

Abstract
Objectives: The composition and structure of site-specific microbiota have been investigated as potential biomarkers for a variety of chronic inflammatory diseases and cancers. While many studies have focused on the changes in the airway microbiota using respiratory specimens from patients with various respiratory diseases, more research is needed to explore the microbial profiles within the distal lung parenchyma in smokers with lung cancer and/or emphysema. Materials and Methods: To describe and contrast lung tissue-associated microbial signatures in smokers with lung cancer and/or emphysema, we employed culture-independent pyrosequencing of 16S rRNA gene hypervariable V4 region and compositional analysis in non-malignant lung tissue samples obtained from 40 heavy smokers, including 10 emphysema-only, 11 lung cancer-only, and 19 with both lung cancer and emphysema. Results and Conclusion: The emphysema-only group presented a lower bacterial community evenness defined by a significantly lower Shannon diversity index compared to the lung cancer patients with or without emphysema (P = 0.006). Furthermore, community compositions of lung cancer patients with or without emphysema were characterized by a significantly lower abundance of Proteobacteria (primary the genera Acinetobacter and Acidovorax) and higher prevalence of Firmicutes (Streptococcus) and Bacteroidetes (Prevotella), compared to emphysema-only patients. In conclusion, the lung microbial composition and communities structures of smokers with lung cancer are distinct from the emphysema-only patients. Although preliminary, our findings suggest that lung microbiome changes could be a biomarker of lung cancer that could eventually be used to help screening for the disease.
Keywords: Lung cancer, emphysema, microbiome, 16S rRNA gene, bacteria
Introduction
The relationship between smoking, airflow obstruction, and lung cancer (LC) is well recognized. We and others have previously shown that tobacco-induced chronic obstructive airways disease (COPD) including both chronic bronchitis and emphysema, is characterized by sustained inflammation in the airways and lung parenchyma [1-4]. COPD is a significant contributor to LC risk in smokers with an observed three- to 10-fold increased risk of LC for smokers with COPD compared to smokers without emphysema or significant airway obstruction [5]. Recent genome-wide association studies have identified overlapping genetic susceptibility loci for LC, smoking behavior, COPD, and pulmonary function [6]. However, there is considerable unexplained inter-individual variation in susceptibility of long-term smokers to developing COPD and/or progressing from COPD to LC.
The human microbiota may alter cancer risk directly through action on local tissue microenvironment, indirectly via exposure to metabolites produced by the commensal bacteria, or by chronic immune stimulation and induction of immune exhaustion that collectively can reduce immune surveillance [7]. Metagenomics tools have been extensively applied to understanding microbial communities in the gut, skin, oral, nasal cavity, genital tract, and more recently, the respiratory tract. The lung had long been considered a sterile environment by culture-based techniques, thus initially excluded from the body sites in the original goals of the Human Microbiome Project (HMP). However, this assumption has been challenged by studies using modern culture-independent techniques (sequencing of bacterial 16S ribosomal RNA [rRNA] gene libraries) that indicate a low-abundance microbiota exists within distal airways of the lung [8-18]. The majority of these strains cannot be detected by routine culture-based technologies and were therefore missed in early studies addressing this topic. Also, there are selective pressures in the lungs for bacterial persistence, colonization and growth that uniquely shape the bacterial community of this body site. Furthermore, several epidemiological and molecular studies have shown serological evidence of an association between bacterium Chlamydia pneumoniae [19,20] and Nontypeable Haemophilus influenzae (NTHi) [21] infections and COPD development and LC promotion.
Due to the difficulty and invasiveness of sampling lung tissue, of the previous LC microbiome studies to date, only one have used the lung tissue of LC patients (the Environment and Genetics in LC Etiology [EAGLE] study) [22], others have relied on respiratory specimens (i.e., expectorated sputum, bronchoscopic brushing, or bronchoalveolar lavage [BAL] fluid) [23,24]; same as most previous research of respiratory disorders (i.e., COPD, asthma and cystic fibrosis) [25-33], few explanted lung tissue [14,25,34,35]. Therefore, the characteristics of lung tissue microbiota remain largely unknown. To address this knowledge gap, we sought to describe and contrast lung microbial signatures in surgically excised tissue samples of smokers with LC and/or emphysema, by using culture-independent 16S rRNA gene sequencing.
Methods
Study population
Eligible subjects were selected from the lung tissue bank collected as part of a COPD and Emphysema Study that enrolled current- or former- smokers at the Houston Methodist Hospital and Michael E. DeBakey Veterans Affairs Medical Center in Houston, Texas, from 2002 to 2012 [36,37]. Demographic and clinical information were collected. Pulmonary function tests (PFT) including forced vital capacity (FVC), forced expiratory volume in 1 second (FEV1), peak expiratory flow, and the ratio of FVC and FEV1, and chest computed tomography (CT) scans were used to determine the clinical phenotype of three groups of subjects: emphysema-only (moderate-to-severe), LC-only, both LC and emphysema (any emphysema). The LC-only patients did not have any evidence for emphysema per CT scan or airflow obstruction by PFT. The Global Obstructive Lung Disease (GOLD) classification score was used to classify the severity of airflow obstruction associated with spirometry.
Patients with history of prior cancer, other significant systemic or respiratory conditions, chronic viral infections (e.g. HIV, Hepatitis B, or C), bacterial infection or antibiotic use within six weeks prior to surgery, and those on immunosuppressive treatment (e.g. chemotherapy, oral steroids, etc.), were excluded. No LC patients had received chemotherapy, radiation therapy, or other treatments for LC before surgery. Cases were selected from lung tissue samples collected from heavy smokers (≥ 20 pack-years, PPY), who were over 50-years of age, and required medically necessary surgery for treatment of LC (LC-only or LC with emphysema), and lung transplant or lung volume reduction for treatment of end-stage emphysema (emphysema-only). The smoking PPY were calculated by multiplying the total number of years smoked by the average number of cigarettes smoked per day divided by 20. All subjects were informed about the nature of the study and provided written informed consent for their participation. The protocol was reviewed and approved by the Committee for the Protection of Human Subjects at Baylor College of Medicine.
Lung tissue sample preparation and microbial DNA isolation
Archived lung tissue was separated into three groups: 1) emphysema that included lung volume reduction surgery or lung transplant; 2) lobectomy for treatment of LC with emphysema; 3) lobectomy for treatment of LC without emphysema. In cases of LC surgery, tumor-free lung tissue was collected at least 10 cm away from the cancer, and in all cases lung tissue were collected sterilely into Hanks balanced salt solution in the operating room and were transported at 4°C to the laboratory ≤ 30 minutes. Small tissue fragments from each sample were embedded in optimal cutting temperature compound, flash frozen in liquid nitrogen, and stored at -80°C. For each subject, usually more than one tissue sample was collected and at least one sample was examined by a pathologist to confirm the absence of tumor nuclei.
Microbial DNA was extracted from tumor-free lung tissue using Powerlyzer UltraClean Microbial DNA Isolation Kit (MO BIO Laboratories, Carlsbad, CA) according to the manufacturer’s instructions. The isolated DNA stored at -20°C to -80°C to prevent degradation prior to amplification steps. To reduce potential procedural contamination, one laboratory technician processed (i.e., aliquoted, frozen, and stored) all the samples and extracted the microbial DNA with the same equipment, kits and reagents. All the tools and materials in contact with the lung tissues were sterile.
16S rRNA gene sequencing and bioinformatics analysis
Bacterial 16S rRNA gene profiling was conducted by The Alkek Center for Metagenomics and Microbiome Research at Baylor College of Medicine as previously described [38]. Briefly, the 16S rRNA gene hypervariable V4 region was amplified and sequenced using the Illumina MiSeq platform. Sequences that did not pass quality checks were removed from the analysis. The read pairs were de-multiplexed based on the unique molecular barcodes, reads were merged using the Ultra-fast Sequence Analysis (USEARCH) [39], and aligned against the SILVA 16S database. We used an in-house pipeline for 16S rRNA analysis. Sequences were assigned into Operational Taxonomic Units (OTUs) at 97% similarity using the UPARSE pipeline [40]. Unclassified sequences were removed from the dataset to exclude non-bacterial sequences.
The taxonomic alpha-diversity (e.g., number of observed OTUs, Shannon diversity index-an indicator of evenness and richness) and relative abundance difference among the three groups were compared using the Kruskal Wallis ANOVA; for difference between the two groups, LC patients (LC-only or LC with emphysema) versus emphysema-only patients, the Wilcoxon rank sum test were performed. Taxonomic betadiversity analysis of bacterial communities was assessed with the weighted UniFrac metric [41] (the calculated value is the sum of the branches weighted by the difference in the number of descendants from each community for each branch), and visualized using principal coordinate analysis (PCoA) of the distance matrix [42] or hierarchical clustering. Between-group differences in beta-diversity were assessed with PERMANOVA [43]. False Discovery Rate (FDR) was used to control for multiple testing [44].
Results
Characteristics of study population
A total of 44 lung tissue samples from 44 heavy smokers were submitted for 16S rRNA gene sequencing, but four samples’ (two LC-only and two patients with both LC and emphysema) sequences didn’t pass the quality control due to suboptimal amplification and read count, bringing our final sample size to 40. The final samples included 10 moderate-to-severe emphysema-only patients, 30 LC patients (11 LC-only and 19 with both LC and emphysema). The demographics and clinical characteristics of the subjects are summarized in Table 1. The vast majority (90%) of patients were male and White. Compare to the emphysema-only patients, the LC patients were heavier smokers (median PPY, 58 versus 55), slightly older (median age, 65 versus 62), and had a significantly higher FEV1 and FEV1/FVC (P < 0.0001). The location of lung tissue in emphysema-only patients were all upper lobe, while the LC patients contained both upper and lower lobes (P = 0.002). Of the LC patients, 43.3% were adenocarcinoma, 46.7% were squamous cell carcinoma, and 9% were small-cell lung carcinoma; most LC patients were early stage (80%).
Table 1.
Basic characteristics of study participants
| Characteristics | Emphysema-only (n = 10) | LC-only (n = 11) | LC with emphysema (n = 19) | P among 3 groups | P between LC (n = 30) and emphysema-only (n = 10) |
|---|---|---|---|---|---|
| Race | 0.056 | 0.956 | |||
| White | 9 | 8 | 19 | ||
| Hispanic and Black | 1 | 3 | - | ||
| Age, years | 0.899 | 0.989 | |||
| Median (range) | 61 (58-78) | 65 (51-80) | 64 (50-77) | ||
| Gender | 0.450 | 0.297 | |||
| Male | 8 | 10 | 18 | ||
| Female | 2 | 1 | 1 | ||
| Smoking status | 0.301 | 0.147 | |||
| Current | - | 2 | 4 | ||
| Former | 10 | 9 | 15 | ||
| Smoking PPY | 0.620 | 0.462 | |||
| Median (range) | 55 (30-120) | 54 (20-129) | 60 (22-160) | ||
| Tissue location | 0.009 | 0.002 | |||
| Lower lobe | - | 5 | 11 | ||
| Upper lobe | 10 | 6 | 8 | ||
| Histology | - | - | |||
| Adenocarcinoma | - | 7 | 6 | ||
| Squamous cell carcinoma | - | 3 | 11 | ||
| Small-cell | - | 1 | 2 | ||
| Tumor stage# | - | - | |||
| Early (T1-T2) | - | 8 | 13 | ||
| Advanced (T3) | - | 1 | 3 | ||
| FEV1 (% predicted) | < 0.0001 | < 0.0001 | |||
| Median (range) | 26 (17-39) | 94 (73-116) | 60 (32-82) | ||
| FEV1/FVC (%) | < 0.0001 | < 0.0001 | |||
| Median (range) | 30.5 (20-39) | 82 (70-100) | 61 (34-71) |
Abbreviations: LC, lung cancer; PPY, pack-year; FEV1, forced expiratory volume in one second; FVC,forced vital capacity.
Numbers do not add up to the column totals due to missing values.
Lung bacterial community richness and alpha diversity
Across all 40 lung tissue samples, 186,972 high-quality 16S (v4) rRNA gene sequences were generated. The dataset was rarefied (normalized) to 1,080 reads and these were classified into 383 OTUs. The number of observed OTUs (richness) did not differ among the three groups (Figure 1A). However, analysis of the Shannon diversity index showed a statistically significant lower index average for the emphysema-only group when compared with the LC patients with or without emphysema (FDR-adjusted P = 0.006; Figure 1B). We found some evidence for a difference in the Shannon diversity index among the three groups (FDR-adjusted P = 0.022). These findings indicate that the emphysema-only group harbored a similar bacterial richness but exhibited a less even community distinct from those with LC-only or both LC and emphysema. These results suggest that samples from the emphysema-only group are dominated by a few bacterial species thus impacting the distribution of the observed OTUs. However, there is no significant difference between the two LC patients groups (LC-only versus LC with emphysema, FDR-adjusted P = 0.47).
Figure 1.
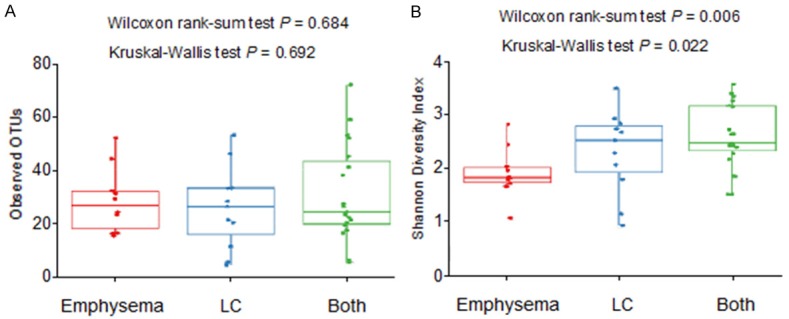
Alpha-diversity of the lung bacterial communities in LC and emphysema patients. A. Number of observed OTUs (operational taxonomic units), a measure of species richness; B. Shannon diversity index, a measure of species richness and evenness. The P values of the Kruskal-Wallis test refer to the comparison among the three groups: emphysema-only (n = 10, red circles), LC only (n = 11, blue circles), and both LC and emphysema (n = 19, green circles). The P values of the Wilcoxon rank-sum test, refer to the comparison between emphysema-only (red circles, n = 10) and LC patients with or without emphysema (blue and green circles, n = 30).
We also examined the difference of bacterial community richness and alpha diversity by epidemiological and clinical variables. There were no significant differences observed in age (continuous), sex, race, and smoking status (former versus current). Although not significant, the number of observed OTUs decreased as the number of smoking PPY (quartiles) increased across all study subjects, with the Kruskal-Wallis test P-trend of 0.36 (Figure 2A); among the three groups, with P-trend of 0.54 for emphysema-only, 0.14 for LC-only, and 0.71 for LC patients with emphysema (Figure 2B). In addition, no significant alpha diversity differences by lung tissue location (upper versus lower lobe), histology (adenocarcinoma versus squamous cell carcinoma), and tumor stage (early versus advanced) in LC patients.
Figure 2.
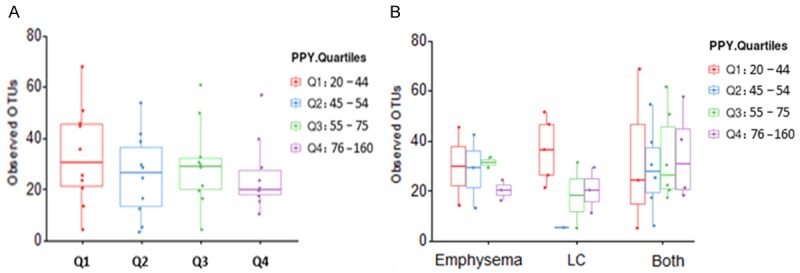
Smoking PPY and observed OTUs. A. In all samples (N = 40): Although not significant, the number of observed OTUs (operational taxonomic units) decreased as the number of PPY (pack-year) increased, with the Kruskal-Wallis test P-trend of 0.36. B. Among the three groups: the Kruskal-Wallis test P-trend were 0.54, 0.14 and 0.71, for emphysema-only (n = 10), LC-only (n = 11), and both LC and emphysema patients (n = 19), respectively.
LC lung bacterial community differed significantly from emphysema-only group
To better discern the relationship between bacterial communities from the same site but different individuals, we performed cluster analysis using weighted UniFrac, which takes into account both phylogenetic relationships and relative abundances of taxa within each community. PCoA based on the weighted UniFrac distance metric revealed two distinct clusters (P-value < 0.001): emphysema-only patients and LC patients (Figure 3). These two components explained 52.6.1% of the variance. The emphysema-only patient clustering was largely attributed to abundance differences in dominant phyla, Proteobacteria, while the LC patient clustering was largely due to Firmicutes. The LC subjects exhibited higher dispersion in the structure of the lung microbiota than emphysema-only patients. The community structures of LC patients, with or without emphysema, clustered together and were indistinguishable, suggesting they share a similar bacterial structure.
Figure 3.
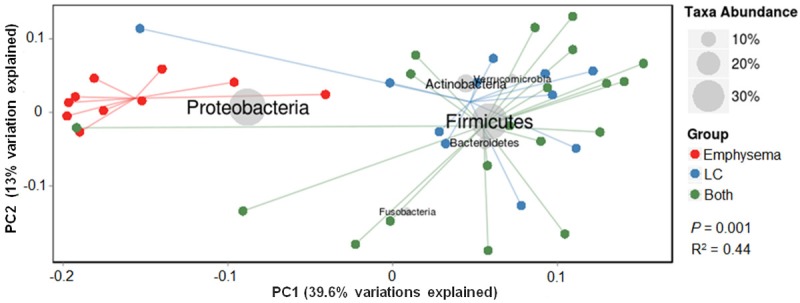
Principal coordinate analysis of lung bacterial communities based on weighted Unifrac distances. Each symbol represents a sample. The proportion of variance explained by each principal component is denoted in the corresponding axis label. These two components explained 52.6.1% of the variance. The lung bacterial communities of the LC patients, with (green circles) or without (blue circles) emphysema, were clustered together, and distinctly separated from the emphysema-only patients (red circles). Both P values from the Kruskal-Wallis test (comparison among three groups) and the Wilcoxon rank-sum test (emphysema-only versus LC patients), were 0.001.
Shift of Proteobacteria and Firmicutes drives the lung bacterial microbiome
The most abundant five bacterial phyla observed in the lung tissue samples were Proteobacteria, Firmicutes, Actinobacteria, Bacteriodetes, and Verrucomicrobia (Table 2; Figures 4A and 5A). Additionally, Fusobacteria, Cyanobacteria and Acidobacteria were present in much lower proportions. Similar to the alpha diversity, the relative abundance of the dominant phyla also varied significantly among the three groups and between the two groups. Patients with emphysema-only were characterized by a substantially higher abundance of Proteobacteria (mean relative abundance 71.7%) and markedly lower prevalence of Firmicutes (12.7%) and Bacteroidetes (2.2%) phyla, compared to LC patients, with FDR-adjusted P values of 0.00003, 0.00003 and 0.013, respectively. In comparison, Firmicutes was the predominant phylum for LC patients (43.7%); whereas Proteobacteria were the subdominant phylum in LC patients (24.7%). The phyla composition of the LC-only patients appeared to be very similar to the LC patients with emphysema (Table 2).
Table 2.
The community composition and relative abundance of non-malignant lung tissue bacterial taxa
| Taxa | Mean relative abundance, %* | P among 3 groups# | P between LC and emphysema-only† | ||
|
| |||||
| Emphysema-only | LC-only | LC with emphysema | |||
|
| |||||
| Phylum | |||||
|
| |||||
| Proteobacteria | 71.7 | 28.9 | 22.3 | 0.0001 | 0.00003 |
| Firmicutes | 12.7 | 43.0 | 44.1 | 0.0002 | 0.00007 |
| Bacteroidetes | 2.2 | 6.93 | 10.4 | 0.0103 | 0.004 |
| Fusobacteria | 0.72 | 0.02 | 2.56 | 0.023 | 0.466 |
| Actinobacteria | 6.82 | 16.6 | 15.7 | 0.063 | 0.021 |
| Verrucomicrobia | 1.09 | 3.60 | 3.62 | 0.314 | 0.199 |
| Cyanobacteria | 2.75 | 0.72 | 1.13 | 0.660 | 0.372 |
| Acidobacteria | 1.98 | - | 0.15 | 0.465 | 0.242 |
|
| |||||
| Genus (Phylum) | |||||
|
| |||||
| Acinetobacter (Proteobacteria) | 55.5 | 6.03 | 4.22 | < 0.00001 | < 0.00001 |
| Acidovorax (Proteobacteria) | 8.23 | 0.41 | 0.15 | < 0.00001 | < 0.00001 |
| Prevotella (Bacteroidetes) | 0.46 | 4.43 | 5.30 | 0.021 | 0.006 |
| Bifidobacterium (Actinobacteria) | 3.41 | 14.2 | 13.0 | 0.027 | 0.009 |
| Streptococcus (Firmicutes) | 0.10 | 1.98 | 4.95 | 0.026 | 0.023 |
| Diaphorobacter (Proteobacteria) | 1.68 | 0.26 | 1.13 | 0.032 | 0.011 |
| Fusobacterium (Fusobacteria) | 0.71 | - | 1.61 | 0.051 | 0.811 |
| Sphingomonas (Proteobacteria) | 2.75 | 0.15 | 0.45 | 0.120 | 0.043 |
| Ruminococcus (Firmicutes) | 1.03 | 6.02 | 3.83 | 0.095 | 0.101 |
| Aggregatibacter (Proteobacteria) | - | 1.86 | 0.47 | 0.201 | 0.077 |
| Bryobacter (Acidobacteria) | 1.97 | - | - | 0.223 | 0.094 |
| Limnobacter (Proteobacteria) | - | 1.46 | - | 0.268 | 0.603 |
| Finegoldia (Firmicutes) | - | 1.51 | 0.26 | 0.282 | 0.183 |
| Akkermansia (Verrucomicrobia) | 1.09 | 3.60 | 3.62 | 0.314 | 0.199 |
| Bartonella (Proteobacteria) | - | - | 1.18 | 0.322 | 0.432 |
| Bacillus (Firmicutes) | - | - | 2.23 | 0.338 | 0.463 |
| Escherichia/Shigella (Proteobacteria) | 0.11 | 2.30 | 2.26 | 0.345 | 0.163 |
| Methylobacterium (Proteobacteria) | - | 6.00 | 0.22 | 0.353 | 0.546 |
| Blautia (Firmicutes) | 0.31 | 4.89 | 1.57 | 0.372 | 0.182 |
| Selenomonas (Firmicutes) | - | 3.81 | 0.20 | 0.386 | 0.183 |
| Pantoea (Proteobacteria) | 0.42 | 3.11 | 0.11 | 0.394 | 0.202 |
| Haemophilus (Proteobacteria) | 0.11 | 1.04 | 2.08 | 0.545 | 0.672 |
| Bacteroides (Bacteroidetes) | 0.74 | 1.60 | 2.24 | 0.574 | 0.778 |
| Faecalibacterium (Firmicutes) | 1.88 | 2.19 | 4.86 | 0.577 | 0.700 |
| Staphylococcus (Firmicutes) | 1.33 | 3.89 | 2.40 | 0.638 | 0.891 |
| Janthinobacterium (Proteobacteria) | - | - | 1.29 | 0.652 | 0.432 |
| Enterobacter (Proteobacteria) | 0.19 | 0.76 | 1.13 | 0.663 | 0.378 |
| Neisseria (Proteobacteria) | 0.11 | 0.16 | 2.31 | 0.691 | 0.423 |
| Dialister (Firmicutes) | 1.00 | 1.63 | 3.21 | 0.757 | 0.621 |
| Sarcina (Firmicutes) | 1.12 | 1.23 | 1.21 | 0.948 | 0.851 |
| Anaerostipes (Firmicutes) | 0.51 | 0.72 | 1.39 | 0.976 | 0.943 |
| Paracoccus (Proteobacteria) | 0.20 | 1.78 | - | 0.999 | 0.999 |
Abbreviations: LC, lung cancer;
To decrease the number of features, we only focused on major taxa and operational taxonomic units (OTUs), defined as those having mean relative abundance > 1% in at least one of the three groups;
“-” indicates relative abundance < 0.1%.
The P values among three groups were determined by the Kruskal-Wallis test with false discovery rate (FDR) adjusted;
The P values between two groups determined by the Wilcoxon rank-sum test with FDR-adjusted.
Figure 4.
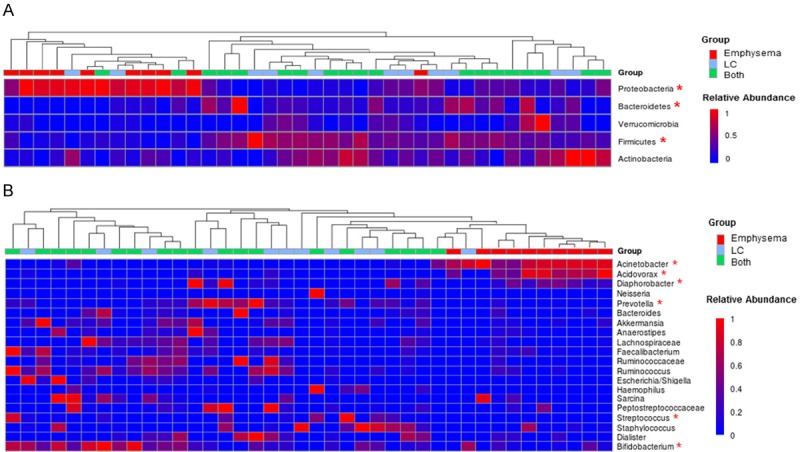
Relative abundance of bacterial genera in the lung communities among the three groups. A. Phylum level; B. Genus level. The colors represent the mean relative abundance (0 to 1, blue to red) of each genus among three groups: emphysema-only (n = 10, red squares), LC-only (n = 11, blue squares), and both LC and emphysema (n = 19, green squares). The asterisks indicates significantly different taxa.
Figure 5.
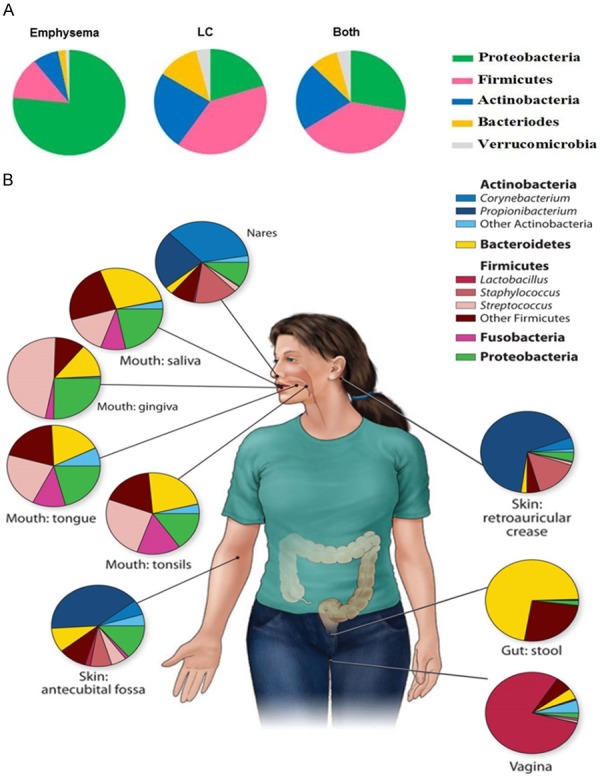
Bacterial distribution patterns in lung tissue and other body sites. A. Phylum-level bacteria in lung tissue among three groups; B. Phylum- and genus-level bacteria in healthy human subjects body sites from HMP (cited from [45]). The proportions of the mean relative abundance of each genus are shown.
Proceeding deeper into the genus composition (Table 2 and Figure 4B), the Proteobacteria-dominant communities in emphysema-only patients were primarily driven by the member of Acinetobacter and Acidovorax genus (FDR-adjusted P < 0.0001), with mean relative abundances 55.5% and 8.2%, respectively. This is equivalent to a 9- and 20-fold increase in these genera, respectively, when compared to the relative abundances of 6.0% or 0.4% in LC patients with emphysema, and equivalent to a 13 and 40-fold increase, respectively, compared to relative abundances of 4.2% and 0.2% in LC-only patients. In contrast, other Proteobacteria that less commonly in emphysema-only patients but more abundant in LC patients include Aggregatibacter, Escherichia/Shigella, Haemophilus and Neisseria. In addition, genera more enriched in LC patients than in the emphysema-only group, including Prevotella (Bacteroidetes), Bifidobacterium (Actinobacteria), Streptococcus (Firmicutes), Ruminococcus (Firmicutes), and Akkermansia (Verrucomicrobia). Among all the major taxa (defined as those having mean relative abundance > 1% in at least one of the three groups), 37% were unique genera and presented in only one or two groups.
Discussion
In this pilot study we carefully analyzed non-malignant lung parenchyma microbiota in three groups of smokers separated by the presence or absence of emphysema and/or LC. While there is a suggestive body of literature to support a role for microbiome in COPD [25-31,34,35], to our knowledge our study is one of only a few studies that has explored the microbiome among heavy smokers with LC and/or emphysema by directly sampling lung tissue, and using modern culture-independent 16S rRNA gene composition analysis. Compared to emphysema-only patients,LC patients harbored a dissimilar and distinct lung bacterial community structure,with significantly increasing Firmicutes and Bacteroidetes while decreasing Proteobacteria. These substantial phyla Proteobacteria-Firmicutes shifts inthe abundance (> 2.5-fold, P < 0.001) between emphysema-only and LC patients imply that specific bacterial populations have the potential to be selectively targeted by the two different diseases.
Our lung tissue bacterial show distinct phylum-level distribution patterns from other body sites of healthy subjects in HMP [45]. The distribution of Firmicutes (mostly Ruminococcus, Staphylococcus, Faecalibacterium, and Streptococcus) and Proteobacteria (primarily Acinetobacter) in the lung tissue in our study (Figure 5A), was most similar to the oral cavity than other sites (i.e., skin, vagina, and gut; Figure 5B). Specifically, in the HMP healthy subjects, Firmicutes (primarily Streptococcus) and Proteobacteria (mostly Neisseria and Haemophilus) dominants the oral cavity; Firmicutes (primarily Lactobacillus) dominates the vagina; Actinobacteria (mostly genera Corynebacterium and Propionibacterium) dominates the nasal cavity; Actinobacteria (primarily Propionibacterium) dominates the skin; Bacteroidetes and Firmicutes dominate the gut. Future research is needed to determine the extent to which these organisms are truly derived from the oral cavity. This would represent a novel approach to preventing and treating conditions associated with LC and emphysema.
In the current study, the most prominent and consistent finding was the dramatically high prevalence of Proteobacteria (primarily driven by Acinetobacter) in emphysema-only patients. Our results are in concordance with several airway microbiome studies in patients with COPD, asthma and cystic fibrosis, that showed increased Proteobacteria (mostly Neisseria, Moraxella, and Haemophilus) [25,28,29,31,35,46-51] and suggested promote lung inflammation and pathology [51]; Similar results were reported by Yu [22] that Proteobacteria(mostly Acinetobacter, Pseudomonas, and Ralstonia) is the dominant phyla (60%) in lung tissue of LC patients with or without other lung diseases (45% with COPD, 10% with emphysema, 15% with pneumonia, and 17% with bronchitis). Several pieces of evidence support the role of genera Acinetobacter in emphysema pathology. Acinetobacter species are ubiquitous in natural environments (e.g. soil and water) and certain species are considered important pathogens due to their multi-drug resistance associated with significant mortality [52-54]. Markedly, it has been shown that Acinetobacter species play an important role in disseminated infections, including the respiratory tract, blood stream, and urinary tract [52,55]. Interestingly, Acinetobacter was detected as one of the 15 major classes of bacteria metagenome of cigarettes [56]. Further detailed investigations of Acinetobacter in emphysema patients will be required.
Another remarkable finding was the higher abundance of genera, Streptococcus (Firmicutes), Prevotella (Bacteroidetes), Bifidobacterium (Actinobacteria), and Escherichia/Shigella and Haemophilus (Proteobacteria) in LC patients than the emphysema-only patients. These findings are consistent with recent studies, where the abundance of Streptococcus and Prevotella are increased in the gut microbiome of patients with colorectal cancer [57,58] and decreased in the BAL of asthmatics or COPD patients [28,49]. While Streptococcusis a proinflammatory bacterium, Bifidobacterium is an anti-inflammatory genus and was commonly used as a vector for anticancer genes in cancer gene therapy [59].
The contribution of the lung microbiome to LC is clearly understudied, in particular, in the lung tissue. To date, only one LC study have explored the role of the microbiota used the lung tissue samples [22] and others have used saliva or buccal specimens as surrogates of the lung [23,24]. Similarly, most of these previous studies in COPD, cystic fibrosis, and asthma have also relied on indirect methods to sample the lung, using the airways specimens such as BAL fluid [14,25-28] and sputum [26,29,30]. Only a few COPD studies have studied surgically acquired lung tissue samples at lung transplantation [25,34,35] and shown microbial communities differed significantly between BAL fluids and lung tissue in relative abundance [60], indicating that analyses of airway specimens may not provide a true representation of the lung microbiota due to contamination by the upper respiratory tract or oral microbiota [8]. Further, it has been demonstrated that there are geographical variations of bacterial composition within the same lung, dependent on the location of the tissue sampling [25], which reflects different habitats in different areas of the lung and possibly localized loci of inflammation that influence the constituents of the surrounding microbiota.
The main strength of this study is the use of surgically excised tissue from a decent number of LC and/or emphysema patients with well-characterized and phenotyped lung function data. The direct sampling of lung tissue in a single center provides a more specific characterization of microbiota than other sampling methods. Also, the uniform processing of all lung tissue specimens and exclusion of a number of confounding conditions (e.g. asthma, atopy, immune suppression, airway infection, antibiotics, or systemic steroids drugs use six weeks prior to sample collection) provided a unique group without large intra-group variations. Other strengths include nonculture-dependent sequencing-based microbiome assessment, which provided a comprehensive survey of the human lung microbiome. However, there are limitations of our study. The major limitation is the lack of validation set but these lung tissue specimens are difficult to obtain. Another limitation is the spirometry-based lung function (such as frequency of exacerbations or severity of airflow obstruction) were not evenly distributed between the emphysema-only and LC patients, this may confound the microbiota composition variance. In addition, given the increased sensitivity of newer molecular tools for microbiota profiling, potential contamination may introduced from specimen processing, laboratory reagents and sequencing.
In summary, our microbial 16S rRNA gene sequencing revealed significant differences of lung microbiome composition and patterns between LC and emphysema patients, in particular, the fundamental shift from microaerophilic Proteobacteria towards many of the obligate anaerobic Firmicutes species, from emphysema-only patients to LC patients. However, whether this bacterial dysbiosis is attributed to disease causation or is a consequence of host selection remains unclear. Future longitudinal studies, with larger sample sizes and control groups, using a metagenomic sequencing techniques will help to define the whole lung microbial community and advance our knowledge of the pathogenesis of LC and emphysema. If our preliminary findings are confirmed, the Proteobacteria (particularly genera Acinetobacter) could become novel microbial screening biomarkers to distinguish smokers at low- and high-risk for both diseases, or to serve as a novel target for therapeutic intervention. Ultimately, manipulation of the lung microbiota together with use of immune modulators such as inhaled TLR4 antagonists may prevent and treat LC and emphysema.
Acknowledgements
This study was supported by the National Institutes of Health (NIH) through grants HL110883, R01HL082487, 1K07CA181480, P30CA125123, P30ES023512 and R01CA127219. We thank Dr. Richard Gibbs, Ms. Donna M. Muzny, and Ms. Ginger A. Metcalf from Human Genome Sequence Center at Baylor College of Medicine for assistance with sequencing. We also thank Ms. Li-Zhen Song, Ms. Sarah Perusich and Ms. Anne Truong for assistance with sample collecting and processing.
Disclosure of conflict of interest
None.
Abbreviations
- LC
lung cancer
- COPD
chronic obstructive airways disease
- HMP
Human Microbiome Project
- rRNA
ribosomal RNA
- BAL
bronchoalveolar lavage
- PFT
pulmonary function tests
- PPY
pack-year
- FEV1
forced expiratory volume in 1 second
- FVC
forced vital capacity
- OTUs
operational taxonomic units
- FDR
false discovery rate
References
- 1.Grumelli S, Corry DB, Song LZ, Song L, Green L, Huh J, Hacken J, Espada R, Bag R, Lewis DE, Kheradmand F. An immune basis for lung parenchymal destruction in chronic obstructive pulmonary disease and emphysema. PLoS Med. 2004;1:e8. doi: 10.1371/journal.pmed.0010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shan M, Cheng HF, Song LZ, Roberts L, Green L, Hacken-Bitar J, Huh J, Bakaeen F, Coxson HO, Storness-Bliss C, Ramchandani M, Lee SH, Corry DB, Kheradmand F. Lung myeloid dendritic cells coordinately induce TH1 and TH17 responses in human emphysema. Sci Transl Med. 2009;1 doi: 10.1126/scitranlsmed.3000154. 4ra10. [DOI] [PubMed] [Google Scholar]
- 3.Barnes PJ, Shapiro SD, Pauwels RA. Chronic obstructive pulmonary disease: molecular and cellular mechanisms. Eur Respir J. 2003;22:672–688. doi: 10.1183/09031936.03.00040703. [DOI] [PubMed] [Google Scholar]
- 4.Barnes PJ. Immunology of asthma and chronic obstructive pulmonary disease. Nat Rev Immunol. 2008;8:183–192. doi: 10.1038/nri2254. [DOI] [PubMed] [Google Scholar]
- 5.Etzel CJ, Kachroo S, Liu M, D’Amelio A, Dong Q, Cote ML, Wenzlaff AS, Hong WK, Greisinger AJ, Schwartz AG, Spitz MR. Development and validation of a lung cancer risk prediction model for African-Americans. Cancer Prev Res (Phila) 2008;1:255–265. doi: 10.1158/1940-6207.CAPR-08-0082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Young RP, Hopkins RJ, Hay BA, Epton MJ, Black PN, Gamble GD. Lung cancer gene associated with COPD: triple whammy or possible confounding effect? Eur Respir J. 2008;32:1158–1164. doi: 10.1183/09031936.00093908. [DOI] [PubMed] [Google Scholar]
- 7.Pflughoeft KJ, Versalovic J. Human microbiome in health and disease. Annu Rev Pathol. 2012;7:99–122. doi: 10.1146/annurev-pathol-011811-132421. [DOI] [PubMed] [Google Scholar]
- 8.Dickson RP, Erb-Downward JR, Freeman CM, McCloskey L, Beck JM, Huffnagle GB, Curtis JL. Spatial variation in the healthy human lung microbiome and the adapted island model of lung biogeography. Ann Am Thorac Soc. 2015;12:821–830. doi: 10.1513/AnnalsATS.201501-029OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dickson RP, Erb-Downward JR, Martinez FJ, Huffnagle GB. The microbiome and the respiratory tract. Annu Rev Physiol. 2016;78:481–504. doi: 10.1146/annurev-physiol-021115-105238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang YJ, Charlson ES, Collman RG, Colombini-Hatch S, Martinez FD, Senior RM. The role of the lung microbiome in health and disease. A national heart, lung, and blood institute workshop report. Am J Respir Crit Care Med. 2013;187:1382–1387. doi: 10.1164/rccm.201303-0488WS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bassis CM, Erb-Downward JR, Dickson RP, Freeman CM, Schmidt TM, Young VB, Beck JM, Curtis JL, Huffnagle GB. Analysis of the upper respiratory tract microbiotas as the source of the lung and gastric microbiotas in healthy individuals. MBio. 2015;6:e00037. doi: 10.1128/mBio.00037-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Charlson ES, Bittinger K, Haas AR, Fitzgerald AS, Frank I, Yadav A, Bushman FD, Collman RG. Topographical continuity of bacterial populations in the healthy human respiratory tract. Am J Respir Crit Care Med. 2011;184:957–963. doi: 10.1164/rccm.201104-0655OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morris A, Beck JM, Schloss PD, Campbell TB, Crothers K, Curtis JL, Flores SC, Fontenot AP, Ghedin E, Huang L, Jablonski K, Kleerup E, Lynch SV, Sodergren E, Twigg H, Young VB, Bassis CM, Venkataraman A, Schmidt TM, Weinstock GM Lung HIV Microbiome Project. Comparison of the respiratory microbiome in healthy nonsmokers and smokers. Am J Respir Crit Care Med. 2013;187:1067–1075. doi: 10.1164/rccm.201210-1913OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang YJ, Nelson CE, Brodie EL, Desantis TZ, Baek MS, Liu J, Woyke T, Allgaier M, Bristow J, Wiener-Kronish JP, Sutherland ER, King TS, Icitovic N, Martin RJ, Calhoun WJ, Castro M, Denlinger LC, Dimango E, Kraft M, Peters SP, Wasserman SI, Wechsler ME, Boushey HA, Lynch SV National Heart, Lung, and Blood Institute’s Asthma Clinical Research Network. Airway microbiota and bronchial hyperresponsiveness in patients with suboptimally controlled asthma. J Allergy Clin Immunol. 2011;127:372–381. e371–373. doi: 10.1016/j.jaci.2010.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang YJ, Lynch SV. The emerging relationship between the airway microbiota and chronic respiratory disease: clinical implications. Expert Rev Respir Med. 2011;5:809–821. doi: 10.1586/ers.11.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sze MA, Hogg JC, Sin DD. Bacterial microbiome of lungs in COPD. Int J Chron Obstruct Pulmon Dis. 2014;9:229–238. doi: 10.2147/COPD.S38932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dickson RP, Martinez FJ, Huffnagle GB. The role of the microbiome in exacerbations of chronic lung diseases. Lancet. 2014;384:691–702. doi: 10.1016/S0140-6736(14)61136-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marsland BJ, Gollwitzer ES. Host-microorganism interactions in lung diseases. Nat Rev Immunol. 2014;14:827–835. doi: 10.1038/nri3769. [DOI] [PubMed] [Google Scholar]
- 19.Littman AJ, White E, Jackson LA, Thornquist MD, Gaydos CA, Goodman GE, Vaughan TL. Chlamydia pneumoniae infection and risk of lung cancer. Cancer Epidemiol Biomarkers Prev. 2004;13:1624–1630. [PubMed] [Google Scholar]
- 20.Zhan P, Suo LJ, Qian Q, Shen XK, Qiu LX, Yu LK, Song Y. Chlamydia pneumoniae infection and lung cancer risk: a meta-analysis. Eur J Cancer. 2011;47:742–747. doi: 10.1016/j.ejca.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 21.Moghaddam SJ, Ochoa CE, Sethi S, Dickey BF. Nontypeable haemophilus influenzae in chronic obstructive pulmonary disease and lung cancer. Int J Chron Obstruct Pulmon Dis. 2011;6:113–123. doi: 10.2147/COPD.S15417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu G, Gail MH, Consonni D, Carugno M, Humphrys M, Pesatori AC, Caporaso NE, Goedert JJ, Ravel J, Landi MT. Characterizing human lung tissue microbiota and its relationship to epidemiological and clinical features. Genome Biol. 2016;17:163. doi: 10.1186/s13059-016-1021-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hosgood HD 3rd, Sapkota AR, Rothman N, Rohan T, Hu W, Xu J, Vermeulen R, He X, White JR, Wu G, Wei F, Mongodin EF, Lan Q. The potential role of lung microbiota in lung cancer attributed to household coal burning exposures. Environ Mol Mutagen. 2014;55:643–651. doi: 10.1002/em.21878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yan X, Yang M, Liu J, Gao R, Hu J, Li J, Zhang L, Shi Y, Guo H, Cheng J, Razi M, Pang S, Yu X, Hu S. Discovery and validation of potential bacterial biomarkers for lung cancer. Am J Cancer Res. 2015;5:3111–3122. [PMC free article] [PubMed] [Google Scholar]
- 25.Erb-Downward JR, Thompson DL, Han MK, Freeman CM, McCloskey L, Schmidt LA, Young VB, Toews GB, Curtis JL, Sundaram B, Martinez FJ, Huffnagle GB. Analysis of the lung microbiome in the “healthy” smoker and in COPD. PLoS One. 2011;6:e16384. doi: 10.1371/journal.pone.0016384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cabrera-Rubio R, Garcia-Nunez M, Seto L, Anto JM, Moya A, Monso E, Mira A. Microbiome diversity in the bronchial tracts of patients with chronic obstructive pulmonary disease. J Clin Microbiol. 2012;50:3562–3568. doi: 10.1128/JCM.00767-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pragman AA, Kim HB, Reilly CS, Wendt C, Isaacson RE. The lung microbiome in moderate and severe chronic obstructive pulmonary disease. PLoS One. 2012;7:e47305. doi: 10.1371/journal.pone.0047305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garcia-Nunez M, Millares L, Pomares X, Ferrari R, Perez-Brocal V, Gallego M, Espasa M, Moya A, Monso E. Severity-related changes of bronchial microbiome in chronic obstructive pulmonary disease. J Clin Microbiol. 2014;52:4217–4223. doi: 10.1128/JCM.01967-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Molyneaux PL, Mallia P, Cox MJ, Footitt J, Willis-Owen SA, Homola D, Trujillo-Torralbo MB, Elkin S, Kon OM, Cookson WO, Moffatt MF, Johnston SL. Outgrowth of the bacterial airway microbiome after rhinovirus exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2013;188:1224–1231. doi: 10.1164/rccm.201302-0341OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rogers GB, Daniels TW, Tuck A, Carroll MP, Connett GJ, David GJ, Bruce KD. Studying bacteria in respiratory specimens by using conventional and molecular microbiological approaches. BMC Pulm Med. 2009;9:14. doi: 10.1186/1471-2466-9-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Millares L, Ferrari R, Gallego M, Garcia-Nunez M, Perez-Brocal V, Espasa M, Pomares X, Monton C, Moya A, Monso E. Bronchial microbiome of severe COPD patients colonised by pseudomonas aeruginosa. Eur J Clin Microbiol Infect Dis. 2014;33:1101–1111. doi: 10.1007/s10096-013-2044-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dickson RP, Huang YJ, Martinez FJ, Huffnagle GB. The lung microbiome and viral-induced exacerbations of chronic obstructive pulmonary disease: new observations, novel approaches. Am J Respir Crit Care Med. 2013;188:1185–1186. doi: 10.1164/rccm.201309-1573ED. [DOI] [PubMed] [Google Scholar]
- 33.Twigg HL 3rd, Morris A, Ghedin E, Curtis JL, Huffnagle GB, Crothers K, Campbell TB, Flores SC, Fontenot AP, Beck JM, Huang L, Lynch S, Knox KS, Weinstock G Lung HIV Microbiome Project. Use of bronchoalveolar lavage to assess the respiratory microbiome: signal in the noise. Lancet Respir Med. 2013;1:354–356. doi: 10.1016/S2213-2600(13)70117-6. [DOI] [PubMed] [Google Scholar]
- 34.Sze MA, Dimitriu PA, Hayashi S, Elliott WM, Mc-Donough JE, Gosselink JV, Cooper J, Sin DD, Mohn WW, Hogg JC. The lung tissue microbiome in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2012;185:1073–1080. doi: 10.1164/rccm.201111-2075OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sze MA, Dimitriu PA, Suzuki M, McDonough JE, Campbell JD, Brothers JF, Erb-Downward JR, Huffnagle GB, Hayashi S, Elliott WM, Cooper J, Sin DD, Lenburg ME, Spira A, Mohn WW, Hogg JC. Host response to the lung microbiome in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2015;192:438–445. doi: 10.1164/rccm.201502-0223OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hesselbacher SE, Ross R, Schabath MB, Smith EO, Perusich S, Barrow N, Smithwick P, Mammen MJ, Coxson H, Krowchuk N, Corry DB, Kheradmand F. Cross-sectional analysis of the utility of pulmonary function tests in predicting emphysema in ever-smokers. Int J Environ Res Public Health. 2011;8:1324–1340. doi: 10.3390/ijerph8051324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu C, Hesselbacher S, Tsai CL, Shan M, Spitz M, Scheurer M, Roberts L, Perusich S, Zarinkamar N, Coxson H, Krowchuk N, Corry DB, Kheradmand F. Autoreactive T cells in human smokers is predictive of clinical outcome. Front Immunol. 2012;3:267. doi: 10.3389/fimmu.2012.00267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, Gormley N, Gilbert JA, Smith G, Knight R. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012;6:1621–1624. doi: 10.1038/ismej.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 40.Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods. 2013;10:996–998. doi: 10.1038/nmeth.2604. [DOI] [PubMed] [Google Scholar]
- 41.Lozupone C, Lladser ME, Knights D, Stombaugh J, Knight R. UniFrac: an effective distance metric for microbial community comparison. ISME J. 2011;5:169–172. doi: 10.1038/ismej.2010.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. 2005;71:8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Anderson MJ. A new method for non-parametric multivariate analysis of variance. Austral Ecology. 2001;26:32–46. [Google Scholar]
- 44.Benjamini Y, Yekutieli D. Quantitative trait Loci analysis using the false discovery rate. Genetics. 2005;171:783–790. doi: 10.1534/genetics.104.036699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grice EA, Segre JA. The human microbiome: our second genome. Annu Rev Genomics Hum Genet. 2012;13:151–170. doi: 10.1146/annurev-genom-090711-163814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang YJ, Nariya S, Harris JM, Lynch SV, Choy DF, Arron JR, Boushey H. The airway microbiome in patients with severe asthma: associations with disease features and severity. J Allergy Clin Immunol. 2015;136:874–84. doi: 10.1016/j.jaci.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang YJ, Kim E, Cox MJ, Brodie EL, Brown R, Wiener-Kronish JP, Lynch SV. A persistent and diverse airway microbiota present during chronic obstructive pulmonary disease exacerbations. OMICS. 2010;14:9–59. doi: 10.1089/omi.2009.0100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cox MJ, Allgaier M, Taylor B, Baek MS, Huang YJ, Daly RA, Karaoz U, Andersen GL, Brown R, Fujimura KE, Wu B, Tran D, Koff J, Kleinhenz ME, Nielson D, Brodie EL, Lynch SV. Airway microbiota and pathogen abundance in agestratified cystic fibrosis patients. PLoS One. 2010;5:e11044. doi: 10.1371/journal.pone.0011044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hilty M, Burke C, Pedro H, Cardenas P, Bush A, Bossley C, Davies J, Ervine A, Poulter L, Pachter L, Moffatt MF, Cookson WO. Disordered microbial communities in asthmatic airways. PLoS One. 2010;5:e8578. doi: 10.1371/journal.pone.0008578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Beck JM, Young VB, Huffnagle GB. The microbiome of the lung. Transl Res. 2012;160:258–266. doi: 10.1016/j.trsl.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Larsen JM, Musavian HS, Butt TM, Ingvorsen C, Thysen AH, Brix S. Chronic obstructive pulmonary disease and asthma-associated Proteobacteria, but not commensal Prevotella spp. , promote Toll-like receptor 2-independent lung inflammation and pathology. Immunology. 2015;144:333–342. doi: 10.1111/imm.12376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shete VB, Ghadage DP, Muley VA, Bhore AV. Multi-drug resistant acinetobacter ventilator-associated pneumonia. Lung India. 2010;27:217–220. doi: 10.4103/0970-2113.71952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Perez F, Endimiani A, Bonomo RA. Why are we afraid of acinetobacter baumannii? Expert Rev Anti Infect Ther. 2008;6:269–271. doi: 10.1586/14787210.6.3.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim UJ, Kim HK, An JH, Cho SK, Park KH, Jang HC. Update on the epidemiology, treatment, and outcomes of carbapenem-resistant acinetobacter infections. Chonnam Med J. 2014;50:37–44. doi: 10.4068/cmj.2014.50.2.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Munoz-Price LS, Weinstein RA. Acinetobacter infection. N Engl J Med. 2008;358:1271–1281. doi: 10.1056/NEJMra070741. [DOI] [PubMed] [Google Scholar]
- 56.Sapkota AR, Berger S, Vogel TM. Human pathogens abundant in the bacterial metagenome of cigarettes. Environ Health Perspect. 2010;118:351–356. doi: 10.1289/ehp.0901201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sobhani I, Tap J, Roudot-Thoraval F, Roperch JP, Letulle S, Langella P, Corthier G, Tran Van Nhieu J, Furet JP. Microbial dysbiosis in colorectal cancer (CRC) patients. PLoS One. 2011;6:e16393. doi: 10.1371/journal.pone.0016393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Abdulamir AS, Hafidh RR, Abu Bakar F. The association of Streptococcus bovis/gallolyticus with colorectal tumors: the nature and the underlying mechanisms of its etiological role. J Exp Clin Cancer Res. 2011;30:11. doi: 10.1186/1756-9966-30-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Forsythe P, Bienenstock J. Immunomodulation by commensal and probiotic bacteria. Immunol Invest. 2010;39:429–448. doi: 10.3109/08820131003667978. [DOI] [PubMed] [Google Scholar]
- 60.Barfod KK, Roggenbuck M, Hansen LH, Schjorring S, Larsen ST, Sorensen SJ, Krogfelt KA. The murine lung microbiome in relation to the intestinal and vaginal bacterial communities. BMC Microbiol. 2013;13:303. doi: 10.1186/1471-2180-13-303. [DOI] [PMC free article] [PubMed] [Google Scholar]