuals have a higher, though non-significant, polygenic score than affected or unaffected family members but it appears that we observe significant consequences of this in that the offspring of an affected married-in parent collectively show significantly increased SCZ:PRS and BPD:PRS. However, it is puzzling we do not see an effect on offspring of two affected parents (which would include a married-in parent), which could indicate this finding to be of limited statistical robustness.
A contribution of the married-in parents to a genetic driven anticipation in age of onset is supported by the increase in SCZ:PRS over generations, although our cross sectional study dataset was less well powered to find an association with age at onset within affected family members. We did observe a trend for association between age at onset and PRS in the youngest generation in this study but not when combining sample across generations. Age at onset can be considered a proxy for severity20,21 and has been previously associated with genetic risk in MDD13,14. However, this variable needs to be interpreted with caution, especially when analyzing patterns over time since it is dependent on context and memory22. Ascertainment bias can be a confounding factor in studies of psychiatric traits, with older generations having less access to psychiatric care and possibly misremembering the onset or nature of their first episode. In addition, although currently classified as “unaffected” or “unknown”, members of the youngest generations can still develop a psychiatric disorder in the future.
Finally, we explored the balance of common and rare risk variation through combining our current PRS results with previously performed linkage analyses. We did not find a decrease in potential rare risk allele genotypes over generations contrasting the increase in SCZ:PRS, and PRS profiles for individuals carrying rare risk genotypes are not significantly different. This indicates that these factors separately confer independent disease risk. We recognize the limitations in sample size of our pedigree and therefore the power to draw statistically robust conclusions, especially in the offspring and combined linkage and PRS analyses. Even though the BBF might not be sufficiently powered, our point is to use this dataset to illustrate our approach and emphasize the unique nature of the family enabling the study of patterns of PRS and the balance of common and rare genetic risk for psychiatric disorders conferred within families. We encourage replication in similar pedigrees including affected married-in individuals when available to fully utilize the potential of PRS in this setting.
In conclusion, our study is an exploration of PRS as a tool for investigating patterns of common genetic risk in a traditional pedigree context. The SCZ and BPD scores appear best suited in our data for teasing apart patterns of assortative mating and anticipation, whereby increased polygenic risk for psychiatric disorders is contributed by affected individuals who married into the family, adding to the already present rare risk variation passed on by the early generations23.
Methods
Subject description
The Brazilian bipolar family (BBF) was ascertained via a 45-year-old female proband who presented with severe Bipolar Type 1 (BPI) disorder and stated there were dozens of cases of mood disorders in the family, most of whom lived in a small village in a rural area of a large state north of São Paulo. Cooperation from the family and a 2003 self-published book about their history was invaluable for our ascertainment. Historically, the entire BBF consists of 960 members. Living family members > 16 years of age underwent semi-structured interviews, using the Portuguese version of the Structured Clinical Interview for DSM-IV Axis I Disorders (SCID-I)24. Members aged 6–16 were assessed using the Portuguese version of Kiddie-SADS-Present and Lifetime Version (K-SADS-PL)25. In total 308 interviews were completed, and 5 eligible members declined an interview. In the rare event of discrepancies, two independent psychiatrists reviewed them and a final consensus diagnosis was assigned. All affected and unaffected adult family members that have been included in the genetic study have given informed consent. Minors have given assent, followed by consulted consent by their parents in accordance with accepted practice in both the U.K. and Brazil. The project was approved by the Brazilian National Ethics Committee (CONEP). Table 1 contains the demographics of the subjects used in the current analysis (n = 243 passed genotype quality control procedures described below). The population control dataset (BRA controls) was collected in Sao Paulo, Brazil, as a control dataset in a genetic study of first-episode psychosis26. They were volunteers who had no abnormal psychiatric diagnoses (SCID) or family history of psychotic illness. The Research Ethics Committee of Federal University of Sao Paulo (UNIFESP) approved the research protocol, and all participants gave informed consent (CEP No. 0603/10). Demographics for n = 57 BRA controls can be found in Table 1.
Table 1.
Demographics of the Brazilian bipolar family members and the Brazilian population control dataset (BRA controls) in the current study
| Diagnosis | n | Male, female | Age (±sd) | Age of onset (±sd) | Married-in | Psychosis |
|---|---|---|---|---|---|---|
| BPI | 17 | 6, 11 | 50.4 (±18.9) | 24.9 (±14.6) | 0 | 13 |
| BPII | 11 | 4, 7 | 38.7 (±15.2) | 24.2 (±13.8) | 1 | 4 |
| BPNOS | 8 | 6, 2 | 29.6 (±19.9) | 17.0 (±18.7) | 0 | 1 |
| rMDD | 17 | 5, 12 | 50.2 (±16.7) | 27.3 (±14.1) | 3 | 4 |
| MDD | 21 | 11, 10 | 43.8 (±17.8) | 34.5 (±15.5) | 6 | 1 |
| SADB | 1 | 0, 1 | 73 | 44 | 0 | 1 |
| Schizophrenia | 1 | 1, 0 | 44 | 36 | 0 | 1 |
| Cyclothymia | 1 | 0, 1 | 40 | 25 | 0 | 0 |
| Dysthymia | 1 | 0, 1 | 52 | — | 1 | 0 |
| Unaffected | 147 | 89, 58 | 36.8 (±20.0) | — | 35 | 0 |
| Unknown | 18 | 14, 4 | 5.7 (±7.1) | — | 0 | — |
| Total | 243 | 136, 107 | 37.3 (±21.0) | 28.3 (±15.5) | 46 | 25 |
| BRA controls | 57 | 33, 24 | 27.1 (±7.2) | — | — | — |
The first column contains the number of individuals affected with the disorder. A breakdown of gender, age, age at onset (with ± sd; standard deviation) is given in the next columns. The married-in column contains the number of individuals in each diagnostic category married-in to the family. The last column contains counts of individuals in each category who have experienced a psychotic episode during their lifetime
Diagnostic categories are BP1 bipolar I, BPII bipolar II, BPNOS bipolar not otherwise specified, rMDD recurrent major depressive disorder, MDD major depressive disorder, SADB schizoaffective disorder, schizophrenia, cyclothymia and dysthymia
Genotype data
Following diagnostic interview, interviewers obtained whole blood in EDTA containing monovettes for adults and lesser amounts or saliva given personal preference or age (DNA Genotek Inc., Ontario, Canada). Genomic DNA was isolated from whole blood and saliva at UNIFESP using standard procedures. Whole-genome genotype data was generated using the Illumina Infinium PsychArray-24 (http://www.illumina.com/products/psycharray.html) for both the BBF and the BRA control dataset at the in-house BRC BioResource Illumina core lab according to manufacturers protocol. Samples were excluded when average call rate was <98%, missingness >1% with additional check for excess heterozygosity, sex, family relationships and concordance rates with previous genotyping assays. SNPs were excluded when missingness > 1%, MAF < 0.01 or HWE < 0.00001 and if showing Mendelian errors for the BBF dataset in Plink v1.0727 and v1.928 or Merlin v1.1.229. The BBF and BRA control datasets were QC’d separately and then merged, applying the same SNP QC thresholds to the merged dataset as well. This quality control procedure resulted in a dataset of 225,235 SNPs for 243 BBF individuals (197 family members and 46 married-in individuals) and 57 BRA controls. Eigensoft v4.230 was used to check for population differences between the BBF family members, married-in individuals and BRA control sets. The BBF members self-reported mixed Southern European ancestry, confirmed by genome-wide principal components analysis showing that family members clustered closely with the Northern and Western European and Tuscan Italian populations in Hapmap3, with a relative lack of African or Native American ancestry (Supplementary Figure 6). The principal components appear to represent within-family structure, with most PCs seemingly separating subfamilies (Supplementary Figures 7 and 8). PRS analyses as described below were also performed to include subfamily as a fixed effect, controlling for household effects (Supplementary Table 3). PC1 and PC2 are significantly correlated to the SCZ:PRS (PC1 r = −0.131, p = 0.023; PC2 r = −0.268, p = 2.611 × 10−6), PC1 to MDD:PRS (PC1 r = −0.251, p = 1.114 × 10−5), and PC1 and PC2 to BPD:PRS (PC1 r = 0.189, p = 9.710 × 10−4; PC2 r = −0.123, p = 0.033). The principal components were not used in subsequent analyses.
Polygenic risk scores
Polygenic risk scores for each family member (n = 243) and population control (n = 57) were generated in the same run using the PRSice v1.25 software31 with the publically available PGC schizophrenia GWAS2 as a base dataset (36,989 SCZ cases, 113,075 controls), in addition to MDD (51,865 MDD cases, 112,200 controls, not including 23andme individuals) and BPD (20,352 BPD cases, 31,358 controls) summary statistics from the latest PGC meta analyses (unpublished data18,19). We performed p-value-informed clumping on the genotype data with a cut-off of r2 = 0.25 within a 200-kb window, excluding the MHC region on chromosome 6 because of its complex linkage disequilibrium structure. Acknowledging the possibility of over-fitting, we selected the PRS thresholds most predictive in discriminating affected from unaffected family members through linear regression in PRSice for SCZ:PRS (p < 0.00055, 1218 SNPs), MDD:PRS (p < 0.0165, 715 SNPs) and BPD:PRS (p < 0.00005, 143 SNPs). PRS showed low to modest correlations (no covariates) amongst each other in our data (SCZ:PRS versus MDD:PRS r = 0.176, p = 0.002, SCZ:PRS versus BPD:PRS r = 0.124, p = 0.032, MDD:PRS versus BPD:PRS r = −0.026, p = 0.660).
Linkage analysis
The main linkage analyses identifying rare genetic risk variation were performed as part of a previous paper on the BBF23 using the Affymetrix 10k linkage genotyping array. In order to explore the balance between common and rare risk variation, we selected the strongest signal for affected versus unaffected family members on chr2p23 (chr2:30000001-36600000, LOD = 3.83). Following the strategy described by Rioux et al.32, we performed a transmission disequilibrium test on the 25 markers in this linkage region in an attempt identify “linkage positive” individuals in n = 300 family members with one or both types of genotype array data. N = 155 individuals overlap with the current study and based on exploration of patterns of PRS in the current study we attempted to answer two questions: (1) with an increase of common risk variation, does rare risk variation become less important over generations, (2) do linkage positive individuals carrying the presumed risk allele show differences in PRS.
Statistical testing
All PRS were standardized mean = 0 and SD = 1. Linear mixed model analyses were selected to be able to model covariates and relatedness within this complicated dataset. The analyses were performed using the Wald conditional F-test33 in ASReml-R software34 with one of the categories of mood disorders or family status as dependent variable and PRS as the independent variable (Supplementary Methods). Age (except for the generation analysis) and sex were fitted as fixed effects in the models. For 7 individuals in the BBF age at collection was missing and imputed to be the mean age of the relevant generation. To account for relatedness in within-family comparisons, an additive genetic relationship matrix was fitted as a random effect. The relationship matrix was constructed using LDAK software35 with weighted predictors and LD correction parameters suited for pedigree data, resulting in pairwise relatedness estimates and inbreeding coefficients on the diagonal. The variance explained by each PRS was calculated using: (var(x × β))/var(y), where x was the standardized PRS, β was the corresponding regression coefficient, and y was the phenotype36. For the analysis of offspring, we defined four spouse pair categories (“both unaffected”, “married-in parent affected”, “family parent affected”, “both affected”). While most spouse pairs contribute 1 or 2 children to the same offspring category (Supplementary Table 1); two “both affected” spouse pairs contribute 7 and 8 children, respectively. To prevent bias in our analysis in the event of more than one child per couple, we calculated the mean PRS for all offspring per spouse pair and entered this in the model as being one representative child for that couple. All p-values reported are uncorrected for multiple testing, since all tests concern overlapping individuals and thus have a complex dependence structure. However, we have performed 42 tests as listed in Supplementary Table 4, and so a conservative Bonferroni threshold for p < 0.05 is 0.001.
Electronic supplementary material
Acknowledgements
We would like to thank the family members for their enthusiastic participation. We thank our ethics consultant Prof. Barbara Prainsack for insightful discussions. This paper represents independent research part-funded by FAPESP (2014/50830-2; 2010/08968-6), the Marie Curie International Research Staff Exchange (FP7-PEOPLE-2011-IRSES/295192), and the National Institute for Health Research (NIHR) Biomedical Research Centre at South London and Maudsley NHS Foundation Trust and King’s College London. SDJ is funded by the European Union’s Horizon 2020 research and innovation programme under Marie Skłodowska-Curie grant IF 658195. S.J.N. is also supported by the National Institute for Health Research (NIHR) University College London Hospitals Biomedical Research Centre, and by awards establishing the Farr Institute of Health Informatics Research at UCLPartners, from the Medical Research Council, Arthritis Research UK, British Heart Foundation, Cancer Research UK, Chief Scientist Office, Economic and Social Research Council, Engineering and Physical Sciences Research Council, National Institute for Health Research, National Institute for Social Care and Health Research, and Wellcome Trust (grant MR/K006584/1). The views expressed are those of the authors and not necessarily those of the EU, the NHS, the NIHR or the Department of Health.
Author contributions
M.J.A.D., A.C.S.R., A.G., R.B.: family phenotyping and sample collection. M.L.S., V.K.O., C.N., R.B., S.I.B.: Brazilian controls phenotyping and sample collection. M.D.D. and B.I.P. working groups of PGC: providing summary statistics. C.C., H.P.: sample processing and genotyping. L.S.H., P.F.O., S.D.J.: statistical analysis and advice. G.B., S.D.J.: study design, drafting manuscript.
Data availability
In order to ensure privacy of the family members and to comply with Brazilian regulations, restrictions apply on availability of the data as determined by the Brazilian National Ethics Committee (CONEP). Data are available upon reasonable request from the corresponding author, pending approval by the BBF ethics committee (CONEP).
Competing Interests
G.B. has been a consultant in preclinical genomics and has received grant funding from Eli Lilly ltd within the last 3 years. A.G. has participated in advisory boards for Janssen-Cilag and Daiichi-Sankyo. The remaining authors declare no competing interests.
Footnotes
A full list of authors and their affiliations is shown at the end of the paper.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Gerome Breen, Email: gerome.breen@gmail.com.
Major Depressive Disorder and Bipolar Disorder Working Groups of the Psychiatric Genomics Consortium:
Naomi R. Wray, Stephan Ripke, Manuel Mattheisen, Maciej Trzaskowski, Enda M. Byrne, Abdel Abdellaoui, Mark J. Adams, Esben Agerbo, Tracy M. Air, Till F. M. Andlauer, Silviu-Alin Bacanu, Marie Bækvad-Hansen, Aartjan T. F. Beekman, Tim B. Bigdeli, Elisabeth B. Binder, Douglas H. R. Blackwood, Julien Bryois, Henriette N. Buttenschøn, Jonas Bybjerg-Grauholm, Na Cai, Enrique Castelao, Jane Hvarregaard Christensen, Toni-Kim Clarke, Jonathan R. I. Coleman, Lucía Colodro-Conde, Baptiste Couvy-Duchesne, Nick Craddock, Gregory E. Crawford, Gail Davies, Ian J. Deary, Franziska Degenhardt, Eske M. Derks, Nese Direk, Conor V. Dolan, Erin C. Dunn, Thalia C. Eley, Valentina Escott-Price, Farnush Farhadi Hassan Kiadeh, Hilary K. Finucane, Andreas J. Forstner, Josef Frank, Héléna A. Gaspar, Michael Gill, Fernando S. Goes, Scott D. Gordon, Jakob Grove, Christine Søholm Hansen, Thomas F. Hansen, Stefan Herms, Ian B. Hickie, Per Hoffmann, Georg Homuth, Carsten Horn, Jouke-Jan Hottenga, David M. Hougaard, Marcus Ising, Rick Jansen, Ian Jones, Lisa A Jones, Eric Jorgenson, James A. Knowles, Isaac S. Kohane, Julia Kraft, Warren W. Kretzschmar, Jesper Krogh, Zoltán Kutalik, Yihan Li, Penelope A. Lind, Donald J. MacIntyre, Dean F. MacKinnon, Robert M. Maier, Wolfgang Maier, Jonathan Marchini, Hamdi Mbarek, Patrick McGrath, Peter McGuffin, Sarah E. Medland, Divya Mehta, Christel M. Middeldorp, Evelin Mihailov, Yuri Milaneschi, Lili Milani, Francis M. Mondimore, Grant W. Montgomery, Sara Mostafavi, Niamh Mullins, Matthias Nauck, Bernard Ng, Michel G. Nivard, Dale R. Nyholt, Hogni Oskarsson, Michael J. Owen, Jodie N. Painter, Carsten Bøcker Pedersen, Marianne Giørtz Pedersen, Roseann E. Peterson, Erik Pettersson, Wouter J. Peyrot, Giorgio Pistis, Danielle Posthuma, Jorge A. Quiroz, Per Qvist, John P. Rice, Brien P. Riley, Margarita Rivera, Saira Saeed Mirza, Robert Schoevers, Eva C. Schulte, Ling Shen, Stanley I. Shyn, Engilbert Sigurdsson, Grant C. B. Sinnamon, Johannes H. Smit, Daniel J. Smith, Hreinn Stefansson, Stacy Steinberg, Fabian Streit, Jana Strohmaier, Katherine E. Tansey, Henning Teismann, Alexander Teumer, Wesley Thompson, Pippa A. Thomson, Thorgeir E. Thorgeirsson, Matthew Traylor, Jens Treutlein, Vassily Trubetskoy, André G. Uitterlinden, Daniel Umbricht, Sandra Van der Auwera, Albert M. van Hemert, Alexander Viktorin, Peter M. Visscher, Yunpeng Wang, Bradley T. Webb, Shantel Marie Weinsheimer, Jürgen Wellmann, Gonneke Willemsen, Stephanie H. Witt, Yang Wu, Hualin S. Xi, Jian Yang, Futao Zhang, Volker Arolt, Bernhard T. Baune, Klaus Berger, Dorret I. Boomsma, Sven Cichon, Udo Dannlowski, E. J. C. de Geus, J. Raymond DePaulo, Enrico Domenici, Katharina Domschke, Tõnu Esko, Hans J. Grabe, Steven P. Hamilton, Caroline Hayward, Andrew C. Heath, Kenneth S. Kendler, Stefan Kloiber, Glyn Lewis, Qingqin S. Li, Susanne Lucae, Pamela A. F. Madden, Patrik K. Magnusson, Nicholas G. Martin, Andrew M. McIntosh, Andres Metspalu, Ole Mors, Preben Bo Mortensen, Bertram Müller-Myhsok, Merete Nordentoft, Markus M. Nöthen, Michael C. O’Donovan, Sara A. Paciga, Nancy L. Pedersen, Brenda W. J. H. Penninx, Roy H. Perlis, David J. Porteous, James B. Potash, Martin Preisig, Marcella Rietschel, Catherine Schaefer, Thomas G. Schulze, Jordan W. Smoller, Kari Stefansson, Henning Tiemeier, Rudolf Uher, Henry Völzke, Myrna M. Weissman, Thomas Werge, Cathryn M. Lewis, Douglas F. Levinson, Anders D. Børglum, Patrick F. Sullivan, Sandra Meier, John Strauss, Wei Xu, John B. Vincent, Keith Matthews, Manuel Ferreira, Colm O’Dushlaine, Shaun Purcell, Soumya Raychaudhuri, Douglas M. Ruderfer, Pamela Sklar, Laura J. Scott, Matthew Flickinger, Margit Burmeister, Jun Li, Weihua Guan, Devin Absher, Robert C. Thompson, Fan Guo Meng, Alan F. Schatzberg, William E. Bunney, Jack D. Barchas, Stanley J. Watson, Richard M. Myers, Huda Akil, Michael Boehnke, Kimberly Chambert, Jennifer Moran, Edward Scolnick, Srdjan Djurovic, Ingrid Melle, Gunnar Morken, Aiden Corvin, Adebayo Anjorin, Radhika Kandaswamy, Jacob Lawrence, Alan W. McLean, Benjamin S. Pickard, Sarah E. Bergen, Vishwajit Nimgaonkar, Mikael Landén, Martin Schalling, Urban Osby, Lena Backlund, Louise Frisén, Niklas Langstrom, Eli Stahl, Amanda Dobbyn, Stéphane Jamain, Bruno Etain, Frank Bellivier, Markus Leber, Anna Maaser, Sascha B. Fischer, Céline S. Reinbold, Sarah Kittel-Schneider, Janice M. Fullerton, Lilijana Oruč, José G. Para, Fermin Mayoral, Fabio Rivas, Piotr M. Czerski, Jutta Kammerer-Ciernioch, Helmut Vedder, Margitta Borrmann-Hassenbach, Andrea Pfennig, Paul Brennan, James D. McKay, Manolis Kogevinas, Markus Schwarz, Peter R. Schofield, Thomas W. Mühleisen, Johannes Schumacher, Michael Bauer, Adam Wright, Philip B. Mitchell, Martin Hautzinger, John R. Kelsoe, Tiffany A. Greenwood, Caroline M. Nievergelt, Paul D. Shilling, Erin N. Smith, Cinnamon S. Bloss, Howard J. Edenberg, Daniel L. Koller, Elliot S. Gershon, Chunyu Liu, Judith A. Badner, William A. Scheftner, William B. Lawson, Evaristus A. Nwulia, Maria Hipolito, William Coryell, John Rice, William Byerley, Francis J. McMahon, Falk W. Lohoff, Peter P. Zandi, Pamela B. Mahon, Melvin G. McInnis, Sebastian Zöllner, Peng Zhang, Szabolcs Szelinger, David St. Clair, Sian Caesar, Katherine Gordon-Smith, Christine Fraser, Elaine K. Green, Detelina Grozeva, Marian L. Hamshere, George Kirov, Ivan Nikolov, David A. Collier, Amanda Elkin, Richard Williamson, Allan H. Young, I. Nicol Ferrier, Vihra Milanova, Martin Alda, Pablo Cervantes, Cristiana Cruceanu, Guy A. Rouleau, Gustavo Turecki, Sara Paciga, Ashley R. Winslow, Maria Grigoroiu-Serbanescu, Roel Ophoff, Rolf Adolfsson, Annelie Nordin Adolfsson, Jurgen Del-Favero, Carlos Pato, Joanna M. Biernacka, Mark A. Frye, Derek Morris, Nicholas J. Schork, Andreas Reif, Jolanta Lissowska, Joanna Hauser, Neonila Szeszenia-Dabrowska, Kevin McGhee, Emma Quinn, Valentina Moskvina, Peter A. Holmans, Anne Farmer, James L. Kennedy, Ole A. Andreassen, Morten Mattingsdal, Michael Gill, Nicholas J. Bass, Hugh Gurling, Andrew McQuillin, René Breuer, Christina Hultman, Paul Lichtenstein, Laura M. Huckins, Marion Leboyer, Mark Lathrop, John Nurnberger, Michael Steffens, Tatiana M. Foroud, Wade H. Berrettini, David W. Craig, and Jianxin Shi
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s42003-018-0155-y.
References
- 1.Purcell SM, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ripke S, et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–427. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sullivan PF. The psychiatric GWAS consortium: big science comes to psychiatry. Neuron. 2010;68:182–186. doi: 10.1016/j.neuron.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ahn K, An SS, Shugart YY, Rapoport JL. Common polygenic variation and risk for childhood-onset schizophrenia. Mol. Psychiatry. 2016;21:94–96. doi: 10.1038/mp.2014.158. [DOI] [PubMed] [Google Scholar]
- 5.Lichtenstein P, et al. Common genetic determinants of schizophrenia and bipolar disorder in Swedish families: a population-based study. Lancet. 2009;373:234–239. doi: 10.1016/S0140-6736(09)60072-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee SH, et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat. Genet. 2013;45:984–994. doi: 10.1038/ng.2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cross-Disorder Group of the Psychiatric Genomics Consortium. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet. 2013;381:1371–1379. doi: 10.1016/S0140-6736(12)62129-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Merikangas KR, Spiker DG. Assortative mating among in-patients with primary affective disorder. Psychol. Med. 1982;12:753–764. doi: 10.1017/S0033291700049059. [DOI] [PubMed] [Google Scholar]
- 9.Nordsletten AE, et al. Patterns of nonrandom mating within and across 11 major psychiatric disorders. JAMA Psychiatry. 2016;73:354–361. doi: 10.1001/jamapsychiatry.2015.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Plomin R, Krapohl E, O’Reilly PF. Assortative mating—a missing piece in the jigsaw of psychiatric genetics. JAMA Psychiatry. 2016;73:323–324. doi: 10.1001/jamapsychiatry.2015.3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robinson MR, et al. Genetic evidence of assortative mating in humans. Nat. Hum. Behav. 2017;1:16. doi: 10.1038/s41562-016-0016. [DOI] [Google Scholar]
- 12.O’Donovan M, Jones I, Craddock N. Anticipation and repeat expansion in bipolar disorder. Am. J. Med. Genet. C. 2003;123C:10–17. doi: 10.1002/ajmg.c.20009. [DOI] [PubMed] [Google Scholar]
- 13.Power Robert A., Tansey Katherine E., Buttenschøn Henriette Nørmølle, Cohen-Woods Sarah, Bigdeli Tim, Hall Lynsey S., Kutalik Zoltán, Lee S. Hong, Ripke Stephan, Steinberg Stacy, Teumer Alexander, Viktorin Alexander, Wray Naomi R., Arolt Volker, Baune Bernard T., Boomsma Dorret I., Børglum Anders D., Byrne Enda M., Castelao Enrique, Craddock Nick, Craig Ian W., Dannlowski Udo, Deary Ian J., Degenhardt Franziska, Forstner Andreas J., Gordon Scott D., Grabe Hans J., Grove Jakob, Hamilton Steven P., Hayward Caroline, Heath Andrew C., Hocking Lynne J., Homuth Georg, Hottenga Jouke J., Kloiber Stefan, Krogh Jesper, Landén Mikael, Lang Maren, Levinson Douglas F., Lichtenstein Paul, Lucae Susanne, MacIntyre Donald J., Madden Pamela, Magnusson Patrik K.E., Martin Nicholas G., McIntosh Andrew M., Middeldorp Christel M., Milaneschi Yuri, Montgomery Grant W., Mors Ole, Müller-Myhsok Bertram, Nyholt Dale R., Oskarsson Hogni, Owen Michael J., Padmanabhan Sandosh, Penninx Brenda W.J.H., Pergadia Michele L., Porteous David J., Potash James B., Preisig Martin, Rivera Margarita, Shi Jianxin, Shyn Stanley I., Sigurdsson Engilbert, Smit Johannes H., Smith Blair H., Stefansson Hreinn, Stefansson Kari, Strohmaier Jana, Sullivan Patrick F., Thomson Pippa, Thorgeirsson Thorgeir E., Van der Auwera Sandra, Weissman Myrna M., Breen Gerome, Lewis Cathryn M. Genome-wide Association for Major Depression Through Age at Onset Stratification: Major Depressive Disorder Working Group of the Psychiatric Genomics Consortium. Biological Psychiatry. 2017;81(4):325–335. doi: 10.1016/j.biopsych.2016.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Power RA, et al. Dissecting the genetic heterogeneity of depression through age at onset. Am. J. Med. Genet. B. 2012;159B:859–868. doi: 10.1002/ajmg.b.32093. [DOI] [PubMed] [Google Scholar]
- 15.Silva MT, Galvao TF, Martins SS, Pereira MG. Prevalence of depression morbidity among Brazilian adults: a systematic review and meta-analysis. Rev. Bras. Psiquiatr. 2014;36:262–270. doi: 10.1590/1516-4446-2013-1294. [DOI] [PubMed] [Google Scholar]
- 16.Machiela Mitchell J., Chanock Stephen J. LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants: Fig. 1. Bioinformatics. 2015;31:3555–3557. doi: 10.1093/bioinformatics/btv402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boies Sébastien, Mérette Chantal, Paccalet Thomas, Maziade Michel, Bureau Alexandre. Polygenic risk scores distinguish patients from non-affected adult relatives and from normal controls in schizophrenia and bipolar disorder multi-affected kindreds. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics. 2017;177(3):329–336. doi: 10.1002/ajmg.b.32614. [DOI] [PubMed] [Google Scholar]
- 18.Wray, N. R. et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat Genet.50, 668–681 (2018). [DOI] [PMC free article] [PubMed]
- 19.Stahl, E. et al. Genomewide association study identifies 30 loci associated with bipolar disorder. bioRxiv 173062. 10.1101/173062 (2017).
- 20.Schulze TG, et al. Further evidence for age of onset being an indicator for severity in bipolar disorder. J. Affect Disord. 2002;68:343–345. doi: 10.1016/S0165-0327(01)00306-8. [DOI] [PubMed] [Google Scholar]
- 21.Schürhoff F, et al. Early and late onset bipolar disorders: two different forms of manic-depressive illness? J. Affect Disord. 2000;58:215–221. doi: 10.1016/S0165-0327(99)00111-1. [DOI] [PubMed] [Google Scholar]
- 22.Alda M, et al. Anticipation in bipolar affective disorder: is age at onset a valid criterion? Am. J. Med. Genet. 2000;96:804–807. doi: 10.1002/1096-8628(20001204)96:6<804::AID-AJMG22>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 23.Diniz, M. J. A. et al. Whole genome linkage analysis in a large Brazilian multigenerational family reveals distinct linkage signals for bipolar disorder and depression. bioRxiv 106260 (2017).
- 24.Del-Ben CM, Rodrigues CR, Zuardi AW. Reliability of the Portuguese version of the structured clinical interview for DSM-III-R (SCID) in a Brazilian sample of psychiatric outpatients. Braz. J. Med. Biol. Res. 1996;29:1675–1682. [PubMed] [Google Scholar]
- 25.Brasil HHA, Bordin IA. Convergent validity of K-SADS-PL by comparison with CBCL in a Portuguese speaking outpatient population. BMC Psychiatry. 2010;10:83. doi: 10.1186/1471-244X-10-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Noto C, et al. Effects of depression on the cytokine profile in drug naïve first-episode psychosis. Schizophr. Res. 2015;164:53–58. doi: 10.1016/j.schres.2015.01.026. [DOI] [PubMed] [Google Scholar]
- 27.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chang CC, et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin--rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 30.Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2:e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Euesden Jack, Lewis Cathryn M., O’Reilly Paul F. PRSice: Polygenic Risk Score software. Bioinformatics. 2014;31(9):1466–1468. doi: 10.1093/bioinformatics/btu848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rioux JD, et al. Genetic variation in the 5q31 cytokine gene cluster confers susceptibility to Crohn disease. Nat. Genet. 2001;29:223–228. doi: 10.1038/ng1001-223. [DOI] [PubMed] [Google Scholar]
- 33.Kenward MG, Roger JH. Small sample inference for fixed effects from restricted maximum likelihood. Biometrics. 1997;53:983–997. doi: 10.2307/2533558. [DOI] [PubMed] [Google Scholar]
- 34.Butler, D. G., Cullis, B. R., Gilmour, A. R. & Gogel, B. J. ASReml-R reference manual 3. Technical Report, Department of Primary Industries Queensland (2002).
- 35.Speed D, Hemani G, Johnson MR, Balding DJ. Improved heritability estimation from genome-wide SNPs. Am. J. Hum. Genet. 2012;91:1011–1021. doi: 10.1016/j.ajhg.2012.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nakagawa S, Schielzeth H. A general and simple method for obtaining R 2 from generalized linear mixed-effects models. Methods Ecol. Evol. 2013;4:133–142. doi: 10.1111/j.2041-210x.2012.00261.x. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
In order to ensure privacy of the family members and to comply with Brazilian regulations, restrictions apply on availability of the data as determined by the Brazilian National Ethics Committee (CONEP). Data are available upon reasonable request from the corresponding author, pending approval by the BBF ethics committee (CONEP).