

Abstract
In this study, through global transcriptional analysis by RNA-Sequencing, we identified the main changes in gene expression that occurred in two functional mutants of the MAPK genes tmk1 and tmk2 in Trichoderma reesei during sugarcane bagasse degradation. We found that the proteins encoded by these genes regulated independent processes, sometimes in a cross-talk manner, to modulate gene expression in T. reesei. In the Δtmk2 strain, growth in sugarcane bagasse modulated the expression of genes involved in carbohydrate metabolism, cell growth and development, and G-protein-coupled receptor-mediated cell signaling. On the other hand, deletion of tmk1 led to decreased expression of the major genes for cellulases and xylanases. Furthermore, TMK1 found to be involved in the regulation of the expression of major facilitator superfamily transporters. Our results revealed that the MAPK signaling pathway in T. reesei regulates many important processes that allow the fungus to recognize, transport, and metabolize different carbon sources during plant cell wall degradation.
Introduction
The filamentous fungus Trichoderma reesei is one of the major sources for industrial cellulases production1. T. reesei has a small repertoire of cellulases and hemicellulases, the smallest among biomass-degrading fungi. However, its cellulolytic enzyme system is the most efficient among filamentous fungi2. Cellulase expression is mainly regulated by the transcription factors XYR1, ACE2, and the Heme Activator Protein complex (HAP2/3/5) as positive regulators, and CRE1 and ACE1 as negative regulators3,4. Recently, other functional cellulase regulators, such as LAE1, VELVET, and ACE3, have been described5–8. However, it remains unclear how exactly the induction signals are delivered. Several signal transduction mechanisms and factors have been reported to influence cellulase expression in T. reesei. Among them, GNA1 and GNA3, two G-alpha subunits, respond to different carbon sources and are involved in regulating cellulolytic enzymes9,10. Additionally, the influence of cAMP levels as well as signal transduction involving light on cellulase gene expression has been discussed11–14.
The MAPK signaling pathway is an evolutionarily conserved cascade involved in transducing extracellular signals, induced by adequate cellular responses15–17. In animals, plants, and fungi, MAPK signaling regulates transcription factor function through MAPK-mediated phosphorylation. In fungi, the MAPK signaling pathway has been well studied in Saccharomyces cerevisiae18, and it controls a range of processes, such as fruiting body development, polarized growth, pathogenicity, circadian rhythm, conidiation, protein biosynthesis and secretion, stress response, and cell wall integrity19–25. In filamentous fungi, three S. cerevisiae homologous MAPKs, Hog1, Slt2, and Fus3, have been identified24,26,27. In the T. reesei database (http://genome.jgi-psf.org/Trire2/Trire2.home.html), three genes are annotated to encode MAPK pathway members (tmk1, tmk2, and tmk3). Recent reports have shown that tmk2, tmk1, and tmk3 signaling pathways may control different processes in this fungus, including cell wall integrity, cellulase production, biomass accumulation, sporulation, and resistance to high osmolarity24,28–30, however, these studies only examined the expression of a small number of genes. In the current study, we conducted a RNA sequencing-based global gene expression analysis to reveal the functions of TMK1 and TMK2 in T. reesei. In addition, our study provides the most complete insight to date about the function of TMK1 and TMK2 in T. reesei and it contributes to the understanding of signal transduction processes that control cellulases production in this important industrial fungus.
Results
tmk1 and tmk2 MAPK Genes Are Induced in T. reesei in the Presence of Sugarcane Bagasse
In silico analysis of the T. reesei genome (http://genome.jgi-psf.org/Trire2/Trire2.home.html) showed that this fungus has 3 genes (tmk1, tmk2, and tmk3) encoding MAPK signaling pathway members. Additionally, several reports have suggested the involvement of this pathway in regulating cellulase expression24,28,30. Thus, we investigated the involvement of these MAPK genes in the regulation of cellulase expression in T. reesei during sugarcane bagasse degradation. First, we profiled MAPK genes expression in the QM6a parental strain grown in the presence of sugarcane bagasse, an inductive carbon source, or glucose, which is a repressive carbon source for cellulase synthesis. As demonstrated in Fig. 1A, the MAPK genes were expressed at low levels in the presence of glucose, with a slight increase at 24 hours for tmk1 and tmk2, and at 48 hours for tmk3. In sugarcane bagasse, MAPK genes expression was analyzed after 24, 48, 72, and 96 hours of cultivation (Fig. 1B). The tmk1 and tmk2 genes showed the highest expression from 48 hours to 96 hours in the presence of sugarcane bagasse, and these levels were significantly higher than the levels observed at 24 hours. No changes in tmk3 expression were observed over time in the presence of sugarcane bagasse (Fig. 1B). Therefore, we selected tmk1 and tmk2 for functional studies. T. reesei Δtmk2 and Δtmk1 strains were constructed by homologous recombination using QM6aΔtmus53Δpyr4, in which the non-homologous end joining pathway is disrupted31, as the parental strain (Additional file 1).
Figure 1.

Expression profile of MAPK genes in the QM6a parental strain grown in sugarcane bagasse and glucose. (A) Relative expression of tmk1, tmk2, and tmk3 upon growth in glucose. Expression levels were calibrated according to the comparative 2−ΔCt method, using the constitutively expressed gene β-actin as an endogenous control (ANOVA followed by Tukey’s pairwise comparison P < 0.05). *Significantly different from 24 h (P < 0.05). (B) Relative expression of tmk1, tmk2, and tmk3 in the presence of sugarcane bagasse. Expression levels were calibrated according to the comparative 2−ΔΔCt method, using the constitutively expressed gene β-actin as an endogenous control and glycerol samples the reference group (ANOVA followed by Tukey’s pairwise comparison P < 0.0001). *Significantly different from 48 h, 72 h, and 96 h (P < 0.0001). These results are based on three replicates of three independent experiments and are expressed as mean ± standard deviation.
Deletion of tmk2 Impairs Growth in Presence of Lactose and Glucose
Phenotypic analysis was conducted for parental, Δtmk1, and Δtmk2 strains by growing these strains on agar plates with different medium compositions (Supplementary Fig. 3A,D). The Δtmk1 strain showed no difference in growth, sporulation, and biomass formation compared to the parental strain (Supplementary Fig. 3B,C). The Δtmk2 did not differ from the parental strain in sporulation patterns either. However, reduction of growth was observed for the Δtmk2 mutant strain when grown in minimal medium supplemented with lactose and glucose (Supplementary Fig. 3D). In addition, we observed that Δtmk2 showed difference in growth profiles (Supplementary Fig. 3E) and similar biomass accumulation after growth on glycerol for 24 hours (Supplementary Fig. 3E).
Gene Regulatory Network Controlled by tmk1 and tmk2 in T. reesei
T. reesei QM6a, Δtmk1, and Δtmk2 strains were cultivated in presence of sugarcane bagasse, glycerol, and glucose as carbon sources, and three biological replicates of each condition were submitted for RNA-Sequencing using an Illumina HiSeq™ 2500 (Additional file 3).
From the RNA-Seq data, regulatory networks were built to identify genes that were specifically expressed in a determined growth condition (Fig. 2). In the Δtmk1 regulatory network (Fig. 2A), 152 genes were exclusively regulated during growth in the presence of sugarcane bagasse (23 up- and 129 downregulated), 187 exclusively in the presence of glucose (146 up- and 41 downregulated), and 15 exclusively in the presence of glycerol (two up- and 13 downregulated) (Supplementary Table 2), while only 1 gene was commonly regulated among the three conditions examined, when compared to the parental strain. Moreover, three genes were common regulated between the sugarcane bagasse and glucose conditions, three between the glucose and glycerol conditions, and eight between the sugarcane bagasse and glycerol conditions, when compared to the parental strain (Fig. 2A). In the Δtmk2 mutant strain, compared to QM6a, 538 genes were exclusively regulated during growth in sugarcane bagasse (308 up- and 230 downregulated), 507 exclusively in glucose (170 up- and 337 downregulated), and 418 exclusively in glycerol (281 up- and 137 downregulated) (Supplementary Table 3), while 78 genes were commonly regulated among the three conditions examined (Fig. 2B). Moreover, 94 genes were common regulated between the sugarcane bagasse and glucose conditions, 68 between the glucose and glycerol conditions, and 277 between the sugarcane bagasse and glycerol conditions (Fig. 2B). Genes differentially expressed in Δtmk1 and Δtmk2 compared to QM6a in the presence of sugarcane bagasse were functionally annotated using Gene Ontology (GO) terms (Supplementary Fig. 9A). The main gene categories included integral to membrane, carbohydrate transport, L-arabinose isomerase activity, sugar:proton symporter activity, and DNA binding activity (Supplementary Fig. 9A). On the other hand, genes involved in transport, nucleic acid binding, hydrolase activity, RNA binding, synthase activity, copper ion transport, chromatin assembly and disassembly and chromatin were the GO terms most enriched in the Δtmk2 mutant strain grown in sugarcane bagasse when compared to the parental strain (Supplementary Fig. 9B). We selected the 90 genes that showed the strongest up- or downregulation (Log2 fold change ≥1 or ≤−1 and adjusted p ≤ 0.05) during growth of Δtmk1 and Δtmk2 in the different carbon sources analyzed (Supplementary Tables 4 and 5). In Δtmk1, when compared to QM6a, the upregulated genes during growth in sugarcane bagasse were mainly related to aromatic compound metabolism, oxidoreductase activity, and the membrane. The most strongly expressed gene encoded FAD-binding domain-containing protein (ID 62463) (13-fold), followed by a gene encoding an unknown protein (ID 105447) (6.5-fold), and one encoding a short chain dehydrogenase/reductase (ID 70520) (6-fold). Two genes encoding major facilitator superfamily (MFS) permeases (ID 65915 and ID 57749) and two genes encoding transcription factors (ID 111446 and ID 54395) were also among the most strongly upregulated genes in Δtmk1 in sugarcane bagasse when compared to the parental strain (Supplementary Table 4). On the other hand, the most strongly downregulated genes in Δtmk1 in presence of sugarcane bagasse when comparing the mutant to the parental strain were mainly related to carbohydrate metabolism and cellulase activity. The most strongly downregulated gene encoded a MFS permease (ID 54005) (70-fold), followed by two xylanase encoding genes (ID 120229 and ID 123818) (34.5 and 28-fold, respectively) and one encoding an unknown protein (ID 39587) (34.5-fold) (Supplementary Table 4).
Figure 2.

Gene Regulatory Network (GRN) of differentially expressed genes in the mutant strains grown in glucose, sugarcane, and glycerol. (A) GRN of 402 differentially expressed genes in the Δtmk1 compared to the QM6a (Δtmk1/QM6a) in the presence of glucose, glycerol, and sugarcane bagasse. Genes are represented as nodes (circles), and interactions are represented as edges (red lines: upregulated interactions, green lines: downregulated interactions), connecting the nodes: 382 interactions, (B) GRN of 2575 differentially expressed genes in the Δtmk2 compared to the QM6a (Δtmk2/QM6a) in the presence of glucose, glycerol, and sugarcane bagasse. Genes are represented as nodes (shown as circles), and interactions are represented as edges (red lines: upregulated interactions, green lines: downregulated interactions), connecting the nodes: 1980 interactions.
In Δtmk2, when compared to QM6a, the most strongly upregulated genes in the presence of sugarcane bagasse encoded a β-lactamase class C (ID 58717) (75-fold), an MFS permease (maltose permease) (ID 65191) (38-fold), and an amidase (ID 78072) (36-fold). Interestingly, the expression of two glycosyl hydrolase-encoding genes (GH105/GH88 glycosyl hydrolase, ID 57179; and GH11 endo-β-1,4-xylanase XYN5, ID 112392) were increased at least 24- and 23-fold in the mutant relative to the parental strain, respectively (Supplementary Table 5). Regarding the downregulated genes in the three carbon sources, 62% were of unknown function. In the presence of sugarcane bagasse, the most strongly downregulated gene (ID 108676) encoded an unknown protein secreted only by Ascomycota, which exhibited a >161-fold decrease in Δtmk2 compared to QM6a. The second-most repressed gene in Δtmk2 upon growth in sugarcane bagasse encoded a transporter belonging to the MFS permease family (ID 103179), showing >117-fold decrease in expression compared to the parental strain (Supplementary Table 5).
To identify potential cross- talk between the TMK1 and TMK2 signaling in the presence of sugarcane bagasse and glucose, we selected the genes whose expression was differentially regulated in both mutant strains when compared to the parental strain. As seen in Fig. 3A, 88 genes were regulated in both Δtmk1 and Δtmk2 in the presence of sugarcane bagasse (approximately 86% of genes were commonly up- or downregulated) (Supplementary Table 6), while 173 genes were regulated in the mutant strains in the presence of glucose in relation to the parental strain (approximately 41% of genes were commonly up- or downregulated) (Supplementary Table 7). GO enrichment analysis (Fig. 3B) showed that the commonly regulated genes in the mutant strains in the presence of sugarcane bagasse mainly encoded proteins with no described function; followed by GO terms related to integral to membrane, zinc ion binding, electron transport, membrane, and oxidoreductase activity.
Figure 3.

Expression pattern of genes commonly expressed between the Δtmk1 and Δtmk2 representing the number of differentially expressed genes in the presence of glucose and sugarcane bagasse. (A) Comparative Venn diagram of commonly expressed genes between the Δtmk1 and Δtmk2 strains in the presence of glucose and sugarcane bagasse. Venn diagram clustering was designed using Venny 2.1 tools. (B) Gene Ontology (GO) enrichment analysis of commonly expressed genes between the Δtmk1 and Δtmk2 strains in the presence of glucose and sugarcane bagasse. The enriched GO terms according to molecular, cellular component, and biological process in T. reesei. Significantly enriched categories (P ≤ 0.05) are shown. The threshold for calling differentially expressed genes was P ≤ 0.05.
TMK1 and TMK2 are Involved in the Regulation Hollocellulase Expression and Activity
An overview of the possible roles for TMK1 and TMK2 in cellulase gene expression is shown in Fig. 4. In the presence of sugarcane bagasse or glucose, CAZyme-encoding genes were significantly up- and downregulated in Δtmk1 when compared to QM6a (Fig. 4B,C and Supplementary Table 13). In the presence of sugarcane bagasse, the deletion of tmk1 negatively affected the expression of the major hollocellulases produced by T. reesei. Only one gene (encoding an oxidoreductase family protein) was upregulated in Δtmk1 in this carbon source as compared to the parental strain (Supplementary Table 13).
Figure 4.

Cellulase gene expression profile of T. reesei QM6a, Δtmk1, and Δtmk2 strains during growth in glucose (gluc), glycerol (glyc), and sugarcane bagasse (SCB). (A) Heatmap of differentially expressed CAZy genes in the Δtmk1 and QM6a strains showing all the conditions of this study. (B) Heatmap of differentially expressed CAZys in Δtmk1 and QM6a strains grown in sugarcane bagasse. (C) Heatmap of differentially expressed CAZys of Δtmk1 and QM6a strains grown in glucose. (D) Heatmap of differentially CAZy genes in the Δtmk2 and QM6a strains showing all the conditions of this study. (E) Heatmap of differentially expressed CAZys in Δtmk2 and QM6a strains grown in sugarcane bagasse. (F) Heatmap of differentially expressed CAZys from Δtmk2 and QM6a strains grown in glucose. (G) Heatmap of differentially expressed CAZys from Δtmk2 and QM6a strains grown in glycerol. The hierarchical clustering was performed using the R pheatmap package. Complete linkage method and Euclidean distance of row centered and scaled TPM values were used for hierarchical clustering of the differentially expressed genes in all analyzed conditions.
The major downregulated holocellulase genes in Δtmk1 in the presence of sugarcane bagasse encoded xylanases XYN3 (ID 120229) (35-fold), XYN2 (ID 123818) (29-fold), and XYN4 (ID 111849) (7-fold) (Fig. 4B). Five genes related to xylan metabolism, encoding CE5 acetyl xylan esterase AXE1 (ID 73632), CE5 acetyl xylan esterase (ID 54219), GH74 xyloglucanase CEL74a (ID 49081), and the L-arabinofuranosidases ABF2 (ID 76210) and ABF3 (55319) were also downregulated in Δtmk1 in this condition (Fig. 4B and Supplementary Table 13). In addition, the two major cellulase genes of T. reesei, cel7a (CBH1; ID 123989), and cel6a (CBH2; ID 72567) also exhibited decreased expression in the presence of sugarcane bagasse (Log2 fold change = −3.93 and −3.69, respectively) (Fig. 4B and Supplementary Table 13). The expression of β-glucosidases and endoglucanases was also altered upon tmk1 deletion in this condition. The genes cel3a (BGL1; ID 76672), cel1a (BGL2; ID 120749), cel5a (EGL2; ID 120312), and cel7b (EGL1; ID 122081) exhibited 19-, 7-, 16-, and 14-fold decreased expression, respectively, in Δtmk1 compared to QM6a grown in sugarcane bagasse (Fig. 4B). Furthermore, three genes coding for lytic polysaccharide monooxygenases (LPMOs) and four genes coding for auxiliary activities (AA1 family) were also downregulated in Δtmk1 in this condition (Fig. 4B and Supplementary Table 13).
In Δtmk2, a different cellulase gene expression profile was observed. In the three carbon sources analyzed, CAZyme-encoding genes were up- or downregulated in Δtmk2 when compared to QM6a (Fig. 4D–G). Notably, in the presence of glucose and glycerol, genes related to cell wall remodeling, such as chitinases, mannanase, galactosidase, glycosyl transferase, and glucanases, were upregulated when compared to the parental strain (Fig. 4F,G). In this way, TMK2 can act both as a positive or negative regulator of cellulase gene expression and it seems to control basal development of T. reesei.
Enzymatic activity analysis was performed to determine whether TMK1 and TMK2 affect the activity of holocellulolytic enzymes in T. reesei during sugarcane bagasse degradation. The endoglucanase (CMCase), β-glucosidase, β-xylosidase and xylanases activities were profiled in Δtmk2, Δtmk1, and QM6a (Fig. 5). Regarding the specific CMCase activity, QM6a and Δtmk2 presented similar profiles, with slightly increased activity in Δtmk2 at 24, 72, and 96 hours. On the other hand, CMCase activity in Δtmk1 decreased over cultivation (Fig. 5A). β-glucosidase activity was higher in Δtmk2 at 24, 48, 72, and 96 hours than in the parental strain and Δtmk1 (Fig. 5B). In the same assay, Δtmk1 presented significantly lower β-glucosidase activity than QM6a at 24 and 48 hours. Both mutants showed significantly lower β-xylosidase activity than the QM6a parental strain, but with different patterns (Fig. 5C). The Δtmk2 mutant presented reduced activity at 72, and 96 hours in comparison to the parental strain. Furthermore, Δtmk1 strain showed reduced β-xylosidase activity over time, when compared to the QM6a strain (Fig. 5C). Regarding the specific xylanase activity, QM6a and Δtmk2 presented similar profiles (Fig. 5D). In the same assay, Δtmk1 presented significantly lower xylanase activity than QM6a at 24, 48, 72 and 96 hours (Fig. 5D). Thus, the MAPKs TMK1 and TMK2 might affect the activity of holocellulolytic enzymes in T. reesei upon exposure to sugarcane bagasse, being this effect due to the regulation of cellulase genes in the mutant strains.
Figure 5.

Holocellulolytic activities of QM6a, Δtmk2, and Δtmk1 grown in sugarcane bagasse. (A) Endoglucanase activity (CMCase), (B) β-glucosidase, (C) β-xylosidase and (D) Xylanase activities from culture supernatant of T. reesei, QM6a parental strain, Δtmk2, and Δtmk1 grown in the presence of sugarcane bagasse for the indicated times. ***Significantly different from the QM6a parental strain (P < 0.001) and *Significantly different from the QM6a parental strain (P < 0.05).
TMK2 Regulates Crosstalk Between Different Signaling Pathways in T. reesei
The RNA-Seq data demonstrated that the expression of distinct genes belonging to various signaling pathways was altered upon tmk2 deletion (Supplementary Table 8). In the presence of sugarcane bagasse, genes encoding G protein–coupled receptors (GPCRs); phospholipases C, A2, and D; kinase proteins; Ras-GTPase; calcium and histidine kinase; calmodulin; calcipressin; GNA1; casein kinase 1; and MAPKKK (Ste11) were differentially expressed in Δtmk2 (Supplementary Table 8). Interestingly, the most strongly differentially regulated genes from different pathways were involved in growth and development, reinforcing the role of TMK2 in the control of basal development of T. reesei. Among the genes upregulated in Δtmk2 in the presence of sugarcane bagasse when compared to the parental strain, we can highlight a gene-encoding conidiospore surface protein 1 (CMP1) (ID 72379) (13.7-fold); a gene (ID 64167) encoding a protein involved in sexual differentiation (10-fold); and three genes encoding GPCR PTH11 (an integral membrane protein required for pathogenicity) isoforms (IDs 109146, 122824, and 27992). These genes were 5-, 3-, and 2-fold increased, respectively (Supplementary Table 8). Additionally, genes encoding other GPCRs (IDs 123806 and 63981) were upregulated in Δtmk2 (4- and 3-fold, respectively) compared to QM6a. Finally, genes encoding a cell cycle protein (ID 64125, 5.8-fold), phospholipase C (ID 21960, 3-fold), and phospholipase D (ID 22331, 2.7-fold) were the most strongly expressed genes in Δtmk2 (Supplementary Table 8) in the same condition.
Among the downregulated genes, GPCR-coding genes were overrepresented (Supplementary Table 8). Eight genes encoding GPCRs showed 1.6–5.25-fold decreases in expression in Δtmk2 in comparison to the QM6a. Moreover, a gene encoding a serine-threonine protein kinase (ID 112669) was repressed at almost 6-fold in the mutant strain in the presence of sugarcane bagasse. Interestingly, our results demonstrated that genes related to calcium signaling were also repressed in Δtmk2 grown in sugarcane bagasse (Supplementary Table 8). In addition, the histidine kinase HHK6 gene and eight other genes encoding kinase proteins were nearly 1.2-fold repressed in this condition. Similarly, the expression of upstream components of MAPK signaling was altered by tmk2 deletion. Genes encoding a mitogen-activated protein kinase kinase kinase (MAPKKK STE11; ID 4945) and its regulator MAPKKK STE50 (ID 105409), were 1.7-fold and 1.6-fold repressed, respectively, in the mutant strain in the presence of sugarcane bagasse (Supplementary Table 8), suggesting the TMK2-dependent regulation of MAPK signaling at various points.
Using the KEGG MAPK signaling pathway for yeast (www.genome.jp/kegg/pathway/sce/sce04011.html) as a reference, we drafted the main MAPK signaling pathways in T. reesei (Fig. 6A). This global view revealed that transcriptional oscillations in genes encoding MAPK pathways occur in a carbon source-dependent manner (Fig. 6B). Regarding the pathway involved in pheromone response, a gene encoding a G protein-coupled receptor (ID 64018) was one of the most strongly repressed genes in Δtmk2, exhibiting a 4-fold decrease in expression in the presence of glycerol. Next, a phosphatase enzyme-coding gene (ID 119697) showed a 2.5-fold decrease in sugarcane bagasse, 4.6-fold in glucose, and 2.3-fold in glycerol compared to the parental strain. Curiously, this gene is involved in both cell wall stress and the starvation response, which seems to be a crucial point of regulation controlled by TMK2. On the other hand, a gene encoding another G protein-coupled receptor (ID 57526) and the G-protein signaling regulator protein (ID 54395) were the most strongly induced in the presence of glycerol, showing 8-fold and 2-fold increases in expression in the mutant strain, respectively. Also, genes with IDs 67982 and 106245, involved in the high osmolarity response, showed 2-fold and 6-fold increased expression in the mutant strain in the presence of glycerol and sugarcane bagasse, respectively, in comparison to the parental strain. Regarding the downregulated genes, the genes encoding a transcription factor Sko1 and two isoforms of Ctt1 protein involved in high osmolarity response, (IDs 103372, 67013, and 73818, respectively) showed 2-fold, 3.3-fold, and 2.6-fold lower expression in Δtmk2 grown in glucose and glycerol (Fig. 6B). Finally, a gene encoding the transcription factor Tec1 (ID 108775) related to the starvation response was expressed 7.7-fold higher in Δtmk2 in response to glycerol. These results suggested that tmk2 deletion might alter up- and downstream signaling through all MAPK pathways in T. reesei, impairing crucial processes in fungal development.
Figure 6.

Global view of the MAPK signaling pathway in Δtmk2 and Δtmk1 mutant strains grown in glucose, glycerol, and sugarcane bagasse. (A) Functional reconstruction of the main MAPK signaling pathways involved in pheromone response, cell wall stress, high osmolarity, and starvation in T. reesei. This analysis was performed using the KEGG MAPK signaling pathway in Yeast (https://www.genome.jp/kegg/pathway/sce/sce04011.html) as reference. (B) Heatmap showing expression of the differentially expressed genes belonging to the MAPK signaling pathway in the T. reesei mutant strains. The hierarchical clustering was performed using the R pheatmap package. Complete linkage method and Euclidean distance of row centered and scaled TPM values were used for hierarchical clustering of the differentially expressed genes in all analyzed conditions. The gene IDs represent the genes differentially expressed in the mutant strains grown in glucose, glycerol, and sugarcane bagasse.
TMK1 and TMK2 Modulate the Expression of Transcription Factors
To understand the transcriptional regulation of cellulases by TMK1 and TMK2, we analyzed the mRNA statuses of transcription factors in the mutant strains. The general expression profile of transcription factors was substantially altered in Δtmk1 and Δtmk2 (Supplementary Tables 9 and 10). Interestingly, none of the transcription factors known to directly regulate cellulase gene expression in T. reesei exhibited altered expression, suggesting an indirect relationship between MAPK and cellulase expression.
The changes in transcription factor gene expression profiles in the Δtmk1 mutant compared to the parental strain were subtler than those observed in the Δtmk2 mutant strain (Supplementary Table 9). In the presence of sugarcane bagasse, only six transcription factors were differentially expressed. The most strongly upregulated transcription factor, a Zn2Cys6 transcriptional regulator (ID 111446), was expressed 2.7-fold higher in Δtmk1 than in QM6a. FlbA (encoding a developmental regulator) (ID 54395) was the second most induced gene in Δtmk1 grown in sugarcane bagasse (2.6-fold). In addition, two other genes encoding Zn2Cys6 transcriptional regulators (IDs 104182 and 79725) were equally upregulated in the presence of sugarcane bagasse, showing 2.11-fold and 2.03-fold increases. PacG/VIB-1 (ID 54675) was the only transcription factor downregulated (2.4-fold) in Δtmk1 in the presence of sugarcane bagasse (Supplementary Table 9).
Thirty-four transcription factor-coding genes were modulated in Δtmk2 in the presence of sugarcane bagasse. Most encoded Zn2Cys6 transcriptional regulators (18 genes), C2H2 (6 genes), bZIP (3 genes), and myb (3 genes) families (Supplementary Table 10). The most strongly transcriptionally upregulated transcription factor in sugarcane bagasse in Δtmk2 compared to the parental strain (ID 105269, 7-fold) was a homolog of the Zn2Cys6 transcription regulator of Aspergillus nidulans AlcR, which is involved in ethanol regulon gene expression regulations. Furthermore, a homolog of the myb gene (ID 4124), previously described as a regulator of nitrogen metabolism in A. nidulans, was expressed 5.6-fold higher in the mutant strain than in QM6a. Curiously, another myb family member was one of the transcriptionally downregulated transcription factors upon growth in sugarcane bagasse (ID 1941, 2-fold) (Supplementary Table 10). This result suggests a refined dual mechanism that might control nitrogen metabolism in T. reesei by modulating transcription factors belonging to the myb family. Moreover, two genes encoding a bZIP transcription factor (ID 72524, 10-fold) and a C2H2 family member (ID 122448, 9.6-fold) were the most repressed genes upon growth in sugarcane bagasse (Supplementary Table 10). Altogether, these results suggest that tmk1 and tmk2 deletion changed the transcriptional response of transcription factors in T. reesei, which might directly or indirectly act to regulate the main transcription factors controlling cellulase gene expression in this fungus.
The gene expression of Sugars, Amino Acids, and Ions transporters is Regulated by the MAPK Signaling Pathway in T. reesei
The RNA-Seq analysis identified distinct transporters that were differentially expressed in Δtmk1 and Δtmk2 in the presence of sugarcane bagasse. Efficient nutrient transport is important for proper fungal development. In the T. reesei genome, approximately 5% (459) of all genes encode proteins with transport function. Among these genes, 6% (30 genes) exhibited altered expression in Δtmk1, while expression of 7% (35 genes) was altered in Δtmk2 when compared to QM6a in the presence of sugarcane bagasse (Supplementary Tables 11 and 12). Our results showed that modulation of transporter expression mediated by MAPK is carbon source-dependent.
In Δtmk1, five genes were upregulated and 17 downregulated exclusively in the presence of sugarcane bagasse (Supplementary Table 11). The largest group of differentially expressed transporters in this condition belonged to the MFS family of permeases. Four MFS permeases were upregulated in Δtmk1, and the gene encoding the most strongly induced MFS permease gene (ID 65915), was expressed at least 3-fold higher in the mutant strain than the parental strain. In addition, 9 genes encoding MFS permeases were downregulated in Δtmk1. The gene with ID 54005 was strongly downregulated in Δtmk1, nearly 70-fold decreased expression when compared to QM6a. Interestingly, one gene encoding copper transporter (IDs 52315) was also repressed in the mutant strain during growth in sugarcane bagasse, indicating the influence of MAPK signaling on cellulose oxidation status. The involvement of copper transporters in the cellulose oxidation status was also suggested by Antonieto et al.37.
The role of TMK2 in the transporter system of T. reesei during sugarcane bagasse degradation was also analyzed. In the presence of sugarcane bagasse, 25 genes were upregulated and 10 were downregulated. In this condition, most differentially regulated genes belonged to the MFS permease family. From the 25 upregulated genes, 21 encoded MFS permeases (Supplementary Table 12). The most strongly induced gene encoded a maltose permease (ID 65191), which was expressed 38-fold higher in the mutant strain. A MFS permease-coding gene (ID 54005) was the most strongly repressed gene in Δtmk2, with 32-fold lower expression in the mutant when compared to the parental strain.
Interestingly, in silico identification of MAPK phosphorylation sites suggested that MFS transporters might be regulated by post-translational modifications such as phosphorylation. Figure 7 shows, the ST (Serine-Threonine), Y (Tyrosine), and ST + Y (Serine-Threonine-Tyrosine) phosphorylation profiles of MFS genes differentially expressed in Δtmk1 and Δtmk2 grown in sugarcane bagasse, when compared to the parental strain. The phosphorylation profiles were correlated with the Log2 fold change values of the MFS genes, and ST and ST + Y were the most conserved phosphorylation profiles among the MFS transporters (Fig. 7). Interestingly, we identified a pattern in the expression profile using Log2 fold change values, where the genes with multiple phosphorylation sites were upregulated in the mutant strains in the presence of sugarcane bagasse, while the genes with only a few MAPK phosphorylation sites were the most repressed in the mutant strains (Fig. 7).
Figure 7.
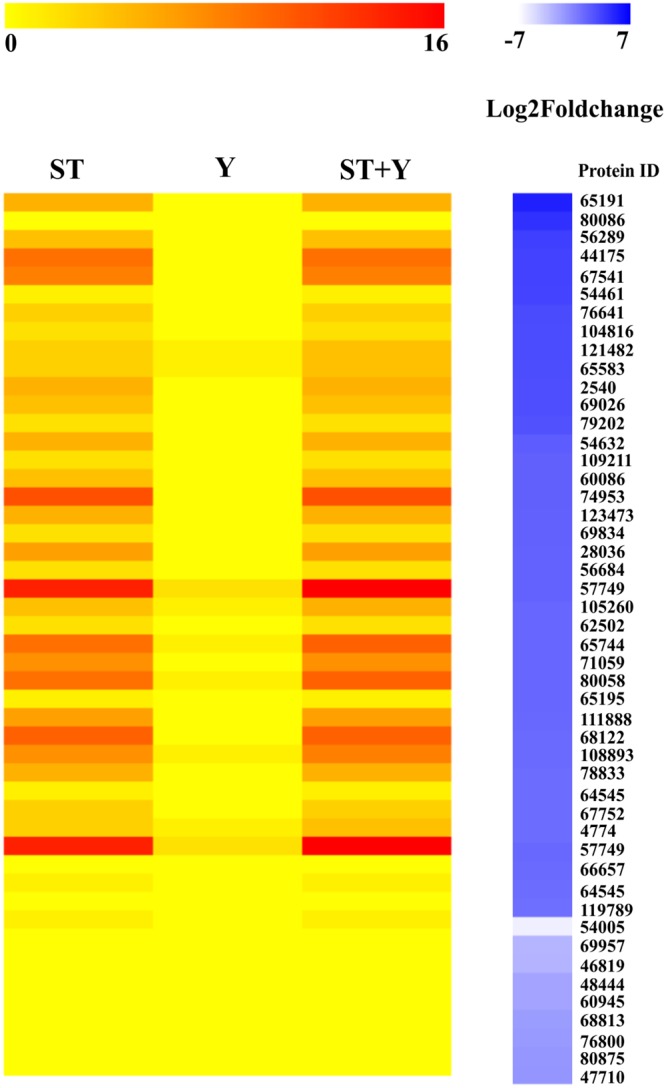
Heat map of predicted MAPK phosphorylation sites of MFS transporters in the Δtmk1 and Δtmk2 strains grown in sugarcane bagasse. ST: serine and threonine residues are phosphorylated; Y: phosphorylation occurs in tyrosine residues and ST + Y: phosphorylation sites in all three residues, serine, threonine, and tyrosine.
Discussion
Here, we described the gene regulatory network of T. reesei Δtmk1 and Δtmk2 grown in the presence of sugarcane bagasse, glucose, or glycerol. The RNA-Seq data showed that deletion of tmk1 and tmk2 affects different processes in T. reesei. In the Δtmk1, we observed the downregulation of the main cellulase genes in T. reesei and the differential expression of genes mostly encoding MFS family transporters. Our results showed that the MAPK genes examined might regulate a range of processes in T. reesei, being some similar processes probably regulated by different mechanisms. The growth of Δtmk2 in the presence sugarcane bagasse and glucose promoted the regulation of cellulase transcription as well as the modulation of genes related to carbohydrate, amino acid, and lipid metabolism, and secondary metabolism. We demonstrated the differential expression of genes related to different signaling pathways such as GPCRs, phospholipases, calcium and MAPK signaling, as well as transporter families like the MFS permeases, and sugar transporters.
Regarding cellulolytic genes, different reports have demonstrated that cellulase expression is regulated at the transcriptional level and in a carbon source-dependent manner2,32. However, the molecular mechanisms that control these processes remain poorly understood33. The involvement of MAPKs in cellulase gene expression was recently suggested. First, Wang et al.24 showed that TMK3 positively regulates both cellulase transcription and production in T. reesei. Additionally, it was demonstrated that there are MAPK phosphorylation sites in the main transcription factors involved in cellulase expression such as XYR1, CRE1, ACE1, and ACE2, suggesting that the MAPK pathway might regulate the activity of these transcription factors that in turn regulate cellulase expression. However, our data did not show the direct modulation of these transcription factors by TMK1 and TMK2, suggesting indirect regulation by other transcription factors in T. reesei34. TMK2 has been suggested to be involved in the control of cell wall integrity, sporulation, and cellulase production28. Deletion of ime2, encoding a MAPK in T. reesei induced the expression of the three main cellulase genes (cbh1, cbh2, and egl1) and repressed the expression of XYR1 and CRE129. Recently, it was shown that deletion of the MAPK tmk1 leads to improved growth and significantly improved cellulase formation in the presence of wheat bran and Avicel30. Our global transcriptional network analysis showed that deletion of tmk1 and tmk2 profoundly affected cellulase expression in T. reesei, but did not affect XYR1 or CRE1. Interestingly, this regulatory mechanism seems to occur in an unusual way, with TMK2 acting as both a positive and negative regulator of cellulase expression, whereas TMK1 was a positive regulator of cellulase expression (Supplementary Tables 13 and 14). Furthermore, our results showed that different CAZymes were differentially expressed between the Δtmk1 and Δtmk2, suggesting a specific pattern of cellulase expression regulation in this organism.
In Δtmk1 grown in the presence sugarcane bagasse, we observed repression of the main cellulase genes of T. reesei, including bgl1, egl2/cel5a, cbh1/cel7a, egl1/cel7b, and cbh2/cel6; the main xylanases xyn3, xyn2 and xyn4; the mannanase man1; lytic polysaccharide monooxygenases cel61b and cel61a; the carbohydrate esterase axe1; and the arabinofuranosidases abf2 and abf3. On the other hand, in the presence of glucose, genes encoding exoglucanases, mannosidases, and galactosidases were de-repressed. This result suggests that TMK1 might be involved in carbon catabolite repression (CCR). This regulation profile was totally different in Δtmk2, in which polysaccharides lyases, glycosyl transferases, chitinases, one xylanase (xyn5), and one β-glucosidase (bgl2/cel1a) were the most upregulated holocellulases upon growth with sugarcane bagasse, while genes encoding glycosyl hydrolases, glycosyl transferases, carbohydrate-binding module enzymes, and carbohydrate esterases were mainly downregulated in this condition. Enzyme activity assays showed that CMCase and β-glucosidase activities were increased in Δtmk2 grown in the presence of sugarcane bagasse, while these enzyme activities were decreased in Δtmk1 when compared to the parental strain. These results are in accordance to our transcriptomic data that pointed to an induction of different cellulolytic genes in Δtmk2. For Δtmk1, decreased CMCase activity might be explained by the downregulation of the main CAZy genes when the mutant strain was grown in the presence of sugarcane bagasse. Similarly, Wang et al.28 demonstrated increased CMCase and β-glucosidase activities when the mutant strain was grown in wheat bran and cellulose. This decrease in β-glucosidase activity was also observed in the Δtmk3 mutant strain grown in cellulose24. In Δtmk1, decreased xylanase activity might be explained by the downregulation of the main xylanase genes when the mutant strain was grown in the presence of sugarcane bagasse. The involvement of MAPK signaling in the cellulase activity has previously been shown in T. reesei24,28,30,35, however, here we showed for the first time that the deletion of a member of MAPK cascade can also regulate xylanase activity in this fungus. Regarding β-xylosidase activity, we observed a decrease in enzyme activity in both mutant strains in the presence of sugarcane bagasse. Curiously, β-xylosidase gene expression was not differentially regulated in either mutant strain when compared to the QM6a parental strain. This effect was also observed upon the deletion of MAPK TMK324. These results suggested that a yet-unknown mechanism might regulate β-xylosidase production and/or induction by MAPK signaling control. Together, our results suggest that TMK2 may play a dual role in regulating cellulase expression, acting as a positive and negative regulator of this process in T. reesei, probably depending on the carbon source.
The involvement of MAPK signaling in regulating cellulase expression was previously demonstrated in Cochliobolus heterostrophus, a pathogenic plant fungus, in which deletion of MAPK gene chk1 modulated the expression of cellobiohydrolase 7 (CBH7) and endoglucanase 6 (EG6) during plant infection36. Accordingly, our data showed that the expression of the main transcription factors involved in the control of cellulase expression did not change in Δtmk1 and Δtmk2 grown in the presence glucose, glycerol, or sugarcane bagasse. Likewise, transcriptional data of Δxyr1 and Δcre1 strains in the presence of cellulose, sophorose, and glucose showed that none of the transcription factors responsible for cellulase expression were differentially expressed in comparison to the expression in the parental strain37–39. Curiously, among the 92 differentially expressed transcription factor-encoding genes in Δtmk2 strain grown in the presence of sugarcane bagasse, glucose, or glycerol, 25 genes were common to transcription factors differentially expressed in the Δxyr1 strain in the presence of cellulose, sophorose, and glucose38. Seven other differentially expressed transcription factors in Δtmk2 were also equally modulated in Δcre1 grown on cellulose and sophorose37,39. These findings suggest an indirect relationship between MAPK signaling and XYR1 and CRE1 activation, that must be elucidated.
Amongst the differentially regulated transcription factors, in Δtmk2 in the presence of sugarcane bagasse, three genes (IDs 4124, 1941, and 34221) encode Myb family transcription factors, which control a range of cellular processes, such as proliferation, apoptosis, differentiation, metabolism, and stress response40–42. In A. nidulans and Fusarium graminearum, Myb transcription factors are important regulators of nitrogen metabolism43,44. The most repressed gene (ID 72524) in Δtmk2 in the presence of sugarcane bagasse encodes a bZIP transcription factor that is also involved in nitrogen metabolism in A. nidulans45. Similarly, four other bZIP members (IDs 65315, 110152, 103372, and 110152) showed 3-fold, 2-fold, and 7-fold lower expression in the mutant strain grown on sugarcane bagasse, glucose, and glycerol, respectively. Therefore, our results suggested that the MAPK signaling pathway might regulate T. reesei nitrogen metabolism and growth in the carbon sources studied. In Δtmk1, only eighteen transcription factors were differentially expressed in the presence of sugarcane bagasse, glucose, and glycerol. The most strongly induced gene in this mutant in the presence of sugarcane bagasse and glucose compared to the parental strain encode Zn2Cys6 and C2H2 transcriptional regulator family members. The transcriptional regulator PacG/VIB-1 (ID 54675), required for the completion of meiosis and sporulation, was the unique downregulated transcription factor in Δtmk1 grown in sugarcane bagasse. Curiously, this gene was also downregulated in Δtmk2 in the same condition, suggesting a possible mechanism of cross-talk between the two MAPK genes in T. reesei.
The transcriptome data of Δtmk2 revealed that the MAPK signaling pathway regulates the expression of different genes encoding other signaling pathways during growth in the presence of sugarcane bagasse. In this strain, genes encoding GPCRs, phospholipases, Ras GTPases, and kinase proteins were overexpressed in the presence of sugarcane bagasse. In contrast, genes related to GPCRs, calcium, histidine kinases, and upstream components of MAPK signaling, such as MAPKKK Ste11 and its regulator Ste50, showed decreased expression in this condition. In Cryphonectria parasitica, G-protein signaling controls the expression of the cellobiohydrolase cbh1 in the presence of cellulose46. Similarly, in T. reesei, the G proteins GNA3, GNB1, and GNG1 positively regulate cellulase expression in the presence of light10,47. The cross-talk between G proteins and MAPK has been described in S. pombe, in which the pheromone response is regulated by the MAPK Spk1, the protein kinase Ras1, and Gα-GTP, being the latter responsible for activating downstream components of this signaling pathway48,49. Regarding GPCRs, we demonstrated that eight PTH11-related genes were the most common GPCRs expressed during the growth of Δtmk2 in sugarcane bagasse. These GPCRs are a new class of receptors described in Magnaporthe grisea involved in pathogenic response50,51. In F. graminearum, the differential expression of PTH11 members seems to be related to fungus-host adaptation52. In Neurospora crassa, genes encoding PTH11 members were differentially expressed upon growth in cellulose, suggesting that these proteins might play a yet-unclear key role in plant cell wall recognition and in the control of holocellulolytic enzyme expression53. Therefore, our results suggested a recognition mechanism for different carbon sources involving PTH11 GPCRs in the activation of MAPK TMK2 during growth of T. reesei in the presence of sugarcane bagasse. Interestingly, it has been demonstrated in the cellulolytic bacterium Ruminococcus flavefaciens that the phosphorylation and dephosphorylation dynamics are important for carbon source recognition and regulation of carbon metabolism54. Similarly, a nutrient-specific mechanism involving G proteins and MAPK signaling was suggested to explain the ability of the fungus Cochliobolus heterostrophus to recognize different carbon sources and survive in diverse environments55. Together, our results pointed to a dual mechanism of carbon source recognition where the differentially expressed genes are components upstream to TMK2 and TMK1 that might be responsible for sensing the initial carbon source and in turn might initiate the degradation signal for these different substrates in T. reesei. However, more studies are necessary to elucidate this mechanism.
Regarding nutrient transport, MFS transporters were the most-modulated classes of transporters in the Δtmk1 and Δtmk2 mutant strains. The MFS permeases represent a large family of secondary transporters carrying small solutes, such as monosaccharides, oligosaccharides, amino acids, peptides, vitamins, and enzyme cofactors56–58. In yeast, transporters belonging to the MFS family have been found to play key roles in sugar uptake59–61. In A. nidulans, two MFS transporters are involved in transporting lactose, xylose, glucose, galactose, and mannose, indicating they accept multiple sugars as a substrate62,63. The lack of specificity of theses transporters could be explained by the similar structure of different sugars, and some transporters can act both as transporters and nutrient sensors. In T. reesei, MFS transporters are highly expressed during growth in cellulose and lactose64. Similarly, the transcriptome profiling of Δxyr1 and Δcre1 revealed that MFS permeases are highly repressed in the presence of cellulose37–39. Moreover, in T. reesei, the MFS protein Ctr1 plays a significant role in cellulolytic signaling, acting as a sophorose transporter65. Another report showed that Ctr1 is upregulated in the presence of lactose, probably acting as a lactose permease66. Our results showed that in Δtmk1, two MFS permease-encoding genes (IDs 48444 and 69957) were specifically downregulated in the presence of sugarcane bagasse. These transporters are highly similar to a putative maltose permease of the human pathogenic fungus Talaromyces marneffeii and may be involved in disaccharide transport67. The maltose permease gene (ID 69957) was also downregulated in the Δxyr1 grown on cellulose38. Recently, Nogueira et al.68 showed that the novel T. reesei 69957-sugar transport system (Tr69957) is capable of transporting xylose, mannose, and cellobiose. The deletion of Tr69957 in T. reesei affected the fungal growth and biomass accumulation, and the sugar uptake in the presence of mannose, cellobiose, and xylose. Additionally, the MFS permease with ID 46819, which was repressed in Δtmk1 in the presence of sugarcane bagasse, is homologous to the putative cellodextrin transporter-like protein CLP1 of N. crassa, which is involved in cellulase induction through a mechanism involving repression of a cellodextrin transporter, in turn inhibiting cellodextrin uptake69. Together, our results suggest that MAPK signaling may regulate the expression of different transporter families in T. reesei, and these transporters seem to play a promiscuous role in signaling pathways involved in cellulase production.
Conclusions
Our study contributes to a better understanding of the role of the MAPK intracellular signaling pathway in the degradation of cellulosic biomass by T. reesei. Transcriptomic analysis showed that several genes exhibited altered expression in a carbon source-dependent manner. The transcript profiles of Δtmk1 and Δtmk2 were drastically altered during growth in the presence of sugarcane bagasse. The most differentially expressed genes included mainly cellulolytic enzymes, genes encoding proteins involved in signaling pathways such as GPCRs and calcium signaling, as well as transporters and transcription factors. We highlighted the modulation of different classes of transporters that seem to be involved in a refined mechanism of sugar recognition. Interestingly, our results revealed a duality of MAPK signaling pathway in the control of metabolic processes and cellulase expression, in which TMK2 seems to be more responsible controlling basal development of T. reesei, while TMK1 showed a specialist role in the regulation of cellulase expression in this fungus.
Methods
Fungal Strain and Growth Conditions
Trichoderma reesei strain QM6aΔtmus53Δpyr431 was used in this study. This strain was obtained from the Institute of Chemical Engineering & Technical Biosciences of Vienna University of Technology, TU Vienna, Austria. The strain was maintained at 4 °C on MEX medium (malt extract 3% (w/v) and agar-agar 2% (w/v)), which was supplemented with 5 mM uridine in the case of the pyr4 deletion strain.
T. reesei QM6aΔtmus53Δpyr4 parental, Δtmk2 and Δtmk1 mutant strains were grown on MEX medium at 28 °C for 7–10 days until complete sporulation. For gene expression assays, a spore suspension containing approximately 107 cells mL−1 was inoculated into 200 mL of Mandels-Andreotti medium10 containing 1% (w/v) sugarcane bagasse, 2% (w/v) glucose, or 1% glycerol as the sole carbon source. The exploded sugarcane bagasse was prepared as previously described70. Briefly, sugarcane bagasse in natura was treated with 14 kg/cm2 water steam, washed exhaustively with distilled water until reducing sugars were not detected by 3,5-dinitrosalicylic acid (DNS)71, and dried at 40 °C for several days. Sugarcane bagasse was kindly donated by Nardini Agroindustrial Ltd., Vista Alegre do Alto, São Paulo, Brazil. The cultures were incubated on an orbital shaker (200 rpm) at 28 °C for 48 hours for the sugarcane bagasse and glucose experiments. For the experiments with sugarcane bagasse, the parental, Δtmk2 and Δtmk1 strains were grown in glycerol 1% (w/v) for 24 hours and then transferred to a medium containing sugarcane bagasse. All experiments were performed in three biological replicates. The resulting mycelia were collected by filtration, frozen in liquid nitrogen, and stored at −80 °C for RNA extraction. For phenotypic assays, the parental and mutant strains were inoculated on minimal medium plates containing 1% of one of the following carbon sources: glucose, starch, sucrose, lactose, glycerol, cellulose (Avicel) MEX, as well as on potato dextrose agar plates24,28.
Vector Construction for Gene Deletion
Deletions of tmk2 (Tr_82351) and tmk1 (Tr_121539) of T. reesei were generated as previously described72. To construct the deletion cassette, the orotidine-5′-phosphate decarboxylase gene of T. reesei (pyr4, Tr_74020) was used as a selection marker. Sequences were obtained from the T. reesei genome database (http://genome.jgi.doe.gov/Trire2/Trire2.home.html). The marker gene and its respective 5′ and 3′ flanking sequences were amplified using the primers described in Additional file 2. The primers were designed and analyzed with the OligoAnalyzer Tool (https://eu.idtdna.com/calc/analyzer). To enable yeast-mediated recombination of the deletion cassette, the external 5′-UTR forward (F) and 3′-UTR reverse (R) primers possessed cohesive ends with the vector pRS42673,74 and the internal primers 5′-UTR R and 3′-UTR F contained cohesive ends with 5′ and 3′sequences of pyr4 gene (Additional file 2). The 50 μL reaction mixture contained 1 U Platinum Taq DNA Polymerase High Fidelity (Thermo Scientific), 1 × High Fidelity PCR Buffer, 0.2 mM dNTPs, 0.3 µM forward and reverse primers, 1 µL T. reesei QM9414 genomic DNA (150 ng/µL) as a template, and nuclease-free water. The PCR fragments were purified using a QIAquick PCR Purification Kit (Qiagen).
For yeast-mediated recombination, the yeast shuttle vector pRS426 (ampR lacZ URA3)73,74 was used, which was digested with EcoRI and XhoI (Thermo Scientific) and was purified with the QIAquick PCR Purification Kit (Qiagen). Yeast transformation was essentially conducted as previously described75–77. An overnight culture (200 rpm, 30 °C) of the yeast S. cerevisiae strain SC9721 (MATα his3-Δ200 URA3-52 leu2Δ1 lys2Δ202 trp1Δ63) (Fungal Genetic Stock Center – www.fgsc.net) was prepared. Then, 1 mL of the overnight culture was added to 50 mL of fresh YPD (1% yeast extract, 2% peptone, 1% glucose) (all from Sigma Aldrich) medium and incubated at 30 °C until OD600 = 1. Thereafter, the cells were centrifuged and resuspended in 100 mM lithium acetate for transformation. To this end, equimolar quantities of both 5′ and 3′ flanking regions, pyr4, and digested pRS426 were mixed and used for yeast transformation through the lithium acetate method78. S. cerevisiae SC9721 transformants were selected for their ability to grow on YPD medium supplemented with lysine, histidine, leucine, and tryptophan, without uracil. S. cerevisiae genomic DNA was extracted by as previously described79,80. The deletion cassettes of Tr_82351 and Tr_121539 were PCR-amplified using TaKaRa Ex Taq DNA Polymerase (Clontech) using 5F and 3R primers (Additional file 2).
Transformation of T. reesei
T. reesei QM6aΔtmus53Δpyr4 was used for transformation to achieve highly efficient homologous integration of the deletion cassette. Protoplast transformation was carried out as previously described81. For transformation, 10 μg of linear purified deletion cassette fragment was used. Transformants were grown on selective minimal medium [1 g/L MgSO4•7H2O, 10 g/L 1% KH2PO4, 6 g/L (NH4)2SO4, 3 g/L trisodium citrate•2H2O, 10 g/L glucose, 20 mL/L 50X trace elements solution (0.25 g/L FeSO4•7H2O, 0.07 g/L ZnSO4•2H2O, 0.1 g/L CoCl2•6H2O, 0.085 g/L MnSO4•H2O), 2% (wt/vol) noble agar lacking uridine] (all from Sigma Aldrich). Cassette integration was verified by PCR using the primer pyr4Sc (inside in the selectable marker gene pyr4) and the gene-specific primer 82351Sc or 121539Sc (outside the transformation cassette) (Additional file 2). The expression profiles of the genes Tr_82351 and Tr_121539 were also analyzed by RT-qPCR.
RT-qPCR Analysis
For expression analysis, total RNA (1 µg) from each sample was digested with DNAse I (Fermentas) to remove genomic DNA. cDNA was synthesized using the Maxima™ First Strand cDNA Synthesis kit (Fermentas) according to the manufacturer’s instructions. The cDNA was diluted 1:50 and used as a template for real-time PCR. Reactions were run in the Bio-Rad CFX96™ by using SsoFast™ EvaGreen® Supermix (Bio-Rad) for detection according to the manufacturer’s instructions. Each reaction (10 µL) contained 5 µL of SsoFast™ EvaGreen® Supermix (Bio-Rad), forward and reverse primers (500 nm each; Additional file 2), cDNA template, and nuclease-free water. PCR cycling conditions were as follows: 10 min at 95 °C, followed by 40 cycles of 10 s at 95 °C and 30 s at 60 °C, and a ramp of 60–95 °C a rate of 0.5 °C/10 s to generate a melting curve to test for primer dimers and nonspecific amplification. β-Actin transcript was used as an internal reference to normalize the amount of total RNA present in each reaction82. For sugarcane bagasse expression analysis, gene expression levels of genes were calculated from the threshold cycle according to the 2−ΔΔCT method83, relative to transcript levels in the parental strain QM6a grown in non-induced condition (glycerol) for 24 h82. For glucose, gene expression levels were calculated from the threshold cycle according to the 2−ΔCT method83, relative to transcript levels of β-Actin. Finally, tmk1 and tmk2 expression levels in the mutant strains were calculated from the threshold cycle according to the 2−ΔΔCT method83, relative to transcript levels in QM6a.
Southern Blotting
The selected transformants were analyzed by Southern hybridization as previously described84, to demonstrate homologous integration of the transformation cassettes at the targeted T. reesei QM6aΔtmus53Δpyr4 loci. For this analysis, 25 µg of total genomic DNA of both parental and mutant strains were digested overnight with EcoRI and HindIII (Δtmk2) and PstI and EcoRV (Δtmk1) (Fermentas) and then, digested DNA was transferred to GE Healthcare Amersham Hybond -N+ Membranes (GE). The probe was produced from a PCR-amplified fragment using 82351-5 F and 82351-5 R (promoter region of tmk2), and 121539-3 F and 121539-3 R (terminator region of tmk1) primer sets (Additional file 2) and was labeled by using a digoxigenin DNA labeling kit (Roche, Mannheim, Germany), following the manufacturer’s instructions. Labeling, hybridization, and immunological detection were carried out with a nonradioactive labeling and immunological detection kit with CDP-Star as the chemiluminescent substrate (Roche, Mannheim, Germany), as previously described85.
Enzyme Activity Assays
CMCase activity was analyzed in microplates (96-well PCR plate) following the protocol described by Xiao et al.86 with some modifications: 30 μL of 1% carboxymethylcellulose (CMC), previously prepared in sodium acetate buffer at pH 4.8, was added to each well with 30 μL of enzyme. After a 30-min incubation at 50 °C, 60 μL of DNS were added, followed by heating at 95 °C for 5 min to allow color development. Then, the samples were transferred to a flat-bottomed microplate and absorbance at 540 nm was read. One enzyme unit was defined as the amount of enzyme capable of releasing 1 μmol of reducing sugar per minute. β-glucosidase and β-xylosidase activities were respectively assayed by abilities to hydrolyze pNPG and pNPX following published protocols2,87,88. Xylanase activity was determined by mixing 25 µL of enzyme solution with 50 µL of xylan from beechwood (1.0 mg mL−1) in 100 mM sodium acetate buffer (pH 5.0) for 30 min at 50 °C. After incubation, 75 µL of DNS were added and heated at 95 °C for 5 min for color development. After this, 100 µL of the solution were transferred to a microliter plate, and the absorbance at 540 nm was read. One unit of enzyme was defined as the amount of enzyme capable of releasing 1 µmol of reducing sugar per minute32.
RNA Extraction, Library Construction, and Sequencing
Total RNA was extracted from mycelia of each sample using a Direct-zol RNA MiniPrep (Zymo Research), according to the manufacturer’s instructions. RNA concentrations were determined by a fluorometric Qubit RNA HS Assay Kit (Thermo Scientific) and RNA integrity was verified by the Agilent 2100 Bioanalyzer. Total RNA of three biological replicates of parental, Δtmk2, and Δtmk1 strains grown in the presence of sugarcane bagasse (48 hours), glucose (48 hours), and glycerol (24 hours), resulting in 27 samples was used for library preparation, using the Illumina TruSeq Stranded mRNA Sample Preparation Kit (Illumina), according to the manufacturer’s instructions. The libraries were quantified using a KAPA Library Quantification Kit Illumina (KAPA Biosystems) and were sequenced at NGS sequencing facility at Laboratório Nacional de Ciência e Tecnologia do Bioetanol (CTBE) (Campinas, São Paulo) on an Illumina Hiseq2500 platform with paired-end 2 × 100-bp reads. RNA-Seq data from the 27 libraries were deposited in the Gene Expression Omnibus (GEO) under accession number GSE100602.
RNA-Seq Data Analysis
In the current study, Illumina Hiseq2500 technology was used for sequencing, resulting in approximately 143 million, 139 million, and 158 million 2 × 100-bp paired-end reads for parental, Δtmk2, and Δtmk1 strains, respectively. Quality-filtered reads (Additional file 3) were mapped to the T. reesei 2.0 reference genome, available at JGI Genome Portal (http://genome.jgi-psf.org/Trire2/Trire2.home.html), using the TopHat2 v2.0.4 aligner89, allowing two mismatches and only unique alignments. After alignment, Samtools version 0.1.1890 was used to process the alignment data, which were visualized using the Integrative Genomics Viewer91. Genes were annotated using the T. reesei 2.0 reference genome, a local database provided by Prof. C. P. Kubicek (TU, Vienna), and the InterPro database (http://www.ebi.ac.uk/interpro/)92,93. The R package DESeq2 version 1.6.394 was used to perform the differential expression analysis using the raw number of reads mapped to each gene in each sample to perform statistical tests, based on the negative binomial distribution. To analyze the expression pattern, we used the following strategy: first, we used the libraries of glycerol to compare the data of parental and mutant strains grown on sugarcane bagasse (QM6a sugarcane bagasse/QM6a glycerol, Δtmk2 glycerol/QM6a glycerol, Δtmk2 sugarcane bagasse/Δtmk2 glycerol, Δtmk1 glycerol/QM6a glycerol, and Δtmk1 sugarcane bagasse/Δtmk1 glycerol). Then, the data of the parental and mutant strains grown on sugarcane bagasse were compared to each other (Δtmk2 sugarcane bagasse/QM6a sugarcane bagasse and Δtmk1 sugarcane bagasse/QM6a sugarcane bagasse). For glucose, the data of the mutant strain were directly compared to those of the parental strain (Δtmk2 glucose/QM6a glucose and Δtmk1 glucose/QM6a glucose). The DESeq2 package was used for normalization, using the median log deviation, and for the differential expression analysis, we applied an adjusted p-value ≤ 0.05 as the threshold. Thus, Log 2-fold change values ≥ 1 and ≤−1 represent up- and downregulation. All statistical analyses were performed using the R package95.
Functional Enrichment and Network Analysis
Differentially expressed transcripts were annotated using Blast2GO96. For functional enrichment analysis, differentially expressed genes were annotated to GO terms using the topGO algorithm97. Significantly enriched GO terms (p ≤ 0.05), were further analyzed using GraphPad Prism v 5.00 Software. To reconstruct the regulatory network of Δtmk2/QM6a and Δtmk1/QM6a under the experimental conditions analyzed, differentially expressed genes in Δtmk2 (2215 in sugarcane bagasse, 1991 in glucose, and 1754 in glycerol in total) and in Δtmk1 (258 in sugarcane bagasse, 803 in glucose, and 32 in glycerol in total) were analyzed using the Cytoscape 3.0.1 software98, following a previously reported procedure38.
Electronic supplementary material
Acknowledgements
We are thankful for Professor Astrid Mach-Aigner from TU-Wien for providing the QM6aΔmus53Δpyr4 strain used in this study. We are also thankful for Professor Fábio M. Squina from Laboratório Nacional de Ciência e Tecnologia do Bioetanol (CTBE) for helping with the RNA-Seq experiments. This work was supported by The State of São Paulo Research Foundation (FAPESP) (proc. 2014/23653-2, 2016/23233-9 and 2017/04206-3).
Author Contributions
Conceived and designed the experiments: R.G.P., G.F.P. and R.N.S. Performed the experiments: R.G.P., C.B.C., D.C.B.L., N.T.A.P. and N.M.M.R. Analyzed the data: R.G.P., G.F.P., R.N.S., R.S.R. Wrote the paper: R.G.P., A.C.C.A., C.B.C. All authors have read and approved the final manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-33383-1.
References
- 1.Schmoll M. The information highways of a biotechnological workhorse-signal transduction in Hypocrea jecorina. BMC Genomics. 2008;9:430. doi: 10.1186/1471-2164-9-430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.dos Santos Castro L, et al. Comparative metabolism of cellulose, sophorose and glucose in Trichoderma reesei using high-throughput genomic and proteomic analyses. Biotechnol Biofuels. 2014;7:41. doi: 10.1186/1754-6834-7-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gupta VK, et al. The Post-genomic Era of Trichoderma reesei: What’s Next? Trends Biotechnol. 2016;0:69–147. doi: 10.1016/j.tibtech.2016.06.003. [DOI] [PubMed] [Google Scholar]
- 4.Shida Y, Furukawa T, Ogasawara W. Deciphering the molecular mechanisms behind cellulase production in Trichoderma reesei, the hyper-cellulolytic filamentous fungus. Biosci. Biotechnol. Biochem. 2016;80:1712–1729. doi: 10.1080/09168451.2016.1171701. [DOI] [PubMed] [Google Scholar]
- 5.Seiboth B, et al. The putative protein methyltransferase LAE1 controls cellulase gene expression in Trichoderma reesei. Mol Microbiol. 2012;84:1150–1164. doi: 10.1111/j.1365-2958.2012.08083.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Degenkolb T, et al. The production of multiple small peptaibol families by single 14-module Peptide synthetases in Trichoderma/Hypocrea. Chem. Biodivers. 2012;9:499–535. doi: 10.1002/cbdv.201100212. [DOI] [PubMed] [Google Scholar]
- 7.Karimi Aghcheh R, et al. The VELVET A orthologue VEL1 of Trichoderma reesei regulates fungal development and is essential for cellulase gene expression. PLoS One. 2014;9:e112799. doi: 10.1371/journal.pone.0112799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hakkinen M, et al. Screening of candidate regulators for cellulase and hemicellulase production in Trichoderma reesei and identification of a factor essential for cellulase production. Biotechnol Biofuels. 2014;7:14. doi: 10.1186/1754-6834-7-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seibel C, et al. Light-dependent roles of the G-protein alpha subunit GNA1 of Hypocrea jecorina (anamorph Trichoderma reesei) BMC Biol. 2009;7:58. doi: 10.1186/1741-7007-7-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schmoll M, Schuster A, Silva Rdo N, Kubicek CP. The G-alpha protein GNA3 of Hypocrea jecorina (Anamorph Trichoderma reesei) regulates cellulase gene expression in the presence of light. Eukaryot Cell. 2009;8:410–420. doi: 10.1128/EC.00256-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tisch D, Schuster A, Schmoll M. Crossroads between light response and nutrient signalling: ENV1 and PhLP1 act as mutual regulatory pair in Trichoderma reesei. BMC Genomics. 2014;15:425. doi: 10.1186/1471-2164-15-425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schuster A, Tisch D, Seidl-Seiboth V, Kubicek CP, Schmoll M. Roles of protein kinase A and adenylate cyclase in light-modulated cellulase regulation in Trichoderma reesei. Appl Env. Microbiol. 2012;78:2168–2178. doi: 10.1128/AEM.06959-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nogueira KMV, et al. Evidence of cAMP involvement in cellobiohydrolase expression and secretion by Trichoderma reesei in presence of the inducer sophorose. BMC Microbiol. 2015;15:195. doi: 10.1186/s12866-015-0536-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang J, Zhang Y, Zhong Y, Qu Y, Wang T. Ras GTPases modulate morphogenesis, sporulation and cellulase gene expression in the cellulolytic fungus Trichoderma reesei. PLoS One. 2012;7:e48786. doi: 10.1371/journal.pone.0048786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hamel L-P, et al. Ancient signals: comparative genomics of plant MAPK and MAPKK gene families. Trends Plant Sci. 2006;11:192–198. doi: 10.1016/j.tplants.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 16.Sinha AK, Jaggi M, Raghuram B, Tuteja N. Mitogen-activated protein kinase signaling in plants under abiotic stress. Plant Signal. Behav. 2011;6:196–203. doi: 10.4161/psb.6.2.14701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singh P, Mohanta TK, Sinha AK. Unraveling the intricate nexus of molecular mechanisms governing rice root development: OsMPK3/6 and auxin-cytokinin interplay. PLoS One. 2015;10:e0123620. doi: 10.1371/journal.pone.0123620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Waltermann C, Klipp E. Signal integration in budding yeast. Biochem Soc Trans. 2010;38:1257–1264. doi: 10.1042/BST0381257. [DOI] [PubMed] [Google Scholar]
- 19.Bussink HJ, Osmani SA. A mitogen-activated protein kinase (MPKA) is involved in polarized growth in the filamentous fungus, Aspergillus nidulans. FEMS Microbiol. Lett. 1999;173:117–25. doi: 10.1111/j.1574-6968.1999.tb13492.x. [DOI] [PubMed] [Google Scholar]
- 20.Mukherjee PK, Latha J, Hadar R, Horwitz BA. TmkA, a mitogen-activated protein kinase of Trichoderma virens, is involved in biocontrol properties and repression of conidiation in the dark. Eukaryot Cell. 2003;2:446–455. doi: 10.1128/EC.2.3.446-455.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paoletti M, et al. Mating Type and the Genetic Basis of Self-Fertility in the Model Fungus Aspergillus nidulans. Curr. Biol. 2007;17:1384–1389. doi: 10.1016/j.cub.2007.07.012. [DOI] [PubMed] [Google Scholar]
- 22.Atoui A, Bao D, Kaur N, Grayburn WS, Calvo AM. Aspergillus nidulans natural product biosynthesis is regulated by mpkB, a putative pheromone response mitogen-activated protein kinase. Appl. Environ. Microbiol. 2008;74:3596–600. doi: 10.1128/AEM.02842-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jain R, et al. The MAP kinase MpkA controls cell wall integrity, oxidative stress response, gliotoxin production and iron adaptation in Aspergillus fumigatus. Mol. Microbiol. 2011;82:39–53. doi: 10.1111/j.1365-2958.2011.07778.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang M, et al. A Mitogen-Activated Protein Kinase Tmk3 Participates in High Osmolarity Resistance, Cell Wall Integrity Maintenance and Cellulase Production Regulation in Trichoderma reesei. PLoS One. 2013;8:e72189. doi: 10.1371/journal.pone.0072189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brown NA, et al. The Aspergillus nidulans signalling mucin MsbA regulates starvation responses, adhesion and affects cellulase secretion in response to environmental cues. Mol. Microbiol. 2014;94:1103–1120. doi: 10.1111/mmi.12820. [DOI] [PubMed] [Google Scholar]
- 26.Gustin MC, Albertyn J, Alexander M, Davenport K. MAP kinase pathways in the yeast Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 1998;62:1264–1300. doi: 10.1128/mmbr.62.4.1264-1300.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao X, Mehrabi R, Xu J-R. Mitogen-activated protein kinase pathways and fungal pathogenesis. Eukaryot. Cell. 2007;6:1701–14. doi: 10.1128/EC.00216-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang M, et al. Identification of the role of a MAP kinase Tmk2 in Hypocrea jecorina (Trichoderma reesei) Sci. Rep. 2014;4:6732. doi: 10.1038/srep06732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen F, et al. An Ime2-like mitogen-activated protein kinase is involved in cellulase expression in the filamentous fungus Trichoderma reesei. Biotechnol. Lett. 2015;37:2055–62. doi: 10.1007/s10529-015-1888-z. [DOI] [PubMed] [Google Scholar]
- 30.Wang M, et al. Role of Trichoderma reesei mitogen-activated protein kinases (MAPKs) in cellulase formation. Biotechnol. Biofuels. 2017;10:99. doi: 10.1186/s13068-017-0789-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Derntl C, Kiesenhofer DP, Mach RL, Mach-Aigner AR. Novel Strategies for Genomic Manipulation of Trichoderma reesei with the Purpose of Strain Engineering. Appl. Environ. Microbiol. 2015;81:6314–23. doi: 10.1128/AEM.01545-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Castro LS, et al. Expression pattern of cellulolytic and xylanolytic genes regulated by transcriptional factors XYR1 and CRE1 are affected by carbon source in Trichoderma reesei. Gene Expr. Patterns. 2014;14:88–95. doi: 10.1016/j.gep.2014.01.003. [DOI] [PubMed] [Google Scholar]
- 33.Nakari-Setälä T, et al. Genetic modification of carbon catabolite repression in Trichoderma reesei for improved protein production. Appl Env. Microbiol. 2009;75:4853–4860. doi: 10.1128/AEM.00282-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Silva-Rocha R, et al. Deciphering the cis-regulatory elements for XYR1 and CRE1 regulators in Trichoderma reesei. PLoS One. 2014;9:e99366. doi: 10.1371/journal.pone.0099366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Z, et al. Functional characterization of the upstream components of the Hog1-like kinase cascade in hyperosmotic and carbon sensing in Trichoderma reesei. Biotechnol. Biofuels. 2018;11:97. doi: 10.1186/s13068-018-1098-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lev S, Horwitz BA. A mitogen-activated protein kinase pathway modulates the expression of two cellulase genes in Cochliobolus heterostrophus during plant infection. Plant Cell. 2003;15:835–44. doi: 10.1105/tpc.010546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Antonieto AC, dos Santos Castro L, Silva-Rocha R, Persinoti GF, Silva RN. Defining the genome-wide role of CRE1 during carbon catabolite repression in Trichoderma reesei using RNA-Seq analysis. Fungal Genet Biol. 2014;73:93–103. doi: 10.1016/j.fgb.2014.10.009. [DOI] [PubMed] [Google Scholar]
- 38.Castro, L. S. et al. Understanding the role of the master regulator XYR1 in Trichoderma reesei by global transcriptional analysis. Front Microbiol175 (2016). [DOI] [PMC free article] [PubMed]
- 39.Antoniêto ACC, et al. Trichoderma reesei CRE1-mediated Carbon Catabolite Repression in Re-sponse to Sophorose Through RNA Sequencing Analysis. Curr. Genomics. 2016;17:119–31. doi: 10.2174/1389202917666151116212901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dubos C, et al. MYB transcription factors in Arabidopsis. Trends Plant Sci. 2010;15:573–581. doi: 10.1016/j.tplants.2010.06.005. [DOI] [PubMed] [Google Scholar]
- 41.Meneses E, et al. The R2R3 Myb protein family in Entamoeba histolytica. Gene. 2010;455:32–42. doi: 10.1016/j.gene.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 42.Ravaglia D, et al. Transcriptional regulation of flavonoid biosynthesis in nectarine (Prunus persica) by a set of R2R3 MYB transcription factors. BMC Plant Biol. 2013;13:68. doi: 10.1186/1471-2229-13-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arratia-Quijada J, Sanchez O, Scazzocchio C, Aguirre J. FlbD, a Myb transcription factor of Aspergillus nidulans, is uniquely involved in both asexual and sexual differentiation. Eukaryot Cell. 2012;11:1132–1142. doi: 10.1128/EC.00101-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim Y, et al. MYT3, a Myb-like transcription factor, affects fungal development and pathogenicity of Fusarium graminearum. PLoS One. 2014;9:e94359. doi: 10.1371/journal.pone.0094359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Polley SD, Caddick MX. Molecular characterisation of meaB, a novel gene affecting nitrogen metabolite repression in Aspergillus nidulans. FEBS Lett. 1996;388:200–5. doi: 10.1016/0014-5793(96)00541-8. [DOI] [PubMed] [Google Scholar]
- 46.Wang P, Nuss DL. Induction of a Cryphonectria parasitica cellobiohydrolase I gene is suppressed by hypovirus infection and regulated by a GTP-binding-protein-linked signaling pathway involved in fungal pathogenesis. Proc. Natl. Acad. Sci. USA. 1995;92:11529–11533. doi: 10.1073/pnas.92.25.11529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tisch D, Kubicek CP, Schmoll M. The phosducin-like protein PhLP1 impacts regulation of glycoside hydrolases and light response in Trichoderma reesei. BMC Genomics. 2011;12:613. doi: 10.1186/1471-2164-12-613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Imai Y, Yamamoto M. The fission yeast mating pheromone P-factor: its molecular structure, gene structure, and ability to induce gene expression and G1 arrest in the mating partner. Genes Dev. 1994;8:328–38. doi: 10.1101/gad.8.3.328. [DOI] [PubMed] [Google Scholar]
- 49.Hughes DA. Control of signal transduction and morphogenesis by Ras. Semin. Cell Biol. 1995;6:89–94. doi: 10.1016/1043-4682(95)90005-5. [DOI] [PubMed] [Google Scholar]
- 50.DeZwaan TM, Carroll AM, Valent B, Sweigard JA. Magnaporthe grisea Pth11p is a novel plasma membrane protein that mediates appressorium differentiation in response to inductive substrate cues. Plant Cell. 1999;11:2013–30. doi: 10.1105/tpc.11.10.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kulkarni RD, Thon MR, Pan H, Dean RA. Novel G-protein-coupled receptor-like proteins in the plant pathogenic fungus Magnaporthe grisea. Genome Biol. 2005;6:R24. doi: 10.1186/gb-2005-6-3-r24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Harris LJ, Balcerzak M, Johnston A, Schneiderman D, Ouellet T. Host-preferential Fusarium graminearum gene expression during infection of wheat, barley, and maize. Fungal Biol. 2016;120:111–23. doi: 10.1016/j.funbio.2015.10.010. [DOI] [PubMed] [Google Scholar]
- 53.Cabrera IE, et al. Global Analysis of Predicted G Protein-Coupled Receptor Genes in the Filamentous Fungus, Neurospora crassa. G3&#58; Genes|Genomes|Genetics. 2015;5:2729–2743. doi: 10.1534/g3.115.020974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vercoe PE, Kocherginskaya SA, White BA. Differential protein phosphorylation-dephosphorylation in response to carbon source in Ruminococcus flavefaciens FD-1. J. Appl. Microbiol. 2003;94:974–80. doi: 10.1046/j.1365-2672.2003.01929.x. [DOI] [PubMed] [Google Scholar]
- 55.Degani O. G protein and MAPK signaling pathways control the ability of Cochliobolus heterostrophus to exploit different carbon sources. Adv. Biol. Chem. 2014;4:40–50. doi: 10.4236/abc.2014.41007. [DOI] [Google Scholar]
- 56.Pao SS, Paulsen IT, Saier MH., Jr. Major facilitator superfamily. Microbiol Mol Biol Rev. 1998;62:1–34. doi: 10.1128/mmbr.62.1.1-34.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Reddy VS, Shlykov MA, Castillo R, Sun EI, Saier MH., Jr. The major facilitator superfamily (MFS) revisited. FEBS J. 2012;279:2022–2035. doi: 10.1111/j.1742-4658.2012.08588.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yan N. Structural Biology of the Major Facilitator Superfamily Transporters. Annu Rev Biophys. 2015;44:257–283. doi: 10.1146/annurev-biophys-060414-033901. [DOI] [PubMed] [Google Scholar]
- 59.Bisson LF, Coons DM, Kruckeberg AL, Lewis DA. Yeast sugar transporters. Crit Rev Biochem Mol Biol. 1993;28:259–308. doi: 10.3109/10409239309078437. [DOI] [PubMed] [Google Scholar]
- 60.Weusthuis RA, Pronk JT, van den Broek PJ, van Dijken JP. Chemostat cultivation as a tool for studies on sugar transport in yeasts. Microbiol Rev. 1994;58:616–630. doi: 10.1128/mr.58.4.616-630.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fan J, Chaturvedi V, Shen SH. Identification and phylogenetic analysis of a glucose transporter gene family from the human pathogenic yeast Candida albicans. J Mol Evol. 2002;55:336–346. doi: 10.1007/s00239-002-2330-4. [DOI] [PubMed] [Google Scholar]
- 62.Fekete E, et al. Identification of a permease gene involved in lactose utilisation in Aspergillus nidulans. Fungal Genet Biol. 2012;49:415–425. doi: 10.1016/j.fgb.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 63.Colabardini AC, et al. Functional characterization of a xylose transporter in Aspergillus nidulans. Biotechnol Biofuels. 2014;7:46. doi: 10.1186/1754-6834-7-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Porciuncula Jde O, et al. Identification of Major Facilitator Transporters Involved in Cellulase Production during Lactose Culture of Trichoderma reesei PC-3-7. Biosci Biotechnol Biochem. 2013;77:8. doi: 10.1271/bbb.120992. [DOI] [PubMed] [Google Scholar]
- 65.Zhang W, et al. Two major facilitator superfamily sugar transporters from Trichoderma reesei and their roles in induction of cellulase biosynthesis. J Biol Chem. 2013;288:32861–32872. doi: 10.1074/jbc.M113.505826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ivanova C, Baath JA, Seiboth B, Kubicek CP. Systems analysis of lactose metabolism in Trichoderma reesei identifies a lactose permease that is essential for cellulase induction. PLoS One. 2013;8:e62631. doi: 10.1371/journal.pone.0062631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Boyce KJ, Andrianopoulos A. Morphogenetic circuitry regulating growth and development in the dimorphic pathogen Penicillium marneffei. Eukaryot Cell. 2013;12:154–160. doi: 10.1128/EC.00234-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nogueira, K. M. V. et al. Characterization of a novel sugar transporter involved in sugarcane bagasse degradation in Trichoderma reesei. Biotechnol. Biofuels11 (2018). [DOI] [PMC free article] [PubMed]
- 69.Cai P, et al. The putative cellodextrin transporter-like protein CLP1 is involved in cellulase induction in Neurospora crassa. J Biol Chem. 2015;290:788–796. doi: 10.1074/jbc.M114.609875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.de Souza WR, et al. Transcriptome analysis of Aspergillus niger grown on sugarcane bagasse. Biotechnol. Biofuels. 2011;4:40. doi: 10.1186/1754-6834-4-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Miller G. Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal Chem. 1959;31:426–8. doi: 10.1021/ac60147a030. [DOI] [Google Scholar]
- 72.Schuster A, et al. A versatile toolkit for high throughput functional genomics with Trichoderma reesei. Biotechnol. Biofuels. 2012;5:1. doi: 10.1186/1754-6834-5-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Christianson TW, Sikorski RS, Dante M, Shero JH, Hieter P. Multifunctional yeast high-copy-number shuttle vectors. Gene. 1992;110:119–122. doi: 10.1016/0378-1119(92)90454-W. [DOI] [PubMed] [Google Scholar]
- 74.Teepe AG, Loprete DM, He Z, Hoggard TA, Hill TW. The protein kinase C orthologue PkcA plays a role in cell wall integrity and polarized growth in Aspergillus nidulans. Fungal Genet. Biol. 2007;44:554–562. doi: 10.1016/j.fgb.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 75.Colot HV, et al. A high-throughput gene knockout procedure for Neurospora reveals functions for multiple transcription factors. Proc. Natl. Acad. Sci. USA. 2006;103:10352–10357. doi: 10.1073/pnas.0601456103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Collopy PD, et al. High-throughput construction of gene deletion cassettes for generation of Neurospora crassa knockout strains. Methods Mol. Biol. 2010;638:33–40. doi: 10.1007/978-1-60761-611-5_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gietz RD, Schiestl RH. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2007;2:31–4. doi: 10.1038/nprot.2007.13. [DOI] [PubMed] [Google Scholar]
- 78.Gietz RD, Schiestl RH. Applications of high efficiency lithium acetate transformation of intact yeast cells using single-stranded nucleic acids as carrier. Yeast. 1991;7:253–63. doi: 10.1002/yea.320070307. [DOI] [PubMed] [Google Scholar]
- 79.Goldman GH, et al. Expressed sequence tag analysis of the human pathogen Paracoccidioides brasiliensis yeast phase: identification of putative homologues of Candida albicans virulence and pathogenicity genes. Eukaryot. Cell. 2003;2:34–48. doi: 10.1128/EC.2.1.34-48.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sambrook, J. & Russell, D. W. Molecular Cloning: ALaboratory Manual. Cold Spring HarborLaboratory 2, (Cold Spring Harbor Laboratory Press, 2001).
- 81.Gruber F, Visser J, Kubicek CP, de Graaff LH. The development of a heterologous transformation system for the cellulolytic fungus Trichoderma reesei based on a pyrG-negative mutant strain. Curr. Genet. 1990;18:71–76. doi: 10.1007/BF00321118. [DOI] [PubMed] [Google Scholar]
- 82.Verbeke J, et al. Transcriptional profiling of cellulase and expansin-related genes in a hypercellulolytic Trichoderma reesei. Biotechnol Lett. 2009;31:1399–1405. doi: 10.1007/s10529-009-0030-5. [DOI] [PubMed] [Google Scholar]
- 83.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 84.Cardoza RE, et al. Identification of loci and functional characterization of trichothecene biosynthesis genes in filamentous fungi of the genus Trichoderma. Appl. Environ. Microbiol. 2011;77:4867–77. doi: 10.1128/AEM.00595-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cardoza RE, et al. Partial silencing of a hydroxy-methylglutaryl-CoA reductase-encoding gene in Trichoderma harzianum CECT 2413 results in a lower level of resistance to lovastatin and lower antifungal activity. Fungal Genet. Biol. 2007;44:269–283. doi: 10.1016/j.fgb.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 86.Xiao Z, Storms R, Tsang A. Microplate-based carboxymethylcellulose assay for endoglucanase activity. Anal. Biochem. 2005;342:176–8. doi: 10.1016/j.ab.2005.01.052. [DOI] [PubMed] [Google Scholar]
- 87.Hideno A, et al. Production and characterization of cellulases and hemicellulases by Acremonium cellulolyticus using rice straw subjected to various pretreatments as the carbon source. Enzyme Microb. Technol. 2011;48:162–168. doi: 10.1016/j.enzmictec.2010.10.005. [DOI] [PubMed] [Google Scholar]
- 88.Lopes FAC, et al. Biochemical and metabolic profiles of Trichoderma strains isolated from common bean crops in the Brazilian Cerrado, and potential antagonism against Sclerotinia sclerotiorum. Fungal Biol. 2012;116:815–24. doi: 10.1016/j.funbio.2012.04.015. [DOI] [PubMed] [Google Scholar]
- 89.Kim D, et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Br. Bioinform. 2013;14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jones P, et al. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014;30:1236–1240. doi: 10.1093/bioinformatics/btu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mitchell A, et al. The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Res. 2015;43:D213–21. doi: 10.1093/nar/gku1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.R Development Core Team, R. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing1, 409 (2011).
- 96.Conesa A, et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 97.Alexa, A. & Rahnenfhrer, J. topGO: Enrichment Analysis for Gene Ontology. R package version 2.18.0 (2010).
- 98.Shannon P, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.