

Abstract
MicroRNAs (miRNAs) are small regulatory non-coding RNAs that have been reported to play an important role in a variety of cellular functions. Recent studies indicated that some miRNAs are involved in regulating endoplasmic reticulum (ER) stress adaptation. However, the miRNAs were still unknown in osteosarcoma. In this study, we demonstrated that miR-1281 induced by ER stress promoted cell apoptosis and decreased ER stress adaptation of osteosarcoma in vitro and in vivo. Further mechanistic studies revealed that p53, an important tumor suppressor, directly bound to the promoter of miR-1281, leading to its increase under ER stress. Additionally, our data suggest that USP39 was the target of miR-1281 and participated in ER stress-induced cell apoptosis. Thus, our findings suggest a new role for miR-1281 in osteosarcoma and suggest that the p53-dependent, miR-1281-mediated USP39 pathway inhibits the survival of human osteosarcoma cells under ER stress.
Keywords: miR-1281, p53, ER stress, osteosarcoma, apoptosis
Introduction
Osteosarcoma (OS) is the most common type of primary bone cancer in childhood and adolescence, and it mainly derives from primitive bone-forming mesenchymal cells [1,2]. Use of surgery along with chemotherapy has improved the overall 5-year survival rate of osteosarcoma patients to some extent [3]. However, chemoresistance is still an obstacle for osteosarcoma treatment. Accumulating evidence indicates that osteosarcoma cells activate the ER stress pathway to regulate cell death with drug treatment and that adaptation to ER stress enhances tumor chemoresistance, leading to tumor recrudescence [4-7]. Thus, understanding the molecular mechanisms of the adaptation to ER stress is essential for osteosarcoma treatment.
The ER is an essential organelle that is critical for the biological processes required for cell survival and death. Many intra- and extracellular stimuli result in an imbalance of ER functions, causing the accumulation of unfolded or misfolded proteins and protein aggregates that are detrimental to cell survival, which has been termed ER stress [8]. Eukaryotic cells have evolved an adaptive response to ER stress, commonly termed the unfolded protein response (UPR), including: 1) general protein translation attenuation, 2) upregulation of ER resident chaperones and 3) activation of a degradative pathway (ER-associated degradation-ERAD) to eliminate unfolded proteins by proteasomal degradation. Recently, many microRNAs were indicated to be involved in ER stress, such as miR-346, which inhibited ER stress-induced apoptosis via activating mitophagy [9]; miR-149* enhanced melanoma adaptation to ER stress by suppressing GSK3β expression [10]; miR-765, a HOXB9-responsive miRNA, promoted melanoma adaptation via targeting FOXA2 [11]. Although many miRNAs were reported to take part in regulating ER stress adaptation in many types of cancer, their role was still unclear in osteosarcoma.
In this study, we found that miR-1281 induced by ER stress facilitated osteosarcoma cell apoptosis in vitro and in vivo. Further mechanistic studies revealed that p53, an important tumor suppressor, directly bound to the promoter of miR-1281, leading to its increase under ER stress. Additionally, we also found that USP39 was the target of miR-1281 and participated in ER stress-induced cell apoptosis. Thus, our findings shed a new light on the role of miR-1281 in osteosarcoma and suggest that the p53-dependent, miR-1281-mediated USP39 pathway impairs the survival of human osteosarcoma cells under ER stress.
Material and method
Cell culture and reagents
U2OS cells were maintained in Dulbecco’s modified Eagle medium. MG63 cells were cultured in Eagle’s Minimum Essential Medium (EMEM). Saos-2 cells were cultured in McCoy’s 5A Medium. These medium were supplemented with 10% fetal bovine serum (FBS) (ExCell Bio, Lot: FSP500), 2 mM L-glutamine, penicillin (100 U/ml), streptomycin (100 μg/ml) and 0.1% SaveltTM (Hanbio Co. LTD 1:1000) in a humidified atmosphere of 5% CO2 maintained at 37°C. The following antibodies were used in this study: antibodies against GAPDH (Santa Cruz Biotechnology, Dallas, TX, USA; SC-25778, 1:1000), PARP (Santa Cruz Biotechnology, SC-8007, 1:1000), p53 (Santa Cruz Biotechnology, SC-126, 1:10 for ChIP, 1:1000 for WB), and USP39 (Proteintech, 23865-1-AP, 1:500).
RNA interference
RNA interference was performed as previously described [12,13]. The targeting sequences of p53 and USP39 were as follows: p53 No. 1: CGGCGCACAGAGGAAGAGAA; No. 2: GTCCAGATGAAGCTCCCAGAA. USP39: No. 1 GCCCGTACTTGGATACCATTA; No. 2: GCCTTATTTGAATTGCTGCAA.
Introduction of microRNA mimics and inhibitors
Mimics and inhibitors of miRNA-1281 were synthesized by the GenePharma Company (Shanghai, People’s Republic of China). For each transfection in a six-well plate, 100 nM miRNA mimics, scramble or inhibitor were used. The transfection of osteosarcoma cells by Oligofectamine (Invitrogen) was performed according to the manufacturer’s instructions. The lentivirus expressing miR-1281 was purchased from GenePharma Company. The cells stably expressing miR-1281 were obtained as previous described [14,15].
Cell viability assay
Osteosarcoma cells were plated in 96-well plates at a density of 800 cells in 200 μl of medium per well 24 hours before the experiment. Following treatments as indicated, the cell viability was determined using a CCK8 kit (Cell Counting Kit-8).
ChIP assay
The ChIP assay was performed as previously described [6].
MiRNA sequencing analysis
U2OS cells were treated with 1 μM TG for indicated times. Total RNA was isolated using Trizol reagent (Invitrogen, Carlsbad, CA, USA). Small RNA sequencing was performed by KangChen Bio-tech using the Illumina Small RNA Sequencing Platform. The TruSeq small RNA library preparation kit (Illumina, San Diego, CA, USA) was used for library preparation. Sequencing was performed using an Illumina HiSeq 2000 sequencing system, and 10 Mb of clean reads were analyzed using routine algorithms (KangChen Bio-tech, Inc., Shanghai, China).
Quantitative real-time polymerase chain reaction assay (q-RT-PCR)
Total RNA was isolated using TRIzol (Invitrogen). One microgram of total RNA was used to synthesize cDNA using the PrimeScriptTM RT reagent kit (Takara, RR047A) according to the manufacturer’s instructions. Pri-miR-1281 levels were normalized to those of β-actin and miR-1281 levels were normalized to U6. Changes in gene expression were determined using the 2-ΔΔCT method. The primers for mature miR-1281 and pri-miR-1281 were purchased from Takara. The primers for β-actin as followed UP: 5-CACCTTCTACAATGAGCTGCGTGTG-3, DN: 5-ATAGCACAGCCTGGATAGCAACGTAC-3.
Promoter reporters and dual-luciferase assay
After transfection into the osteosarcoma cells, luciferase activity was measured in a 1.5 ml Eppendorf tube with the Promega Dual-Luciferases Reporter Assay kit (Promega E1980) according to the manufacturer’s protocols. Relative Renilla luciferase activity was normalized to firefly luciferase activity. The assay was performed as previously described [16-18].
Statistics and data analyses
Data are expressed as the mean ± SD, and statistical evaluation was performed using one-way analysis of variance (ANOVA). Values of P < 0.05 were considered statistically significant.
Results
ER stress induces increased miR-1281 expression in osteosarcoma
To investigate ER stress-altered miRNA expression, the osteosarcoma cell line U2OS was treated with thapsigargin (TG), which is a non-competitive inhibitor of the sarco/endoplasmic reticulum Ca2+ ATPase and was used to induce ER stress. Then, the cells were processed for miRNA sequencing analysis. Among the changes in miRNAs triggered by thapsigargin, a dramatic increase in miR-10400-5p, miR-7704, miR-200a-5p, miR-1281, miR-302-5p, miR-12128, miR-9718 were observed (Figure 1A-C and Table S1). In these increased miRNAs, we found that miR-1281 was also increased in p53 overexpressed Saos2 cells (Table S2). Thus, we chose miR-1281 as the potential candidate. Subsequently, the elevation of miR-1281 was confirmed in U2OS cells after thapsigargin treatment with different doses and times (Figure 1D and 1F). GRP78 was used as the ER stress marker (Figure 1E and 1G). To further determine whether the increase of miR-1281 was dependent on ER stress, U2OS cells were treated with tunicamycin (TM), another ER stress inducer that induces ER stress by inhibition of glycosylation. Of note, miR-1281 expression was augmented with increasing times and doses of tunicamycin treatment (Figure 1H-K).
Figure 1.
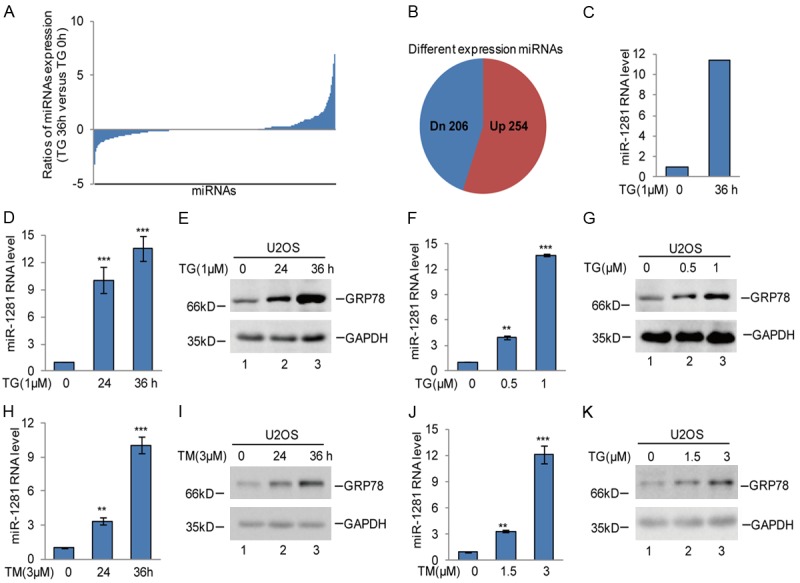
ER stress triggered miR-1281 upregulation in osteosarcoma cells. A, B. U2OS cells were treated with 1 μM TG at indicated times, and the cells were subjected to miRNA sequencing analysis. C. The miR-1281 expression level was obtained from RNA sequencing analysis. D-G. U2OS cells were treated with 1 μM TG as indicated. miR-1281 RNA levels were analyzed using q-RT-PCR. GRP78 was used as the ER stress marker. The data represent the means ± SD of three independent experiments; **P < 0.01, ***P < 0.001 vs. control. H-K. U2OS cells were treated with 3 μM TM as indicated. The expression levels of miR-1281 were measured using q-RT-PCR. The data represent the means ± SD of three independent experiments; **P < 0.01, ***P < 0.001 vs. control.
p53 transcriptionally upregulates miR-1281 expression under ER stress
Based on the increase of miR-1281 in U2OS (p53 wild type) under ER stress, we then focused on examination the expression of miR-1281 in other osteosarcoma cell lines, MG63 (p53 mutation) and Saos-2 (p53 null), under ER stress treatment. Interestingly, we found that the upregulation of miR-1281 in U2OS cells disappeared in MG63 and Saos-2 cells, implying that WT p53 may contribute to miR-1281 expression increase under ER stress (Figure 2A). To further confirm this, we first knocked down endogenous p53 using two independent shRNAs in U2OS cells. As shown in Figure 2B and 2C, inhibition of p53 significantly impaired miR-1281 elevation in response to ER stress treatment. In contrast, overexpression of p53 in Saos-2 cells enhanced miR-1281 expression under ER stress (Figure 2D, 2E).
Figure 2.
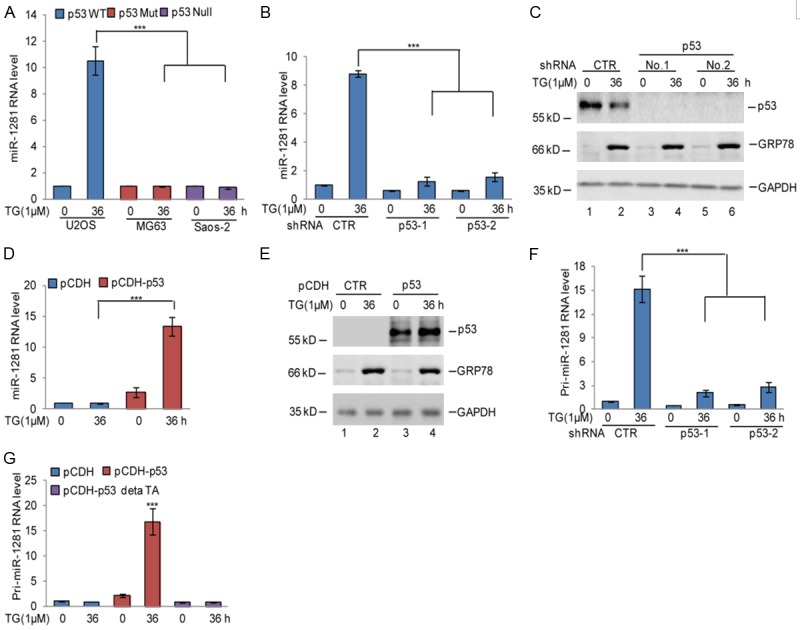
p53 upregulated miR-1281 expression under ER stress. A. U2OS, MG63 and Saos-2 cells were treated with 1 μM TG at indicated times. The expression levels of miR-1281 were measured using q-RT-PCR. The data represent the means ± SD of three independent experiments; ***P < 0.001 vs. control. B, C. p53 was knocked down in U2OS cells using pLKO.1 expressing vector. The cells were then treated with 1 μM TG at indicated times. MiR-1281 expression levels were measured using q-RT-PCR. The protein levels of p53 and GRP78 were analyzed using western blotting assay. GAPDH was used as the loading control. The data represent the means ± SD of three independent experiments; ***P < 0.001 vs. control. D, E. p53 was overexpressed in Saos-2 cells using the pCDH expressing vector. The cells were then treated with 1 μM TG at indicated times. MiR-1281 expression levels were measured using q-RT-PCR. The protein levels of p53 and GRP78 were analyzed using western blotting assay. GAPDH was used as the loading control. The data represent the means ± SD of three independent experiments; ***P < 0.001 vs. control. F. The expression levels of pri-miR-1281 were measured using q-RT-PCR in U2OS cells with or without p53 knockdown after 1 μM TG treatment. The data represent the means ± SD of three independent experiments; ***P < 0.001 vs. control. G. Wild type p53 or a p53 mutant lacking the transactivation domain (p53ΔTA) were transfected into Saos-2 cells, and the cells were treated with 1 μM TG as indicated. The expression level of pri-miR-1281 was measured using q-RT-PCR. The data represent the means ± SD of three independent experiments; ***P < 0.001 vs. control.
There is evidence that p53 regulates miRNA expression by both transcription-dependent and -independent means [19]. Next, we sought to determine whether p53 regulates miR-1281 expression at the transcriptional or post-transcriptional level. Expression of pri-miR-1281 was measured using q-RT-PCR analysis in U2OS cells with or without p53 knockdown in response to ER stress treatment. Interestingly, treatment with thapsigargin notably increased the levels of pri-miR-1281, and the increase was abolished when p53 was knocked down (Figure 2F). After that, we overexpressed wild type p53 or a p53 mutant lacking the transactivation domain (p53ΔTA) into Saos-2 cells and then treated them with thapsigargin as indicated. The expression level of pri-miR-1281 was measured using q-RT-PCR. As shown in Figure 2G, wild type p53 not the p53 mutant enhanced pri-miR-1281 expression under ER stress. Collectively, these data suggest that p53 transcriptionally up-regulates miR-1281 expression in response to ER stress treatment.
p53 directly binds to the promoter of miR-1281
To identify the potential p53-binding regions on the miR-1281 promoter, we first cloned the upstream sequence of miR-1281 and different truncations by PCR. Then, we inserted them into pGL3-based luciferase reporter plasmids, which were named P1-P4 (Figure 3A). We subsequently transfected them into U2OS cells with or without 1 μM TG treatment. As shown in Figure 3B, the luciferase activities of P1, but not P2, P3 and P4, were increased in U2OS cells under ER stress treatment, indicating that the region (-2000 to -15000 bp) was a key region for the promotion of miR-1281 by p53 under ER stress. To further confirm this, P1 was transfected into U2OS cells with or without p53 knockdown, and then the cells were treated with 1 μM TG at indicated times. As shown in Figure 3C, we found that the loss of p53 abolished the increase of the luciferase activity of P1 under ER stress.
Figure 3.
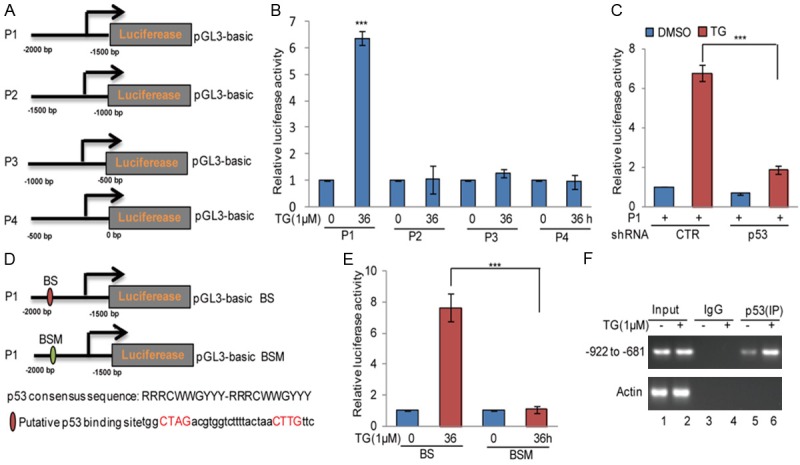
p53 bound to the promoter of miR-1281 under ER stress. A. Schematic illustration of pGL3-based reported constructs that were used in luciferase assays to examine the transcriptional activity of miR-1281. B. The promoters of miR-1281, named P1, P2, P3, and P4, were individually transfected into U2OS cells with or without TG treatment. The luciferase activity was measured. The data represent the means ± SD of three independent experiments; ***P < 0.001 vs. control. C. P1 was transfected into U2OS cells with or without p53 knockdown, and the cells were treated with 1 μM TG as indicated. The luciferase activity was measured. The data represent the means ± SD of three independent experiments; ***P < 0.001 vs. control. D. The potential p53-binding sites were inspected by JASPAR. A schematic illustration of p53 wild type binding site (WT) and the matching mutant (Mut) that were used in the luciferase assays is shown. E. p53 wild type binding site (BS) and the matching mutant (BSM) were individually transfected into U2OS cells with or without TG treatment. The luciferase activity was measured. The data represent the means ± SD of three independent experiments; ***P < 0.001 vs. control. F. ChIP analysis showed the binding of p53 to the promoter of miR-1281 in U2OS cells in response to 1 μM TG treatment. An isotype-matched IgG was used as a negative control.
To seek the potential p53-binding sites, we inspected the sequence of P1 using JASPAR software and found one putative p53-binding site on the miR-1281 promoter. To verify that the potential p53-binding site was indeed responsive to p53, a series of pGL3-based luciferase reporter plasmids named wild type binding site (BS) and binding site mutant (BSM) were generated (Figure 3D). The two plasmids were individually transfected into U2OS cells with or without 1 μM TG treatment. We found that the luciferase activity of BS, but not BSM, was significantly increase in p53WT U2OS cells under ER stress treatment. However, the increase disappeared when p53 was knocked down (Figure 3E). In addition, the subsequent chromatin immunoprecipitation (ChIP) assays showed that the chromatin fragments corresponding to the putative p53-binding sites were specifically present in anti-p53 immunoprecipitates from U2OS cells, and the binding was increased under ER stress (Figure 3F). Taken together, these data indicate that p53 can directly bind to the promoter of miR-1281, leading to its increase in response to ER stress treatment.
miR-1281 enhances ER stress-induced apoptosis
To investigate the biologic role of miR-1281 in osteosarcoma cells, we applied anti-miR-1281, a synthesized inhibitor oligo, to decrease the endogenous miR-1281 expression and observed that inhibition of miR-1281 in U2OS cells strongly suppressed cell apoptosis, as indicated by the decrease in cleaved PARP and the increase in cell viability (Figure 4A-C). Conversely, miR-1281 overexpression accelerated cell apoptosis (Figure 4D-F).
Figure 4.
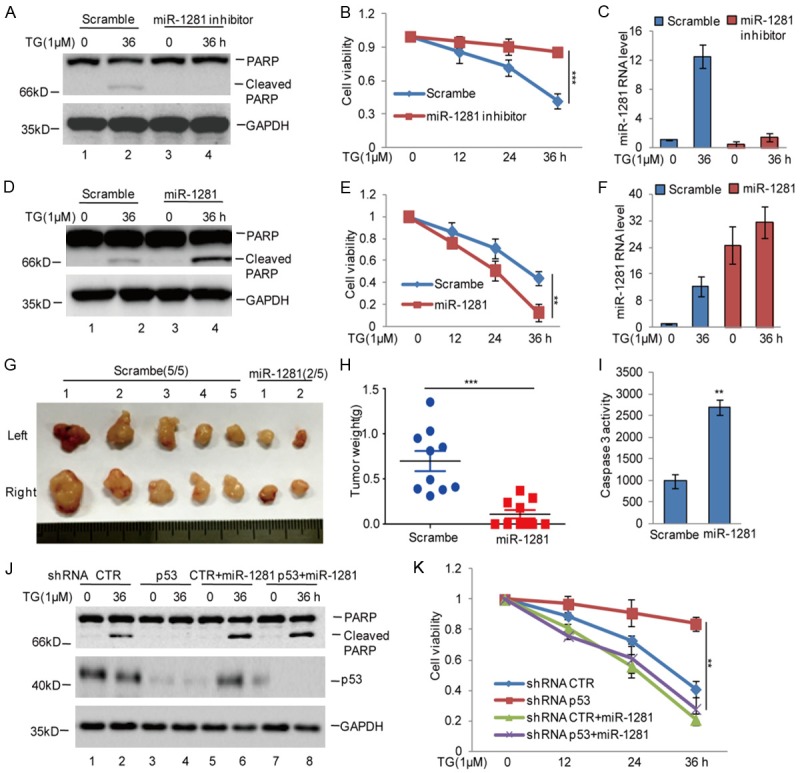
Elevated miR-1281 enhanced ER stress-induced apoptosis. A-C. miR-1281 inhibitor or the negative control was introduced into U2OS cells, and the cells were then treated with 1 μM TG at indicated times. Cell apoptosis was detected using western blotting assay. Cell viability was measured using CCK8 assay. The expression levels of miR-1281 were analyzed using q-RT-PCR. The data represent the means ± SD of three independent experiments; ***P < 0.001 vs. control. D-F. miR-1281 or the negative control was introduced into U2OS cells, and the cells were then treated with 1 μM TG at indicated times. Cell apoptosis was detected using western blotting assay. Cell viability was measured using CCK8 assay. The expression levels of miR-1281 was analyzed using q-RT-PCR. The data represent the means ± SD of three independent experiments; **P < 0.01 vs. control. G-I. miR-1281 was stably expressed in U2OS cells, and the cells then were subcutaneously injected into nude mice (n = 5 in each group) for tumor formation (2×107 cells per mouse, 6 weeks). Representative bright-field imaging of the tumors in the mice implanted with the indicated cells are shown. After 6 weeks, mice receiving transplants of indicated cells were sacrificed. The tumor weight was calculated. Caspase-3 activity was measured by a luciferase activity assay; **P < 0.01, ***P < 0.001 vs. control. J, K. miR-1281 was introduced into U2OS cells with or without p53 knockdown, and then the cells were treated with 1 μM TG at indicated times. Cell apoptosis was detected using western blotting assay. Cell viability was measured using CCK8 assay. The data represent the means ± SD of three independent experiments; **P < 0.01 vs. control.
To further identify the effects of miR-1281 on ER stress-induced apoptosis in vivo, we stably overexpressed miR-1281 in U2OS cells, and then, the cells were subcutaneously injected into nude mice. Compared with control cells, the overexpression of miR-1281 decreased tumor growth and increased cell apoptosis (Figure 4G-I).
Considering our data showing that p53 promotes miR-1281 expression and that the latter functions in thapsigargin-induced apoptosis in U2OS cells, we hypothesized that p53 exerted its pro-apoptotic function through upregulating miR-1281 upon thapsigargin exposure. Accordingly, we overexpressed miR-1281 in p53 stable knockdown U2OS cells, with which we carried out immunoblotting and cell viability experiments. Compared to the control, p53 knockdown significantly decreased cell apoptosis; however, the decrease was reversed by miR-1281 overexpression (Figure 4J, 4K). Taken together, these data indicate that miR-1281 is an important regulator in ER stress-induced apoptosis, and the effect of p53 on ER stress-induced apoptosis is dependent on miR-1281.
miR-1281 inhibits USP39 expression in osteosarcoma
Having identified p53 as the upstream transcriptional factor of miR-1281 in osteosarcoma under ER stress, we next sought to investigate downstream targets of miR-1281. In our study, we found that ER stress induced USP39 downregulation in U2OS cells, and the downregulation did not occur in MG63 and Saos-2 cells, while it was negatively associated with miR-1281 expression in U2OS cells (Figure 5A). Thus, we hypothesized that USP39 was a potential target of miR-1281. To test this hypothesis, we first searched the TargetScan database for potential targets of miR-1281. Among the candidates identified, the 3’UTR of USP39 contains a putative region (nucleotides 56-62) that matched perfectly to the miR-1281 “seed” region (Figure 5B). To examine whether USP39 was indeed downregulated by miR-1281, we introduced luciferase reporter plasmids containing the wild type 3’UTR (WT) or the matched mutant (MUT) of USP39 into U2OS cells, and then the cells were treated with or without the miR-1281 mimics. We found that overexpression of miR-1281 markedly suppressed the report activity of wild type 3’UTR of USP39, but not the mutant, and led to a decrease in endogenous USP39 (Figure 5C, 5D). Furthermore, treatment with the miR-1281 inhibitor resulted in the report activity of wild type 3’UTR elevation and USP39 expression increase (Figure 5E, 5F). Subsequently, we also found that miR-1281 inhibitor reversed the downregulation of USP39 induced by ER stress (Figure 5G). In contrast, overexpression of miR-1281 promoted USP39 downregulation in Saos-2 cells under ER stress (Figure 5H). Thus, these data indicate that USP39 is a putative target of miR-1281.
Figure 5.
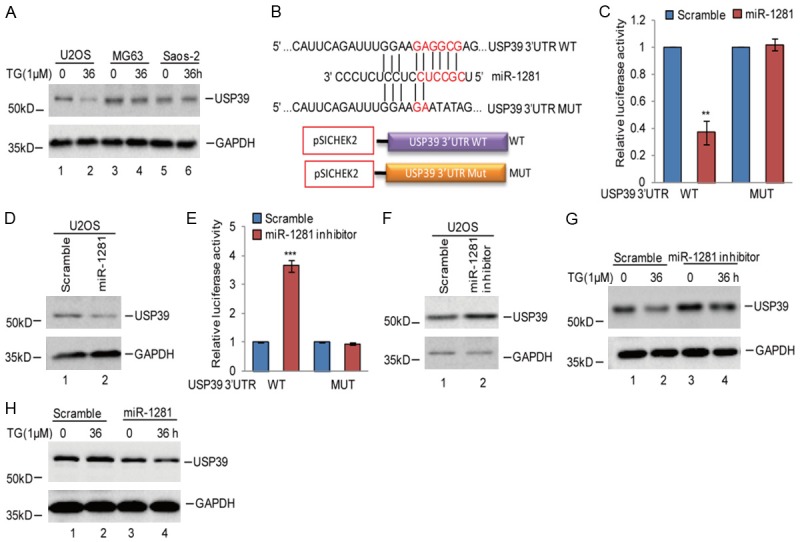
miR-1281 suppressed USP39 expression under ER stress. A. U2OS, MG63 and Saos-2 cells were treated with 1 μM TG at indicated times. The expression levels of USP39 were detected using western blotting assay. B. The potential binding region of miR-1281 on USP39 was predicted by TargetScan. The sequences of the USP39 3’UTR containing the wild type miR-1281-binding site (WT) or the mutant (MUT) were constructed in a pSICHECK2 vector, where the red indicates the mutated region. C, D. The wild type of the USP39 3’UTR (WT) or the mutant (MUT) was transfected into U2OS cells with or without miR-1281 overexpression. The luciferase activities were measured. The protein levels of USP39 were measured using western blotting assay. Data represent the mean ± SD of three independent experiments; **P < 0.01 vs. control. E, F. The wild type of the USP39 3’UTR (WT) or the mutant (MUT) was transfected into U2OS cells with or without miR-1281 inhibitors. The luciferase activities were measured. The protein levels of USP39 were measured using western blotting assay. Data represent the mean ± SD of three independent experiments. ***P < 0.001 vs. control. G. miR-1281 inhibitor or the negative control was introduced into U2OS cells, and then the cells were treated with 1 μM TG at indicated times. The protein levels of USP39 were detected using western blotting assay. H. miR-1281 or the negative control was transfected into Saos-2 cells, and the cells then treated with 1 μM TG at indicated times. The protein levels of USP39 were detected using western blotting assay.
The p53-miR-1281-USP39 signaling pathway enhances ER stress-induced apoptosis
To determine the functional significance of the p53-miR-1281-USP39 pathway on the adaptation to ER stress in osteosarcoma, we first assessed the effect of USP39 on miR-1281-induced cell apoptosis in response to ER stress and found that USP39 knockdown recovered ER stress-induced apoptosis, which was decreased by the miR-1281 inhibitor (Figure 6A, 6B). After that, we investigated the association between p53 and USP39. We found that inhibition of p53 upregulated USP39 expression and abolished ER stress-induced downregulation of USP39. Additional, USP39 knockdown recovered ER stress-induced cell apoptosis, which was abolished by p53 knockdown (Figure 6C, 6D). Thus, our data suggest that USP39 is an important downstream regulator in the p53-dependent, miR-1281-mediated survival of osteosarcoma under ER stress.
Figure 6.
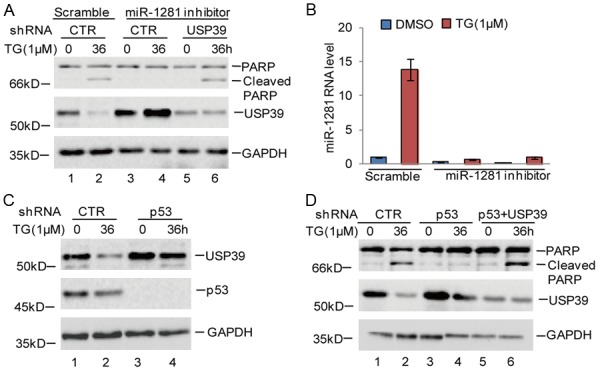
p53 enhanced cell apoptosis via regulating the miR-1281-USP39 axis. A, B. miR-1281 inhibitor or the negative control was transfected into U2OS cells with or without USP39 knockdown, and the cells were treated with 1 μM TG at indicated times. Cell apoptosis was measured using western blotting assay. The expression levels of miR-1281 were analyzed using q-RT-PCR. C. U2OS cells with or without p53 knockdown were treated with 1 μM TG at indicated times. Cell lysates were analyzed using the indicated antibodies. D. USP39 was knocked down in U2OS cells with or without p53 knockdown, and the cells were treated with 1 μM TG at indicated times. Cell lysates were analyzed using the indicated antibodies.
Discussion
It is becoming clear that miRNAs are essential for the ER stress process and affect ER stress-induced apoptosis. However, their roles in osteosarcoma are largely unknown. In this study, we found that miR-1281 was induced in response to ER stress treatment and promoted cell apoptosis by targeting USP39 in osteosarcoma cells. Subsequently, our data revealed that p53 directly bound to the promoter of miR-1281 and contributed to miR-1281 upregulation under ER stress. Thus, our finding suggests that the p53-miR-1281-USP39 axis is a novel ER stress responsive pathway and plays an important role in osteosarcoma adaptation to ER stress.
Previous studies indicated that miR-1281 was decreased in bladder cancer [20,21], and miR-1281 was shown to suppress platelet-derived growth factor-induced proliferation and migration of pulmonary artery smooth muscle cells by targeting HDAC4, an important oncogene, implying that miR-1281 may be a potential tumor suppressor [22-25]. Here, our data provided a series of evidence supporting a tumor suppressive role of miR-1281 in osteosarcoma. First, ER stress dramatically triggered miR-1281 upregulation and an accompanying increase in cell apoptosis. Second, miR-1281 ablation markedly inhibited ER stress-induced apoptosis. Furthermore, overexpression of miR-1281 promoted cell apoptosis. Third, a miR-1281 inhibitor significantly decreased tumor growth and increased cell apoptosis in vivo.
Interestingly, we found that upregulation of miR-1281 under ER stress only occurred in p53 WT, not p53 mutant or null, osteosarcoma cells, indicating that p53 may facilitate the increase of miR-1281. Accumulated evidence has revealed that p53 regulates miRNA expression by both transcription-dependent and -independent means. Here, we found that p53 could directly bind to the promoter of miR-1281, leading to miR-1281 upregulation in a transcription-dependent manner. Furthermore, we found that p53 expression was inconsistent with the increase of miR-1281 under TG treatment. The protein levels of p53 were upregulated after TM treatment for 36 h or TG treatment for 24 h. However, the increase was abolished when U2OS cells were treated with TG for 36 h. In order to assess whether the transcription activity of p53 was increased after TG treatment for 36 h, we used p21 as a positive control. Amazingly, we found that the activity of p21 promoter was increased, indicating that the transcription activity of p53 was elevated under TG treatment (Figure S1A-E). However, the molecular mechanism was still illuminated in our future work.
USP39 is a deubiquitinating enzyme and has been reported to promote tumor growth [26,27]. A recent study indicated that USP39 plays an important role in osteosarcoma, and USP39 knockdown inhibited cell growth and enhanced cell apoptosis [28]. In our study, we found that ER stress induced USP39 downregulation in U2OS cells following miR-1281 increase. Our data further indicated that miR-1281 inhibited USP39 expression, resulting in cell apoptosis under ER stress. However, the downstream pathway regulated by USP39 will need to be illuminated by further study.
In summary, our findings shed light on a new role of miR-1281 in osteosarcoma and suggested that the p53-dependent, miR-1281-mediated USP39 pathway impairs the survival of human osteosarcoma cells under ER stress.
Acknowledgements
This research was supported by the National Nature Science Foundation of China (No. 81271719).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Cortini M, Avnet S, Baldini N. Mesenchymal stroma: role in osteosarcoma progression. Cancer Lett. 2017;405:90–99. doi: 10.1016/j.canlet.2017.07.024. [DOI] [PubMed] [Google Scholar]
- 2.Savage SA, Mirabello L, Wang Z, Gastier-Foster JM, Gorlick R, Khanna C, Flanagan AM, Tirabosco R, Andrulis IL, Wunder JS, Gokgoz N, Patino-Garcia A, Sierrasesumaga L, Lecanda F, Kurucu N, Ilhan IE, Sari N, Serra M, Hattinger C, Picci P, Spector LG, Barkauskas DA, Marina N, de Toledo SR, Petrilli AS, Amary MF, Halai D, Thomas DM, Douglass C, Meltzer PS, Jacobs K, Chung CC, Berndt SI, Purdue MP, Caporaso NE, Tucker M, Rothman N, Landi MT, Silverman DT, Kraft P, Hunter DJ, Malats N, Kogevinas M, Wacholder S, Troisi R, Helman L, Fraumeni JF Jr, Yeager M, Hoover RN, Chanock SJ. Genomewide association study identifies two susceptibility loci for osteosarcoma. Nat Genet. 2013;45:799–803. doi: 10.1038/ng.2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harrison DJ, Geller DS, Gill JD, Lewis VO, Gorlick R. Current and future therapeutic approaches for osteosarcoma. Expert Rev Anticancer Ther. 2018;18:39–50. doi: 10.1080/14737140.2018.1413939. [DOI] [PubMed] [Google Scholar]
- 4.Shimizu T, Kamel WA, Yamaguchi-Iwai S, Fukuchi Y, Muto A, Saya H. Calcitriol exerts an anti-tumor effect in osteosarcoma by inducing the endoplasmic reticulum stress response. Cancer Sci. 2017;108:1793–1802. doi: 10.1111/cas.13304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sun X, Wei Q, Cheng J, Bian Y, Tian C, Hu Y, Li H. Enhanced Stim1 expression is associated with acquired chemo-resistance of cisplatin in osteosarcoma cells. Hum Cell. 2017;30:216–225. doi: 10.1007/s13577-017-0167-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang L, Wang Y, Li X, Xia X, Li N, He R, He H, Han C, Zhao W. ZBTB7A enhances osteosarcoma chemoresistance by transcriptionally repressing lncRNALINC00473-IL24 activity. Neoplasia. 2017;19:908–918. doi: 10.1016/j.neo.2017.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cho SW, Na W, Choi M, Kang SJ, Lee SG, Choi CY. Autophagy inhibits cell death induced by the anti-cancer drug morusin. Am J Cancer Res. 2017;7:518–530. [PMC free article] [PubMed] [Google Scholar]
- 8.Helenius A. How N-linked oligosaccharides affect glycoprotein folding in the endoplasmic reticulum. Mol Biol Cell. 1994;5:253–265. doi: 10.1091/mbc.5.3.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guo J, Yang Z, Yang X, Li T, Liu M, Tang H. miR-346 functions as a pro-survival factor under ER stress by activating mitophagy. Cancer Lett. 2018;413:69–81. doi: 10.1016/j.canlet.2017.10.030. [DOI] [PubMed] [Google Scholar]
- 10.Jin L, Hu WL, Jiang CC, Wang JX, Han CC, Chu P, Zhang LJ, Thorne RF, Wilmott J, Scolyer RA, Hersey P, Zhang XD, Wu M. MicroRNA-149*, a p53-responsive microRNA, functions as an oncogenic regulator in human melanoma. Proc Natl Acad Sci U S A. 2011;108:15840–15845. doi: 10.1073/pnas.1019312108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin J, Zhang D, Fan Y, Chao Y, Chang J, Li N, Han L, Han C. Regulation of cancer stem cell self-renewal by HOXB9 antagonizes endoplasmic reticulum stress-induced melanoma cell apoptosis via the miR-765-FOXA2 axis. J Invest Dermatol. 2018;138:1609–1619. doi: 10.1016/j.jid.2018.01.023. [DOI] [PubMed] [Google Scholar]
- 12.Jiang R, Zhang C, Liu G, Gu R, Wu H. MicroRNA-101 inhibits proliferation, migration and invasion in osteosarcoma cells by targeting ROCK1. Am J Cancer Res. 2017;7:88–97. [PMC free article] [PubMed] [Google Scholar]
- 13.Mei Y, Du Z, Hu C, Greenwald NF, Abedalthagafi M, Agar NYR, Dunn GP, Bi WL, Santagata S, Dunn IF. Osteoglycin promotes meningioma development through downregulation of NF2 and activation of mTOR signaling. Cell Commun Signal. 2017;15:34. doi: 10.1186/s12964-017-0189-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gomez Zubieta DM, Hamood MA, Beydoun R, Pall AE, Kondapalli KC. MicroRNA-135a regulates NHE9 to inhibit proliferation and migration of glioblastoma cells. Cell Commun Signal. 2017;15:55. doi: 10.1186/s12964-017-0209-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang P, Shao G, Lin X, Liu Y, Yang Z. MiR-338-3p inhibits the growth and invasion of non-small cell lung cancer cells by targeting IRS2. Am J Cancer Res. 2017;7:53–63. [PMC free article] [PubMed] [Google Scholar]
- 16.Ma B, Yuan Z, Zhang L, Lv P, Yang T, Gao J, Pan N, Wu Q, Lou J, Han C, Zhang B. Long noncoding RNA AC023115.3 suppresses chemoresistance of glioblastoma by reducing autophagy. Biochim Biophys Acta. 2017;1864:1393–1404. doi: 10.1016/j.bbamcr.2017.05.008. [DOI] [PubMed] [Google Scholar]
- 17.Yang X, Du T, Wang X, Zhang Y, Hu W, Du X, Miao L, Han C. IDH1, a CHOP and C/EBPbeta- responsive gene under ER stress, sensitizes human melanoma cells to hypoxia-induced apoptosis. Cancer Lett. 2015;365:201–210. doi: 10.1016/j.canlet.2015.05.027. [DOI] [PubMed] [Google Scholar]
- 18.Keremu A, Maimaiti X, Aimaiti A, Yushan M, Alike Y, Yilihamu Y, Yusufu A. NRSN2 promotes osteosarcoma cell proliferation and growth through PI3K/Akt/MTOR and Wnt/betacatenin signaling. Am J Cancer Res. 2017;7:565–573. [PMC free article] [PubMed] [Google Scholar]
- 19.Gurtner A, Falcone E, Garibaldi F, Piaggio G. Dysregulation of microRNA biogenesis in cancer: the impact of mutant p53 on Drosha complex activity. J Exp Clin Cancer Res. 2016;35:45. doi: 10.1186/s13046-016-0319-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pignot G, Cizeron-Clairac G, Vacher S, Susini A, Tozlu S, Vieillefond A, Zerbib M, Lidereau R, Debre B, Amsellem-Ouazana D, Bieche I. microRNA expression profile in a large series of bladder tumors: identification of a 3-miRNA signature associated with aggressiveness of muscle-invasive bladder cancer. Int J Cancer. 2013;132:2479–2491. doi: 10.1002/ijc.27949. [DOI] [PubMed] [Google Scholar]
- 21.Farabaugh SM, Chan BT, Cui X, Dearth RK, Lee AV. Lack of interaction between ErbB2 and insulin receptor substrate signaling in breast cancer. Cell Commun Signal. 2016;14:25. doi: 10.1186/s12964-016-0148-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Y, Li L, Qian Z, Lin B, Chen J, Luo Y, Qu J, Raj JU, Gou D. Phosphatidylinositol 3-Kinase-DNA methyltransferase 1-miR-1281-histone deacetylase 4 regulatory axis mediates platelet-derived growth factor-induced proliferation and migration of pulmonary artery smooth muscle cells. J Am Heart Assoc. 2018:7. doi: 10.1161/JAHA.117.007572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao SB, Li KL, Qiu H, Zhu LY, Pan CB, Zhao Y, Wei SH, Shi S, Jin GH, Xue LX. Enhancing chemotherapy sensitivity by targeting PcG via the ATM/p53 pathway. Am J Cancer Res. 2017;7:1874–1883. [PMC free article] [PubMed] [Google Scholar]
- 24.Gruber W, Scheidt T, Aberger F, Huber CG. Understanding cell signaling in cancer stem cells for targeted therapy - can phosphoproteomics help to reveal the secrets? Cell Commun Signal. 2017;15:12. doi: 10.1186/s12964-017-0166-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jarvelin AI, Noerenberg M, Davis I, Castello A. The new (dis)order in RNA regulation. Cell Commun Signal. 2016;14:9. doi: 10.1186/s12964-016-0132-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fraile JM, Manchado E, Lujambio A, Quesada V, Campos-Iglesias D, Webb TR, Lowe SW, Lopez-Otin C, Freije JM. USP39 deubiquitinase is essential for KRAS oncogene-driven cancer. J Biol Chem. 2017;292:4164–4175. doi: 10.1074/jbc.M116.762757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yuan X, Sun X, Shi X, Jiang C, Yu D, Zhang W, Guan W, Zhou J, Wu Y, Qiu Y, Ding Y. USP39 promotes the growth of human hepatocellular carcinoma in vitro and in vivo. Oncol Rep. 2015;34:823–832. doi: 10.3892/or.2015.4065. [DOI] [PubMed] [Google Scholar]
- 28.Gan Z, Han K, Lin S, Hu H, Shen Z, Min D. Knockdown of ubiquitin-specific peptidase 39 inhibited the growth of osteosarcoma cells and induced apoptosis in vitro. Biol Res. 2017;50:15. doi: 10.1186/s40659-017-0121-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.