

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is an aggressive disease, which is characterized by its high invasiveness, rapid progression, and profound resistance to therapy. Gemcitabine is the first-line treatment option for pancreatic cancer patients, but the overall survival is quite low. Therefore, it is an urgent issue to identify new molecules for improved therapies, with better efficacy and less toxicity. Our previous data indicated that Euchromatic histone-lysine N-methyltransferase 2 (EHMT2) functions as a therapeutic target to override GEM resistance and promote metastasis in the treatment of pancreatic cancer. Here, we screened a small-molecule library of 143 protein kinase inhibitors, to verify cytotoxicity of different inhibitors in EHMT2-depleted cells. We determined that the EHMT2 plays a promising modulating role for targeted PI3K/mTOR inhibition. Our data revealed that EHMT2 down-regulates p27 expression, and this contributes to tumor growth. The depletion of EHMT2, ectopic expression of methyltransferase-dead EHMT2, or treatment with an EHMT2 inhibitor decreases H3K9 methylation of p27 promoter and induces G1 arrest in PANC-1 pancreatic cancer cells. Consistent with these findings, in vivo tumor xenograft models, primary tumors, and the Oncomine database utilizing bioinformatics approaches, also show a negative correlation between EHMT2 and p27. We further demonstrated that low EHMT2 elevated BEZ235 sensitivity through up-regulation of p27 in PDAC cells; high levels of SKP2 decrease BEZ235 responsiveness in PDAC cells. Altogether, our results suggest the EHMT2-p27 axis as a potential marker to modulate cell response to dual PI3K/mTOR inhibition, which might provide a strategy in personalized therapeutics for PDAC patients.
Keywords: EHMT2, SKP2, p27, pancreatic cancer, cell cycle
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is one of the most aggressive cancer types with a five-year survival rate of less than 8% [1]. More than 80% of patients were diagnosed with unresectable or metastatic PDAC due to unnoticed clinical symptoms and insufficient specific examinations [2]. There are dismal results from several trials of PDAC, even in novel therapeutic agents [3]. Gemcitabine, the standard treatment for advanced pancreatic cancer since 1997, displays modest effects with median overall survival times ranging from 5 to 8 months [4,5]. Combination with tyrosine kinase inhibitor, erlotinib, reveals limited but significant improvement in overall survival [6], and combination chemotherapy with FOLFIRINOX (5-fluorouracil, oxaliplatin, irinotecan, leucovorin) in metastatic disease shows superior survival but moderate toxicity [7]. In addition, the MPACT study demonstrated improvement of overall survival in nearly one year by using nab-paclitaxel plus gemcitabine versus gemcitabine alone [8].
The human EHMT2 (Euchromatic histone-lysine N-methyltransferase) gene is located at the chromosome band 6p21.33. The EHMT2 gene encodes a methyltransferase (EHMT2, also known as G9a) that methylates lysine residues of histone H3 and functions as recruitment of additional epigenetic regulators and repressors of transcription. Our previous study demonstrated that EHMT2 induces down-regulation of histone demethylase KDM7A, which inhibits de-methylation of H3K9m2 and H3K27m3 of the E-cadherin promoter, inhibiting E-cadherin expression, and enhancing epithelial-mesenchymal transition, migration, and invasion [9]. In addition, EHMT2 mediates interleukin-8 expression to elevate gemcitabine resistance and trans-endothelial invasion [10]. However, more studies are needed to precisely elucidate how the multiple properties of EHMT2 contribute to tumorigenesis.
p27, a cell cycle negative regulator, inhibits cyclin-dependent kinase (CDK) complexes to cause cell cycle arrest. Overexpression of p27 prevents CDK2 activation and entry from the late G1 into the S phase of the cell cycle [11]. In several types of cancers, the expression of p27 represents poor prognosis, including pancreatic cancer [12-15]. Some studies reveal that p27 is regulated via post-translational modifications [16]. SKP2 is a component of the SCF ubiquitin ligase complex, which mediates the degradation of p27 [17,18] and participates in G2-M progression [19]. Several investigations indicated that p27 expression is lost frequently in pancreatic cancer patients [20-24].
To date, the association between EHMT2 and p27 is still uncertain in pancreatic cancer. In our study, we found that the depletion of EHMT2 halts the cell cycle at G1 phase by activating p27 post-translational modifications. p27 up-regulation ceases cancer cell replication and proliferation. The inverse relation between EHMT2 and p27 is established in vivo and in vitro. In addition, combination with a dual PI3K/mTOR inhibitor significantly improves the therapeutic effect, as evidenced by the p27 expression levels in pancreatic cancer cell line. p27 expression in pancreatic cancer is significant because it may be a valuable disease biomarker and therapeutic strategy.
Materials and methods
Cell lines, reagents and plasmids
PANC-1 and Mia PaCa-2 cells were cultured in Dulbecco’s modified Eagles medium (DMEM) containing 10% fetal bovine serum (FBS). sh-EHMT2 plasmids were from the National RNAi Core Facility (Academia Sinica, Taiwan). sh-EHMT2 #1 sequence: GCTCCAGGAATTTAACAAGAT; sh-EHMT2 #2 sequence: CTCCAGGAATTTAA CAAGATT. Antibodies againsy EHMT2, p21, Tubulin, Actin, PCNA were purchased from Genetex (SanAntonio, TX, USA). p27, p57, H3K9m2, H3K4m3, H3K27m3, Ki67 were purchase from Abcam (Cambridge, MA). UNC0638 was purchased from Cayman Chemical (Ann Arbor, MI, USA). Cycloheximide (C4859, CHX, 100 μg/ml) was obtained from Sigma.
Quantitative reverse transcriptional PCR (RT-qPCR)
Total RNA extraction was extracted using the RNeasy mini kit (qiagen, Valencia, CA), according to the manufacturer’s instructions. Equal amounts of RNA were used to synthesize first-strand cDNA using the RT2 first strand kit (Qiagene). RT-PCR was performed using SYBR Green on Real-Time PCR System (Applied Biosystems, Foster City, CA). The primers of p27 were (forward) 5’-AAGGGCCAACAGAACAGAAG-3’ and (reverse) 5’-GGATGTCCATTCAATGGAGTC-3’. p27 promoter chip sequence were (forward) 5’-ACTCGCCGTGTCAATCATTT-3’ and (reverse) 5’-AACACCCCGAAAAGACGAG-3’.
Flow cytometry for cell cycle analysis
PANC-1 and Mia PaCa-2 cells were transfected with sh-EHMT2 and harvested by trypsinization and fixed with 70% ice-cold ethanol for overnight at -20°C. Next day, the cell pellet was re-suspended in PI-staining buffer (50 μl/ml PI, RNAse A, Beckman Coulter, Brea, CA) and was incubated for 15 min at 37°C for further cell cycle analysis. Cell cycle distribution was analyzed by FACS Calibur (BD Biosciences, San Diego, CA) using ModFit software.
Cell viability test
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was used for assessment of cell viability. 3000 cells were seeded in 96-well plates. Cells were treated as indicated drugs for 72 h. After inoculation, cells were then incubated with 0.5 mg/ml of MTT at 37°C for 2 h. Medium was replaced by 100 μl DMSO per well to dissolve the precipitates. Colorimetric analysis using a 96-well micro-plate reader (BioTek Instruments) was performed at wavelength 490 nm.
Xenograft of tumor-bearing SCID mice
Parental PANC-1 and two sh-EHMT2 PANC-1cells (1 * 106 cells) were suspended in 30 μL of HBSS and injected subcutaneously into the left hide leg of each 7-wk-old nonobese diabetic/servere combined immunodeficient mice. After 4 weeks, eight mice of each group were sacrificed for further analysis. The tumor volume was calculated using the equation: tumor volume = (length × width2)/2. All experiments were performed in accordance with the animal care and use guideline of Kaohsiung Medical university (Taiwan) approved by the Animal Care and Use Committee of Kaohsiung Medical University.
Immunohistochemistry
Mouse tumor samples were cut 4 μ m-thick sections and deparaffined in xylene as described [25]. Sections were stained with mouse anti-PCNA Ab (1:20000), anti-p27 Ab (1:1000), anti-Ki67 Ab (1:150), anti-EHMT2 (1:1000), anti-H3K9m2 (1:1000) following the manufacturer’s protocol. After incubation for 30 min at 25°C with secondary antibodies and Envission system (Dako, Denmark). Finally, sections were counterstained with hematoxylin and analyzed under a microscope. Besides, we used pancreatic cancer tissue microarrays (PA721a, Biomax Inc and Super Bio Chips Laboratories). Staining protocol is the same as mouse tumor tissue.
Oncomine data analysis
Oncomine (http://www.oncomine.org) is a cancer microarray database and integrated data-mining platform. Pancreatic cancer data set GSE15471 (sample size = 78) and GSE16515 (sample size = 52) was used to analyze the correlation between EHMT2 and p27 expression.
Statistics
All experiments were performed in triplicate. Data are expressed as mean ± SD, and were used to compare the differences between experimental groups. Multiple comparisons were evaluated by one-way ANOVA, and differences between groups were calculated by two-tailed Student’s t-test.
Results
Inhibition of EHMT2 retards cell proliferation and tumor growth through G1 arrest
Our previous data indicated that EHMT2 is involved in cancer stemness, drug resistance, and metastasis in pancreatic cancer. To further investigate the role of EHMT2 in pancreatic tumor growth in vivo, studies were performed in SCID mice. As shown in Figure 1A, knockdown of EHMT2 significantly reduced tumor size compared with that of PANC-1 parental cells. Proliferating-cell nuclear antigen (PCNA) plays a role in facilitating DNA replication; therefore, it was used as a marker of cell proliferation. Consistent with the image of tumor size, a decrease in PCNA protein level was observed in EHMT2-depleted cells, compared with parental cells (Figure 1B). These data suggest that down-regulation of EHMT2 hinders cell proliferation and replication. In order to clarify how the EHMT2 interferes in tumor growth, we verified alterations in cell cycle via flow cytometry. Knockdown of EHMT2 leads to considerable increase in the number of cells in the G1 (white bars) phase, accompanied by a remarkable decrease in the number of S (black bars) and G2/M phase (gray bars) cells, indicating G1 arrest in PANC-1 cells (Figure 1C). Consistent with this observation, increase of G1 phase was performed in Mia PaCa-2 cells (Figure S1). Overall, these results indicate that knockdown of EHMT2 negatively regulates cell proliferation in pancreatic cancer cell lines.
Figure 1.
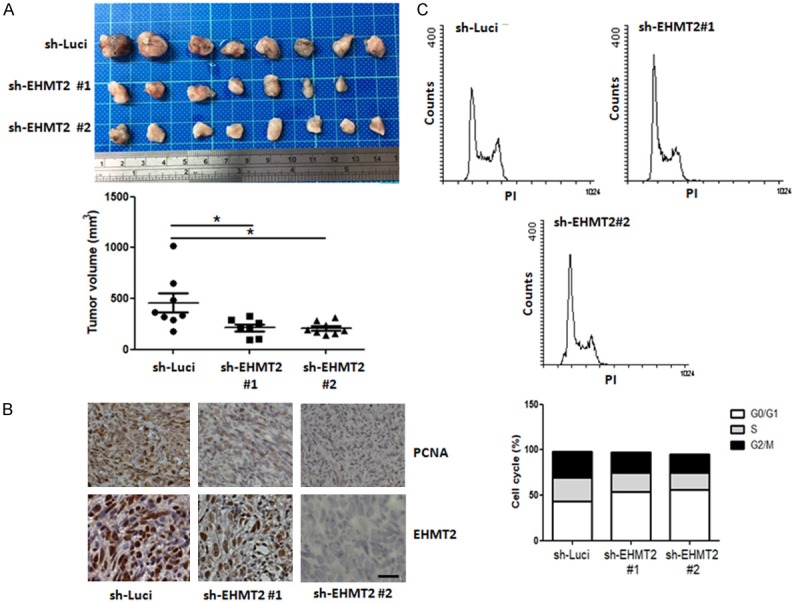
Knockdown of EHMT2 leads to the reduction of tumor growth and the accumulation of cells in G1 arrest. A. Top panel, representative images of pancreatic cancer tissue with high or low expression of EHMT2 in SCID mice. Bottom panel, quantitative analysis of tumor size after injection in designated cells at 4 weeks. Statistical comparisons were evaluated by one-way ANOVA. (*P < 0.05). B. Representative staining of PCNA, EHMT2 in tumors of different treatment groups. Original magnification: × 40, scale bar: 10 μm. C. Expression profiles of cell cycle in PANC-1 and PANC-1 EHMT2 deficient (sh-EHMT2) cell lines were analyzed by flow cytometry, and the percentage of the cell population at different stages of the cell cycle were calculated.
EHMT2 effects in the cell cycle of pancreatic cancer cells
Next, to study the molecular mechanisms responsible for EHMT2-induced G1 arrest, we examined the levels of G1 checkpoint-associated proteins in EHMT2 depleted cells. As showed in Figure 2A, knockdown of EHMT2 resulted in increased level of p27, but not p21 or p57, in PANC-1, and Mia PaCa-2 cells. To confirm the effect of EHMT2 in p27 expression, cells were treated with UNC0638, an EHMT2 inhibitor, for 3 days. We obtained a similar result as that for knockdown of EHMT2, elevated p27 protein level in both PANC-1 cells and Mia PaCa-2 cells (Figure 2B). We also evaluated the levels of p27 in an in vivo mouse model. Consistent with the in vitro cell line model, UNC0638 treatment elevated p27 expression and reduced levels of PCNA, Ki67, and H3K9m2 (Figure 2C). Likewise, the induction of p27 was also observed in EHMT2-depleted cells in vivo (Figure 2D). Results indicate that inhibition of EHMT2 suppressed cell replication and proliferation, and negatively regulated G1 cell cycle progression by p27 tumor suppressor. Altogether, these data demonstrate that EHMT2 is an important mediator of p27 expression in pancreatic cancer.
Figure 2.
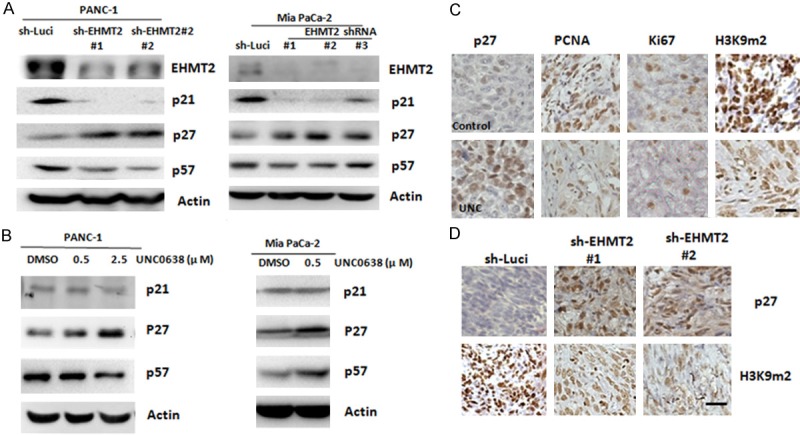
Depletion of EHMT2 increases the expression of p27. A. Expression of EHMT2, p21, p27, and p57 proteins in PANC-1 cells and Mia PaCa-2 with EHMT2 deficiency were determined by western blot analysis. B. PANC-1 cells and Mia PaCa-2 were continuously incubated with the indicated concentrations of UNC0638 for 3 days. Expression of p21, p27, and p57 was detected by western blot analysis. C. Representative staining for p27, PCNA, Ki67, and H3K9m2 in tumors with mock and UNC0638 (UNC) treatment groups. Original magnification: × 40, scale bar: 10 μm. D. Representative staining of p27 and H3K9m2 in tumors with different EHMT2 expression. Original magnification: × 40, scale bar: 10 μm.
Knockdown of EHMT2 up-regulates p27 expression in a methyltransferase-dependent manner
To better understand these events in the context of protein metabolism homeostasis, we used cycloheximide (CHX), a protein synthesis inhibitor, to measure the degradation of the protein after blocking its biosynthesis. We showed that p27 stabilization is affected in EHMT2 depleted cells for the indicated periods of time. We found that knockdown of EHMT2 did not decelerate the degradation of p27 in pancreatic cancer cell lines (Figure 3A). These data suggest that EHMT2 down-regulates the levels of p27 in a non-post-translational manner. EHMT2 is a well-known H3K9 methyltransferase, with an important role in gene silencing. Therefore, we next investigated the role of EHMT2 in p27 gene expression. As showed in Figure 3B, knockdown of EHMT2 significantly increased p27 mRNA. Ectopic expression of methyltransferase-dead EHMT2 also increased p27 mRNA expression by threefold (Figure 3C). Chromatin immunoprecipitation-quantitative polymerase chain reaction (ChIP-qPCR) assay further demonstrated that inhibition of EHMT2 suppressed the di-methylation of H3K9, indicating attenuation of p27 transcriptional repression in cells (Figures 3D and S2). Depletion of EHMT2 also reduced H3K9 methylation of p27 gene promoter, resulting in enhancement of gene activation (Figures 3E and S3). Collectively, these data suggest that EHMT2 depletion directly down-regulates H3K9 methylation on p27 promoter to increase its transcription.
Figure 3.
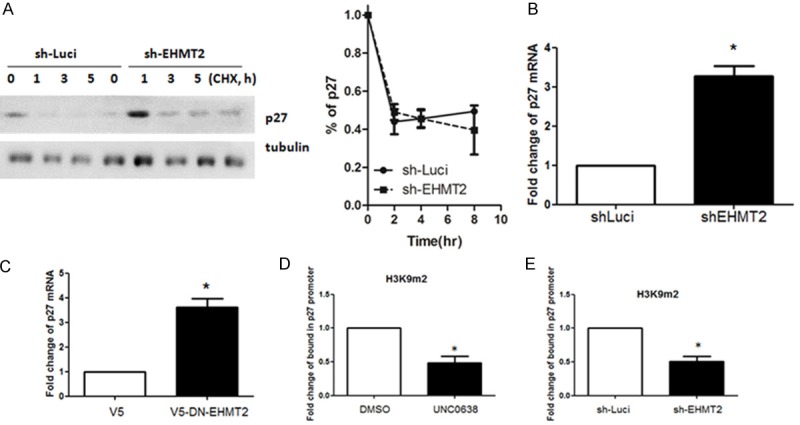
Knockdown of EHMT2 up-regulates p27 expression in a methyltransferase-dependent manner. A. Protein stability of p27 was detected in PANC-1 cell or PANC-1 sh-EHMT2 cells. Densitometry was utilized to quantify p27 protein levels after normalization with tubulin to obtain the percentage of p27 degradation (mean ± SD; n = 3). Error bars indicate SD. B. Expression of p27 in parental PANC-1 (sh-Luci) and EHMT2-deficient (sh-EHMT2) cells was determined by RT-qPCR analysis. Columns represent the mean of triplicate PCR assays, normalized to GAPDH (*P < 0.05). C. RT-qPCR analysis was used to investigate the expression of p27 in PANC-1 cells transfected with control or overexpressed methyltransferase-dead EHMT2 (DN-EHMT2). Columns represent the mean of triplicate PCR assays, normalized to GAPDH (*P < 0.05). D. PANC-1 cells were treated with DMSO or UNC0638. ChIP-qPCR analysis was performed to determine the status of H3K9m2 in the p27 gene promoter. Experiments were performed in triplicate. (*P < 0.05). E. ChIP-qPCR analysis was used to determine the status of H3K9m2 in the p27 gene promoter in PANC-1 or EHMT2 knockdown cells. The experiments were performed in triplicate (*P < 0.05).
Inhibition of the PI3K/mTOR pathway combined with depletion of EHMT2 increases cytotoxicity in pancreatic cancer
To identify pathways involved in EHMT2-mediated oncogenic signaling, we tested the cytotoxicity of EHMT2 inhibition with small molecules reported in the Library of Pharmacologically Active Compounds, in a cell viability assay. We evaluated 143 protein kinase inhibitors, including 31 in ongoing clinical trials. We found that dual PI3K/mTOR inhibitor (NVP-BEZ235, BEZ235) had a more potent repressive effect in EHMT2-depleted cells, compared with other kinase inhibitors and we selected them for in vitro testing (Figures 4A and S4). Notably, the expression of p27 is considerably elevated in EHMT2-deficient cells treated with both dual PI3K/mTOR inhibitors (BEZ235; Cay10626, Cay), possibly due to a synergistic effect (Figure 4B). To further identify the role of EHMT2-mediated p27 as a regulator of PI3K/mTOR inhibitor sensitivity, we reduced the expression of p27 by increasing SKP2 expression in EHMT2 depleted cells. Elevated SKP2 led to reduction in p27 expression in EHMT2 knockdown cells (Figure 4C). Overexpression of SKP2 could reverse the cytotoxicity of dual PI3K/mTOR inhibition in EHMT2-depleted cells (Figures 4D and S5). Altogether, knockdown of EHMT2 reinforced the cytotoxic effect of dual PI3K/mTOR inhibition in pancreatic cancer cells.
Figure 4.
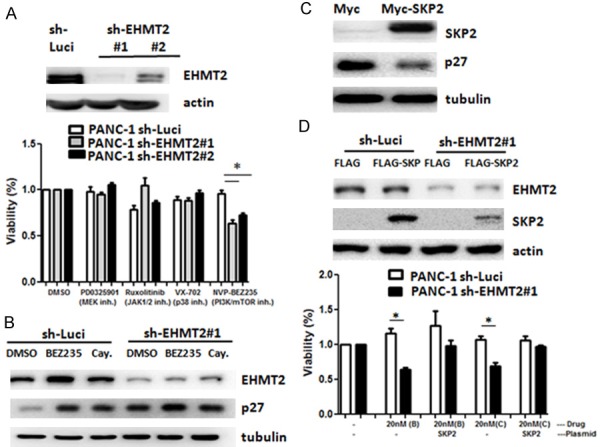
Knockdown of EHMT2 enhances the cytotoxicity of BEZ235 in pancreatic cancer cells. A. Parental PANC-1 cells (sh-Luci) and EHMT2-depleted cells (sh-EHMT2 #1 and sh-EHMT2 #2) were treated with different compounds for 72 h. Cell viability was determined by MTT assay (*P < 0.05). Protein levels of EHMT2 were detected by western blot analysis. B. Expression levels of p27 in parental PANC-1 (sh-Luci) and EHMT2-deficient (sh-EHMT2) cells treated with dual PI3K/mTOR inhibitor (NVP-BEZ235, BEZ235, and Cay10626, Cay) for 72 h were determined by western blot assay. C. PANC-1 cells were transfected with Myc or Myc-SKP2 expression plasmid, and the levels of SKP2 and p27 were detected by western blot assay. Tubulin was used as a loading control. D. Parental PANC-1 (sh-Luci) and EHMT2-deficient (sh-EHMT2) cells were transfected with FLAG or FLAG-SKP2 plasmids and treated with dual PI3K/mTOR inhibitor (NVP-BEZ235, B and Cay10626, C) for 72 h. Cell viability was determined by MTT assay. The protein level of EHMT2 was detected by western blot analysis. Statistical comparisons were evaluated by a one-way ANOVA (*P < 0.05).
In vivo evidence supports the negative correlation of EHMT2 and p27 in pancreatic cancer tissue
To determine the association between EHMT2 and p27 in vivo, cancer tissue analysis and a bioinformatics approach were adopted. Immunohistochemistry of a tissue microarray indicated that EHMT2 expression is inversely associated with p27 expression in pancreatic cancer tissue (Figure 5A). Next, we analyzed the correlation of these genes in two different public datasets (GSE15471 and GSE16515, Oncomine). Results show that the expression of EHMT2 is negatively associated with p27 in both public datasets (Figure 5B). Taken together, these data support the results of our cell-based study, and suggest that EHMT2 may regulate p27, controlling cell cycle and the induction of cell proliferation.
Figure 5.
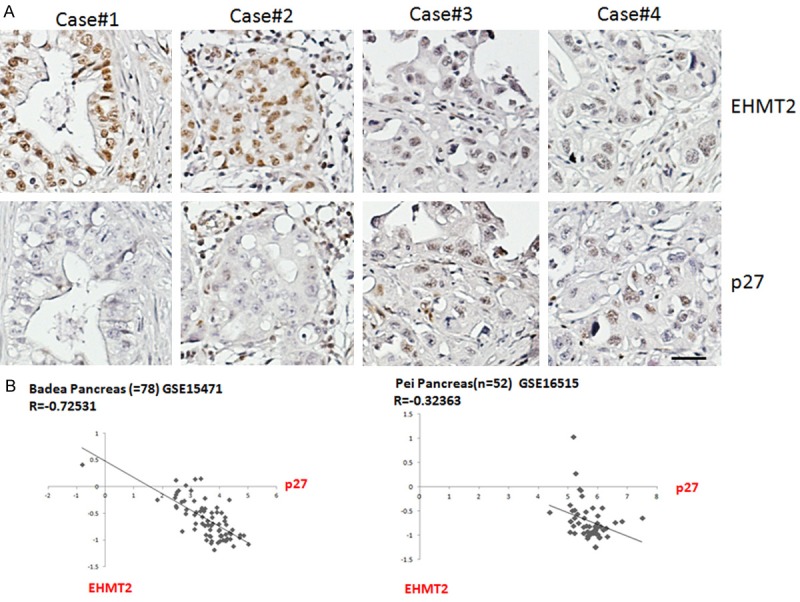
EHMT2 expression inversely correlates with p27 in pancreatic cancer. A. IHC representative images of EHMT2 and p27 in four pancreatic cancer tissue sections. B. Correlations between EHMT2 and p27 were determined from two independent ONCOMINE datasets, GSE15471 and GSE16515. Correlation coefficient and P-value are shown. Original magnification: x 40, scale bar: 10 μm.
Discussion
In the United States, pancreatic cancer is a fatal disease and is the fourth-leading cause of cancer-related deaths [26], and its poor prognosis is attributed to the lack of response to conventional cytotoxic chemotherapy, advanced disease status, and poor survival rates. Another reason for dismal prognosis is drug resistance [27,28]. Therefore, identification of effective biomarkers and development of novel therapeutic agents are currently a priority.
p27, a tumor suppressor, is actively involved in regulating and interrupting cell cycle progression at the G1/S checkpoint by inhibiting CDK2, thus regulating cell proliferation, motility, and apoptosis via diverse pathways. In cancer, there are several approaches to inactivating p27, including impaired synthesis, accelerated degradation, and mislocalization [29]. As previously reported, SKP2 possesses oncogenic potential via degradation of p27 protein [30]. Activation of the PI3K/Akt pathway modifies the pathway downstream of p27, causing tumorigenesis and therapeutic resistance in many cancers [31-33]. Loss of p27 is connected with mutational inactivation of PTEN tumor suppressor gene in several human cancers [34]. Similarly, reduced PTEN staining is associated with lower expression of p27 protein in breast cancers [35]. Nevertheless, unlike p53 and KRAS, which are frequently mutated in pancreatic cancer, the patterns of mutation and mechanisms of p27 are still inconclusive [36].
EHMT2, a histone methyltransferase, mediates mono- and di-methylation of H3K9, and participates in multiple epigenetic regulatory and developmental pathways [37]. EHMT2 expression and methyltransferase activity have been associated with tumor progression hallmarks, such as tumor proliferation, invasion, and transformation of stem cell phenotypes, and even with therapeutic resistance [38,39]. High EHMT2 protein expression has been reported in different types of cancer, such as breast, bladder, and head and neck cancers [40-42]. High expression levels of EHMT2 are predictive for poor prognosis in some malignant diseases [43,44]. EHMT2 induces downstream effector interleukin-8, and promotes the resistance to chemotherapeutic drugs by changing cancer cells and microenvironment simultaneously [10]. EHMT2 depletion retards cancer progression by inducing cell cycle arrest and DNA damage response, triggering apoptosis or inducing autophagy, both pharmaceutically and genetically [45,46]. In our study, we identified a novel mechanism of EHMT2 controlling p27 expression directly in a SKP2-independent pathway, which promoted cancer cell proliferation and replication. Therefore, for the first time, we revealed an inverse correlation between EHMT2 and p27, which shows that EHMT2 accelerates the di-methylation of H3K9 on p27 promoter. In addition, inhibition of EHMT2 slows down the degradation of p27, which suppresses tumor growth.
Pancreatic cancer is characterized by frequent KRAS mutations and dysregulation of PI3K/AKT signaling, which mediate the activation of mTOR kinase through TORC1 and TORC2 complexes [47,48]. BEZ235, a synthetic low molecular mass compound belonging to the class of imidazoquinolines, binds to the ATP-binding clefts of the class I PI3K/mTOR kinase, to delay PI3K signaling and attenuate TORC1 and TORC2 activity [49-51]. In diverse cancer cell types, monotherapy and combination therapy with BEZ235 shows potential anti-proliferative responses and facilitates G0/G1 cell cycle arrest [49,51,52]. Monotherapy with BEZ235 in pancreatic cancer exerts anti-proliferative and antiangiogenic response in cancer cell lines and murine xenografts [53,54]. In pancreatic cancer in vitro and in vivo, a combination of BEZ235 with gemcitabine, the standard first-line chemotherapeutic, is most effective as sequential administration of gemcitabine followed by BEZ235 [55]. However, identification of predictive biomarkers that translate the mechanisms of resistance to therapy is required.
In this context, we identify the important role of p27 in cell proliferation of pancreatic cancer via direct EHMT2 transcriptional regulation. We also observed that a combination of p27 modifications and inhibition of the PI3K/mTOR pathway strengthen cytotoxicity of pancreatic cancer cells. Thus, our experiments might provide a new perspective on the role of EHMT2 and p27 in tumorigenesis. The possible therapeutic role of the EHMT2-p27 axis and dual PI3K/mTOR inhibition in the regulation of pancreatic cancer is a crucial feature that merits future research.
Acknowledgements
This study was supported by grants from the Ministry of Science and Technology of Republic of China [106-2314-B-037-049-MY3]; Research Center for Environmental Medicine, Kaohsiung Medical University [KMU-TP105A15]; Kaohsiung Medical University [106CM-KMU-08]; KMU-KMUH Co-Project of Key Research, grant No.KMU-DK107013; Taiwan Ministry of Health and Welfare [MOHW106-TDU-B-212-144007, MOHW107-TDU-B-212-114020] and Health and Welfare surcharge of tobacco products. The authors also thank the Center for Research Resources and Development in Kaohsiung Medical University for the assistance in flow cytometry analysis.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 2.Li D, Xie K, Wolff R, Abbruzzese JL. Pancreatic cancer. Lancet. 2004;363:1049–1057. doi: 10.1016/S0140-6736(04)15841-8. [DOI] [PubMed] [Google Scholar]
- 3.Oettle H, Post S, Neuhaus P, Gellert K, Langrehr J, Ridwelski K, Schramm H, Fahlke J, Zuelke C, Burkart C, Gutberlet K, Kettner E, Schmalenberg H, Weigang-Koehler K, Bechstein WO, Niedergethmann M, Schmidt-Wolf I, Roll L, Doerken B, Riess H. Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: a randomized controlled trial. JAMA. 2007;297:267–277. doi: 10.1001/jama.297.3.267. [DOI] [PubMed] [Google Scholar]
- 4.Burris HA 3rd, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo AM, Tarassoff P, Nelson R, Dorr FA, Stephens CD, Von Hoff DD. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J. Clin. Oncol. 1997;15:2403–2413. doi: 10.1200/JCO.1997.15.6.2403. [DOI] [PubMed] [Google Scholar]
- 5.Louvet C, Labianca R, Hammel P, Lledo G, Zampino MG, Andre T, Zaniboni A, Ducreux M, Aitini E, Taieb J, Faroux R, Lepere C, de Gramont A Gercor and Giscad. Gemcitabine in combination with oxaliplatin compared with gemcitabine alone in locally advanced or metastatic pancreatic cancer: results of a GERCOR and GISCAD phase III trial. J. Clin. Oncol. 2005;23:3509–3516. doi: 10.1200/JCO.2005.06.023. [DOI] [PubMed] [Google Scholar]
- 6.Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, Au HJ, Murawa P, Walde D, Wolff RA, Campos D, Lim R, Ding K, Clark G, Voskoglou-Nomikos T, Ptasynski M, Parulekar W National Cancer Institute of Canada Clinical Trials Group. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 2007;25:1960–1966. doi: 10.1200/JCO.2006.07.9525. [DOI] [PubMed] [Google Scholar]
- 7.Conroy T, Desseigne F, Ychou M, Bouché O, Guimbaud R, Bécouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de la Fouchardière C, Bennouna J, Bachet JB, Khemissa-Akouz F, Péré-Vergé D, Delbaldo C, Assenat E, Chauffert B, Michel P, Montoto-Grillot C, Ducreux M Groupe Tumeurs Digestives of Unicancer; PRODIGE Intergroup. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364:1817–1825. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 8.Ramanathan RK, Goldstein D, Korn RL, Arena F, Moore M, Siena S, Teixeira L, Tabernero J, Van Laethem JL, Liu H, McGovern D, Lu B, Von Hoff DD. Positron emission tomography response evaluation from a randomized phase III trial of weekly nab-paclitaxel plus gemcitabine versus gemcitabine alone for patients with metastatic adenocarcinoma of the pancreas. Ann Oncol. 2016;27:648–653. doi: 10.1093/annonc/mdw020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pan MR, Hsu MC, Chen LT, Hung WC. G9a orchestrates PCL3 and KDM7A to promote histone H3K27 methylation. Sci Rep. 2015;5:18709. doi: 10.1038/srep18709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pan MR, Hsu MC, Luo CW, Chen LT, Shan YS, Hung WC. The histone methyltransferase G9a as a therapeutic target to override gemcitabine resistance in pancreatic cancer. Oncotarget. 2016;7:61136–61151. doi: 10.18632/oncotarget.11256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abukhdeir AM, Park BH. P21 and p27: roles in carcinogenesis and drug resistance. Expert Rev Mol Med. 2008;10:e19. doi: 10.1017/S1462399408000744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Loda M, Cukor B, Tam SW, Lavin P, Fiorentino M, Draetta GF, Jessup JM, Pagano M. Increased proteasome-dependent degradation of the cyclin-dependent kinase inhibitor p27 in aggressive colorectal carcinomas. Nat Med. 1997;3:231–234. doi: 10.1038/nm0297-231. [DOI] [PubMed] [Google Scholar]
- 13.Masciullo V, Sgambato A, Pacilio C, Pucci B, Ferrandina G, Palazzo J, Carbone A, Cittadini A, Mancuso S, Scambia G, Giordano A. Frequent loss of expression of the cyclin-dependent kinase inhibitor p27 in epithelial ovarian cancer. Cancer Res. 1999;59:3790–3794. [PubMed] [Google Scholar]
- 14.Kamai T, Takagi K, Asami H, Ito Y, Oshima H, Yoshida KI. Decreasing of p27(Kip1)and cyclin E protein levels is associated with progression from superficial into invasive bladder cancer. Br J Cancer. 2001;84:1242–1251. doi: 10.1054/bjoc.2000.1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rabbani F, Koppie TM, Charytonowicz E, Drobnjak M, Bochner BH, Cordon-Cardo C. Prognostic significance of p27Kip1 expression in bladder cancer. BJU Int. 2007;100:259–263. doi: 10.1111/j.1464-410X.2007.06927.x. [DOI] [PubMed] [Google Scholar]
- 16.Singh K, Dong Q, TimiriShanmugam PS, Koul S, Koul HK. Tetrandrine inhibits deregulated cell cycle in pancreatic cancer cells: differential regulation of p21(Cip1/Waf1), p27(Kip1) and cyclin D1. Cancer Lett. 2018;425:164–173. doi: 10.1016/j.canlet.2018.03.042. [DOI] [PubMed] [Google Scholar]
- 17.Carrano AC, Eytan E, Hershko A, Pagano M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat Cell Biol. 1999;1:193–199. doi: 10.1038/12013. [DOI] [PubMed] [Google Scholar]
- 18.Sutterluty H, Chatelain E, Marti A, Wirbelauer C, Senften M, Muller U, Krek W. p45SKP2 promotes p27Kip1 degradation and induces S phase in quiescent cells. Nat Cell Biol. 1999;1:207–214. doi: 10.1038/12027. [DOI] [PubMed] [Google Scholar]
- 19.Nakayama K, Nagahama H, Minamishima YA, Miyake S, Ishida N, Hatakeyama S, Kitagawa M, Iemura S, Natsume T, Nakayama KI. Skp2-mediated degradation of p27 regulates progression into mitosis. Dev Cell. 2004;6:661–672. doi: 10.1016/s1534-5807(04)00131-5. [DOI] [PubMed] [Google Scholar]
- 20.Lu CD, Morita S, Ishibashi T, Hara H, Isozaki H, Tanigawa N. Loss of p27Kip1 expression independently predicts poor prognosis for patients with resectable pancreatic adenocarcinoma. Cancer. 1999;85:1250–1260. [PubMed] [Google Scholar]
- 21.Hu YX, Watanabe H, Li P, Wang Y, Ohtsubo K, Yamaguchi Y, Sawabu N. An immunohistochemical analysis of p27 expression in human pancreatic carcinomas. Pancreas. 2000;21:226–230. doi: 10.1097/00006676-200010000-00002. [DOI] [PubMed] [Google Scholar]
- 22.Juuti A, Nordling S, Louhimo J, Lundin J, von Boguslawski K, Haglund C. Loss of p27 expression is associated with poor prognosis in stage I-II pancreatic cancer. Oncology. 2003;65:371–377. doi: 10.1159/000074651. [DOI] [PubMed] [Google Scholar]
- 23.Fukumoto A, Ikeda N, Sho M, Tomoda K, Kanehiro H, Hisanaga M, Tsurui Y, Tsutsumi M, Kato JY, Nakajima Y. Prognostic significance of localized p27Kip1 and potential role of Jab1/CSN5 in pancreatic cancer. Oncol Rep. 2004;11:277–284. [PubMed] [Google Scholar]
- 24.Diersch S, Wenzel P, Szameitat M, Eser P, Paul MC, Seidler B, Eser S, Messer M, Reichert M, Pagel P, Esposito I, Schmid RM, Saur D, Schneider G. Efemp1 and p27(Kip1) modulate responsiveness of pancreatic cancer cells towards a dual PI3K/mTOR inhibitor in preclinical models. Oncotarget. 2013;4:277–288. doi: 10.18632/oncotarget.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu CC, Pan MR, Wei YC, Lin CH, Yang SF, Tsai HP, Luo CW, Chai CY. CHD4 as a potential biomarker in differentiating between cellular schwannoma and malignant peripheral nerve sheath tumor. Appl Immunohistochem Mol Morphol. 2017 doi: 10.1097/PAI.0000000000000522. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 26.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 27.Chand S, O’Hayer K, Blanco FF, Winter JM, Brody JR. The landscape of pancreatic cancer therapeutic resistance mechanisms. Int J Biol Sci. 2016;12:273–282. doi: 10.7150/ijbs.14951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ireland L, Santos A, Ahmed MS, Rainer C, Nielsen SR, Quaranta V, Weyer-Czernilofsky U, Engle DD, Perez-Mancera PA, Coupland SE, Taktak A, Bogenrieder T, Tuveson DA, Campbell F, Schmid MC, Mielgo A. Chemoresistance in pancreatic cancer is driven by stroma-derived insulin-like growth factors. Cancer Res. 2016;76:6851–6863. doi: 10.1158/0008-5472.CAN-16-1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chu IM, Hengst L, Slingerland JM. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat Rev Cancer. 2008;8:253–267. doi: 10.1038/nrc2347. [DOI] [PubMed] [Google Scholar]
- 30.Sistrunk C, Kim SH, Wang X, Lee SH, Kim Y, Macias E, Rodriguez-Puebla ML. Skp2 deficiency inhibits chemical skin tumorigenesis independent of p27(Kip1) accumulation. Am J Pathol. 2013;182:1854–1864. doi: 10.1016/j.ajpath.2013.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee-Hoeflich ST, Pham TQ, Dowbenko D, Munroe X, Lee J, Li L, Zhou W, Haverty PM, Pujara K, Stinson J, Chan SM, Eastham-Anderson J, Pandita A, Seshagiri S, Hoeflich KP, Turashvili G, Gelmon KA, Aparicio SA, Davis DP, Sliwkowski MX, Stern HM. PPM1H is a p27 phosphatase implicated in trastuzumab resistance. Cancer Discov. 2011;1:326–337. doi: 10.1158/2159-8290.CD-11-0062. [DOI] [PubMed] [Google Scholar]
- 32.Wu CY, Carpenter ES, Takeuchi KK, Halbrook CJ, Peverley LV, Bien H, Hall JC, DelGiorno KE, Pal D, Song Y, Shi C, Lin RZ, Crawford HC. PI3K regulation of RAC1 is required for KRASinduced pancreatic tumorigenesis in mice. Gastroenterology. 2014;147:1405–1416. e1407. doi: 10.1053/j.gastro.2014.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tang Q, Zheng X, Zhang J. Long non-coding RNA CRNDE promotes heptaocellular carcinoma cell proliferation by regulating PI3K/Akt/beta-catenin signaling. Biomed Pharmacother. 2018;103:1187–1193. doi: 10.1016/j.biopha.2018.04.128. [DOI] [PubMed] [Google Scholar]
- 34.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 35.Tsutsui S, Inoue H, Yasuda K, Suzuki K, Tahara K, Higashi H, Era S, Mori M. Inactivation of PTEN is associated with a low p27Kip1 protein expression in breast carcinoma. Cancer. 2005;104:2048–2053. doi: 10.1002/cncr.21471. [DOI] [PubMed] [Google Scholar]
- 36.Bryant KL, Mancias JD, Kimmelman AC, Der CJ. KRAS: feeding pancreatic cancer proliferation. Trends Biochem Sci. 2014;39:91–100. doi: 10.1016/j.tibs.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kramer JM. Regulation of cell differentiation and function by the euchromatin histone methyltranserfases G9a and GLP. Biochem Cell Biol. 2016;94:26–32. doi: 10.1139/bcb-2015-0017. [DOI] [PubMed] [Google Scholar]
- 38.Lee JY, Lee SH, Heo SH, Kim KS, Kim C, Kim DK, Ko JJ, Park KS. Novel function of lysine methyltransferase G9a in the regulation of sox2 protein stability. PLoS One. 2015;10:e0141118. doi: 10.1371/journal.pone.0141118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang T, Zhang P, Li W, Zhao T, Zhang Z, Chen S, Yang Y, Feng Y, Li F, Shirley Liu X, Zhang L, Jiang G, Zhang F. G9A promotes tumor cell growth and invasion by silencing CASP1 in nonsmall-cell lung cancer cells. Cell Death Dis. 2017;8:e2726. doi: 10.1038/cddis.2017.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dong C, Wu Y, Yao J, Wang Y, Yu Y, Rychahou PG, Evers BM, Zhou BP. G9a interacts with Snail and is critical for Snail-mediated E-cadherin repression in human breast cancer. J Clin Invest. 2012;122:1469–1486. doi: 10.1172/JCI57349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li F, Zeng J, Gao Y, Guan Z, Ma Z, Shi Q, Du C, Jia J, Xu S, Wang X, Chang L, He D, Guo P. G9a inhibition induces autophagic Cell Death via AMPK/mTOR pathway in bladder transitional cell carcinoma. PLoS One. 2015;10:e0138390. doi: 10.1371/journal.pone.0138390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ren A, Qiu Y, Cui H, Fu G. Inhibition of H3K9 methyltransferase G9a induces autophagy and apoptosis in oral squamous cell carcinoma. Biochem Biophys Res Commun. 2015;459:10–17. doi: 10.1016/j.bbrc.2015.01.068. [DOI] [PubMed] [Google Scholar]
- 43.Alves-Silva JC, de Carvalho JL, Rabello DA, Serejo TRT, Rego EM, Neves FAR, Lucena-Araujo AR, Pittella-Silva F, Saldanha-Araujo F. GLP overexpression is associated with poor prognosis in Chronic Lymphocytic Leukemia and its inhibition induces leukemic cell death. Invest New Drugs. 2018;36:955–960. doi: 10.1007/s10637-018-0613-x. [DOI] [PubMed] [Google Scholar]
- 44.Qin J, Li Q, Zeng Z, Wu P, Jiang Y, Luo T, Ji X, Zhang Q, Hao Y, Chen L. Increased expression of G9A contributes to carcinogenesis and indicates poor prognosis in hepatocellular carcinoma. Oncol Lett. 2018;15:9757–9765. doi: 10.3892/ol.2018.8572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li KC, Hua KT, Lin YS, Su CY, Ko JY, Hsiao M, Kuo ML, Tan CT. Inhibition of G9a induces DUSP4-dependent autophagic cell death in head and neck squamous cell carcinoma. Mol Cancer. 2014;13:172. doi: 10.1186/1476-4598-13-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang J, He P, Xi Y, Geng M, Chen Y, Ding J. Down-regulation of G9a triggers DNA damage response and inhibits colorectal cancer cells proliferation. Oncotarget. 2015;6:2917–2927. doi: 10.18632/oncotarget.2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Agbunag C, Bar-Sagi D. Oncogenic K-ras drives cell cycle progression and phenotypic conversion of primary pancreatic duct epithelial cells. Cancer Res. 2004;64:5659–5663. doi: 10.1158/0008-5472.CAN-04-0807. [DOI] [PubMed] [Google Scholar]
- 48.Venkannagari S, Fiskus W, Peth K, Atadja P, Hidalgo M, Maitra A, Bhalla KN. Superior efficacy of co-treatment with dual PI3K/mTOR inhibitor NVP-BEZ235 and pan-histone deacetylase inhibitor against human pancreatic cancer. Oncotarget. 2012;3:1416–1427. doi: 10.18632/oncotarget.724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maira SM, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C, Brachmann S, Chene P, De Pover A, Schoemaker K, Fabbro D, Gabriel D, Simonen M, Murphy L, Finan P, Sellers W, Garcia-Echeverria C. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther. 2008;7:1851–1863. doi: 10.1158/1535-7163.MCT-08-0017. [DOI] [PubMed] [Google Scholar]
- 50.Serra V, Markman B, Scaltriti M, Eichhorn PJ, Valero V, Guzman M, Botero ML, Llonch E, Atzori F, Di Cosimo S, Maira M, Garcia-Echeverria C, Parra JL, Arribas J, Baselga J. NVPBEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res. 2008;68:8022–8030. doi: 10.1158/0008-5472.CAN-08-1385. [DOI] [PubMed] [Google Scholar]
- 51.Markman B, Dienstmann R, Tabernero J. Targeting the PI3K/Akt/mTOR pathway--beyond rapalogs. Oncotarget. 2010;1:530–543. doi: 10.18632/oncotarget.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, Maira M, McNamara K, Perera SA, Song Y, Chirieac LR, Kaur R, Lightbown A, Simendinger J, Li T, Padera RF, Garcia-Echeverria C, Weissleder R, Mahmood U, Cantley LC, Wong KK. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14:1351–1356. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cao P, Maira SM, Garcia-Echeverria C, Hedley DW. Activity of a novel, dual PI3-kinase/mTor inhibitor NVP-BEZ235 against primary human pancreatic cancers grown as orthotopic xenografts. Br J Cancer. 2009;100:1267–1276. doi: 10.1038/sj.bjc.6604995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Awasthi N, Yen PL, Schwarz MA, Schwarz RE. The efficacy of a novel, dual PI3K/mTOR inhibitor NVP-BEZ235 to enhance chemotherapy and antiangiogenic response in pancreatic cancer. J Cell Biochem. 2012;113:784–791. doi: 10.1002/jcb.23405. [DOI] [PubMed] [Google Scholar]
- 55.Maute L, Wicht J, Zoernig M, Niederhagen M, Bergmann L. Effect of the combination of the dual mTOR/pI3K inhibitor NVP-BEZ235 with gemcitabine on growth inhibition in pancreatic cancer cells in vitro and in vivo. Journal of Clinical Oncology. 2013;31:e15070–e15070. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.