

Abstract
Hepatocellular carcinoma (HCC) is the most common type of primary liver cancer, and is the third most frequent cause of cancer-related deaths worldwide. The development of safe new anti-tumor agents has become increasingly important due to the steady rise in drug-resistant tumors. After assessing the efficacy of several candidate compounds that could inhibit hepatocellular carcinoma, we focused on atovaquone, an FDA-approved anti-malarial drug. In the present study, we found that atovaquone significantly inhibited hepatoma cell proliferation via S phase cell cycle arrest and both extrinsic and intrinsic apoptotic pathway induction associated with upregulation of p53 and p21. Molecular investigations demonstrated that atovaquone inhibits hepatoma cell proliferation by inducing double-stranded DNA breaks, leading to sustained activation of ataxia-telangiectasia mutated (ATM) and its downstream molecules such as cell cycle checkpoint kinase-2 (CHK2) and H2AX. In addition, we found that atovaquone also induced apoptosis, inhibited both cell proliferation and angiogenesis in vivo, and prolonged the survival time of tumor-bearing mice, without any obvious side effects. In conclusion, our data indicate that atovaquone is a safe and effective candidate drug that could be rapidly repurposed for HCC treatment.
Keywords: Hepatocellular carcinoma, atovaquone, cell cycle arrest, apoptosis, DNA double-strand breaks
Introduction
Hepatocellular carcinoma (HCC)-the most common type of primary liver cancer worldwide-is the third most frequent cause of cancer-related deaths, with an estimated 782,500 new cases and 745,500 deaths annually [1]. The global incidence of HCC continues to grow and contribute to an increasingly significant health burden in many countries, especially in Asia and Africa [2]. Abundant evidence demonstrates that most HCC cases are associated with chronic liver disease, which could be due to several well-known underlying risk factors, such as chronic hepatitis B or C viral infections, diabetes, or alcohol abuse [3].
Existing effective therapeutic HCC strategies depend on curative treatments-surgical resection, liver transplantation, and ablation-which have high positive response rates; however, most HCC patients are diagnosed in the intermediate-advanced stage, and thus palliative treatments become the only available option [4]. Conventional chemotherapy plays an important role in tumor therapy but many common anti-tumor drugs have significant drawbacks, including poor efficacy, negative side effects, and a rise in chemotherapy resistance [4]. Therefore, it has become increasingly urgent to develop novel safe and effective anti-tumor drugs.
The development pipeline for any novel anti-tumor drug is lengthy and expends multiple resources. Fortunately, several recent studies have found that old drugs such as flavonoids, metformin, and aspirin could have new life in the hands of the oncologist [5]. Atovaquone (ATO), a hydroxyl-1,4-naphthoquinone (Figure 1A), was approved in 1995 by the US Food and Drug Administration (FDA) for the treatment of primary malaria and pneumocystis pneumonia [6]. Mechanistically, ATO acts as a competitive inhibitor of co-enzyme Q10, and in mitochondria isolated from Plasmodium falciparum, specially inhibits the mitochondrial electron transport chain at mitochondrial complex III [7,8]. Interestingly, by inhibiting mitochondrial complex III, ATO reduces the oxygen consumption rate and thus alleviates tumor hypoxia in combination with radiation, delays tumor growth [9]. Moreover, ATO can also eradicate cancer stem cells via the same mechanism in concert with oxidative phosphorylation [10]. In view of the above, ATO already presents anti-tumor activity derived from its ability to inhibit mitochondrial complex III activity. However, it is unclear whether ATO is also able to inhibit tumor growth through other molecules or pathways.
Figure 1.
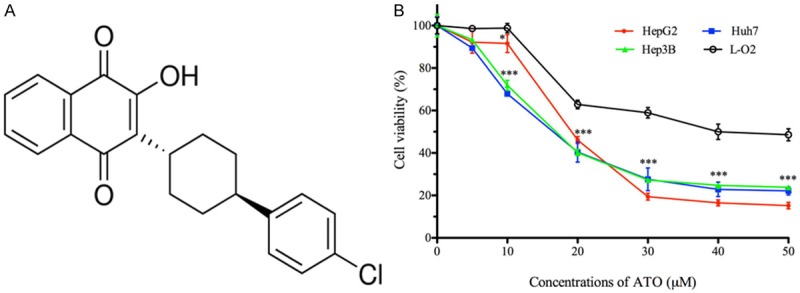
ATO significantly inhibits HCC cell growth in vitro. A. Chemical structure of ATO. B. HepG2, Hep3B, and Huh7 cells were treated with ATO at the indicated concentrations for 48 h, and then analyzed with a CCK-8 kit. ATO inhibited HepG2, Hep3B, and Huh7 cell growth with an IC50 of 19.82±0.32 μM, 17.88±0.67 μM, and 16.84±0.74 μM, respectively. In addition, L-O2 cell proliferation was inhibited by ATO with an IC50 of 40.95±0.17 μM. All data shown represent the mean ± SD from three independent experiments. Significant differences compared to L-O2 cell viability are denoted by * for P < 0.05 and *** for P < 0.001.
In the present study, we determined that ATO significantly inhibited hepatoma cell proliferation in vitro. Moreover, ATO induced S phase cell cycle arrest and both extrinsic and intrinsic apoptosis pathway induction by activating caspase 3, 8, and 9 associated with upregulation of p53 and p21 in HepG2 cells. Molecular investigations demonstrated that ATO’s negative effect on cell proliferation depended on DNA damage with increased ATM-induced phosphorylation of CHK2 and H2AX. Altogether, we determined that in vivo, ATO induces apoptosis, and inhibits both cell proliferation and angiogenesis, thus prolonging the survival time of tumor-bearing mice without any obvious side effects. In conclusion, we have described a novel DNA-damage-based ATO anti-tumor mechanism, and in doing so suggest that ATO could be used as a safe and effective anti-tumor agent to treat HCC patients.
Materials and methods
Ethics statements
This study was carried out in strict accordance with the recommendations of the Xuzhou Medical University Laboratory Animal Ethics Committee Care and Use of Laboratory Animals guidelines. The protocol was approved by the Xuzhou Medical University Laboratory Animal Ethics Committee (Permit Number: 201547).
Cells and animals
The human hepatoma cell lines including HepG2, Hep3B, and Huh7, as well as the normal human hepatic cell line L-O2 were purchase from American Type Culture Collections and cultured in DMEM medium supplemented with 10% heat-inactivated fetal bovine serum purchased from Gibco (Life technologies, Carlsbad, CA, USA). Cultures were maintained at 37°C in a humidified chamber with 5% CO2. All cell lines were splitted no more than 20 passages. Female 4-6-week-old Balb/c nude mice (16-18 g/mouse body weight) were purchased from the Beijing Vital River Laboratory Animal Technology Co., Ltd (Beijing, China) and maintained at the Xuzhou Medical University Animal Center (Xuzhou, China).
Atovaquone, inhibitors, and antibodies
Atovaquone (Sigma Aldrich, Saint Louis, MO, USA), was dissolved in DMSO at a concentration of 12.5 mM, and then stored at -20°C until use. KU-55933 (ATMi, an ATM inhibitor purchased from Selleck Chemicals, Houston, TX, USA), was used at a final concentration of 10 μM, and added to culture medium 2-4 h before ATO treatment. Pifithrin-α hydrobromide (PFT, a p53 inhibitor purchased from Med-ChemExpress, Monmouth, NJ, USA), was used at a final concentration of 20 μM, and added to culture medium 2-3 h before ATO treatment. The details of antibodies associated with the cell cycle (p21, p53, CDK2, and CyclinA2), apoptosis (PARP, caspase-3, caspase-8, caspase-9, Bcl-2, and Bax), DNA damage (ATM, phosphorylated-ATM, CHK2, phosphorylated-CHK2, H2AX, and phosphorylated-H2AX [γH2A-X]), CD-31, KI-67, β-Actin, and GAPDH were all purchased from Cell Signaling Technology (Danvers, MA, USA).
Cell proliferation assays
Cells were cultured in 96-well plates and incubated overnight. Medium was then replaced with fresh medium containing ATO at the indicated concentrations, and incubated for a further 48 h. Finally, cell viability was detected with a CCK-8 assay kit (Dojindo Molecular Technologies, Japan) according to the manufacturer’s instructions. Half maximal inhibitory concentration (IC50) values were calculated from dose-response curves using GraphPad Prism 5.0 (GraphPad Software, Inc., USA).
Cell cycle analysis
Cells were treated with ATO at the indicated concentrations for 48 h, and were then typsinized, washed with PBS, and fixed with pre-chilled 70% ethanol at 4°C for 30 min. Fixed cells were washed with cold PBS, incubated with 100 mg/ml RNase A, and then stained with PI (BD Biosciences, USA) at 37°C for 30 min in the dark. Finally, cells were detected with a flow cytometer and data analyzed with FlowJo 7.6.5 (FlowJo LLC, USA).
Apoptosis analysis
Cultured cells were treated with ATO at the indicated concentrations for 48 h, then collected after trypsinization and washed with cold PBS. The cells were then stained with a FITC-labeled Annexin V apoptosis detection kit (BD Biosciences, USA) according to the manufacturer’s instructions. Flow cytometry was used to detect early and late cellular apoptosis.
Protein preparation and Western blotting
Cells were cultured with or without ATO at the indicated concentrations and for the specified time periods, and were then collected by scraping and lysed in RIPA buffer + protease inhibitors on ice for 30 min. The supernatant was collected after centrifuging at 13,000 g for 15 min, and protein content was determined with a BCA protein concentration assay kit (Beyotime, Shanghai, China). A Western-blotting assay was performed to analyze protein expression. For Western-blotting, equal quantities of protein samples were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride (PVDF) membranes. After blocking in Tris-buffered saline with 0.1% Tween-20 and 5% non-fat dry milk at room temperature for 1 h, the membranes were incubated with primary antibodies overnight at 4°C. Then membranes were incubated with secondary antibodies at room temperature for 1 h. Immunoreactive bands were visualized using a Tanon imaging system (Tanon Science & Technology Co., Ltd, China).
HepG2 xenograft mouse model
Twenty female Balb/c nude mice were subcutaneously inoculated in the flank with HepG2 cells (5 × 106 cells). When tumor volume reached 80 mm3 in all mice, the mice were randomly divided into two groups. For the ATO treatment group, mice received a daily gavage dose of 200 mg/kg ATO in castor oil for 28 days, thus mimicking the clinical administration of ATO as an oral suspension [11]. For the control group, the mice were given an equal volume of castor oil under the same conditions. The length and width of the tumors were measured with a caliper every four days, and tumor volumes were calculated as: length × width2/2. Two tumor-bearing mice from each group were for euthanasia, and tumors resected and fixed with 10% neutral formalin buffering solution then embedded in paraffin. Meanwhile, the lung, heart, liver, spleen, and kidney were also resected, fixed, and embedded. The remaining mice in each group were maintained for the survival study.
Hematoxylin-eosin (HE) staining
The fixed and embedded lung, heart, liver, spleen, and kidney organs were sectioned into 4-μm sections and HE stained to evaluate the effects of ATO treatment. Tissue sections were observed under a microscope, and at least three fields per section were analyzed and necrotic or apoptotic cells were counted using Image-Pro Plus 7.0 (Media Cybernetics, Inc., USA).
Immmunohistochemical (IHC) staining
Paraffin-embedded tumors were sectioned into 4-μm-thick sections. After dewaxing and rehydration, tumor sections were blocked with 1% BSA, then incubated with anti-CD31 (marker of de novo blood vessels) or anti-KI67 (cell proliferation marker) antibodies overnight at 4°C, followed by incubation with a horseradish peroxidase (HRP)-labeled secondary antibody 1 h at room temperature, and then stained by diaminobenzidine (DAB). At least three fields per section were analyzed and positively-stained cells were counted using Image-Pro Plus 7.0.
Terminal dexynucleotidyl transferase dUTP nick ending labeling (TUNEL) assay
A one-step TUNEL apoptosis assay kit (KeyGen Biotech, China) was used to detect cell apoptosis in tumor tissue sections according to the manufacturer’s protocol. Apoptotic cells presented as red under a fluorescence microscope, and three fields per section were analyzed.
Statistical analysis
All in vitro experiments were repeated at least three independent times, and statistical analysis was performed using two-tailed unpaired student’s t tests. Values were presented as mean ± standard deviation (SD) and considered significant at P < 0.05. Survival analysis of the tumor xenograft mice was performed using the Kaplan-Meier method. All of the data analysis was performed with Graphpad Prism 5.0 (GraphPad Software, Inc., USA).
Results
ATO significantly inhibits hepatoma cell growth in vitro
In order to determine whether ATO impedes cell growth, hepatoma and normal hepatic cell lines were treated with a range of ATO concentrations for 48 h. As shown in Figure 1B, ATO significantly inhibited the proliferation of HepG2, Hep3B, and Huh7 cells compared to L-O2 cells at equivalent concentrations.
ATO induces cellcycle arrest at S phase and apoptosis in hepatoma cells
In order to investigate whether cell cycle arrest and apoptosis may contribute to the reduced cell viability following ATO treatment, both aspects were analyzed in HepG2 and Hep3B cells treated with different ATO concentrations for 48 h. In these hepatoma cell lines, 20 μM ATO significantly induced cell cycle arrest at the S phase compared to untreated controls (Figure 2A and 2B). ATO also significantly induced apoptosis in a concentration-dependent manner compared to untreated controls in the two hepatoma cell lines (Figure 2C and 2D).
Figure 2.

ATO induces cell cycle arrest at S phase and cell apoptosis in hepatoma cells. HepG2 and Hep3B cells were both treated with 20 μM ATO for 48 h. Cells were collected and stained with PI or a FITC Annexin V apoptosis detection kit, and then analyzed with a flow cytometer. A and B. ATO treatment induced S phase cell cycle arrest compared to the untreated controls in both HepG2 and Hep3B cell lines. HepG2 and Hep3B cells were treated with different ATO concentrations (0 μM, 10 μM, 20 μM, 30 μM) for 48 h. Cells were collected and stained with a FITC Annexin V apoptosis detection kit, and then analyzed with a flow cytometer. C and D. ATO induced cell apoptosis in a concentration-dependent manner in both HepG2 and Hep3B cell lines. All data shown represent the mean ± SD from three independent experiments, significant differences compared to the control are denoted by ** for P < 0.01 and *** for P < 0.001.
ATO regulates cell cycle- and apoptosis-associated protein expression
To elucidate the mechanisms of ATO-induced cell cycle arrest, HepG2 cells were treated with 20 μM ATO for 48 h, then collected and analyzed by Western-blotting. Results indicated that ATO treatment down-regulated both CyclinA2 and CDK2, key S phase regulators. Moreover, we observed that ATO treatment induced p53 and p21 protein expression (Figure 3A).
Figure 3.
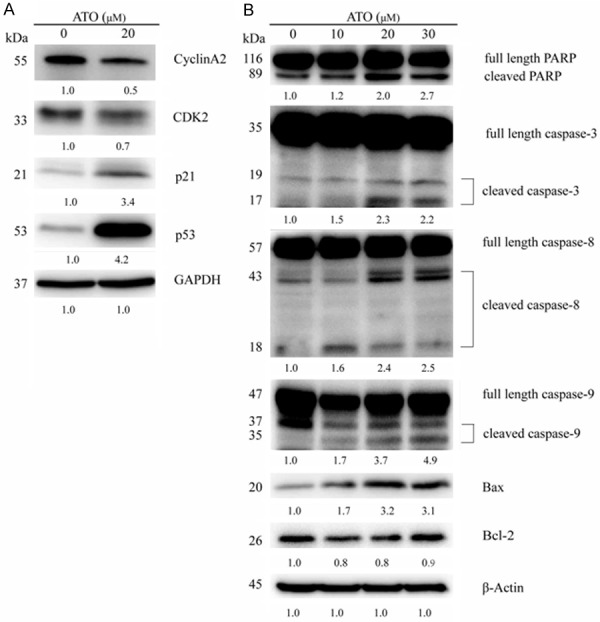
ATO regulates the expression of cell cycle and apoptosis related proteins. A. Lysates of HepG2 cells which had been treated with 20 µM ATO for 48 h, were analyzed via Western-blotting and specific antibodies. ATO treatment downregulated expression of CyclinA2 and CDK2, and upregulated expression of p21 and p53. B. Lysates of HepG2 cells which had been treated with varying ATO concentrations (0 μM, 10 μM, 20 μM, 30 μM) for 48 h, were analyzed by Western-blotting and specific antibodies. ATO treatment induced cleavage of PARP, caspase-3, caspase-8, and caspase-9, upregulated Bax, and downregulated Bcl-2.
Apoptosis-associated protein expression in ATO-treated HepG2 cells was also analyzed with Western-blotting to identify ATO-induced apoptosis mechanisms. As shown in Figure 3B, ATO induced cleavage of the classical apoptosis markers PARP and caspase-3 in a dose-dependent manner. Moreover, the classical apoptosis markers caspase-8 and caspase-9 cleavage, indicating that ATO activates both extrinsic and intrinsic apoptosis pathway. Additionally, ATO upregulated the pro-apoptosis protein Bax in a dose-dependent manner, and downregulated the anti-apoptosis protein Bcl-2 (Figure 3B).
ATO-induced DNA damage plays a key role in inhibiting cell proliferation
To further characterize ATO anti-tumor mechanisms in hepatoma cells, γH2AX protein expression (a marker of DNA damage) was determined. ATO upregulated γH2AX without affecting total protein expression in both HepG2 and Hep3B cells, confirming the occurrence of DNA damage (Figure 4A). Thus, cell cycle arrest and subsequent apoptosis is likely dependent on ATO-induced DNA damage.
Figure 4.
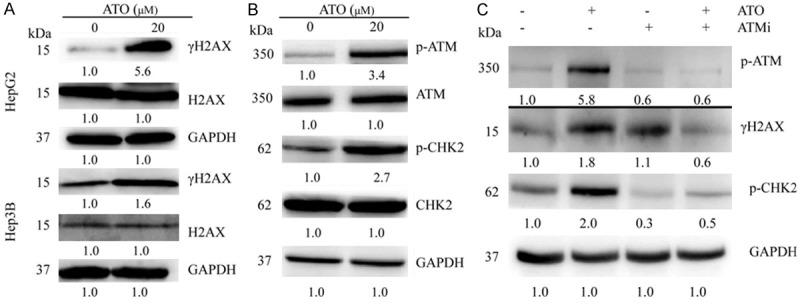
DNA damage is positively associated with ATO-induced cell proliferation inhibition. HepG2 and Hep3B cells were treated with 20 μM ATO for 48 h. Cell lysates were analyzed with Western-blotting and specific antibodies. A. ATO treatment increased γH2AX expression in both HepG2 and Hep3B cell lines. B. ATO treatment also increased the expression of phosphorylated ATM and CHK2 in HepG2 cells. C. ATM was then inhibited with KU55933 (ATMi) which decreased expression of phosphorylated ATM, phosphorylated CHK2, and γH2AX in ATO-treated HepG2 cells.
In response to DNA damage, ATM activation initiates a signaling cascade by phosphorylating many key proteins. In HepG2 cells, ATO activated phosphorylated ATM, and upregulated phosphorylated CHK2 without affecting total protein expression (Figure 4B). To investigate whether ATM activation is involved in ATO-induced cell proliferation inhibition, HepG2 cells were treated with ATO in the presence or absence of ATMi (an ATM inhibitor). ATMi inhibited both γH2AX and phosphorylated ATM expression in ATO-treated HepG2 cells (Figure 4C). In addition, ATMi also significantly inhibited ATO-induced S phase cell cycle arrest and apoptosis in HepG2 cells (Figure 5A and 5B). Moreover, ATMi treatment also inhibited ATO-induced upregulation of p53, p21, and cleavage of PARP, as well as downregulation of CDK2 (Figure 5C).
Figure 5.

Inhibition of ATM significantly decreased ATO-induced cell cycle arrest and apoptosis. HepG2 cells were pre-cultured with 10 μM ATMi for 3 h, then treated with 20 μM ATO for 48 h. Cells were collected and stained with PI or a FITC-Annexin V apoptosis detection kit, and then analyzed with a flow cytometer. In addition, cell lysates were analyzed with Western-blotting and specific antibodies. A. ATMi decreased ATO-induced S phase cell cycle arrest in HepG2 cells. B. ATMi decreased ATO-induced apoptosis in HepG2 cells. C. ATMi increased ATO-induced downregulation of CDK2, but decreased ATO-induced upregulation of p21, p53 and cleavage of PARP. All data shown represent the mean ± SD from three independent experiments, significant differences compared to the control are denoted by ** for P < 0.01 and *** for P < 0.001.
It is known that the p53 tumor suppressor protein plays an essential role in cell cycle arrest and apoptosis after diverse stresses, including DNA damage. To investigate p53 activation is involved in ATO-induced cell proliferation inhibition, HepG2 cells were treated with ATO in the presence or absence of PFT (a p53 inhibitor). Our results indicated that PFT significantly inhibited ATO-induced S phase cell cycle arrest and apoptosis in HepG2 cells (Figure 6A and 6B). Moreover, PFT treatment also inhibited ATO-induced upregulation of p53, p21, and cleavage of PARP, as well as downregulation of CDK2 (Figure 6C).
Figure 6.

Inhibition of p53 significantly decreased ATO-induced cell cycle arrest and apoptosis. HepG2 cells were pre-cultured with 20 μM PFT (a p53 inhibitor) for 2 h, then treated with 20 μM ATO for 48 h. Cells were collected and stained with PI or a FITC Annexin V apoptosis detection kit, and then analyzed with a flow cytometer. In addition, cell lysates were analyzed with Western-blotting and specific antibodies. A. PFT decreased ATO-induced S phase cell cycle arrest in HepG2 cells. B. PFT also decreased ATO-induced apoptosis in HepG2 cells. C. PFT increased ATO-induced downregulation of CDK2, but decreased ATO-induced upregulation of p21, p53, and cleavage of PARP. All data shown represent the mean ± SD from three independent experiments, significant differences compared to the control are denoted by * for P < 0.05.
ATO inhibits HCC tumor growth in vivo
In order to characterize the anti-tumor activity of ATO in vivo, a subcutaneous xenograft tumor mouse model was successfully established with HepG2 cells. Compared to the controls (castor oil), ATO significantly inhibited tumor growth in the xenograft tumor mice (Figure 7A and 7B). Moreover, ATO treatment significantly prolonged the survival time of the tumor-bearing mice compared to the controls (Figure 7C). ATO treatment also induced endonucleolytic chromatin cleavage and massive necrosis at tumor edges (Figure 8A and 8B). Interestingly, ATO also inhibited angiogenesis and cell proliferation in vivo, which was confirmed by characterizing the expression of CD31 (de novo vessel marker) and KI67 (cell proliferation marker) (Figure 8B), respectively. Mouse body weight and internal organs including lung, heart, liver, spleen, and kidney of ATO-treated tumor-bearing mice showed no differences compared to control mice, implying that ATO has no obvious toxicity in experimental animals (Figure 9A and 9B).
Figure 7.
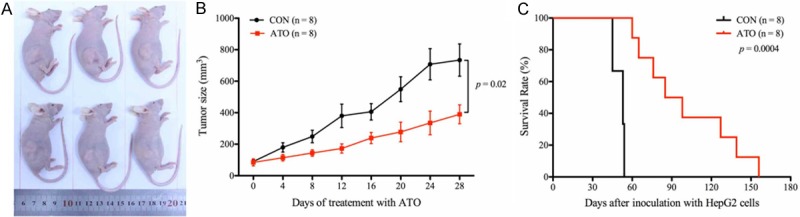
ATO inhibits tumor growth in vivo and prolongs the survival time of tumor-bearing mice. HepG2 xenograft tumor mice were treated with ATO or castor oil (CON) for 28 days. A and B. ATO treatment significantly inhibited tumor growth in vivo compared to the control. C. ATO treatment prolonged the survival time of the tumor-bearing mice compared to the control. All data shown represent the mean ± SD from eight independent mice.
Figure 8.
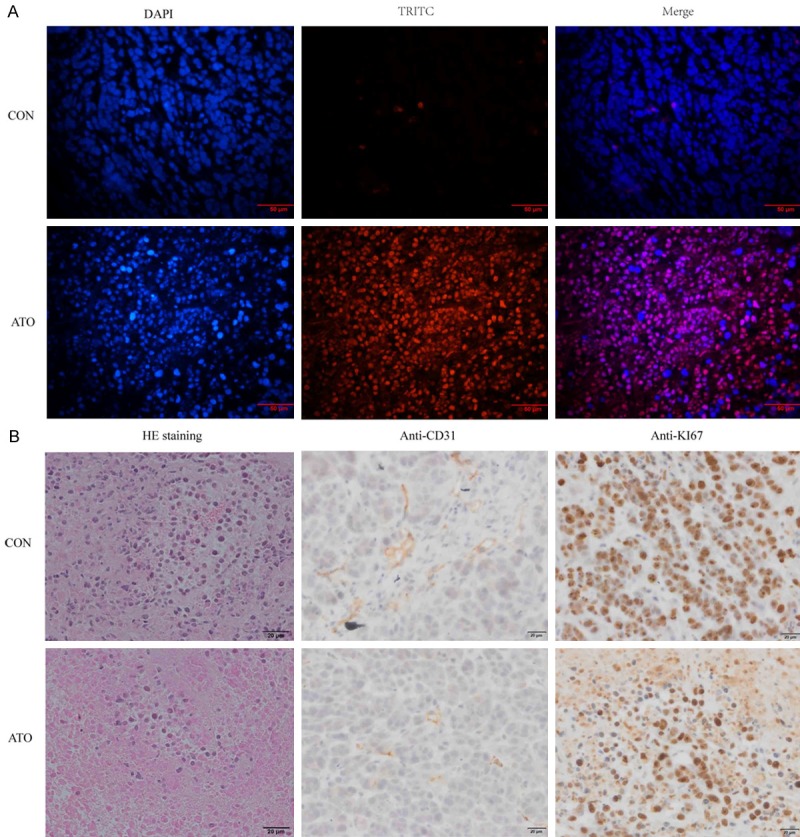
ATO inhibits tumor growth in vivo by inducing cell apoptosis, and inhibiting angiogenesis and cell proliferation. A. TUNEL staining showed that ATO treatment induced endonucleolytic chromatin cleavage. B. HE staining showed that ATO treatment induced massive tumor necrosis, and IHC staining with anti-CD31 and anti-KI67 antibodies showed that ATO treatment inhibited angiogenesis and cell proliferation, respectively.
Figure 9.
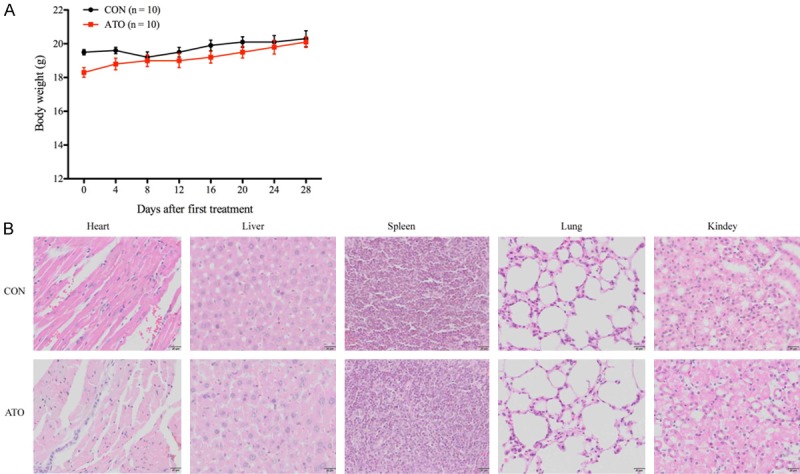
ATO treatment does not affect the body weight and internal organs of tumor-bearing mice. A. The body weight of ATO-treated or castor oil-treated (CON) tumor-bearing mice was recorded every four days, all data shown represent the mean ± SD from ten independent mice. B. Following treatment with either ATO or castor oil (CON) for 28 days, the lung, heart, liver, spleen, and kidney from the tumor-bearing mice were HE stained. No differences were seen in the organs between ATO-treatment and control tumor-bearing mice.
Discussion
HCC is the most common malignant hepatobiliary disease, and has the highest incidence in Asia and Africa, consistent with high hepatitis B and C viral infection prevalence [3]. In view of HCC’s high mortality, rapid progression, and high recurrence rate, the race to identify novel therapeutic targets and develop safe effective anti-tumor drugs is becoming increasingly critical. ATO, an FDA-approved anti-parasitic drug, hasa good safety profile with few side effects in normal mammalian cells. ATO is easily absorbed and is rapidly eliminated by the liver, and is not metabolized in vivo to a significant extent [6]. ATO’s primary anti-parasitic mechanism is via the inhibition of mitochondrial complex III, and studies have indicated that ATO can also inhibit cancer cell viability by targeting the mitochondrial complex III [7-10]. In the present study, our experimental data indicated that ATO also inhibits hepatoma cells proliferation both in vitro and in vivo by inducing DNA damage in cancer cells, thus revealing a novel ATO anti-tumor mechanism.
ATO, an established anti-malaria drug, has already been repurposed for its novel anti-tumor effects in cancer cells [9,10,12]. However, there are no studies, which demonstrate ATO’s anti-tumor activity in hepatoma cells. In the present study, we first performed in vitro experiments to investigate ATO’s anti-tumor activity in hepatoma cells. Our results demonstrated that ATO significantly inhibits HepG2, Hep3B, and Huh7 cell proliferation compared to L-O2 cells (Figure 1B). In addition, it should be noted that hepatoma cell proliferation inhibition occurred with 20 μM ATO, corresponding to a concentration of 7.33 μg/mL. This is nearly two-thirds lower than the average steady-state plasma concentration of 21.0±4.9 μg/mL in HIV-infected volunteers who had been treated with ATO suspended in food at the standard regimen of 750 mg twice daily [13].
Chemotherapeutic drugs that cause cell cycle arrest are known to be effective inhibitors of tumor cell proliferation [14]. Interestingly, our results showed that ATO significantly inhibited HepG2 and Hep3B tumor cell proliferation by inducing S phase arrest (Figure 2A and 2B). The cell cycle consists of G1, S, G2, and M phases, which are tightly modulated by cyclins (regulatory subunits) and cyclin-dependent kinases (CDKs, catalytic subunits) [15]. Cyclin A and CDK2 play an essential role in modulating DNA synthesis and S phase progression [16,17]. Furthermore, upregulation of p21 in cancer cells can inhibit the expression of cell cycle regulatory proteins such as cyclin A and CDK2 [18]. Here, we showed that ATO-induced S phase cell cycle arrest in HepG2 cells was accompanied by downregulated expression of CyclinA2 and CDK2, and upregulated expression of p53 and p21 (Figure 3A). In addition, p53 plays a key role in cell cycle arrest and apoptosis; activated p53 can hold the cell cycle arrest in S phase though activating expression of p21, and down-regulates CyclinA/CDK2 [19].
Apoptosis, or programmed cell death, is crucial in maintaining cellular homeostasis, and also plays an important role in tumor proliferation, drug resistance, and metastasis [20]. Normally, apoptosis is triggered by a highly-specific cascade of extrinsic or intrinsic ligands, and is tightly regulated by a series of anti-apoptotic and pro-apoptotic signaling pathways [20,21]. Here, we found that ATO induced apoptosis in HepG2 and Hep3B cells (Figure 2C and 2D), and caused PARP and caspase-3 cleavage, both classical apoptosis markers (Figure 3B). Mechanistic analysis revealed that ATO upregulated the pro-apoptotic protein Bax and downregulated the anti-apoptotic protein Bcl-2 (Figure 3B). Interestingly, ATO treatment also resulted in the cleavage of caspase-8 and caspase-9 (Figure3B), suggesting that ATO may activate both extrinsic and intrinsic apoptosis pathway.
DNA integrity is vital for proper cellular function and proliferation, such that high levels of DNA damage and delayed DNA damage repair will result in cell cycle arrest and subsequent apoptosis [22]. If DNA lesions occur during the S phase of the cell cycle, they can inhibit replication fork progression and cause replication-associated DNA double-strand breaks (DSBs), which are among the most toxic of all DNA lesions [23]. At nascent DSBs sites, the histone protein H2AX becomes rapidly phosphorylated to form γH2AX, and thus can act as a sensitive DSBs marker [24]. In this study, we found that γH2AX was strongly upregulated in ATO-treated hepatoma cells compared to control (Figure 4A), suggesting that ATO treatment could induce DSBs in hepatoma cells. DSBs are a cause of cancer and paradoxically, they are also an effective cancer treatment. Currently, DSBs-causing therapeutic agents-such as radiation and chemotherapy-play essential roles in the elimination of cancer cells to decrease tumor burden during treatment [22]. It has previously been reported that 1,4-Naphthoquinone and its derivatives can inhibit cancer cell viability and proliferation by inducing DNA damage [25,26]. In the present study, we provided strong evidence that ATO, a hydroxy-1,4-naphthoquinone, exhibits anti-tumor activity by inducing DSBs in hepatoma cells.
Many current cancer therapies exploit the DSBs introduction mechanism to activate cell death pathways in cancer cells [27]. This process occurs via ATM, a serine/threonine protein kinase, which is recruited and activated in the presence of DSBs [28,29]. Following activation, ATM phosphorylates several proteins including CHK2 and H2AX, which then triggers activation of the DNA damage checkpoint, leading to either cell cycle arrest, DNA repair, and/or cell apoptosis [28,29]. Our results showed that phosphorylated ATM and phosphorylated CHK2increased in ATO-treated HepG2 cells (Figure 4B). In addition, γH2AX and phosphorylated-CHK2 were obviously decreased in ATO-treated HepG2 cells that had been pre-treated with an ATM inhibitor (Figure 4C). In addition, cell cycle arrest and apoptosis, as well as their associated protein expression were remarkably inhibited in ATO-treated HepG2 cells that had been pre-treated with ATM inhibitor (Figure 5). Altogether, these results strongly suggest that ATM plays an essential role in HepG2 cell cycle arrest and apoptosis independent of ATO-induced DSBs.
p53, the downstream signal molecule of ATM, takes a central role for controlling cell cycle arrest and apoptosis in response to diverse stresses, including DNA damage [28]. In the present study, our results confirmed that ATM inhibitor downregulated ATO-induced p53 expression (Figure 5C). Furthermore, to confirm p53 activation is involved in ATO-induced cell cycle arrest and apoptosis in HepG2 cells, cells were pre-treated with PFT (a p53 inhibitor). The results showed that PFT cotreatment significantly reduced the proportion of cells in the S-phase cell cycle arrest and apoptosis compared to that of HepG2 cells treated with ATO alone (Figure 6A and 6B). Consistently, PFT reversed the effect of ATO on cell cycle and apoptosis related protein expressions (Figure 6C). Taken together, our results demonstrate that p53 plays a considerable role in HepG2 cell cycle arrest and apoptosis induced by ATO.
Furthermore, our results also show that ATO treatment inhibits tumor growth and prolongs the survival time of tumor-bearing mice in vivo (Figure 7). Our data indicated that ATO significantly inhibited HepG2 xenograft mice tumor growth by inducing cell apoptosis, decreasing cell proliferation, and inhibiting angiogenesis (Figure 8). Tumor survival, growth and metastasis cannot occur without sufficient nutrients and oxygen [30]. As these vital factors are transported by blood vessels, angiogenesis plays a significant role during tumor progression, and consequently is a promising cancer treatment target [31,32]. However, the underlying mechanism by which ATO inhibits angiogenesis remains to be elucidated.
ATO presents with a well-tolerated safety profile, with no evident side effects in normal mammalian cells [6]. Moreover, ATO is easily absorbed and eliminated by the liver in a short period of time [6]. Our in vivo data showed that ATO dramatically reduced tumor growth in pre-clinical xenograft models without any detectable toxicity (Figure 9). ATO has already been approved by the FDA for use in humans, and has shown anti-tumor activity in a variety of cancer cells, including colorectal cancer, breast cancer, and acute myeloid leukemia [9,10,12]. Based on these observations, it appears that ATO could probably directly enter into phase II clinical trials for HCC, potentially saving significant developmental time and resources.
Conclusion
Our data indicate that ATO is an excellent candidate for therapeutic HCC treatment. ATO inhibits hepatoma cell proliferation by inducing DNA double-strand breaks, leading to sustained activation of ATM and its downstream molecules (Figure 10). More importantly, ATO also exhibits an anti-tumor effect in vivo and prolongs the survival time of tumor-bearing mice without any evidence of toxicity. In summary, we suggest that repurposing ATO could be an effective and safe candidate for clinical HCC treatment.
Figure 10.
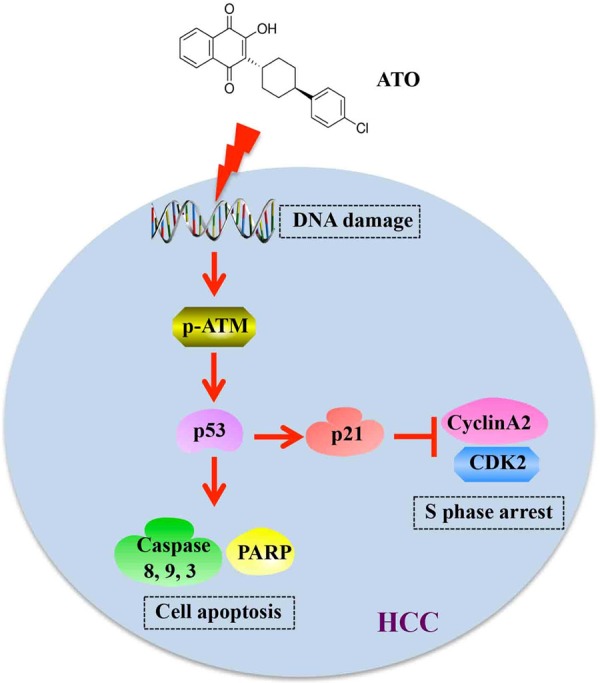
Schematic illustration of ATO exhibiting anti-tumor effects via inducing DNA damage. In brief, ATO induces double-stranded DNA breaks, which lead to upregulation of phosphorylated-ATM and p53. On one hand, cell cycle was arrested at S phase by inhibition of CyclinA2 and CDK2 expression via upregulation of p21; on the other hand, cell apoptosis was activated by inducing cleavage of caspase-8, caspase-9, caspase-3, and PARP.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 81673008, 81802389 and 81502221), the China Postdoctoral Science Foundation (Grant No. 2016M590504), the Natural Science Foundation of Jiangsu Province (Grant No. BK20150212 and BK20150216), the Research Foundation of Xuzhou Medical University (Grant No. D2015006 and D2015009), the Jiangsu Province Postdoctoral Science Foundation (Grant No. 1501088B), the Social development project of Jiangsu Province (Grant No: BE2017642), the Science and Technology Plan Projects of Xuzhou (Grant No: KC17012), the Project of Invigorating Health Care through Science, Technology and Education (NO. CXTDA2017034) and A Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Disclosure of conflict of interest
None.
References
- 1.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 2.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 3.Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet. 2012;379:1245–1255. doi: 10.1016/S0140-6736(11)61347-0. [DOI] [PubMed] [Google Scholar]
- 4.Schlachterman A, Craft WW Jr, Hilgenfeldt E, Mitra A, Cabrera R. Current and future treatments for hepatocellular carcinoma. World J Gastroenterol. 2015;21:8478–8491. doi: 10.3748/wjg.v21.i28.8478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang F, Li M, Wang J, Liang X, Su Y, Wang W. Finding new tricks for old drugs: tumoricidal activity of non-traditional antitumor drugs. AAPS PharmSciTech. 2016;17:539–552. doi: 10.1208/s12249-016-0518-y. [DOI] [PubMed] [Google Scholar]
- 6.Nixon GL, Moss DM, Shone AE, Lalloo DG, Fisher N, O’Neill PM, Ward SA, Biagini GA. Antimalarial pharmacology and therapeutics of atovaquone. J Antimicrob Chemother. 2013;68:977–985. doi: 10.1093/jac/dks504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fry M, Pudney M. Site of action of the antimalarial hydroxynaphthoquinone, 2-[trans-4-(4’-chlorophenyl) cyclohexyl]-3-hydroxy-1,4-naphthoquinone (566C80) Biochem Pharmacol. 1992;43:1545–1553. doi: 10.1016/0006-2952(92)90213-3. [DOI] [PubMed] [Google Scholar]
- 8.Mather MW, Darrouzet E, Valkova-Valchanova M, Cooley JW, McIntosh MT, Daldal F, Vaidya AB. Uncovering the molecular mode of action of the antimalarial drug atovaquone using a bacterial system. J Biol Chem. 2005;280:27458–27465. doi: 10.1074/jbc.M502319200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ashton TM, Fokas E, Kunz-Schughart LA, Folkes LK, Anbalagan S, Huether M, Kelly CJ, Pirovano G, Buffa FM, Hammond EM, Stratford M, Muschel RJ, Higgins GS, McKenna WG. The anti-malarial atovaquone increases radiosensitivity by alleviating tumour hypoxia. Nat Commun. 2016;7:12308. doi: 10.1038/ncomms12308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fiorillo M, Lamb R, Tanowitz HB, Mutti L, Krstic-Demonacos M, Cappello AR, Martinez-Outschoorn UE, Sotgia F, Lisanti MP. Repurposing atovaquone: targeting mitochondrial complex III and OXPHOS to eradicate cancer stem cells. Oncotarget. 2016;7:34084–34099. doi: 10.18632/oncotarget.9122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Comley JC, Sterling AM. Effect of atovaquone and atovaquone drug combinations on prophylaxis of Pneumocystis carinii pneumonia in SCID mice. Antimicrob Agents Chemother. 1995;39:806–811. doi: 10.1128/aac.39.4.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xiang M, Kim H, Ho VT, Walker SR, Bar-Natan M, Anahtar M, Liu SH, Toniolo PA, Kroll Y, Jones N, Giaccone ZT, Heppler LN, Ye DQ, Marineau JJ, Shaw D, Bradner JE, Blonquist T, Neuberg D, Hetz C, Stone RM, Soiffer RJ, Frank DA. Gene expression-based discovery of atovaquone as a STAT3 inhibitor and anticancer agent. Blood. 2016;128:1845–1853. doi: 10.1182/blood-2015-07-660506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Falloon J, Sargent S, Piscitelli SC, Bechtel C, LaFon SW, Sadler B, Walker RE, Kovacs JA, Polis MA, Davey RT Jr, Lane HC, Masur H. Atovaquone suspension in HIV-infected volunteers: pharmacokinetics, pharmacodynamics, and TMP-SMX interaction study. Pharmacotherapy. 1999;19:1050–1056. doi: 10.1592/phco.19.13.1050.31598. [DOI] [PubMed] [Google Scholar]
- 14.Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432:316–323. doi: 10.1038/nature03097. [DOI] [PubMed] [Google Scholar]
- 15.Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9:153–166. doi: 10.1038/nrc2602. [DOI] [PubMed] [Google Scholar]
- 16.Solomon VR, Pundir S, Le HT, Lee H. Design and synthesis of novel quinacrine-[1,3] - thiazinan-4-one hybrids for their anti-breast cancer activity. Eur J Med Chem. 2018;143:1028–1038. doi: 10.1016/j.ejmech.2017.11.097. [DOI] [PubMed] [Google Scholar]
- 17.Qin JL, Shen WY, Chen ZF, Zhao LF, Qin QP, Yu YC, Liang H. Oxoaporphine metal complexes (Co(II), Ni(II), Zn(II)) with high antitumor activity by inducing mitochondria-mediated apoptosis and s-phase arrest in HepG2. Sci Rep. 2017;7:46056. doi: 10.1038/srep46056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Song X, Li L, Shi Q, Lehmler HJ, Fu J, Su C, Xia X, Song E, Song Y. Polychlorinated biphenyl quinone metabolite promotes p53-dependent DNA damage checkpoint activation, S-phase cycle arrest and extrinsic apoptosis in human liver hepatocellular carcinoma hepG2 cells. Chem Res Toxicol. 2015;28:2160–2169. doi: 10.1021/acs.chemrestox.5b00320. [DOI] [PubMed] [Google Scholar]
- 19.Kastenhuber ER, Lowe SW. Putting p53 in context. Cell. 2017;170:1062–1078. doi: 10.1016/j.cell.2017.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ouyang L, Shi Z, Zhao S, Wang FT, Zhou TT, Liu B, Bao JK. Programmed cell death pathways in cancer: a review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 2012;45:487–498. doi: 10.1111/j.1365-2184.2012.00845.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature. 2012;481:287–294. doi: 10.1038/nature10760. [DOI] [PubMed] [Google Scholar]
- 23.Cheung-Ong K, Giaever G, Nislow C. DNAdamaging agents in cancer chemotherapy: serendipity and chemical biology. Chem Biol. 2013;20:648–659. doi: 10.1016/j.chembiol.2013.04.007. [DOI] [PubMed] [Google Scholar]
- 24.Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S, Pommier Y. GammaH2AX and cancer. Nat Rev Cancer. 2008;8:957–967. doi: 10.1038/nrc2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Farias MS, Pich CT, Kviecinski MR, Bucker NC, Felipe KB, Da Silva FO, Gunther TM, Correia JF, Rios D, Benites J, Valderrama JA, Calderon PB, Pedrosa RC. Substituted 3-acyl-2-phenylamino-1,4-naphthoquinones intercalate into DNA and cause genotoxicity through the increased generation of reactive oxygen species culminating in cell death. Mol Med Rep. 2014;10:405–410. doi: 10.3892/mmr.2014.2160. [DOI] [PubMed] [Google Scholar]
- 26.Ourique F, Kviecinski MR, Felipe KB, Correia JF, Farias MS, Castro LS, Grinevicius VM, Valderrama J, Rios D, Benites J, Calderon PB, Pedrosa RC. DNA damage and inhibition of akt pathway in mcf-7 cells and ehrlich tumor in mice treated with 1,4-naphthoquinones in combination with ascorbate. Oxid Med Cell Longev. 2015;2015:495305. doi: 10.1155/2015/495305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer. 2008;8:193–204. doi: 10.1038/nrc2342. [DOI] [PubMed] [Google Scholar]
- 28.Ditch S, Paull TT. The ATM protein kinase and cellular redox signaling: beyond the DNA damage response. Trends Biochem Sci. 2012;37:15–22. doi: 10.1016/j.tibs.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith J, Tho LM, Xu N, Gillespie DA. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res. 2010;108:73–112. doi: 10.1016/B978-0-12-380888-2.00003-0. [DOI] [PubMed] [Google Scholar]
- 30.De Bock K, Mazzone M, Carmeliet P. Antiangiogenic therapy, hypoxia, and metastasis: risky liaisons, or not? Nat Rev Clin Oncol. 2011;8:393–404. doi: 10.1038/nrclinonc.2011.83. [DOI] [PubMed] [Google Scholar]
- 31.Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rajabi M, Mousa SA. The role of angiogenesis in cancer treatment. Biomedicines. 2017:5. doi: 10.3390/biomedicines5020034. [DOI] [PMC free article] [PubMed] [Google Scholar]