

Abstract
Aberrant glycosylation affects the malignant progression of cancers. Here, we report that N-acetyl-galactosaminyltransferase 2 (GALNT2), an enzyme that initiates the mucin type-O glycosylation, suppresses malignant phenotypes in gastric adenocarcinoma (GCA) cells by modifying epidermal growth factor receptor (EGFR) activity. GALNT2 was knocked down using siRNA in AGS and MKN28 cells. The expression of phosphorylated EGFR (pEGFR), phosphorylated Akt (pAkt) and Tn antigen were detected by western blotting. Proliferation, migration and invasion of cells with/without GLANT2-knockdown were assessed. Expression of pEGFR in the resected gastric cancer tissue was analyzed by Immunohistochemical staining, and was correlated with clinicopathological factors. The results showed that GALNT2 knockdown enhanced phosphorylation of EGFR and decreased expression of the Tn antigen on EGFR. Inhibiting EGFR activity with Gefitinib decreased the migration/invasion abilities and reversed the increase pAkt caused by GALNT2 knockdown in GCA cells. The addition of MK2206 (Akt inhibitor) mitigated the migration and invasion abilities of the GALNT2-knockdown cells. Patients with increased expressions of pEGFR in their cancer tissues were associated more metastasis, advanced stage and recurrence after surgical resection. Our results indicate that GALNT2 suppresses the malignant potential of GCA cells through the EGFR-Akt signaling pathway. The significance of O-glycosylation in receptor tyrosine kinases activities and GCA progression deserve further studies.
Keywords: GALNT2, galnac-transferase, o-glycosylation, epidermal growth factor receptor, receptor tyrosine kinase, gastric cancer
Introduction
Gastric cancer (GC) is the fourth most common cancer and the third leading cause of cancer-related deaths worldwide, with an estimated 951,600 new gastric cancer cases and 723,100 deaths occurred in 2012 [1]. The prognosis in gastric cancer remains poor, owing in part to frequent lymphatic metastasis when diagnosed [2]. The 5-year recurrence-free survival for lymph node-positive GC is only 53% [3,4]. Researches exploring the mechanisms underlying gastric cancer progression are pertinent.
Glycosylation is the most common post-translational modification of proteins, in which a carbohydrate is attached to a protein, lipid, or other organic compound [5]. There are two major types of glycosylation: N-linked and O-linked glycosylation. The most common type of O-glycosylation is mucin-type O-glycosylation, which is initiated by the transfer of N-acetylgala-ctosamine from the sugar donor to the hydroxyl residue on serine or threonine. The first glycosylation process is enzymatically catalyzed by a large family of polypeptide N-acetylgalactosami-nyltransferases (GALNTs). There are at least 20 known GALNT members in humans, namely GALNT1 to 20 [6]. Glycans have been found to participate in numerous fundamental biological processes, including inflammation, immune surveillance, cell-cell adhesion, cell-matrix interaction, inter- and intracellular signaling, and cellular metabolism [7]. Aberrant glycosylation occurring in tumorigenesis and malignant progression is a new focus of researches in cancer biology. The glycosylation abnormalities occur often owing to under-/overexpression of glycosyltransferases or mis-localization of glycosyltransferases [8].
Abnormal expression of GALNTs has been associated with cancer progression. For instance, GALNT6 was upregulated in breast cancer and might contribute to mammary carcinogenesis through aberrant glycosylation and stabilization of MUC1 [9]. GALNT14 was overexpressed in colorectal carcinoma and pancreatic cancer, and was associated with altered sensitivity to TRAIL-induced apoptosis through modulation of the O-glycosylation of death receptors on these tumor cells [10]. Our previous study showed that low GALNT2 expression in gastric cancers correlated with increased tumor depth, lymph node metastasis, advanced TNM stage and shorter disease-free survival of patients [11]. Besides, the downregulation of GALNT2 enhances the progression of gastric cancers through increasing MET (Hepatocyte growth factor receptor) phosphorylation [11]. Epidermal growth factor receptor (EGFR) as well as MET are members of the receptor tyrosine kinases (RTKs), which are promising targets for cancer treatment. Deng et al. showed that RTK/RAS genomic amplifications, including FGFR2, EGFR, Her2, and MET, occurred in approximately 37% of gastric cancer patients [12]. Since the RTK array in our previous study also showed that GALNT2 knockdown increased phosphorylation of EGFR in AGS cells [11], we would like to explore whether dysregulation of GALNT2 contributes to the malignant progression of gastric cancer by modifying another RTK member-EGFR.
The epidermal growth factor receptor is a 170 kDa transmembrane receptor tyrosine kinase in the ErbB family. The EGFR is activated by its specific ligand binding that promotes receptor dimer formation. EGFR dimerization leads to the phosphorylation and activation of the kinase domain, which stimulates diverse downstream signaling pathways [13,14]. These downstream signal transductions involve principally the MAPK and PI3K-AKT pathways that may modulate cell migration, invasion and proliferation [14]. Therefore, dysregulated EGFR activity is one of the driving forces for malignant progression.
Dysregulation of EGFR has been observed in a variety of cancers, including breast cancer [15,16], colorectal cancer [17] and lung cancer [18], etc. Studies also showed that increased EGFR expression was detected in 14-44% in gastric cancers [19-22]. Besides, EGFR expression was correlated with poor clinical outcome in gastric cancer [19]. However, the correlation between EGFR expression and clinicopathological characteristics was controversial. Increased EGFR expression has been shown correlated with advanced TNM stage, lymph node metastasis, vascular invasion and shorter progression-free-survival (PFS) in GC patients [19-23]. On the other hand, Fuse et al. found that there was no correlation between EGFR expression and overall survival rate [24]. Aside from studying the correlation of EGFR expression and cancer progression, some studies investigated the significance of phosphorylated-EGFR (pEGFR) expression in cancers. As a receptor tyrosine kinase, EGFR becomes phosphorylated when it is activated. The prognostic significance of pEGFR has been reported in cancers including breast cancer [25] and non-small cell lung cancer [18,26]. The reports about the prognostic impact of pEGFR in gastric cancer remain limited.
This study aims to investigate whether GALNT2 could modify the malignant behaviors of gastric cancer cells by affecting the glycosylation and activation of EGFR. In addition, the clinicopathological correlation of pEGFR expression in gastric cancer progression was investigated.
Materials and methods
Cell line
Human gastric cancer cell lines AGS and MKN28 were maintained in RPMI medium with 10% fetal bovine serum, 2% sodium bicarbonate, 2 mM L-Glutamine, and 1% penicillin, 1% streptomycin, and 1% amphotericin at 37°C with 5% CO2 in a 95% humidified atmosphere.
Western blot analysis
Total cell lysates from cultured AGS cells or MKN28 were used. Protein concentration was determined using Bio-Rad protein assay (Bio-Rad). Equal amounts (30 μg) of extracted protein were resolved on SDS-PAGE by electrophoresis, transferred and blocked with 5% BSA in TBST (50 mM Tris, 150 mM NaCl, 0.05% Tween 20, pH 7.5). The polyvinylidine difluoride membrane was incubated with primary antibody overnight at 4°C, followed by horseradish peroxidase conjugated secondary antibody (mouse IgG, 1:1000, GeneTex; Rabbit IgG, 1:1000, Genetex) for 1 hour. The primary antibodies used were GALNT2 (1:1000, Sigma), EGFR (1:1000, Cell signaling technology), phospho-EGFR (P-Tyr1068) (1:1000, Cell signaling technology), Akt (1:1000, abcam), phospho-Akt (1:1000, abcam), ERK (1:1000, Cell signaling technology), phospho-ERK (1:1000, abcam) and GAPDH (1:1000, Novus Biologicals). Immunoreactive proteins were developed with Luminata Crescendo Western HRP Substrate (Millipore). Protein signals were quantified by GelPro software and normalized to GAPDH expression.
Immunohistochemistry
For immunohistochemical staining, the 5-μm sections of the Paraffin-embedded tissue blocks were probed with GALNT2 polyclonal antibody (1:200, Sigma) and phospho-EGF Receptor (P-Tyr1068) (mouse mAb, 1:250, Cell signaling technology) diluted with 5% BSA/TBS overnight at 4°C. Signals were detected employing UltraVision Quanto Detection System HRP (Thermo) and visualized by DAB quanto (Thermo). All sections were counterstained with hematoxylin. The GALNT2 or pEGFR staining was quantified by one pathologist using a semi-quantitative immunoreactivity scoring (IRS) system. The intensities of immunostaining (I) were graded as 0 (no staining), 1 (weak staining), 2 (moderate staining), and 3 (strong staining). The percentage of immuno-reactive cells (P) was graded as 0 (none), 1 (< 10%), 2 (11-50%), 3 (51-80%), and 4 (> 80%). Multiplication of I and P resulted in an IRS ranging from 0 to 12 for each tumor. Scoring was performed for four random distinct fields per slide, and then 4 scores were averaged. We used a grouping algorithm (raw scores, negative [IRS 0-4] vs positive [IRS 5-12]) to test the correlation between pEGFR expression and clinicopathologic features in gastric carcinoma patients.
Real-time reverse transcription PCR (RT-PCR)
Total RNA was isolated from gastric cancer tissues using Trizol reagent (Invitrogen, Life Technologies). Reverse transcription was performed using 2 μg of total RNA and the High Capacity cDNA Reverse Transcription Kits (Applied bio-system) according to the manufacturer’s protocol. The cDNA was subjected to real-time PCR using quantitative PCR System Mx-3000P (Stratagene). Primers for GALNT2 were 5-AAGGAGAAGTCGGTGAAGCA-3 and 5-TTGAGCGTGAACTTCCACTG-3. Primers for GAPDH were 5-ACAGTCAGCCGCATCTTCTT-3 and 5-GACAAGCTTCCCGTTCTCAG-3. Relative quantity of mRNA expression normalized to GAPDH was analyzed with MxPro Software (Stratagene).
SiRNA knockdown of GALNT2 expression
In transient knockdown experiments, a siRNA oligonucleotides against GALNT2 (5-siRNA-1: CAGCAGGGAACUAACUGCCUCGACA-3 and siRNA-2: 5-UGUCGAGGCAGUUAGUUCCCUGCUG) and a non-targeting siRNA control were synthesized by Invitrogen. The AGS or MKN28 cells (1.2×106 cells) were transfected with siRNA-2 (5-UGUCGAGGCAGUUAGUUCCCUGCUG) using Lipofectamine RNAiMAX Reagent (Invitrogen) with the final concentration of 10 nM for 24 hours. The procedure is briefly described as below. Serum-free RPMI-1640 (500 µl) containing siRNA was mixed with 500 µl serum-free RPMI-1640 containing 10 µl Lipofectamine RNAiMAX Reagent. The resultant mixture was kept at room temperature for 20 min, and then was added into the culture plates. The plates were incubated at 37°C for 24 hours.
MTT assay
The cell viability was assessed by measuring the ability of cells to reduce 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) to the dark blue formazan product. According to the manufacturer’s instructions (Cayman Chemical, Ann Arbor, MI), AGS or MKN28 cells were seeded at a density of 2×103 cells per well and incubated with MTT for 3 hours at 37°C. Absorbance was read at 570 nm. Results are expressed as the percentage of absorbance compared with that of the control cells. For assessing the effect of gefitinib (ApexBio Technology), cells were incubated with 10% FBS containing DMSO (0.1%, Sigma) or gefitinib (1 μM). The effect of MK2206 (AdooQ BioScience) was assessed by adding DMSO (0.1%, Sigma) or MK2206 (1 μM) to the wells.
Transwell migration assay
Cell migration was assessed using BD FalconTM Cell Culture Inserts (8.0-μm pore size; BD Bioscience, Bedford, MA) in 24-well culture plates. The lower chamber was filled with RPMI containing 10% FBS (PAA laboratory). The siGLANT2-transfected cells (3×104) were resuspended in serum-free RPMI containing EGF (50 ng/ml, Sigma) and then added to the upper chamber. After 24 hours, the cells that migrated to the lower surface of the filter were stained with 0.5% (wt/vol) crystal violet (Sigma) and counted under a phase contrast microscope. Four random fields were examined and analyzed at 100× magnification. In selected groups, the transfected cells were treated with gefitinib (1 μM) or MK2206 (1 μM) in the upper chamber.
Matrigel invasion assay
Cell invasion were assessed by using BD FalconTM Cell Culture Inserts (8.0-μm pore size; BD Bioscience, Bedford, MA) in 24-well culture plates. The upper surface of the insert was coated with MatrigelTM Basement Membrane Matrix (BD Biosciences, San Jose, CA, USA) diluted with serum-free RPMI (1:4) at 37°C overnight the day before experiment. The cells and agents tested were the same as in the migration assay.
Lectin pull down assay
Vicia Villosa Lectin (VVA) agarose beads (Vector Laboratories) were used to detect the Tn antigen on glycoproteins, as reported. The cell lysates (0.5 mg) were incubated with 30 μl VVA-conjugated agarose beads at 4°C for 16 hours. The lectin/glycoprotein complexes were collected by centrifugation (10,000 rpm, 1 min). Glycoproteins were released from the complexes after boiled in 5 μl of 5× sample buffer for 5 minutes. The precipitated proteins were subjected to Western blotting to detect the amount of EGFR. The EGFR in total lysates was served as the internal control.
Statistical analyses
Statistical analyses were performed using Prism6. In vitro tumor cell viability migration and invasion data were analyzed by one way analysis of variance (ANOVA). The disease-free survival data by Kaplan-Meier log rank tests. Student t test was used for other experiments. Data are presented as means ± SD. P < 0.05 or less was considered to be statistically significant, and all experiments were performed in triplicate to verify reproducibility.
Results
Knockdown of GALNT2 increased epidermal growth factor receptor (EGFR) phosphorylation and decreased EGFR O-glycosylation
To investigate the effect of GALNT2-knockdown on EGFR phosphorylation, AGS cells and MKN28 cells were transfected with siGALNT2 or non-targeting siRNA control (SiC) for 24 hours. The transfected cells were starved for 6 hours and then stimulated by EGF for 10 minutes. Efficiency of GALNT2 knockdown was confirmed by western blot analysis (Figure S1). When compared with that in the siC group, the increased expressions of pEGFR in the siGALNT2 cells were significant, either without EGF treatment (P = 0.014 in AGS cells, P < 0.01 in MKN28 cells) or with EGF treatment (P = 0.018 in AGS cell, P = 0.013 in MKN28 cells) (Figure 1A and 1B).
Figure 1.
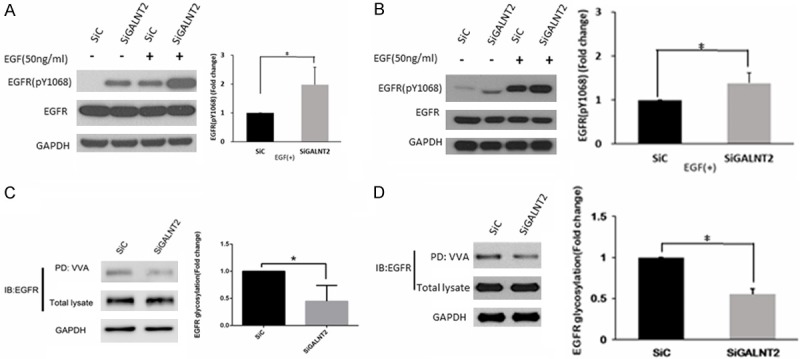
GALNT2 modifies the activity and O-glycosylation of EGFR. GALNT2 modulated EGF-induced phosphorylation of EGFR. Control and GALNT2-knockdown AGS (A) or MKN28 (B) cells were treated with/without EGF (50 ng/ml), and lysates were analyzed by Western blotting. The expression of pEGFR was quantified and normalized to GAPDH. Knockdown of GALNT2 decreased VVA binding to EGFR in AGS (C) or MKN28 (D) cells. The lysates were incubated with VVA-conjugated agarose beads. Proteins pulled down by VVA were analyzed by immunoblotting with anti-EGFR antibody. The expression of VVA-bound EGFR was quantified and normalized to total EGFR. The results are represented as mean ± S.D. from three independent experiments. *P < 0.05.
To verify whether GALNT2 could modify the O-glycosylation of EGFR, a VVA lectin pull-down assay was performed to detect the expression of Tn antigen (GalNAc-O-Ser/Thr) on EGFR in siC and siGALNT2 group. The expression of VVA-bound EGFR was quantified and normalized to total EGFR. As shown in the Figure 1C, 1D, knockdown of GALNT2 reduced VVA binding to EGFR (P < 0.01 in AGS cells, P = 0.048 in MKN28 cells), which indicated that knockdown of GALNT2 modified the O-glycosylation of EGFR.
Knockdown of GALNT2 enhanced the malignant phenotypes of gastric cancer through increasing EGFR phosphorylation in vitro
To investigate whether GALNT2-knockdown enhances the malignant phenotypes of GC through the activation of EGFR, siC and siGALNT2-transfected cells were treated with Gefitinib (EGFR inhibitor, 1 μΜ) or DMSO (0.1%). As shown in Figure 2A, there were no differences in cell viability between siC and siGALNT2 group, either treated with DMSO or Gefitinib. The number of migrated cells of siGALNT2-transfected group was 2.8 fold higher than that of siC group, the addition of Gefitinib markedly suppressed the migration of siGALNT2-transfected cells (Figure 2B). Similarly, the enhanced invasion observed in siGALNT2-transfected cells was suppressed when the cells were treated with Gefitinib (Figure 2C). Similar findings were noted in MKN28 cells (Figure 2D-F). Besides, when treated with Geftinib, the migrated AGS cells were 0.76±0.25 for siC and 0.91±0.29 for si GALNT2 (P = 0.805). The invaded AGS cells under Geftinib treatment was 0.58±0.13 for siC and 1.25±0.55 for siGALNT2, P = 0.225). When treated with Geftinib, the migrated MKN cells were 0.47±0.09 in siC group and 0.79±0.12 in si GALNT2 group (P = 0.630). The invaded MKN cells under Geftinib treatment was 0.71±0.02 for siC and 0.80±0.03 for siGALNT2 (P = 0.716). It seemed that EGFR inhibition fully rescued siGALNT2-induced migration and invasion in AGS/MKN cells.
Figure 2.

Effects of EGFR inhibitor (Gefitinib) on malignant phenotypes in GALNT2-knockdown AGS cells. Cell viability was analyzed by MTT assay in AGS cells or MKN28 cells. The results were graphed after standardization by siC group treated with DMSO (Day 1) to 1.0. There were no significant differences of cell viability between siGALNT2 and siC group treated with either DMSO or gefitinib. GALNT2-knockdown increased the number of migrated (B, E)/invaded (C, F) cells compared with that of siC group, but the addition of Gefitinib (1 μΜ) mitigated these changes. The number of migrated/invaded cells was calculated at 4 random fields per experiment (×100) and expressed as fold change of siC without gefitinib treatment. Results are represented as mean ± S.D. from three independent experiments. *P < 0.05. (A-C, AGS cells; D-F, MKN28 cells).
Knockdown of GALNT2 enhanced the malignant phenotypes of gastric cancer through increasing EGFR-Akt pathway in vitro
The activation of EGFR induces the subsequent transduction of EGFR-ERK and EGFR-Akt signaling, which are important for cellular proliferation, differentiation and migration. The effect of EGFR phosphorylation on the Akt and ERK signaling in gastric cancer cells was shown in Figure 3. Knockdown of GALNT2 was associated with the increased expression of pAkt. The addition of Gefitinib (EGFR inhibitor) significantly mitigated the increase of pAkt in GALNT2-knockdown cells. The result suggested the signaling link between GALNT2, EGFR and Akt. On the other hands, the expressions of total or phosphorylated ERK were not changed significantly between SiC and GALNT2-knockdown cells. Though Gefitinib treatment decreased the expressions of pERK in siC and GALNT2-knockdown cells, but the differences were not significant between groups.
Figure 3.
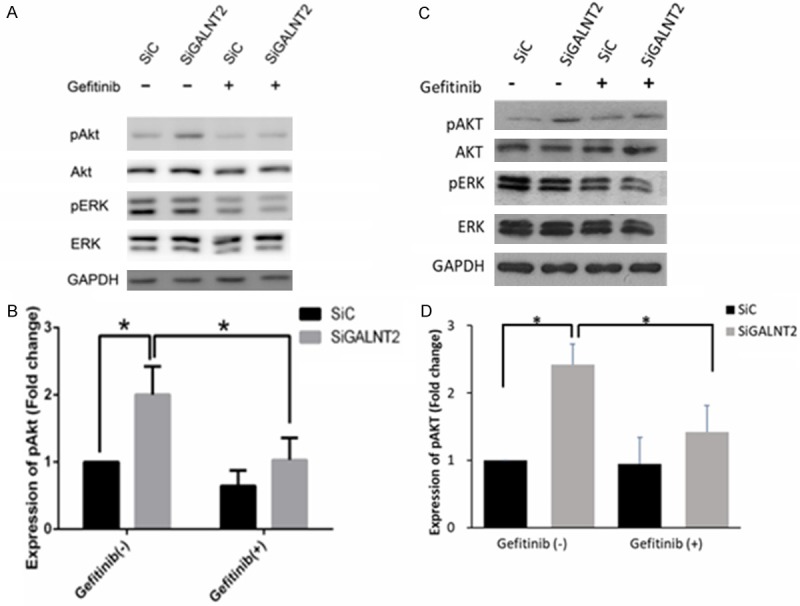
Effects of EGFR inhibitor on Akt and ERK1/2 phosphorylation. Enhanced phosphorylation of Akt mediated by GALNT2 knockdown was reversed by EGFR inhibitor. Control and GALNT2-knockdown AGS cells (A, B) or MKN28 cells (C, D) were treated with/without Gefitinib (1 μM) followed by stimulation of EGF (50 ng/ml) for 10 minutes, and lysates were analyzed by Western blotting. Expression of pAkt significantly increased in the siGALNT2 group compared to the siC group, and this effect was reduced by the addition of Gefitinib. There were no significant differences in the expressions of total ERK, total Akt and pERK between siC and siGALNT2 treated with/without Gefitinib. Expression levels of pAkt and pERK were quantified and normalized to GAPDH. Results are represented as mean ± S.D. from three independent experiments. *P < 0.05.
To explore the effect of EGFR-Akt signaling on malignant phenotypes in gastric cancers, the SiC and siGALNT2 groups were treated with either MK2206 (Akt inhibitor, 1 μΜ) or DMSO (control). As shown in Figure 4, the addition of MK2206 did not significantly alter the viabilities of siGALNT2 cells when compared with that of the DMSO treatment (A, D). In contrast to DMSO treatment, the addition of MK2206 significantly mitigated the migration and invasion abilities of the siGALNT2 cells (B and C for AGS cells, E and F for MKN28 cells). Besides, the migrated AGS cells treated with MK2206 were 0.06±0.08 for siC and 0.98±0.57 for siGALNT2 (P = 0.548). The invaded AGS cells treated with MK2206 was 0.58±0.13 for siC and 1.30±0.55 for siGALNT2, P = 0.901). The migrated MKN cells treated with MK2206 were 0.57±0.04 for siC and 0.45±0.03 for si GALNT2 (P = 0.073). The invaded MKN cells treated with MK2206 was 0.51±0.04 for siC and 0.55±0.06 for siGALNT2 (P = 0.067). It seemed that MK2206 fully rescued siGALNT2-induced migration and invasion in AGS/MKN cells.
Figure 4.
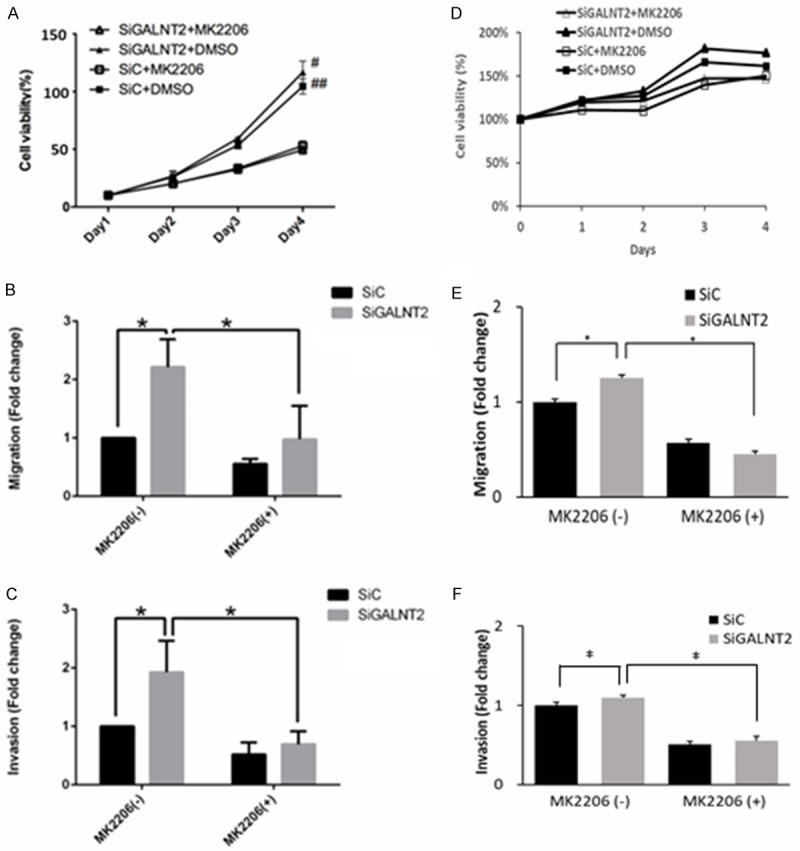
Effects of Akt inhibitor on malignant phenotypes in GALNT2-knockdown cells. Cell viability was analyzed by MTT assay (A-C, AGS cells; D-F, MKN28 cells). The results were graphed after standardization by siC group treated with DMSO (Day 1) to 1.0. There were no differences in cell viabilities between siGALNT2 and siC group in MKN28 cells. However, cell viability was significantly lower in groups treated with MK2206 than groups treated with DMSO in AGS cells at day 4 of the culture. GALNT2-knockdown increased the number of migrated/invaded cells compared with that of siC group, but the addition of MK2206 (1 μΜ) mitigated these changes. Results are represented as mean ± S.D. from three independent experiments. *P < 0.05; #P < 0.05, siG2+DMSO compared to siG2+MK2206; ##P < 0.05, siC+DMSO compared to siC+MK2206.
Clinicopathological correlation of pEGFR expression in gastric cancers
Our In-vitro experiments demonstrated that GALNT2 knockdown enhanced malignancy of gastric cancer via the modulation of EGFR activities. To examine the correlation of pEGFR expression with clinicopathological characteristics, the expression of pEGFR in gastric cancers was evaluated by immunohistochemical staining. The representative staining was shown in Figure 5A. Of all 78 patients, 38 (49%) had positive pEGFR staining (the pEGFR (+) group), while 40 (51%) had negative pEGFR staining (the pEGFR (-) group) in their cancer tissues. Patients with pEGFR (+) were associated with metastasis, advanced stage and recurrence after surgical resection (P = 0.011, P = 0.037, P = 0.041, respectively). The expression of pEGFR was not significantly correlated with age, cell differentiation, size, lympho-vascular invasion (Table 1). For the 64 patients with follow-up time longer than 24 months, the 5-year progression-free survival (PFS) in pEGFR (+) group was worse than that of pEGFR (-) group. (38.7% vs 76.7%, P = 0.013) (Figure 5B).
Figure 5.
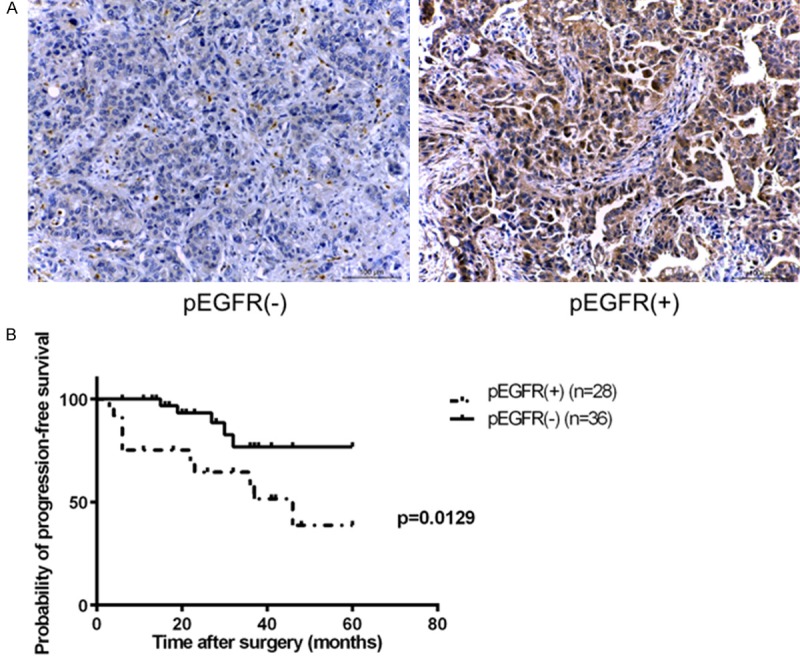
Clinicopathological correlation of pEGFR expression in gastric cancers. A. Representative immunohistochemical staining of phosphorylated EGFR (pEGFR). B. Of the 64 patients, 28 (44%) were pEGFR positive and 36 (56%) were pEGFR negative. The 5-year PFS rate for pEGFR (+) and pEGFR (-) group were 38.7% and 76.7% respectively. (P = 0. 013).
Table 1.
Clinicopathological correlation of pEGFR expression in gastric cancers
| pEGFR | |||
|---|---|---|---|
|
|
|||
| Positive (n = 38) | Negative (n = 40) | p value | |
| Age, mean (range) | 70.5 (38-88) | 71.2 (43-89) | 0.7836 |
| Differentiation | 0.8908 | ||
| Well-differentiation | 10 (26%) | 10 (25%) | |
| Mod-differentiation | 22 (58%) | 25 (62%) | |
| Poor-differentiation | 6 (16%) | 5 (13%) | |
| Size | 0.3798 | ||
| < 5 cm | 21 (55%) | 26 (65%) | |
| ≥ 5 cm | 17 (45%) | 14 (35%) | |
| Lymphovascular invasion | 0.3409 | ||
| Absent | 14 (37%) | 19 (48%) | |
| Present | 24 (63%) | 21 (52%) | |
| Neural invasion | 0.1699 | ||
| Absent | 15 (39%) | 22 (55%) | |
| Present | 23 (61%) | 18 (45%) | |
| Primary tumor | 0.0916 | ||
| T1 | 9 (24%) | 10 (25%) | |
| T2 | 5 (13%) | 14 (35%) | |
| T3 | 12 (32%) | 10 (25%) | |
| T4 | 12 (31%) | 6 (15%) | |
| Regional lymph node | 0.4767 | ||
| N0 | 13 (34%) | 17 (43%) | |
| N1 | 4 (11%) | 7 (18%) | |
| N2 | 8 (21%) | 8 (20%) | |
| N3 | 13 (34%) | 8 (20%) | |
| Distant metastasis | 0.0104 | ||
| M0 | 30 (79%) | 39 (98%) | |
| M1 | 8 (21%) | 1 (2%) | |
| Stage | 0.0373 | ||
| I | 11 (29%) | 20 (50%) | |
| II | 8 (21%) | 6 (15%) | |
| III | 11 (29%) | 13 (33%) | |
| IV | 8 (21%) | 1 (2%) | |
| Recurrence | 0.0409 | ||
| No | 18 (64%) | 31 (86%) | |
| Yes | 10 (36%) | 5 (14%) | |
Paired IHC staining of GALNT2 and pEGFR was performed in gastric cancer tissues, and IHC scores for GALNT2 and pEGFR staining were correlated using a linear regression model (Figure 6). The Pearson’s correlation coefficient between the GALNT2 and pEGFR IHC scores was -0.46 (P < 0.005).
Figure 6.
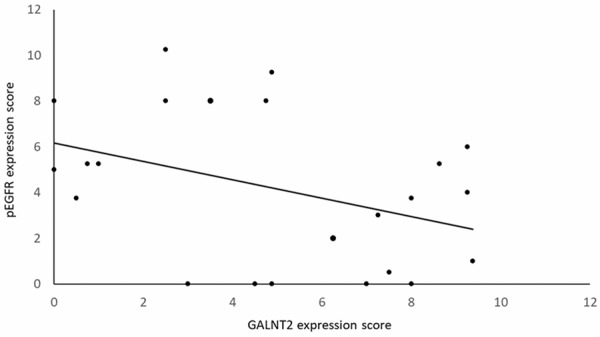
Correlations between GALNT2 and pEFGR expression in gastric cancer tissues. IHC staining of GALNT2 and pEGFR was done in paired gastric cancer tissues. The IHC scores for GALNT2 and pEGFR staining were correlated using a linear regression model. The Pearson’s correlation coefficient between the GALNT2 and pEGFR IHC scores was -0.46 (P < 0.005).
Discussion
In this study, we showed that knockdown of GALNT2 in gastric cancer cells increased the activation of EGFR. GALNT2 downregulation promoted migration and invasion of gastric cancer cells by increasing EGFR phosphorylation and downstream Akt activation. Clinically, the patients with increased pEGFR expression in their cancer tissues were associated with more advanced disease and shorter progression-free survival after surgical treatment.
GalNAc-transferases (GALNTs) are a family of crucial O-glycosyltransferases that initiate the formation of mucin-type O-glycan. Abnormal expression of glycosyltransferases alters the expression of glycans, which plays a critical role in cancer progression. In our previous studies, GALNT2 was shown to be the most highly expressed GALNTs in the stomach tissue, and downregulation of GALNT2 in gastric cancers was correlated with malignant progression through the activation of hepatocyte growth factor receptor (MET), one of the important receptor tyrosine kinases (RTKs) [11]. In this study, we found that downregulation of GALNT2 could also contribute to cancer progression by activating EGFR, another important RTK. Therefore, the present and our previous study showed that GALNT2 downregulation could affect gastric cancer phenotypes through the activation of at least two RTKs, cMET and EGFR. Our works implicate that targeting GALNT2 is an alternative strategy to reduce the activation of multiple RTKs in gastric cancers, which works by reducing redundant and compensatory RTK signals.
RTKs are important biological receptors with ligand-binding activity and protein kinase activity. Among them, EGFR was the first reported [27] and most well studied RTK, for its roles in signal transduction and cancer progression. The modulation of glycosylation of RTKs can lead to conformational changes and significantly alter their biological activities [28]. When the glycosylation of EGFR was modulated, their binding affinities to their ligand (EGF) would be different [29]. Besides, blocking the N-glycosylation of EGFR mitigated the cell surface transport of EGFR, phosphorylation of EGFR, and increased the senescence of RTK-driven tumor growth [30]. The EGFR can be activated in cancer cells by either ligand-depend or ligand-independent pathway [31]. One study showed that the modification of O-glycosylation of MET in hepatocellular carcinoma cells could enhance its ligand-induced dimerization and activation of MET [32]. In our experiment, the knockdown of GALNT2 enhanced EGFR phosphorylation irrespective of EGF treatment. This implied that the EGFR activation through GALNT2 knockdown might be ligand-independent.
In the present study, the GALNT2-EGFR activation pathway seems affecting the migratin/invasion abilities more than the cell proliferation of gastric cancer cells. As shown, the O-glycosylation influenced the cell migration by modifying key proteins involved in endothelia-mesenchymal transition (EMT) [33]. Besides, EGF could also induce aberrant N-glycosylation and affect EMT [34]. Further studies of the GALNT2-EGFR signaling on the EMT deserve more investigations. Besides, when treated with Geftinib, the enhanced migration or invasion of gastric cancer cells by siGALNT2 treatment was rescued when compared to that of siC group. In our previous study, the MET inhibitor PHA665752 also fully rescued the phenotypes induced by GALNT2 knock-down. Multiple oncogenic drivers often co-exist in cancers, which necessitates multiple targeting in their treatment. Knowing GALNT2 modulated both the cMET and EGFR activities, it might be an alternative to target GALNT2 directly for these drives in cancer treatment.
In our previous study [11], GALNT2 knockdown increased the viability of the gastric cancer cell lines. In the present experiment, there were no differences in cell viability between siC and siGALNT2 group, either treated with DMSO or Gefitinib. The discrepancy on the viability was due to the different dose of siRNA used. We used 10 nM in the present study rather than 100 nM (in the previous experiment). The smaller dose knock-downed the expression of GALNT2 to a significant efficiency, though the effect may not as powerful (as that large dose) to affect the viability of cells.
In summary, our study showed that GALNT2 modifies the O-glycosylation of EGFR, and affects the migration/invasion phenotypes of gastric cancer cells through the activation of EGFR-Akt signaling. Targeting GALNT2 to redfuce broad RTK activations seems to be an alternative strategy for gastric cancer treatment.
Acknowledgements
This study was supported by Ministry of Science and Technology R.O.C. The authors acknowledge the grant support from the Excellent Translational Medicine Research Projects of National Taiwan University College of Medicine, and National Taiwan University Hospital (107C101-23). The authors acknowledge the grant support from the Ministry of Science and Technology (106-2320-B-002-034).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 2.Maruyama K, Gunvén P, Okabayashi K, Sasako M, Kinoshita T. Lymph node metastases of gastric cancer: general pattern in 1931 patients. Ann Surg. 1989;210:596–602. doi: 10.1097/00000658-198911000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sasako M, Sakuramoto S, Katai H, Kinoshita T, Furukawa H, Yamaguchi T, Nashimoto A, Fujii M, Nakajima T, Ohashi Y. Five-year outcomes of a randomized phase III trial comparing adjuvant chemotherapy with S-1 versus surgery alone in stage II or III gastric cancer. J. Clin. Oncol. 2011;29:4387–93. doi: 10.1200/JCO.2011.36.5908. [DOI] [PubMed] [Google Scholar]
- 4.Noh SH, Park SR, Yang HK, Chung HC, Chung IJ, Kim SW, Kim HH, Choi JH, Kim HK, Yu W, Lee JI, Shin DB, Ji J, Chen JS, Lim Y, Ha S, Bang YJ CLASSIC trial investigators. Adjuvant capecitabine plus oxaliplatin for gastric cancer after D2 gastrectomy (CLASSIC): 5-year follow-up of an open-label, randomized phase 3 trial. Lancet Oncol. 2014;15:1389–96. doi: 10.1016/S1470-2045(14)70473-5. [DOI] [PubMed] [Google Scholar]
- 5.Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME. Essentials of Glycobiology. 2nd edition. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 2009. [PubMed] [Google Scholar]
- 6.Bennett EP, Mandel U, Clausen H, Gerken TA, Fritz TA, Tabak LA. Control of mucin-type O-glycosylation: a classification of the polypeptide GalNAc-transferase gene family. Glycobiology. 2012;22:736–756. doi: 10.1093/glycob/cwr182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pinho SS, Reis CA. Glycosylation in cancer: mechanisms and clinical implications. Nat Rev Cancer. 2015;15:540–555. doi: 10.1038/nrc3982. [DOI] [PubMed] [Google Scholar]
- 8.Pinho SS, Carvalho S, Marcos-Pinto R, Magalhães A, Oliveira C, Gu J, Dinis-Ribeiro M, Carneiro F, Seruca R, Reis CA. Gastric cancer: adding glycosylation to the equation. Trends Mol Med. 2013;19:664–676. doi: 10.1016/j.molmed.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 9.Park JH, Nishidate T, Kijima K, Ohashi T, Takegawa K, Fujikane T, Hirata K, Nakamura Y, Katagiri T. Critical roles of mucin 1 glycosylation by transactivated polypeptide N-acetylgalactosaminyltransferase 6 in mammary carcinogenesis. Cancer Res. 2010;70:2759–2769. doi: 10.1158/0008-5472.CAN-09-3911. [DOI] [PubMed] [Google Scholar]
- 10.Wagner KW, Punnoose EA, Januario T, Lawrence DA, Pitti RM, Lancaster K, Lee D. Death-receptor O-glycosylation controls tumorcell sensitivity to the proapoptotic ligand Apo2L/TRAIL. Nat Med. 2007;13:1070–1077. doi: 10.1038/nm1627. [DOI] [PubMed] [Google Scholar]
- 11.Liu SY, Shun CT, Hung KY, Juan HF, Hsu CL, Huang MC, Lai IR. Mucin glycosylating enzyme GALNT2 suppresses malignancy in gastric adenocarcinoma by reducing MET phosphorylation. Oncotarget. 2016;7:11251–62. doi: 10.18632/oncotarget.7081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deng N, Goh LK, Wang H, Das K, Tao J, Tan IB, Zhang S, Lee M, Wu J, Lim KH, Lei Z, Goh G, Lim QY, Tan AL, Sin Poh DY, Riahi S, Bell S, Shi MM, Linnartz R, Zhu F, Yeoh KG, Toh HC, Yong WP, Cheong HC, Rha SY, Boussioutas A, Grabsch H, Rozen S, Tan P. A comprehensive survey of genomic alterations in gastric cancer reveals systematic patterns of molecular exclusivity and co-occurrence among distinct therapeutic targets. Gut. 2012;61:673–84. doi: 10.1136/gutjnl-2011-301839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klapper LN, Kirschbaum MH, Sela M, Yarden Y. Biochemical and clinical implications of the ErbB/HER signaling network of growth factor receptors. Adv Cancer Res. 2000;77:25–79. [PubMed] [Google Scholar]
- 14.Yewale C, Baradia D, Vhora I, Patil S, Misra A. Epidermal growth factor receptor targeting in cancer: a review of trends and strategies. Biomaterials. 2013;34:8690–8707. doi: 10.1016/j.biomaterials.2013.07.100. [DOI] [PubMed] [Google Scholar]
- 15.Magkou C, Nakopoulou L, Zoubouli C, Karali K, Theohari I, Bakarakos P, Giannopoulou I. Expression of the epidermal growth factor receptor (EGFR) and the phosphorylated EGFR in invasive breast carcinomas. Breast Cancer Res. 2008;10:R49–56. doi: 10.1186/bcr2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Masuda H, Zhang D, Bartholomeusz C, Doihara H, Hortobagyi GN, Ueno NT. Role of epidermal growth factor receptor in breast cancer. Breast Cancer Res Treat. 2012;136:331–345. doi: 10.1007/s10549-012-2289-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Porebska I, Harlozinska A, Bojarowski T. Expression of the tyrosine kinase activity growth factor receptors (EGFR, ERB B2, ERB B3) in colorectal adenocarcinomas and adenomas. Tumour Biol. 2000;21:105–115. doi: 10.1159/000030116. [DOI] [PubMed] [Google Scholar]
- 18.Siegelin MD, Borczuk AC. Epidermal growth factor receptor mutations in lung adenocarcinoma. Lab Invest. 2014;94:129–137. doi: 10.1038/labinvest.2013.147. [DOI] [PubMed] [Google Scholar]
- 19.Lieto E, Ferraraccio F, Orditura M, Castellano P, Mura AL, Pinto M, Zamboli A, De Vita F, Galizia G. Expression of vascular endothelial growth factor (VEGF) and epidermal growth factor receptor (EGFR) is an independent prognostic indicator of worse outcome in gastric cancer patients. Ann Surg Oncol. 2008;15:69–79. doi: 10.1245/s10434-007-9596-0. [DOI] [PubMed] [Google Scholar]
- 20.Nagatsuma AK, Aizawa M, Kuwata T, Doi T, Ohtsu A, Fujii H, Ochiai A. Expression profiles of HER2, EGFR, MET and FGFR2 in a large cohort of patients with gastric adenocarcinoma. Gastric Cancer. 2015;18:227–238. doi: 10.1007/s10120-014-0360-4. [DOI] [PubMed] [Google Scholar]
- 21.Kurokawa Y, Matsuura N, Kawabata R, Nishikawa K, Ebisui C, Yokoyama Y, Shaker MN, Hamakawa T, Takahashi T, Takiguchi S, Mori M, Doki Y. Prognostic impact of major receptor tyrosine kinase expression in gastric cancer. Ann Surg Oncol. 2014;21(Suppl 4):S584–590. doi: 10.1245/s10434-014-3690-x. [DOI] [PubMed] [Google Scholar]
- 22.Gao M, Liang XJ, Zhang ZS, Ma W, Chang ZW, Zhang MZ. Relationship between expression of EGFR in gastric cancer tissue and clinicopathological features. Asian Pac J Trop Med. 2013;6:260–264. doi: 10.1016/S1995-7645(13)60054-1. [DOI] [PubMed] [Google Scholar]
- 23.Galizia G, Lieto E, Orditura M, Castellano P, Mura AL, Imperatore V, Pinto M, Zamboli A, De Vita F, Ferraraccio F. Epidermal growth factor receptor (EGFR) expression is associated with a worse prognosis in gastric cancer patients undergoing curative surgery. World J Surg. 2007;31:1458–1468. doi: 10.1007/s00268-007-9016-4. [DOI] [PubMed] [Google Scholar]
- 24.Fuse N, Kuboki Y, Kuwata T, Nishina T, Kadowaki S, Shinozaki E, Ohtsu A. Prognostic impact of HER2, EGFR, and c-MET status on overall survival of advanced gastric cancer patients. Gastric Cancer. 2016;19:183–191. doi: 10.1007/s10120-015-0471-6. [DOI] [PubMed] [Google Scholar]
- 25.Nieto Y, Nawaz F, Jones RB, Shpall EJ, Nawaz S. Prognostic significance of overexpression and phosphorylation of epidermal growth factor receptor (EGFR) and the presence of truncated EGFRvIII in locoregionally advanced breast cancer. J. Clin. Oncol. 2007;25:4405–4413. doi: 10.1200/JCO.2006.09.8822. [DOI] [PubMed] [Google Scholar]
- 26.Wang F, Wang S, Wang Z, Duan J, An T, Zhao J, Bai H, Wang J. Phosphorylated EGFR expression may predict outcome of EGFR-TKIs therapy for the advanced NSCLC patients with wild type EGFR. J Exp Clin Cancer Res. 2012;31:65–74. doi: 10.1186/1756-9966-31-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cohen S. The epidermal growth factor (EGF) Cancer. 1983;51:1787–91. doi: 10.1002/1097-0142(19830515)51:10<1787::aid-cncr2820511004>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 28.Liu YC, Yen HY, Chen CY, Chen CH, Cheng PF, Juan YH, Chen CH, Khoo KH, Yu CJ, Yang PC, Hsu TL, Wong CH. Sialylation and fucosylation of epidermal growth factor receptor suppress its dimerization and activation in lung cancer cells. Proc Natl Acad Sci U S A. 2011;108:11332–11337. doi: 10.1073/pnas.1107385108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stowell SR, Ju T, Cummings RD. Protein glycosylation in cancer. Annu Rev Pathol. 2015;10:473–510. doi: 10.1146/annurev-pathol-012414-040438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lopez-Sambrooks C, Shrimal S, Khodier C, Flaherty DP, Rinis N, Charest JC, Gao N, Zhao P, Wells L, Lewis TA, Lehrman MA, Gilmore R, Golden JE, Contessa JN. Oligosaccharyltransferase inhibition induces senescence in RTKdriven tumor cells. Nat Chem Biol. 2016;12:1023–1030. doi: 10.1038/nchembio.2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guo G, Gong K, Wohlfeld B, Hatanpaa KJ, Zhao D, Habib AA. Ligand-Independent EGFR Signaling. Cancer Res. 2015;75:3436–3441. doi: 10.1158/0008-5472.CAN-15-0989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu YM, Liu CH, Huang MJ, Lai HS, Lee PH, Hu RH, Huang MC. C1GALT1 enhances proliferation of hepatocellular carcinoma cells via modulating MET glycosylation and dimerization. Cancer Res. 2013;73:5580–90. doi: 10.1158/0008-5472.CAN-13-0869. [DOI] [PubMed] [Google Scholar]
- 33.Park SY, Kim HS, Kim NH, Ji S, Cha SY, Kang JG, Ota I, Shimada K, Konishi N, Nam HW, Hong SW, Yang WH, Roth J, Yook JI, Cho JW. Snail1 is stabilized by O-GlcNAc modification in hyperglycaemic condition. EMBO J. 2010;29:3787–3796. doi: 10.1038/emboj.2010.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu Q, Qu C, Wang W, Gu J, Du Y, Song L. Specific N-glycan alterations are coupled in epithelial-mesenchymal transition induced by EGF in GE11 epithelial cells. Cell Biol Int. 2016;41:124–133. doi: 10.1002/cbin.10707. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.