

Abstract
β-catenin is not only a key component of adherens junctions but also a transcriptional co-activator downstream of canonical Wnt signaling. The Wnt/β-catenin pathway plays critical roles in animal development and tissue homeostasis, while mutation or overexpression of β-catenin often leads to tumorigenesis and metastasis. Ubiquitination-mediated proteasomal degradation of β-catenin is a key molecular event in the Wnt/β-catenin pathway. Because deubiquitination of β-catenin can stabilize β-catenin and activate Wnt/β-catenin signaling, targeting the β-catenin deubiquitinase may provide a strategy for treating β-catenin-driven cancers. Here, by screening a human deubiquitinase library, we identified USP2a as a deubiquitinase that binds, deubiquitinates, and stabilizes β-catenin protein. USP2a promotes the nuclear accumulation and transcriptional activity of β-catenin, leading to elevated expression of Wnt/β-catenin target genes. Importantly, either genetic knockdown or pharmacological inhibition of USP2a leads to β-catenin destabilization. These findings suggest that USP2a may serve as a therapeutic target for targeting the cancer-promoting protein β-catenin.
Keywords: USP2, β-catenin, deubiquitinase, deubiquitination, ubiquitination
Introduction
β-catenin, a key downstream effector of the Wnt signaling pathway, plays important roles in animal development and human diseases [1,2]. The cellular level of β-catenin protein is controlled by ubiquitin-dependent proteasomal degradation. In the canonical Wnt pathway, glycogen synthase kinases 3α and 3β (GSK3α and GSK3β), casein kinase 1 (CK1), adenomatous polyposis coli (APC), and Axin form a complex for β-catenin destruction. Phosphorylation of β-catenin by this complex promotes the binding of β-catenin to a ubiquitin E3 ligase, β-transduction repeat-containing protein (β-TrCP), leading to ubiquitination and proteasomal degradation of β-catenin [3]. Activation of Wnt signaling inhibits the phosphorylation and proteolytic ubiquitination of β-catenin, resulting in β-catenin accumulation and nuclear translocation, which in turn activates TCF (T-cell factor)/LEF (lymphoid enhancer factor)-dependent transcription of Wnt target genes [1-3]. Mutation or overexpression of β-catenin often leads to tumor formation and metastatic progression [4-6]. Thus, the Wnt pathway has become an attractive therapeutic target for anticancer drug development [7]. Since ubiquitination-mediated β-catenin degradation plays a key role in regulating β-catenin protein stability, we hypothesize that identification of the β-catenin deubiquitinase may reveal a new therapeutic target.
Deubiquitinases (also known as deubiquitinating enzymes/DUBs) are proteases that remove ubiquitin from proteins [8] and play important roles in regulating protein stability, activity, and subcellular localization [9]. Since DUBs regulate many key pathways in cancer cells and are potentially druggable, interests in developing DUB inhibitors as antitumor drugs have increased substantially [9-11]. In the present study, we identified USP2a as a deubiquitinase that deubiquitinates and stabilizes β-catenin, and found that inhibition of USP2a by a small-molecule inhibitor led to downregulation of β-catenin in cancer cells.
Materials and methods
Cell lines
The HEK293T, Hela, HCC1806, and BT549 cell lines were from the American Type Culture Collection, and LM2 (a subline of the MDA-MB-231 cell line) cells were from Dr. Xiang Zhang (Baylor College of Medicine). HEK293T, Hela, and BT549 cells were cultured in DMEM medium supplemented with 10% FBS and 1% penicillin and streptomycin, and HCC1806 cells were cultured in RPMI medium supplemented with 10% FBS and 1% penicillin and streptomycin. All cell lines were authenticated at the Characterized Cell Line Core at the MD Anderson Cancer Center.
Chemicals
The USP2 inhibitor ML364 was from Dr. Anton Simeonov (National Institutes of Health). Cycloheximide was from Cayman Chemical. MG132 was from Sigma.
Plasmids and shRNAs
68 SFB-tagged human deubiquitinase ORFs were described previously [12-15]. The full-length β-catenin ORF and its deletion mutants were subcloned into a MYC-tagged expression vector using the Gateway system. The full-length USP2a ORF and its deletion mutant were subcloned into an SFB-tagged expression vector (a gift from Dr. Junjie Chen at MD Anderson Cancer Center). The full-length USP2a and β-catenin ORFs were also subcloned into the pDEST-GST vector (Invitrogen) and the SFB-tagged expression vector, respectively. The catalytically inactive mutant of USP2a (USP2aC276A) was generated using the QuikChange Site-Directed Mutagenesis Kit (Agilent). pLOC-USP2a and the control pLOC vector were from Dharmacon. The primer sequences used for USP2aC276A are 5’-TCGAAACCTTGGGAACACGGCCTTCATGAACTCAATTCTG-3’ and 5’-CAGAATTGAGTTCATGAAGGCCGTGTTCCCAAGGTTTCGA-3’. HA-ubiquitin, TOPflash, and FOPflash constructs were from Addgene (plasmid number: 17608, 12456, and 12457). The pRL Renilla luciferase plasmid was from Promega (E2241). Human USP2 shRNA constructs were from Sigma (TRCN0000007277 for shRNA #1, and TRCN0000415688 for shRNA #2).
Luciferase reporter assay
HEK293T cells were plated in triplicate in 96-well plates with white polystyrene upper structure (Nunc). Next day, 16.67 ng of the firefly luciferase vector (TOPflash or FOPflash), 0.1 ng of the Renilla luciferase vector, and 50 ng of each SFB-tagged deubiquitinase construct were co-transfected per well. 2 days post transfection, firefly and Renilla luciferase activities were measured using a Dual-Luciferase Reporter Assay system (Promega) on a Gen5 Microplate Reader (BioTek) according to the manufacturer’s protocol. Firefly luciferase activity was normalized to Renilla luciferase activity. For ML364 treatment: 24 hours post transfection, DMSO or 3 µM ML364 was used to treat cells for 24 hours.
Immunoprecipitation and pulldown assays
General procedures including pulldown of SFB-tagged proteins were performed as described previously [15]. S-protein beads were from EMD Millipore. The MYC-tagged proteins were immunoprecipitated using anti-MYC agarose affinity gel beads (Sigma, A7470) or anti-MYC magnetic beads (ThermoFisher, 88842), and mouse IgG (isotype control)-magnetic beads (MBL, M076-9) were used as the control. For immunoprecipitation of HA-tagged ubiquitinated proteins, an HA-specific antibody (Abcam, ab9110) was used after conjugation with agarose beads (Santa Cruz Biotechnology, sc-2003). Normal rabbit IgG (Santa Cruz Biotechnology, sc-2027) was used as a control. DMSO or the USP2 inhibitor ML364 was used to treat cells at the indicated concentrations for 24 hours, and then cells were harvested and subjected to immunoprecipitation and pulldown assays.
Total RNA isolation, reverse transcription, and quantitative PCR
Cells were harvested in TRIzol reagent (Invitrogen). Total RNA was isolated using the RNeasy Mini Kit and RNase-free DNase Set (Qiagen) and reverse-transcribed using the iScript cDNA Synthesis Kit (Bio-Rad) according to the manufacturer’s protocol. qPCR was performed using SYBR Green reagent (Bio-Rad) and gene-specific primers as follows: CTNNB1-forward, 5’-GTTCAGTTGCTTGTTCGTGC-3’; CTNNB1-reverse, 5’-GTTGTGAACATCCCGAGCTAG-3’; BIRC5-forward, 5’-CAAGGAGCTGGAAGGCTG-3’; BIRC5-reverse, 5’-TTCTTGGCTCTTTCTCTGTCC-3’; CUBN-forward, 5’-TCTCAGCAATCCAAATCAGGG-3’; CUBN-reverse, 5’-ACCACAAAGTCTCCACATCAG-3’; FGF7-forward, 5’-CCTGAGGATCGATAAAAGAGGC-3’; FGF7-reverse, 5’-CACTTTCCACCCCTTTGATTG-3’; CDON-forward, 5’-GGAGTGGTACTTACAGATTCCTC-3’; CDON-reverse, 5’-TGAAGGCAGTGATTGGAGAAC-3’; BTRC-forward, 5’-CTTAAATGGACACAAACGAGGC-3’; BTRC-reverse, 5’-CAACGCACCAATTCCTCATG-3’; ANTXR1-forward, 5’-CCTTTATTGTTTTCTCCACCCG-3’; ANTXR1-reverse, 5’-TCACTGGCCCTTTCAAATCC-3’; IGF2-forward, 5’-AGACGTACTGTGCTACCCC-3’; IGF2-reverse, 5’-TGCTTCCAGGTGTCATATTGG-3’.
GST fusion protein purification
GST protein was purchased from Sigma. BL21 competent cells (NEB) were transformed with GST-GFP, GST-USP2a, or GST-USP2aC276A and cultured in LB medium. GST fusion proteins were induced by IPTG (Ambion). Bacterial cells were harvested in lysis buffer (5 mM EDTA, 1 mM PMSF, and 1 mM dithiothreitol in PBS) with protease inhibitor and sonicated. 1% Triton X-100 was added and incubated to fully lyse the cells. Glutathione sepharose beads (ThermoFisher, 16100) were added and incubated. GST fusion proteins were eluted in elution buffer (50 mM Tris at pH 8.0 with 10 mM reduced glutathione, Sigma) and concentrated using the 100 K MWCO Protein Concentrator (ThermoFisher, 88523).
In vitro binding assay using GST fusion proteins
FLAG-tagged β-catenin was expressed in HEK293T cells and purified with anti-FLAG agarose beads (Sigma, M8823) and 3 × FLAG peptides (Sigma, F4799) according to the manufacturer’s protocol. 1 µg of GST, GST-USP2a, or GST-USP2aC276A was mixed with 1 µg of purified FLAG-β-catenin and glutathione sepharose beads and incubated at 4°C overnight. The beads were washed and the bound proteins were eluted by boiling in Laemmli buffer, followed by Western blot analysis with the indicated antibodies.
In vivo deubiquitination assay
HEK293T cells were co-transfected with MYC-β-catenin, SFB-tagged deubiquitinases, and HA-ubiquitin and treated with the proteasome inhibitor MG132 (10 µM) for 6 hours [16,17]. Cells were lysed in RIPA buffer (50 mM Tris-HCl at pH 7.4, 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 1 mM EDTA). For denaturing, lysates were heated at 95°C for 5 minutes in the presence of 1% SDS, followed by 10-fold dilution with lysis buffer (to 0.1% SDS) and sonication, as described previously [18]. β-catenin was immunoprecipitated with anti-MYC beads and immunoblotted with the indicated antibodies.
In vitro deubiquitination assay
HEK293T cells were co-transfected with MYC-β-catenin and HA-ubiquitin. After 2 days, cells were treated with 10 µM MG132 for 6 hours and then harvested. Cell pellet was lysed in NETN buffer (20 mM Tris-HCl at pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5% Nonidet P-40) and ubiquitinated MYC-β-catenin was immunoprecipitated with anti-MYC agarose beads (Sigma, A7470). After wash with NETN buffer, beads were washed once with deubiquitination buffer (50 mM Tris-HCl at pH 8.0, 50 mM NaCl, 1 mM EDTA, 10 mM dithiothreitol, 5% glycerol). The in vitro deubiquitination reaction was performed as described previously [14]. Briefly, ubiquitinated β-catenin protein (bound to beads) was incubated with 1 µg of purified GST-GFP, GST-USP2a, or GST-USP2aC276A in deubiquitination buffer at 37°C for 2 hours. After the reaction, the beads were washed with deubiquitination buffer, and the bound proteins were eluted by boiling in Laemmli buffer and subjected to Western blot analysis with the indicated antibodies.
Immunoblotting
Immunoblotting was performed as described previously [15] and the following antibodies were used: antibodies against FLAG (1:5,000, Sigma, F7425), MYC (1:1,000, Santa Cruz Biotechnology, SC-40), HA (1:5,000, Santa Cruz Biotechnology, SC-7392), ubiquitin (1:1,000, Invitrogen, 13-1600), β-catenin (1:1,000, Cell Signaling Technology, 9582), β-actin (1:2,000, Santa Cruz Biotechnology, SC-1615), cyclophilin B (1:5,000, Thermo, PA1-027A), HSP90 (1:5,000, BD Biosciences, 610419), Lamin B1 (1:1,000, Cell Signaling Technology, 12586), and USP2 (1:1,000, Cell Signaling Technology, 8036).
Lentiviral production and transduction
HEK293T cells were co-transfected with a lentiviral vector (pLKO.1 control, pLKO.1-USP2 shRNA, pLOC control, or pLOC-USP2a), an envelope plasmid (pCMV-VSV-G, Addgene, 8454), and a packaging plasmid (pCMV-dR8.2 dvpr, Addgene, 8455). Viral supernatant was harvested 2 days post transfection, filtered through a 0.45 µm filter, and added to the target cells. Antibiotics (1.5 µg/ml puromycin or 10 µg/ml blasticidin) was added to select stably transduced cells.
Cytoplasmic and nuclear fractionation
HEK293T cells were transfected with the control SFB or SFB-USP2a vector and harvested after 2 days. Fractionation of nuclear and cytoplasmic proteins was done by using the NE-PER Nuclear and Cytoplasmic Extraction Kit (ThermoFisher Scientific, 78833) according to the manufacturer’s protocol. After fractionation, Western blot analysis was performed to analyze USP2a and β-catenin in the cytoplasm and the nucleus. HSP90 and Lamin B1 were used as markers of the cytoplasm and nucleus, respectively.
Human Wnt signaling targets PCR array
The 96-well format Human Wnt signaling targets RT2 Profiler PCR Array (Qiagen, PAHS-243Z), consisting of 84 key genes responsive to Wnt signal activation, was used to profile USP2a-overexpressing HEK293T cells. Total RNA was extracted from control and USP2a-overexpressing HEK293T cells using the RNeasy Mini Kit (Qiagen) and DNase I treatment. Reverse transcription and qPCR were performed according to the manufacturer’s protocol. Briefly, after reverse transcription, the cDNA was combined with a SYBR Green reagent (Qiagen), and then equal aliquots of this mixture were added to each well of the PCR Array plate that contains the pre-dispensed gene-specific primer sets. Real-time PCR and data collection were performed on a CFX96 instrument (Bio-Rad).
Statistical analysis
Data are presented as mean ± S.D. or mean ± S.E.M. (as specified in the figure legends), and a two-tailed unpaired t-test was used to compare two groups of independent samples. P < 0.05 was considered statistically significant. *: P < 0.05; **: P < 0.01; ***: P < 0.001; n.s.: not significant.
Results
USP2a upregulates β-catenin protein and promotes its transcriptional activity
To identify the β-catenin deubiquitinase in an unbiased manner, we screened an open-reading-frame (ORF) library of 68 human deubiquitinases [12,15] by a pulldown assay (Figure 1A). We transiently co-transfected SFB (a triple-epitope tag containing S-protein, FLAG tag, and streptavidin-binding peptide)-tagged deubiquitinases with MYC-tagged β-catenin into HEK293T cells and pulled down the deubiquitinases with S-protein beads. Immunoblotting assays showed that β-catenin was pulled down by 33 deubiquitinases (Figure 1A). Since nearly half of the 68 deubiquitinases interacted with β-catenin in the pulldown assay, we performed a second screen using the TOPflash/FOPflash reporter assay. If a deubiquitinase stabilizes β-catenin, it should specifically increase the activity of the TOPflash reporter containing tandem TCF/LEF binding sites, relative to that of the control FOPflash reporter containing mutated TCF/LEF binding sites [19]. We found that in HEK293T cells, overexpression of six deubiquitinases, including USP2a, USP26, USP36, USP42, OTUD7B, and DUB3, specifically elevated β-catenin activity by more than 8-fold (z score > 2, Figure 1B).
Figure 1.
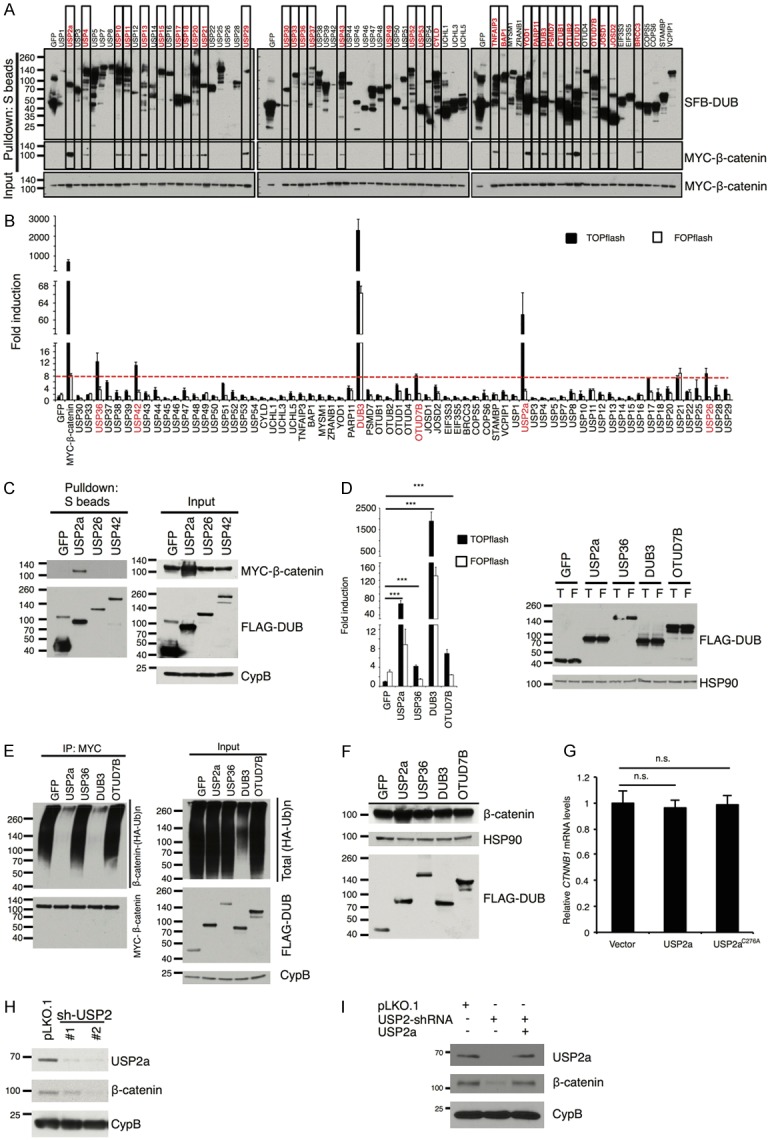
USP2a upregulates β-catenin protein and promotes its transcriptional activity. A. Each SFB-tagged deubiquitinase was co-transfected with MYC-tagged β-catenin into HEK293T cells, followed by pulldown with S-protein beads and immunoblotting with antibodies against FLAG and MYC. B. Either β-catenin-responsive TOPflash or its mutant FOPflash construct was co-transfected with each SFB-tagged deubiquitinase and Renilla luciferase into HEK293T cells. A dual luciferase assay was used to determine β-catenin activity. Firefly luciferase activity was normalized to Renilla luciferase activity. MYC-tagged β-catenin serves as a positive control. Error bars are S.D. C. SFB-tagged GFP, USP2a, USP26, and USP42 were co-transfected with MYC-tagged β-catenin into HEK293T cells, and pulled down with S-protein beads. Antibodies against MYC and FLAG were used to detect β-catenin and DUBs. CypB (cyclophilin B) serves as the loading control. D. Left panel: luciferase reporter assay validating that the 4 candidate deubiquitinases (USP2a, USP36, DUB3, and OTUD7B) promote the transcriptional activity of β-catenin. Error bars are S.D. ***: P < 0.001. Right panel: immunoblotting of HSP90 and SFB-tagged GFP, USP2a, USP36, DUB3, and OTUD7B in HEK293T cells. HSP90 serves as the loading control. T: TOPflash; F: FOPflash. E. Each SFB-tagged candidate deubiquitinase was co-transfected with HA-tagged ubiquitin and MYC-tagged β-catenin into HEK293T cells. After MG132 treatment for 6 hours, β-catenin was immunoprecipitated with a MYC-specific antibody, followed by immunoblotting with antibodies against HA and MYC. F. Immunoblotting of β-catenin, FLAG, and HSP90 in HEK293T cells transfected with SFB-tagged GFP, USP2a, USP36, DUB3, or OTUD7B. G. qPCR of CTNNB1 (the gene that encodes β-catenin) in HEK293T cells transfected with the empty vector, wild-type USP2a, or the catalytically inactive mutant (C276A). Error bars are S.D. n.s.: not significant. H. Immunoblotting of USP2a, β-catenin, and CypB in BT549 cells transduced with USP2 shRNAs. I. Immunoblotting of USP2a, β-catenin, and CypB in BT549 cells transduced with USP2 shRNA with or without ectopic expression of USP2a.
Because USP26 and USP42 did not interact with β-catenin (Figure 1C), we investigated whether USP2a, USP36, DUB3, and OTUD7B are β-catenin deubiquitinases. First, we validated that all of them did activate β-catenin using the TOPflash/FOPflash reporter assay (Figure 1D). Consistent with the screening result (Figure 1B), USP2a and DUB3 exhibited a much stronger effect than USP36 and OTUD7B (Figure 1D). Next, to determine whether these four deubiquitinases can promote β-catenin deubiquitination in cells, we co-transfected the deubiquitinases with MYC-β-catenin and HA-ubiquitin into HEK293T cells and treated the cells with MG132, a proteasome inhibitor. β-catenin was immunoprecipitated by anti-MYC beads and its polyubiquitination was detected by an HA-specific antibody. Whereas both USP2a and DUB3 drastically reduced the polyubiquitination of β-catenin (Figure 1E), DUB3, but not other candidate deubiquitinases, exhibited a strong inhibitory effect on global protein ubiquitination (Figure 1E, input). In addition, when the four candidate deubiquitinases were ectopically expressed in HEK293T cells by transient transfection, only USP2a increased the protein level of endogenous β-catenin (Figure 1F), without altering its mRNA level (Figure 1G). Conversely, shRNA-mediated stable knockdown of USP2a in the BT549 human breast cancer cell line decreased endogenous β-catenin protein level (Figure 1H), and re-expression of USP2a reversed the downregulation of β-catenin caused by USP2 shRNA (Figure 1I), demonstrating that the shRNA effect was specific to USP2a. Therefore, USP2a is a β-catenin-interacting deubiquitinase that positively regulates β-catenin protein level and activity.
USP2a binds and deubiquitinates β-catenin
We asked whether USP2a directly interacts with β-catenin and acts as a bona fide β-catenin deubiquitinase. First, SFB-USP2a or SFB-GFP was transiently transfected with or without MYC-β-catenin into HEK293T cells, and SFB-USP2a or SFB-GFP was pulled down by S-protein beads. We found that SFB-USP2a was able to pull down both exogenous MYC-β-catenin (Figure 2A) and endogenous β-catenin (Figure 2B). Reciprocally, when MYC-β-catenin was immunoprecipitated by anti-MYC beads, SFB-USP2a could be co-immunoprecipitated (Figure 2C), indicating that USP2a and β-catenin co-exist in a protein complex in vivo. Next, to demonstrate direct interaction between USP2a and β-catenin, we performed an in vitro GST-pulldown assay using purified GST-tagged USP2a and FLAG-tagged β-catenin, and found that both wild-type USP2a and its catalytically inactive mutant C276A [20,21], but not the GST control, were able to directly bind β-catenin under a cell-free condition (Figure 2D).
Figure 2.
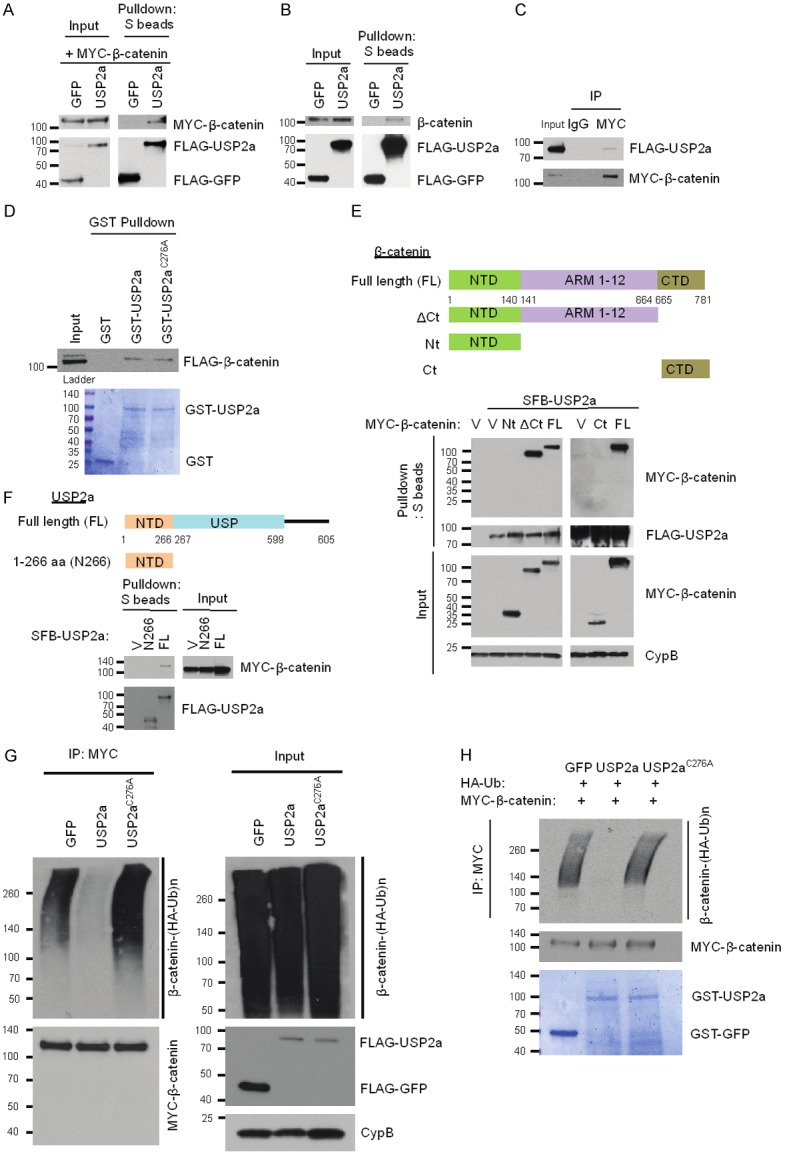
USP2a directly binds and deubiquitinates β-catenin. A. HEK293T cells were co-transfected with SFB-USP2a and MYC-β-catenin, followed by pulldown with S-protein beads and immunoblotting with antibodies against MYC and FLAG. B. HEK293T cells were transfected with SFB-USP2a, followed by pulldown with S-protein beads and immunoblotting with antibodies against β-catenin and FLAG. C. HEK293T cells were co-transfected with SFB-USP2a and MYC-β-catenin, followed by immunoprecipitation with a MYC-specific antibody and immunoblotting with antibodies against FLAG and MYC. D. In vitro binding between purified GST-USP2a fusion protein and FLAG-tagged β-catenin. Top panel: 1 μg of GST, GST-USP2a, or GST-USP2aC276A purified from bacteria was mixed with 1 μg of SFB-β-catenin purified from HEK293T cells, followed by pulldown with glutathione sepharose beads and immunoblotting with a FLAG-specific antibody. Bottom panel: purified recombinant proteins were analyzed by SDS-PAGE and Coomassie blue staining. Ladder: pre-stained protein ladder. E. Top panel: schematic representation of full-length β-catenin and its various deletion mutants. NTD, N-terminal domain; ARM, Armadillo repeat domain; CTD, C-terminal domain. Bottom panel: HEK293T cells were co-transfected with SFB-USP2a and MYC-tagged full-length β-catenin or its deletion mutants, followed by pulldown with S-protein beads and immunoblotting with antibodies against MYC and FLAG. V, empty vector; Nt: β-catenin aa 1-140; ΔCt: β-catenin aa 1-664; FL: β-catenin full length; Ct: β-catenin aa 665-781. F. Top panel: schematic representation of full-length USP2a and its truncation mutant. NTD, N-terminal domain; USP, ubiquitin-specific protease domain. Bottom panel: HEK293T cells were co-transfected with MYC-β-catenin and SFB-tagged full-length USP2a or its deletion mutant, followed by pulldown with S-protein beads and immunoblotting with antibodies against MYC and FLAG. V, empty vector; N266, USP2a aa 1-266; FL, USP2a full length. G. USP2a, but not USP2aC276A, deubiquitinates β-catenin in vivo. HEK293T cells were co-transfected with HA-ubiquitin, MYC-β-catenin, and SFB-tagged GFP, USP2a, or USP2aC276A. After MG132 treatment for 6 hours, β-catenin was immunoprecipitated with a MYC-specific antibody, followed by immunoblotting with antibodies against HA and MYC. H. USP2a, but not USP2aC276A, deubiquitinates β-catenin in vitro. Top panel: ubiquitinated MYC-β-catenin was purified with anti-MYC beads from HEK293T cells co-transfected with MYC-β-catenin and HA-ubiquitin, and was incubated with GST-tagged GFP, USP2a, or USP2aC276A purified from bacteria. After the in vitro deubiquitination reaction, the bound proteins were eluted by boiling in Laemmli sample buffer and immunoblotted with antibodies against HA and MYC. Bottom panel: purified recombinant proteins were analyzed by SDS-PAGE and Coomassie blue staining.
β-catenin protein consists of the N-terminal domain (NTD), the C-terminal domain (CTD), and the central ARM domain (Armadillo repeats 1-12, which mediate the binding of β-catenin to APC). To map the domain that is critical for the interaction with USP2a, we transiently transfected HEK293T cells with SFB-USP2a and wild-type MYC-β-catenin or its fragments, including the NTD (aa 1-140) and the C-terminal domain deletion mutant (ΔCt, aa 1-664). We pulled down SFB-USP2a by S-protein beads. Both full-length β-catenin and the ΔCt mutant (= NTD + ARM 1-12), but not the NTD fragment, could bind to USP2a (Figure 2E), suggesting that the ARM domain and/or the C-terminal domain are essential for the interaction with USP2a. To further identify the binding domain, we performed the pulldown assay using the CTD fragment (aa 665-781), which was unable to bind USP2a (Figure 2E). Thus, the ARM domain of β-catenin mediates its interaction with USP2a.
USP2a protein consists of an N-terminal domain and a catalytic domain (USP domain). We transiently transfected MYC-β-catenin with full-length SFB-USP2a or its N-terminal domain (N266, aa 1-266) into HEK293 cells, and pulled down SFB-USP2a by S-protein beads. Only full-length USP2a, but not its N-terminal domain, could bind β-catenin (Figure 2F), suggesting that the USP domain of USP2a is responsible for the binding of USP2a to β-catenin.
We sought to determine whether USP2a deubiquitinates β-catenin through its deubiquitinase activity using both in vivo and in vitro deubiquitination assays (Figure 2G, 2H). First, we transiently transfected HEK293T cells with HA-ubiquitin, MYC-β-catenin, and SFB-USP2a or its catalytically inactive mutant C276A, and treated cells with 10 µM MG132. β-catenin was immunoprecipitated by anti-MYC beads and its polyubiquitination was detected by an HA-specific antibody. As shown in Figure 2G, wild-type USP2a, but not the C276A mutant, which is still capable of binding to β-catenin (Figure 2D), abrogated the polyubiquitination of β-catenin, suggesting that the enzymatic activity of USP2a is essential for the deubiquitination of β-catenin by USP2a. Next, to determine whether β-catenin is a direct substrate of USP2a, we purified GST-USP2a and ubiquitinated β-catenin and then incubated them in a cell-free system. Wild-type USP2a purified from bacteria, but not its enzyme-dead mutant C276A, strongly deubiquitinated β-catenin in vitro (Figure 2H). Taken together, these data suggest that USP2a directly binds and deubiquitinates β-catenin.
USP2a stabilizes β-catenin
To determine whether USP2a stabilizes β-catenin protein, we transiently transfected HEK293T cells with MYC-β-catenin and SFB-USP2a or SFB-GFP, treated the cells with 100 μg/ml cycloheximide (CHX), an inhibitor of protein synthesis, and determined MYC-β-catenin levels at different time points post CHX treatment by Western blotting. In HEK293T cells, overexpression of USP2a increased both the steady-state level and the half-life of ectopically expressed β-catenin protein (Figure 3A).
Figure 3.
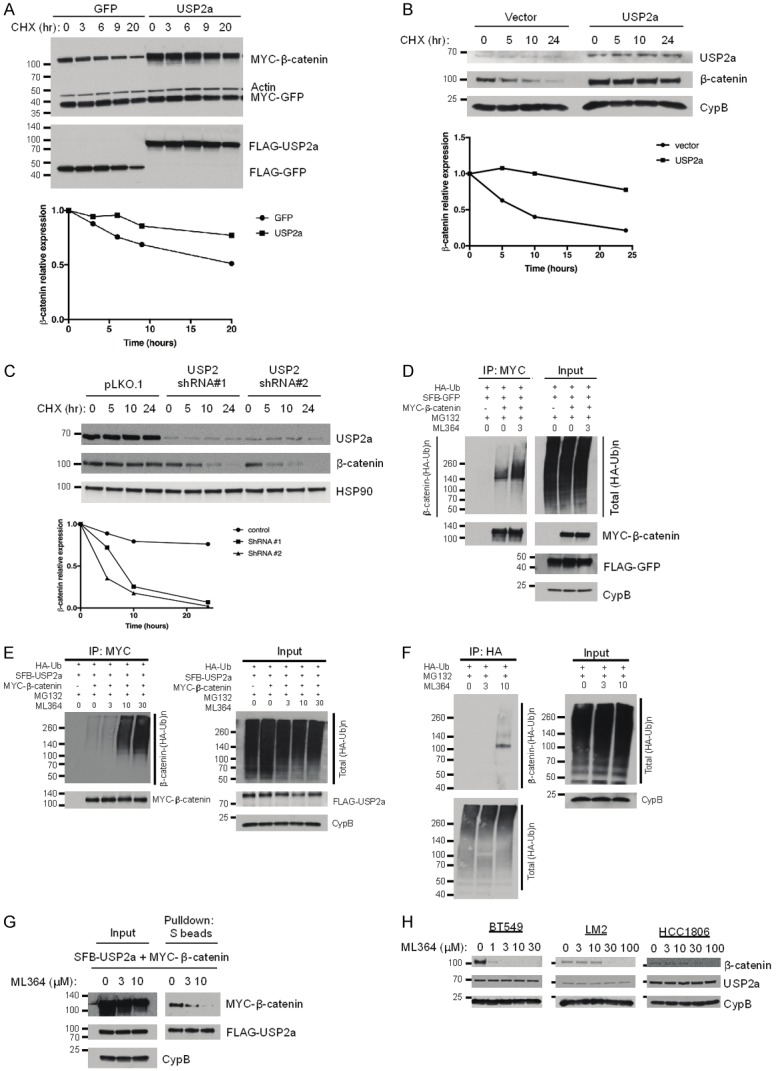
USP2a stabilizes β-catenin. (A) Top panel: HEK293T cells were co-transfected with MYC-β-catenin, MYC-GFP, and SFB-tagged GFP or USP2a, treated with 100 µg/ml cycloheximide (CHX), harvested at different time points, and then immunoblotted with antibodies against MYC, β-actin, and FLAG. MYC-GFP serves as the control for transfection. Bottom panel: quantification of β-catenin protein levels (normalized to MYC-GFP). (B) Top panel: MDA-MB-231 cells were transfected with the control vector (pLOC-RFP) or USP2a (pLOC-USP2a), treated with 100 µg/ml cycloheximide (CHX), harvested at different time points, and then immunoblotted with antibodies against USP2a, β-catenin, and CypB. Bottom panel: quantification of β-catenin protein levels (normalized to CypB). (C) Top panel: BT549 cells were transfected with USP2 shRNA (#1 or #2), treated with 100 µg/ ml CHX, harvested at different time points, and then immunoblotted with antibodies against USP2, β-catenin, and HSP90. Bottom panel: quantification of β-catenin protein levels (normalized to HSP90). (D and E) HEK293T cells were co-transfected with HA-ubiquitin, MYC-β-catenin, and SFB-tagged GFP (D) or USP2a (E). Cells were pretreated with ML364 (0, 3, 10, or 30 µM) for 4 hours and then treated with 10 µM MG132 in the presence of ML364 for 6 hours. β-catenin was immunoprecipitated with anti-MYC beads, followed by immunoblotting with antibodies against HA and MYC. (F) HEK293T cells were transfected with HA-ubiquitin, pretreated with 0, 3, or 10 µM ML364 for 24 hours, and then treated with 10 µM MG132 in the presence of ML364 for 6 hours. Polyubiquitinated proteins were immunoprecipitated with anti-HA beads, followed by immunoblotting with antibodies against β-catenin and HA. (G) HEK293T cells were co-transfected with SFB-USP2a and MYC-β-catenin and treated with ML364 (0, 3, or 10 µM) for 24 hours, followed by pulldown with S-protein beads and immunoblotting with antibodies against MYC and FLAG. (H) Immunoblotting of β-catenin, USP2a, and CypB in BT549, LM2, and HCC1806 cells treated with ML364 at the indicated doses.
To determine whether USP2a regulates endogenous β-catenin protein level and its half-life, we transiently transfected the MDA-MB-231 human breast cancer cell line with USP2a and found that ectopic expression of USP2a increased the stability of endogenous β-catenin three days after transfection (Figure 3B). Moreover, we transiently transfected BT549 breast cancer cells with USP2 shRNA and four days after transfection, we found that the stability of endogenous β-catenin was substantially decreased by two independent USP2 shRNAs (Figure 3C).
Recently, a small-molecule inhibitor, ML364, was identified as a specific USP2 inhibitor [22]. We co-transfected HEK293T cells with MYC-β-catenin and HA-ubiquitin, treated the cells with 3 µM ML364 and the proteasome inhibitor MG132, and examined the ubiquitination status of β-catenin. We found that 3 µM ML364 treatment increased β-catenin polyubiquitination (Figure 3D); when we overexpressed USP2a in this system, a higher concentration of ML364 (10 µM) was needed to increase β-catenin ubiquitination (Figure 3E), suggesting that the increased ubiquitination of β-catenin was due to inhibition of USP2. In addition, to determine the effect on the ubiquitination of endogenous β-catenin, we transfected HEK293T cells with HA-ubiquitin and treated the cells with MG132. Total ubiquitinated proteins were immunoprecipitated by anti-HA beads and ubiquitinated β-catenin was detected by a β-catenin-specific antibody. We found that ML364 treatment increased the level of ubiquitinated β-catenin (Figure 3F). Interestingly, when we used S-protein beads to perform a pulldown assay in HEK293T cells transfected with SFB-tagged USP2a and MYC-tagged β-catenin, we found that ML364 treatment inhibited the interaction between USP2a and β-catenin in a dose-dependent manner (Figure 3G). Notably, in breast cancer cell lines BT549, LM2, and HCC1806, chemical inhibition of USP2 with ML364 decreased β-catenin protein levels in a dose-dependent manner without altering the expression of USP2a (Figure 3H), suggesting that pharmacological inhibition of USP2 could be a new strategy for destruction of β-catenin in cancer cells.
USP2a promotes the activity of Wnt/β-catenin signaling
We asked whether USP2a regulates Wnt/β-catenin signaling. First, we transiently transfected HEK293T cells with SFB-USP2a and performed a fractionation assay. In control HEK293T cells, β-catenin protein was localized in the cytoplasm, and overexpression of USP2a resulted in not only upregulation of total β-catenin but also accumulation of nuclear β-catenin (Figure 4A). Next, we validated the effect of USP2a on β-catenin activity using the TOPflash/FOPflash assay and found that wild-type USP2a, but not the C276A mutant, markedly induced the TOPflash luciferase activity (Figure 4B). Moreover, we transfected HEK293T cells with USP2a, treated the cells with the USP2 inhibitor ML364, and measured β-catenin activity. Consistently, ML364 treatment decreased the TOPflash (relative to FOPflash) luciferase activity in HEK293T cells with or without USP2a overexpression (Figure 4C).
Figure 4.
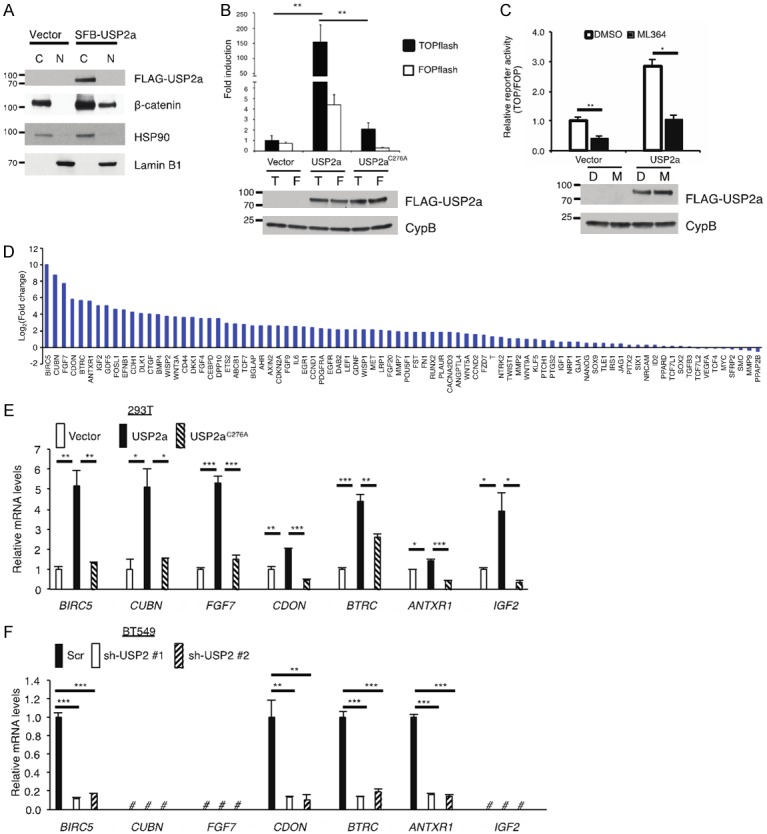
USP2a activates Wnt/β-catenin signaling. A. Immunoblotting of FLAG-USP2a, β-catenin, HSP90 (cytoplasmic marker), and Lamin B1 (nuclear marker) in cytoplasmic and nuclear fractions of HEK293T cells transfected with the empty vector or SFB-USP2a. B. Top panel: either β-catenin-responsive TOPflash or its mutant FOPflash construct was co-transfected with SFB-tagged GFP, USP2a, or USP2aC276A and Renilla luciferase into HEK293T cells. A dual luciferase assay was used to determine β-catenin activity. Firefly luciferase activity was normalized to Renilla luciferase activity. Error bars are S.D. **: P < 0.01. Bottom panel: immunoblotting of SFB-USP2a, SFB-USP2aC276A, and CypB in HEK293T cells. T: TOPflash; F: FOPflash. C. Top panel: either β-catenin-responsive TOPflash or its mutant FOPflash construct was co-transfected with the SFB vector or SFB-USP2a and Renilla luciferase into HEK293T cells. Cells were treated with DMSO or 3 µM ML364 for 24 hours. A dual luciferase assay was used to determine β-catenin activity. Firefly luciferase activity was normalized to Renilla luciferase activity, and then TOPflash reads were normalized to FOPflash reads. Error bars are S.E.M. *: P < 0.05; **: P < 0.01. Bottom panel: immunoblotting of SFB-USP2a and CypB in HEK293T cells. D: DMSO; M: ML364. D. Human Wnt signaling targets PCR array analysis of HEK293T cells transfected with SFB-USP2a. Gene expression levels in SFB-USP2a-transfected cells were compared to those in empty vector-transfected cells (log2 scale). E. qPCR of BIRC5, CUBN, FGF7, CDON, BTRC, ANTXR1, and IGF2 in HEK293T cells transfected with the empty vector, SFB-USP2a, or SFB-USP2aC276A. Error bars are S.E.M. *: P < 0.05; **: P < 0.01; ***: P < 0.001. F. qPCR of BIRC5, CUBN, FGF7, CDON, BTRC, ANTXR1, and IGF2 in BT549 cells transduced with USP2 shRNAs or a scramble control (Scr). Error bars are S.E.M. *: P < 0.05; **: P < 0.01; ***: P < 0.001; #: not detectable.
To further investigate the effect of USP2a on Wnt/β-catenin signaling, we determined whether USP2a regulates the expression of β-catenin target genes by using a qPCR array, which profiles the expression of 84 key genes responsive to Wnt/β-catenin pathway activation. We transfected HEK293T cells with USP2a and found that 57 out of 84 genes on the array were upregulated by more than 2-fold (Figure 4D). We then validated the effect of USP2a on the top seven upregulated genes from the qPCR array analysis, including BIRC5, CUBN, FGF7, CDON, BTRC, ANTXR1, and IGF2. To do so, we transiently transfected HEK293T cells with USP2a or its enzyme-dead mutant C276A and determined the expression of these seven genes by individual qPCR assays. We found that wild-type USP2a, but not the C276A mutant, significantly increased the mRNA levels of these seven genes (Figure 4E). In addition, we stably knocked down USP2a in BT549 cells and measured the expression of these seven genes by qPCR. Among these seven β-catenin targets, three genes (CUBN, FGF7, and IGF2) were not detectable in BT549 cells, while the mRNA levels of BIRC5, CDON, BTRC, and ANTXR1 were significantly decreased upon knockdown of USP2a (Figure 4F). Taken together, our data suggest that USP2a not only stabilizes β-catenin but also promotes the activity of Wnt/β-catenin signaling.
Discussion
The canonical Wnt signaling pathway is activated in multiple types of cancer, including colorectal cancer and breast cancer [1]. The Wnt pathway effector β-catenin can be stabilized in tumors via overexpression of Wnt ligands, downregulation of Wnt antagonists, loss of the APC tumor suppressor, and other mechanisms [23]. Upon its stabilization, β-catenin protein accumulates in the nucleus where it activates gene expression by associating with the TCF family of transcription factors. In human breast cancer where Wnt/β-catenin signaling is often activated, mutations in APC, Axin, or β-catenin are usually absent [24]. One possibility is that dysregulation of proteolytic ubiquitination in the absence of genetic alterations may play a major role in β-catenin signaling activation in breast cancer.
It has been recently reported that three deubiquitinases, USP47, USP9X, and USP20, promote β-catenin deubiquitination [25-27]. In our interaction screen, USP47 did not stand out as a β-catenin-interacting deubiquitinase (Figure 1A). In addition, compared with the six candidate deubiquitinases identified in our reporter screen, USP47 had a minor effect on β-catenin activity without Wnt stimulation (Figure 1B), suggesting that USP47 may not play a major role in regulating basal β-catenin activity in our screening system, or that the regulation of β-catenin by USP47 may be cell-type specific. Yang et al. identified USP9X as a deubiquitinase that stabilizes β-catenin, enhances Wnt signaling, and promotes glioma cell proliferation [26]. However, the role of USP9X in cancer is highly tissue-type dependent. On one hand, USP9X stabilizes MCL1 and promotes lymphoma cell survival [28]. On the other hand, USP9X is a tumor suppressor in pancreatic cancer [29,30], raising caution about targeting USP9X as a therapeutic strategy. USP20 was recently reported as a β-catenin deubiquitinase that promotes tumorigenesis and chemoresistance [27]. In our screen, we also found that USP20 interacted with β-catenin (Figure 1A); however, USP20 did not stood out in our reporter screen, because it increased the TOPflash signal by only ~4-fold while increasing the FOPflash signal by ~2-fold (Figure 1B). A possible explanation is that different screening systems, including assays, cell lines, and data analysis methods, may yield different top candidates.
USP2 is overexpressed in human breast tumors and has been shown to promote migration and invasion of triple-negative breast cancer (TNBC) cells [31]. Several proteins have previously been identified as the USP2 substrates. For instance, USP2a can stabilize MDM2 [32], fatty acid synthase [33], EGFR [34], RIP1 [35], and TRAF2 [36]. Because USP2a regulates key signaling molecules in cancer, it has attracted growing interests for its therapeutic potential [22,37]. In the present study, we identified USP2a as a bona fide β-catenin deubiquitinase that deubiquitinates and stabilizes β-catenin. Whether USP2a promotes breast tumorigenesis and metastasis through stabilization of β-catenin warrants future investigation. Importantly, here we found that genetic depletion or pharmacological inhibition of USP2 in TNBC cells destabilized β-catenin, suggesting that USP2 may serve as a novel therapeutic target of Wnt/β-catenin signaling in certain cancers such as TNBC.
Acknowledgements
We thank MD Anderson’s shRNA and ORFeome Core and Characterized Cell Line Core Facility for technical assistance. We thank Dr. Anton Simeonov (National Institutes of Health) for providing the USP2 inhibitor ML364. We are grateful to all members of the Ma Lab for discussion and to Baochau Ton for critical reading of the manuscript. L.M. is supported by US National Institutes of Health (NIH) grants R01CA166051 and R01CA181029, a Cancer Prevention and Research Institute of Texas (CPRIT) grant RP150319, and a Stand Up To Cancer Innovative Research Grant (award number: 403235).
Disclosure of conflict of interest
None.
References
- 1.Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149:1192–1205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 2.Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nat Rev Cancer. 2008;8:387–398. doi: 10.1038/nrc2389. [DOI] [PubMed] [Google Scholar]
- 3.Gao C, Xiao G, Hu J. Regulation of Wnt/beta-catenin signaling by posttranslational modifications. Cell Biosci. 2014;4:13. doi: 10.1186/2045-3701-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhan T, Rindtorff N, Boutros M. Wnt signaling in cancer. Oncogene. 2017;36:1461–1473. doi: 10.1038/onc.2016.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Polakis P. Wnt signaling in cancer. Cold Spring Harb Perspect Biol. 2012:4. doi: 10.1101/cshperspect.a008052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Polakis P. The many ways of Wnt in cancer. Curr Opin Genet Dev. 2007;17:45–51. doi: 10.1016/j.gde.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 7.Polakis P. Drugging Wnt signalling in cancer. EMBO J. 2012;31:2737–2746. doi: 10.1038/emboj.2012.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wilkinson KD. Regulation of ubiquitin-dependent processes by deubiquitinating enzymes. FASEB J. 1997;11:1245–1256. doi: 10.1096/fasebj.11.14.9409543. [DOI] [PubMed] [Google Scholar]
- 9.Xiao Z, Zhang P, Ma L. The role of deubiquitinases in breast cancer. Cancer Metastasis Rev. 2016;35:589–600. doi: 10.1007/s10555-016-9640-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davis MI, Simeonov A. Ubiquitin-specific proteases as druggable targets. Drug Target Rev. 2015;2:60–64. [PMC free article] [PubMed] [Google Scholar]
- 11.D’Arcy P, Wang X, Linder S. Deubiquitinase inhibition as a cancer therapeutic strategy. Pharmacol Ther. 2015;147:32–54. doi: 10.1016/j.pharmthera.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 12.Yao F, Zhou Z, Kim J, Hang Q, Xiao Z, Ton BN, Chang L, Liu N, Zeng L, Wang W, Wang Y, Zhang P, Hu X, Su X, Liang H, Sun Y, Ma L. SKP2-and OTUD1-regulated non-proteolytic ubiquitination of YAP promotes YAP nuclear localization and activity. Nat Commun. 2018;9:2269. doi: 10.1038/s41467-018-04620-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang J, Zhang P, Wei Y, Piao HL, Wang W, Maddika S, Wang M, Chen D, Sun Y, Hung MC, Chen J, Ma L. Deubiquitylation and stabilization of PTEN by USP13. Nat Cell Biol. 2013;15:1486–1494. doi: 10.1038/ncb2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang P, Wei Y, Wang L, Debeb BG, Yuan Y, Zhang J, Yuan J, Wang M, Chen D, Sun Y, Woodward WA, Liu Y, Dean DC, Liang H, Hu Y, Ang KK, Hung MC, Chen J, Ma L. ATM-mediated stabilization of ZEB1 promotes DNA damage response and radioresistance through CHK1. Nat Cell Biol. 2014;16:864–875. doi: 10.1038/ncb3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou Z, Zhang P, Hu X, Kim J, Yao F, Xiao Z, Zeng L, Chang L, Sun Y, Ma L. USP51 promotes deubiquitination and stabilization of ZEB1. Am J Cancer Res. 2017;7:2020–2031. [PMC free article] [PubMed] [Google Scholar]
- 16.Stenoien DL, Patel K, Mancini MG, Dutertre M, Smith CL, O’Malley BW, Mancini MA. FRAP reveals that mobility of oestrogen receptor-alpha is ligand- and proteasome-dependent. Nat Cell Biol. 2001;3:15–23. doi: 10.1038/35050515. [DOI] [PubMed] [Google Scholar]
- 17.Wang YJ, Bian Y, Luo J, Lu M, Xiong Y, Guo SY, Yin HY, Lin X, Li Q, Chang CCY, Chang TY, Li BL, Song BL. Cholesterol and fatty acids regulate cysteine ubiquitylation of ACAT2 through competitive oxidation. Nat Cell Biol. 2017;19:808–819. doi: 10.1038/ncb3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhong B, Liu X, Wang X, Chang SH, Liu X, Wang A, Reynolds JM, Dong C. Negative regulation of IL-17-mediated signaling and inflammation by the ubiquitin-specific protease USP25. Nat Immunol. 2012;13:1110–1117. doi: 10.1038/ni.2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Veeman MT, Slusarski DC, Kaykas A, Louie SH, Moon RT. Zebrafish prickle, a modulator of noncanonical Wnt/Fz signaling, regulates gastrulation movements. Curr Biol. 2003;13:680–685. doi: 10.1016/s0960-9822(03)00240-9. [DOI] [PubMed] [Google Scholar]
- 20.Shi Y, Solomon LR, Pereda-Lopez A, Giranda VL, Luo Y, Johnson EF, Shoemaker AR, Leverson J, Liu X. Ubiquitin-specific cysteine protease 2a (USP2a) regulates the stability of Aurora-A. J Biol Chem. 2011;286:38960–38968. doi: 10.1074/jbc.M111.231498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bozza WP, Liang Q, Gong P, Zhuang Z. Transient kinetic analysis of USP2-catalyzed deubiquitination reveals a conformational rearrangement in the K48-linked diubiquitin substrate. Biochemistry. 2012;51:10075–10086. doi: 10.1021/bi3009104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davis MI, Pragani R, Fox JT, Shen M, Parmar K, Gaudiano EF, Liu L, Tanega C, McGee L, Hall MD, McKnight C, Shinn P, Nelson H, Chattopadhyay D, D’Andrea AD, Auld DS, DeLucas LJ, Li Z, Boxer MB, Simeonov A. Small molecule inhibition of the ubiquitin-specific protease USP2 accelerates cyclin D1 degradation and leads to cell cycle arrest in colorectal cancer and mantle cell lymphoma models. J Biol Chem. 2016;291:24628–24640. doi: 10.1074/jbc.M116.738567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.King TD, Suto MJ, Li Y. The Wnt/betacatenin signaling pathway: a potential therapeutic target in the treatment of triple negative breast cancer. J Cell Biochem. 2012;113:13–18. doi: 10.1002/jcb.23350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Howe LR, Brown AM. Wnt signaling and breast cancer. Cancer Biol Ther. 2004;3:36–41. doi: 10.4161/cbt.3.1.561. [DOI] [PubMed] [Google Scholar]
- 25.Shi J, Liu Y, Xu X, Zhang W, Yu T, Jia J, Liu C. Deubiquitinase USP47/UBP64E regulates beta-catenin ubiquitination and degradation and plays a positive role in wnt signaling. Mol Cell Biol. 2015;35:3301–3311. doi: 10.1128/MCB.00373-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang B, Zhang S, Wang Z, Yang C, Ouyang W, Zhou F, Zhou Y, Xie C. Deubiquitinase USP9X deubiquitinates beta-catenin and promotes high grade glioma cell growth. Oncotarget. 2016;7:79515–79525. doi: 10.18632/oncotarget.12819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu C, Luo K, Zhao F, Yin P, Song Y, Deng M, Huang J, Chen Y, Li L, Lee S, Kim J, Zhou Q, Tu X, Nowsheen S, Luo Q, Gao X, Lou Z, Liu Z, Yuan J. USP20 positively regulates tumorigenesis and chemoresistance through betacatenin stabilization. Cell Death Differ. 2018 doi: 10.1038/s41418-018-0138-z. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schwickart M, Huang X, Lill JR, Liu J, Ferrando R, French DM, Maecker H, O’Rourke K, Bazan F, Eastham-Anderson J, Yue P, Dornan D, Huang DC, Dixit VM. Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature. 2010;463:103–107. doi: 10.1038/nature08646. [DOI] [PubMed] [Google Scholar]
- 29.Toloczko A, Guo F, Yuen HF, Wen Q, Wood SA, Ong YS, Chan PY, Shaik AA, Gunaratne J, Dunne MJ, Hong W, Chan SW. Deubiquitinating enzyme USP9X suppresses tumor growth via LATS kinase and core components of the hippo pathway. Cancer Res. 2017;77:4921–4933. doi: 10.1158/0008-5472.CAN-16-3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pérez-Mancera PA, Rust AG, van der Weyden L, Kristiansen G, Li A, Sarver AL, Silverstein KA, Grützmann R, Aust D, Rümmele P, Knösel T, Herd C, Stemple DL, Kettleborough R, Brosnan JA, Li A, Morgan R, Knight S, Yu J, Stegeman S, Collier LS, ten Hoeve JJ, de Ridder J, Klein AP, Goggins M, Hruban RH, Chang DK, Biankin AV, Grimmond SM Australian Pancreatic Cancer Genome Initiative. Wessels LF, Wood SA, Iacobuzio-Donahue CA, Pilarsky C, Largaespada DA, Adams DJ, Tuveson DA. The deubiquitinase USP9X suppresses pancreatic ductal adenocarcinoma. Nature. 2012;486:266–270. doi: 10.1038/nature11114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qu Q, Mao Y, Xiao G, Fei X, Wang J, Zhang Y, Liu J, Cheng G, Chen X, Wang J, Shen K. USP2 promotes cell migration and invasion in triple negative breast cancer cell lines. Tumour Biol. 2015;36:5415–5423. doi: 10.1007/s13277-015-3207-7. [DOI] [PubMed] [Google Scholar]
- 32.Stevenson LF, Sparks A, Allende-Vega N, Xirodimas DP, Lane DP, Saville MK. The deubiquitinating enzyme USP2a regulates the p53 pathway by targeting Mdm2. EMBO J. 2007;26:976–986. doi: 10.1038/sj.emboj.7601567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Graner E, Tang D, Rossi S, Baron A, Migita T, Weinstein LJ, Lechpammer M, Huesken D, Zimmermann J, Signoretti S, Loda M. The isopeptidase USP2a regulates the stability of fatty acid synthase in prostate cancer. Cancer Cell. 2004;5:253–261. doi: 10.1016/s1535-6108(04)00055-8. [DOI] [PubMed] [Google Scholar]
- 34.Liu Z, Zanata SM, Kim J, Peterson MA, Di Vizio D, Chirieac LR, Pyne S, Agostini M, Freeman MR, Loda M. The ubiquitin-specific protease USP2a prevents endocytosis-mediated EGFR degradation. Oncogene. 2013;32:1660–1669. doi: 10.1038/onc.2012.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mahul-Mellier AL, Datler C, Pazarentzos E, Lin B, Chaisaklert W, Abuali G, Grimm S. Deubiquitinating proteases USP2a and USP2c cause apoptosis by stabilising RIP1. Biochim Biophys Acta. 2012;1823:1353–1365. doi: 10.1016/j.bbamcr.2012.05.022. [DOI] [PubMed] [Google Scholar]
- 36.Mahul-Mellier AL, Pazarentzos E, Datler C, Iwasawa R, AbuAli G, Lin B, Grimm S. De-ubiquitinating protease USP2a targets RIP1 and TRAF2 to mediate cell death by TNF. Cell Death Differ. 2012;19:891–899. doi: 10.1038/cdd.2011.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tomala MD, Magiera-Mularz K, Kubica K, Krzanik S, Zieba B, Musielak B, Pustula M, Popowicz GM, Sattler M, Dubin G, Skalniak L, Holak TA. Identification of small-molecule inhibitors of USP2a. Eur J Med Chem. 2018;150:261–267. doi: 10.1016/j.ejmech.2018.03.009. [DOI] [PubMed] [Google Scholar]