

Abstract
Previous studies demonstrated that live Mycobacterium leprae (M. leprae) infection promoted macrophage differentiation toward the M2 type, with elevated interleukin (IL)-10 production. The underlying mechanism is not entirely clear. In this study, we treated macrophages with primary M. leprae strains isolated from both lepromatous leprosy (L-lep) and tuberculoid leprosy (T-lep) patients. We found that infection by live M. leprae, regardless of the primary strain, resulted in M2 skewing in the infected macrophage. This skewing was associated with downregulated IRGM expression, a core organizer protein in the autophagy assembly and reduced autophagosome formation, and with lower annexin V staining and lower caspase 3 and caspase 9 activity. Moreover, live M. leprae-infected macrophages prevented efficient phagocytosis by uninfected bystander macrophages. As a result, the phagocytes secreted less pro-inflammatory cytokines, and preferentially primed anti-inflammatory T cell responses. Together, these results suggested that live M. leprae could employ a strain-independent mechanism to suppress inflammation, possibly involving the inhibition of autophagy and apoptosis in the infected macrophages.
Keywords: Apoptosis, macrophage, Mycobacterium leprae
Introduction
Mycobacterium leprae (M. leprae) is the causative pathogen of human leprosy and infects macrophages and Schwann cells [1]. As an obligate intracellular pathogen, clearance of M. leprae is dependent on efficient T cell-mediated clearance of infected host cells. However, studies by others and us have found that live M. leprae-infected macrophages, but not heat-killed M. leprae-treated macrophages, preferentially exhibit the regulatory M2 type, which is characterized by high interleukin 10 (IL-10) and low IL-12 expression [2-5]. As antigen presenting cells (APCs), these infected macrophages preferentially induce regulatory T cell responses instead of proinflammatory and cytotoxic T cell responses [2]. These features are thought to contribute to the persistence of M. leprae in the host and result in worse infection outcomes. Indeed, patients with lepromatous leprosy (L-lep), a progressive disease with numerous lesions and plenty of intracellular bacteria, are found to have weak cellular immunity and elevated Foxp3+ regulatory T cells at the lesion site [6-9], while patients with tuberculoid leprosy (T-lep), a self-containing disease with fewer lesions and less bacteria, are found with robust Th1-skewing immunity [10,11]. These results raised the question of how live, but not dead, M. leprae promoted M2 macrophage skewing and whether there were strain differences between L-lep and T-lep patients.
Internalization of cytosolic contents through autophagy is crucial for the homeostasis of cellular organelles. During intracellular infection, autophagy provides a defense mechanism through which antigens of intracellular pathogens can be internalized in lysosomes, and then presented by MHC molecules [12,13]. Programmed cell death through apoptosis represents another defense mechanism during which the dying infected cell keeps its cellular membrane intact and prevents the release of intracellular pathogens [14]. In another mycobacterium infection by virulent M. tuberculosis, it has been shown that inhibition of autophagy and apoptosis is a strategy to evade antigen presentation and facilitate bacterial release [15]. Similarly, mutations in NOD2, a bacterial peptidoglycan sensor that initiates autophagy, is associated with susceptibility to leprosy [16-18]. Our previous study has also identified that polymorphisms in immunity-related GTPase (IRGM), an organizer of the core autophagy machinery [19], is associated with leprosy risk [20]. Moreover, infection of murine macrophages by live M. leprae inhibits apoptosis [21]. Whether live M. leprae infection promotes M2 macrophage differentiation and inhibits proinflammatory T cell immunity through the inhibition of apoptosis and autophagy is unclear.
In this study, we investigated the underlying mechanism that M. leprae promoted M2 macrophage differentiation and inhibited proinflammatory T cell immunity. We found that live M. leprae, but not killed M. leprae, could inhibit autophagy and apoptosis in infected macrophages. The effectiveness of this function depended on the concentration but not on whether the strain was harvested from L-lep or T-lep patients.
Materials and methods
Human samples
The source of uninfected peripheral blood mononuclear cells (PBMCs) was a healthy female individual who did not have leprosy, autoimmune disorders, other systemic diseases or leprosy in a family member (within third-degree relatives). All L-lep and T-lep participants were diagnosed by the consensus of two dermatologists based on Ridley and Jopling’s criteria [6]. They were also free of M. tuberculosis infection and other chronic infections except M. leprae. A 6-mm skin biopsy specimen was obtained by standard punch technique. All participants provided written informed consent. The study was approved by the ethics committee of Shanghai Skin Disease Hospital.
Harvest and growth of primary M. leprae
Collection of primary M. leprae was attempted using the skin biopsy samples from six L-lep and two T-lep individuals, using a method adopted from digesting mice foot pads with slight modifications [22]. Briefly, the tissue was minced in Hanks’ balanced salt solution (HBSS, Thermo Fisher) containing 0.05% Tween 80 (Sigma Aldrich), and then centrifuged at 150 g for 10 min. The supernatant was treated with 0.05% trypsin at 37°C for 1 hour. The bacilli was then pelleted by centrifuging for 20 min at 4000 g, resuspended in HBSS, and treated with 1% sodium hydroxide for 15 min at 33°C. The bacilli was then washed and counted for resuspension at a final concentration of 2 × 107 per mL [23]. The amount of 0.05 mL suspension was injected into the foot pads of BALB/c athymic NU/NU mice for further amplification. Three primary strains, including L1 and L5 from two L-lep subjects and T2 from one T-lep subject were successfully obtained. M. leprae viability was determined by viability staining [24]. More than 85% viability was used as a standard for live M. leprae. Equivalent amount of live M. leprae was heated in 80°C for 30 min to obtain heat-killed M. leprae. All M. leprae samples were used within 24 h of harvest.
Cell isolation
Macrophage was derived from peripheral blood monocytes, which was obtained by using Human Monocyte Isolation Kit II (Miltenyi) on PBMCs from the healthy control individual. A total of 106 per mL monocytes were then cultured in RPMI 1640 with L-glutamine, Pen Strep (Thermo Fisher) and 10% autologous serum for 6 days at 37°C and 5% CO2, with media replacement every two days. At the end, the plate was swirled gently and the upper level medium was carefully removed to leave only live adherent macrophages at the bottom. Macrophage identity was then confirmed by microscopic examination and CD16 straining. Autologous naive CD45RA+ T cells was obtained by applying Naive Pan T Cell Isolation Kit (Miltenyi) and confirmed by CD45RA staining.
M. leprae treatment of macrophages
Live or heat-killed M. leprae was added to adherent macrophages at an MOI of 20:1 and 100:1 for 6 h at 33°C. The plates were then washed twice to remove excess bacteria, following a previously established method [21].
Flow cytometry
For the detection of autophagosomes, live or heat-killed M. leprae-treated macrophages were washed twice and incubated with prepared Cyto-ID Green Dye (Enzo Life Sciences) for 30 min at 37°C, and then washed again and incubated with Violet Dead Cell Stain (Invitrogen) and APC-conjugated anti-CD16 antibody (BioLegend) for 15 min in 4°C. For identification of apoptotic cells, the Annexin V-FITC Apoptosis Detection Kit (Abcam) was used. Samples were acquired in a BD FACS Canto cytometer and analyzed in FlowJo (Tree Star).
Coculture with T cells
We followed a previously published protocol [2]. Briefly, naive T cells were cocultured with M. leprae-treated macrophages for 6 days, and then purified using Human T Cell Enrichment Kit (Stemcell) and cultured alone for an additional 72 h in the presence of anti-human CD3 (OKT3, BD) and anti-human CD28 (CD28.2, eBioscience).
ELISA
Macrophages after 12 h incubation, or T cells after 72 h incubation were centrifuged at 300 g for 5 min. 100 µl supernatant was taken per 2 × 105 cells. Human interferon (IFN) gamma, IL-12 p70, and IL-10 ELISA kits (eBioscience) were used.
Quantitative RT-PCR
cDNA from M. leprae-treated macrophages was obtained by coupling the RNeasy Mini Kit and the QuantiTect Reverse Transcription Kit (Qiagen). IRGM expression was measured by quantitative real-time PCR using the using the LightCycler SYBR Green System (Roche). 5’-GCAGATCAGAGAAAATGTCCTGGAA-3’ foward primer and 5’-TGGCTAGCTGTTGAATATCCTGAGC-3’ reverse primer were used under the following condition: 1 min at 94°C, 45 cycles of 30 s at 94°C, 30 s at 55°C, and 30 s 72°C, and 7 min at 72°C.
Caspase assay
A total of 106 M. leprae-treated macrophages were lysed on ice and the caspase 3 and caspase 9 activities were measured using Caspase-3 and Caspase-9 Colorimetric Assay Kit (R&D Systems), following manufacturer’s instructions. For all experiments, a separate macrophage culture with the pan-caspase inhibitor z-VAD-FMK (100 µM, Santa Cruz Biotechnology) was used as a negative control for the colorimetric assays.
Phagocytosis assay
Uninfected macrophages (phagocytes) were labeled with FITC-anti-human CD16 (BioLegend) and added to pHrodo-labeled M. leprae-treated macrophages at 4:1 phagocyte to target cell ratio. pHrodo-labeling was completed using pHrodo Red AM Intracellular pH Indicator (Thermo Fisher) following manufacturer’s instructions. FITC-labeled cells was acquired in FACS Canto for measuring the level of pHrodo fluorescence or sorted in FACS Aria for subsequent coculture with T cells.
Statistical analyses
All results were expressed as mean ± SEM. Differences between live or heat-killed M. leprae treatment were examined by Welch’s t test or one-way ANOVA followed by Tukey’s test per experiment. Two-tailed P < 0.05 was considered significant.
Results
Low proinflammatory/regulatory T cell ratio after priming by live M. leprae-infected macrophage
We previously reported that live, but not dead, M. leprae Thai 53 strain induced M2-type macrophage differentiation. These live M. leprae-infected macrophages preferentially primed regulatory T cell responses with elevated IL-10 secretion, lower IFN-γ secretion, and reduced cytotoxicity [2]. However, monocytes from T-lep patients were significantly less IL-10-producing and more IL-12-producing than monocytes from L-lep patients [2]. These previous observations raised the question of whether strain differences induced different macrophage responses, such that the M. leprae strain from T-lep subjects promoted M1-type differentiation while M. leprae strain from L-lep subjects promoted M2-type differentiation. To answer this, we cultured primary M. leprae isolates from two L-lep patients, named strain L1 and strain L5, and from one T-lep patient, named strain T2. These primary strains were then cultured with macrophages derived from peripheral blood monocytes from healthy volunteers, in live or heat-killed form. After M. leprae treatment, the IL-10 and IL-12 mRNA transcription levels in macrophages, as well as the secreted IL-10 and IL-12 concentrations in the supernatant, were measured. We found that in general, macrophages incubated with live and killed strains presented similar levels of IL-10 secretion, but macrophages incubated with live strains presented very low IL-12 expression, while macrophages incubated with killed strains presented significantly elevated IL-12 expression (Figure 1A and 1B). Macrophages incubated with live L1 and T2 strains presented significantly higher IL-10/IL-12 concentration ratio compared to macrophages incubated with killed L1 and T2 strains. Macrophages incubated with live L1, L5 and T2 strains also expressed lower MHC class II than those incubated with killed strains (Figure 1C). A fraction of macrophages were washed and cocultured with CD45RA+ naive T cells. The T cells were isolated again after 6 days and cultured alone for 72 additional hours in the presence of anti-CD3/CD28 stimulation. Cytokine secretion in the supernatant was measured by ELISA. We found that T cells incubated with live L1, L5 and T2-treated macrophages presented lower IFN-γ/IL-10 secretion than T cells incubated with killed L1, L5 and T2-treated macrophages (Figure 1D). Together, these data demonstrated that primary M. leprae isolates recapitulated our previous finding in the Thai 53 strain [2]. Moreover, the primary M. leprae strain T2 from T-lep patients was not different from the primary L1 and L5 strains from L-lep patients in terms of converting macrophages toward a more regulatory type.
Figure 1.

Macrophages treated with primary live M. leprae strains tended to present M2-type functionality. Macrophages were derived from peripheral blood monocytes from a healthy volunteer and were incubated with live (black bars) or heat-killed (white bars) primary L1 and L5 strains from two L-lep patients and primary T2 strain from one T-lep patient. A. IL-10 and IL-12 mRNA levels, as well as the IL-10/IL-12 mRNA ratio, in macrophages, as measured by qPCR. B. Secreted IL-10 and IL-12 protein concentrations, as well as the IL-10/IL-12 protein concentration ratio, by macrophages in the supernatant, as measured by ELISA. C. MHC class II expression level by macrophages after treatment with live or killed M. leprae. The mean fluorescence intensity (MFI) was shown. D. Cytokine expression by autologous naive CD45RA+ T cells after incubation with live or killed M. leprae-treated macrophages. Naive CD45RA+ T cells were first incubated with macrophages for 6 days, and then purified and incubated separately with anti-CD3/CD28 stimulation for 72 hours. IFN-γ and IL-10 concentration was measured by ELISA. The ratio of IFN-γ to IL-10 level is shown. All experiments were performed in triplicates. Welch’s t test. *P < 0.05. **P < 0.01. ***P < 0.001. NS: not significant.
Live M. leprae infection selectively inhibited macrophage autophagy
IRGM is necessary for defense against intracellular pathogens, such as Listeria, Toxoplasma, and M. tuberculosis, by inducing autophagy [25], a process through which macrophage degrades cytoplasmic components and presents them on the MHC molecules. Previous studies by others and us have shown that polymorphisms in IRGM is associated with susceptibility to tuberculosis and leprosy [2,20,26,27]. We therefore investigated whether live or heat-killed M. leprae-treatment affected the expression of IRGM. At high multiplicity of infection (MOI = 100:1), killed L5 strain-treated macrophages expressed significantly higher IRGM than live L5-strain treated macrophages, while the difference in IRGM expression between live or killed L1 and T5 treatment were not significant (Figure 2A). At low MOI (20:1), however, live L1, L5, and T2-treated macrophages expressed significantly lower levels of IRGM than killed L1, L5 and T2-treared macrophages (Figure 2B). Since IRGM acts as the core protein for autophagy assembly, we also investigated the level of autophagy in live or killed M. leprae-treated macrophages by flow cytometry (Figure 2C). At high MOI, the level of autophagy in live L5-treated macrophages was significantly higher than that in killed L5-treated macrophages, while at low MOI, the level of autophagy was significantly lower in all macrophages treated with live strains (Figure 2C). Together, these results suggest that autophagy was not effectively upregulated when live, but not killed, M. leprae was added at low concentrations.
Figure 2.

IRGM was preferentially expressed in killed M. leprae-treated macrophages. Macrophages were incubated with live (black bar) or heat-killed (white bar) M. leprae at various MOI levels. The macrophages were then harvested for IRGM mRNA expression by quantitative RT-PCR. A. IRGM expression in macrophages when M. leprae was added at 100:1 or at 20:1 MOI level. B. Detection of autophagy by Cyto-ID Green Autophagy Dye was first verified in control macrophages (incubated in full-nutrient medium), starved macrophages (incubated in amino acid-deprived medium for 18 h), and recovered macrophages (starved macrophages replaced in full-nutrient medium for 4 h). The level of autophagy was then examined in live or killed M. leprae-treated macrophages. C. The level of autophagy in macrophages when M. leprae was added at 100:1 or at 20:1 MOI level. All experiments were performed in triplicates. *P < 0.05. **P < 0.01. ***P < 0.001. NS: not significant.
Live M. leprae infection at low multiplicity of infection (MOI) inhibited caspase 3 and caspase 9 activation
Apoptosis represents another mechanism through which the host controls intracellular infections. It is thought that rupture of infected macrophages in necrosis can result in bacterial dispersal to uninfected neighboring cells, while apoptosis keeps the membrane intact and promotes the uptake of dying cell by other phagocytes, which can then present intracellular bacterial antigen. Indeed, M. tuberculosis was previously shown to block the formation of apoptotic envelope in infected macrophages and resulted in reduced mycobacterial antigen presentation [15,28]. Here we show that M. tuberculosis phagosomes are secluded from the cytosolic MHC-I processing pathway and that mycobacteria-infected cells lose their antigen-presenting capacity. We also show that mycobacteria induce apoptosis in macrophages, causing the release of apoptotic vesicles that carry mycobacterial antigens to uninfected antigen-presenting cells (APCs. Here, we investigated the effect of live or killed M. leprae treatment in macrophage apoptosis at high (100:1) and low (20:1) MOI by annexin V staining (Figure 3A). No significant difference was observed between live or killed M. leprae-treated macrophages at high MOI, but at low MOI, the killed M. leprae-treated macrophages presented significantly higher apoptosis than live M. leprae-treated macrophages (Figure 3B). The caspase 3 and caspase 9 activation in live or killed M. leprae-treated macrophages was also examined and was expressed as a ratio compared to untreated control macrophages. We found that at high MOI, live and killed M. leprae-treated macrophages presented comparable levels of caspase 3 and caspase 9 activation, which were higher than the levels in controls (Figure 3C). At low MOI, live L1, L5, and T2 strains induced lower caspase 3 activation than killed L1, L5 and T2 strains; live L1 and T2 strains also induced lower caspase 9 activation than killed L1 and T2 strains. Interestingly, live M. leprae treatment at low MOI actually inhibited apoptosis, since the ratios of caspase 3 and caspade 9 activation when compared to controls were smaller than 1, suggesting that M. leprae might employ anti-apoptotic machinery similar to that in M. tuberculosis [15].
Figure 3.
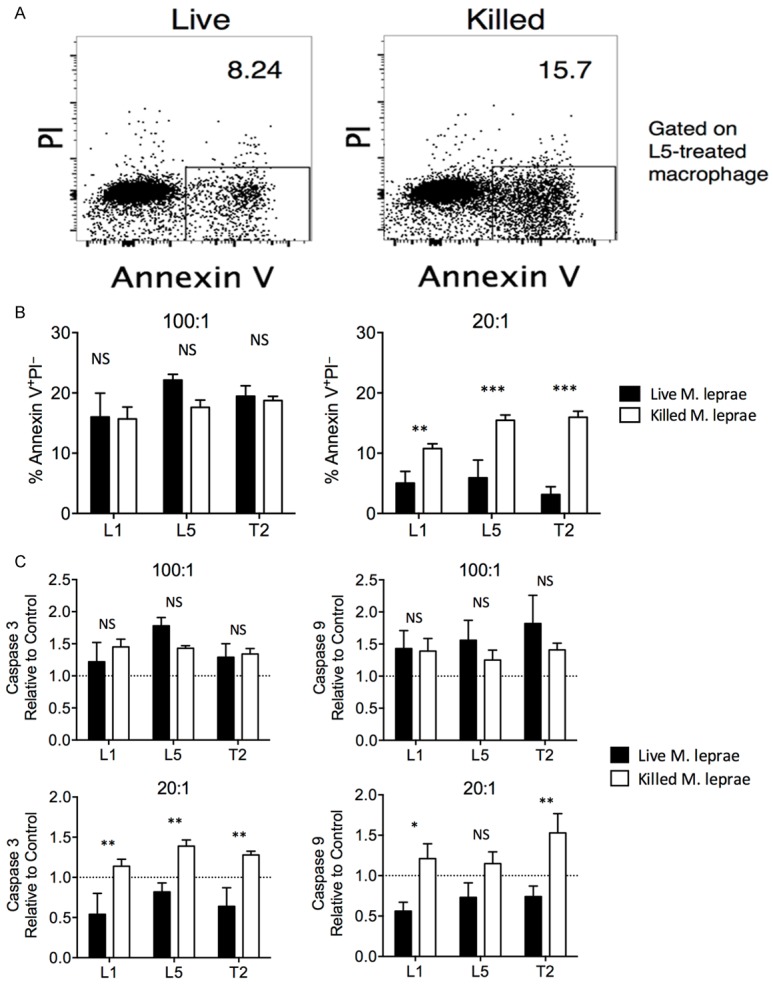
Apoptosis was higher in killed M. leprae-treated macrophages at low MOI. Macrophages were incubated with either live (black bar) or heat-killed (white bar) M. leprae. A. Representative gating of apoptotic cells (annexin V+PI-) in L5-treated macrophages. B. The percentage of annexin V+PI- cells in live or killed M. leprae-treated macrophages at high (100:1) or low (20:1) MOI levels. C. The induction of caspase 3 and caspase 9 was examined. Untreated macrophages were used as a control. The results were expressed as experiment to control ratio. A. The caspase 3 and caspase 9 activation by macrophages at high (100:1) or low (20:1) MOI levels. All experiments were performed in triplicates. Welch’s t test. **P < 0.01. *P < 0.05. NS: not significant.
Lack of apoptosis prevented antigen transfer to bystander antigen presenting cells, thus preventing proinflammatory T cell priming
The results above suggest that live M. leprae infection could inhibit autophagy and apoptosis, but how this leads to higher levels of M2 type macrophages and lower levels of proinflammatory T cell inflammation is unknown. We hypothesized that reduction in autophagy and apoptosis in live M. leprae-infected macrophages might prevent them from being digested by uninfected macrophages, thus preventing effective antigen presentation. To investigate this hypothesis, we labeled live or killed M. leprae-treated macrophages with pHrodo, a dye that is only weakly fluorescent at neutral pH, such as in the medium or cytosol, but is increasingly fluorescent as the pH value drops in phagosomes and endosomes [29]. The pHrodo-labeled macrophages were then cultured with untreated macrophages, which were then examined in flow cytometry for engulfment analysis. pHrodo-labeled irradiated macrophages were used as a positive control while pHrodo-labeled untreated macrophages were used as negative controls. We found that killed M. leprae-treated macrophages were engulfed more efficiently than live M. leprae-treated macrophages (Figure 4A). These phagocytic macrophages were then stained for intracellular cytokine secretion, and also sorted and cocultured with naive CD45RA+ T cells. We found that uninfected macrophages that were cocultured with killed M. leprae-treated macrophages had significantly lower IL-10/IL-12 ratio than uninfected macrophages that were cocultured with live M. leprae-treated macrophages. T cells incubated with macrophages that were cocultured with killed M. leprae-treated macrophages also present significantly elevated IFN-γ/IL-10 ratio (Figure 4B). Together, these results demonstrated that live M. leprae-treated macrophages were not engulfed effectively by uninfected macrophages, which resulted in less proinflammatory cytokine secretion by both macrophages and T cells.
Figure 4.
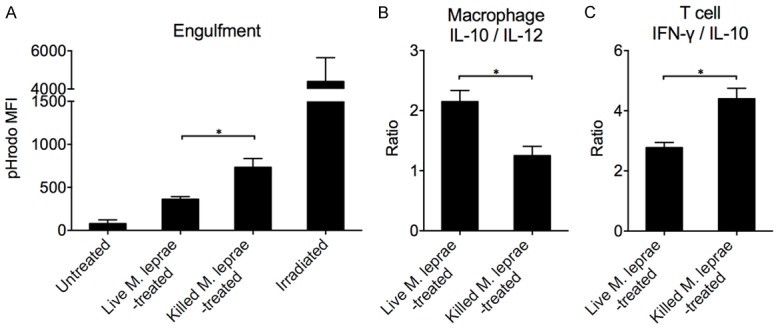
Live M. leprae-treated macrophages were not as effectively ingested as heat-killed M. leprae-treated macrophages. Live or heat-killed M. leprae-treated macrophages (MOI = 20:1) were labeled with pHrodo and cultured with FITC-anti-CD16-labeled uninfected macrophages. The amount of macrophages being ingested was then quantified by flow cytometry. A. The level of engulfment expressed as the MFI of pHrodo fluorescence under acidic pH. One-way ANOVA followed by Tukey’s test. *P < 0.05. B. The IL-10 to IL-12 expression ratio by uninfected macrophages after incubation with live or killed M. leprae-treated macrophages. The uninfected macrophages (phagocytes) were isolated by FITC sorting and cultured separately for 24 hours. The supernatant was harvested for ELISA measurement. C. The IFN-γ to IL-10 expression ratio by naive CD45RA+ T cells after coculture with FITC-sorted uninfected macrophages (phagocytes). Naive CD45RA+ T cells were first incubated with phagocytes for 6 days, and then purified and incubated separately with anti-CD3/CD28 stimulation for 72 hours. IFN-γ and IL-10 concentration was measured in the supernatant by ELISA. Welch’s t test. * P < 0.05.
Discussion
This study has two major findings. First, we found that no significant difference was observed between primary M. leprae isolates from L-lep and T-lep patients, suggesting that the different infection outcome was not primarily due to strain differences. To demonstrate this, primary L1 and L5 strains from two L-lep patients and primary T2 strain from one T-lep subject were harvested. When used to treat macrophages from a healthy volunteer, all three live M. leprae isolates preferentially promoted M2 macrophage differentiation, inhibited proinflammatory T cell activity, and suppressed autophagy and apoptosis, regardless of whether the strain was obtained from L-lep or T-lep subject. Second, we found that the M2 macrophage differentiation and inhibition of T cell activity was associated with autophagy and apoptosis. At low MOI (20:1), we found that macrophages treated by heat-killed M. leprae upregulated IRGM, an inducer of the autophagy assembly, while macrophages treated by live M. leprae failed to do so. This reduction in autophagy, coupled with lower MHC class II expression in live M. leprae-treated macrophages (Figure 1B), likely results in reduced M. leprae antigen presentation on the cell surface and limited T cell stimulation. Moreover, we found that at high MOI (100:1), both live and heat-killed M. leprae-treated macrophages exhibited higher caspase 3 and caspase 9 activity than control untreated macrophages. However, at low MOI (20:1), live M. leprae-treated macrophages actually downregulated the caspase 3 and caspase 9 activity compared to untreated control macrophages, suggesting that live M. leprae infection could actually inhibit apoptosis. Overall, these results confirmed earlier findings in murine macrophages [21], and discovered that primary live, but not heat-killed, M. leprae isolates from both L-lep and T-lep subjects promoted M2 macrophage skewing and inhibited autophagy and apoptosis.
Interestingly, differences between live or killed M. leprae were more pronounced at low MOI (20:1) than at high MOI (100:1). It is possible that at high MOI, the amount of dead bacteria in the live M. leprae sample (viability ≥ 85%) was concentrated enough to stimulate an inflammatory response like the heat-killed M. leprae.
The inhibition of apoptosis could assist the persistence of M. leprae through several mechanisms. First, M. leprae is an obligate intracellular pathogen that depends on the macrophage fatty acid metabolism for the synthesis of microbial lipids [30], thus requires the host cells to persist. Second, the failure of apoptosis could induce necrosis, another cell death modality in which the cell membrane is fragmented, resulting in the release of viable M. leprae for spreading infection. Indeed, it was shown that virulent M. tuberculosis actively prevented the formation of a stable apoptotic envelope matrix in macrophages, which resulted in membrane disintegration and necrosis [15]. And third, uptake of apoptotic cells enables antigen presentation to T cells by the uninfected phagocyte, which can then initiate an immune response against infected macrophages [28]. Failure to induce apoptosis may prevent efficient uptake of M. leprae-infected macrophages. We specifically examined the third possibility by using pHrodo to label M. leprea-treated macrophages and FITC-anti-human CD16 to label uninfected macrophages, which were used as phagocytes. We found that heat-killed M. leprea-treated macrophages were more efficiently ingested by phagocytes than live M. leprae-treated macrophages. Furthermore, we showed that FITC-labeled macrophages that were cocultured with live M. leprae-treated macrophages were skewed toward M2 type and preferentially primes regulatory T cell responses. Together, these results demonstrated an additional mechanism through which the suppression of apoptosis by live M. leprae resulted in reduced proinflammatory immune responses.
Our study has a couple of unresolved issues. First, in this study, no obvious differences between the L-lep strains and T-lep strains were found, but this conclusion is limited by the lack of more primary T-lep and L-lep strains. Therefore, whether the differences in the inflammatory responses between L-lep and T-lep patients are more host-dependent or strain-dependent still requires further investigations. Second, the mechanisms by which live M. leprae inhibited macrophage autophagy and apoptosis is still unclear. It has been demonstrated by several reports that M. tuberculosis and M. bovis Bacillus Calmette Guérin could evade macrophage apoptosis [31,32], involving several virulence genes and alterations in the expression of pro-survival transcription factors, such as bcl-2 [33,34]. Whether M. leprae employs a shared mechanism or has unique virulence factors is still unclear. Also, Th1 polarization tended to induce macrophage autophagy while Th2 polarization tended to inhibit it [35]. Since a reduction in T cell-secreted IFN-γ/IL-10 ratio was observed following Live M. leprae treatment, it might further inhibit autophagy through a positive feedback loop. The molecular mechanisms for the inhibition of autophagy and apoptosis in M. leprae still requires further investigations.
Acknowledgements
This study was supported by National Natural Science Foundation of China (81470144), and Shanghai Municipal Training Project of Excellent Young Medical Talents (XYQ2011005).
Disclosure of conflict of interest
None.
References
- 1.Scollard DM, Adams LB, Gillis TP, Krahenbuhl JL, Truman RW, Williams DL. The continuing challenges of leprosy. Clin Microbiol Rev. 2006;19:338–381. doi: 10.1128/CMR.19.2.338-381.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang D, Shui T, Miranda JW, Gilson DJ, Song Z, Chen J, Shi C, Zhu J, Yang J, Jing Z. Mycobacterium leprae-Infected macrophages preferentially primed regulatory T cell responses and was associated with lepromatous leprosy. PLoS Negl Trop Dis. 2016;10:e0004335. doi: 10.1371/journal.pntd.0004335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Montoya D, Cruz D, Teles RM, Lee DJ, Ochoa MT, Krutzik SR, Chun R, Schenk M, Zhang X, Ferguson BG, Burdick AE, Sarno EN, Rea TH, Hewison M, Adams JS, Cheng G, Modlin RL. Divergence of macrophage phagocytic and antimicrobial programs in leprosy. Cell Host Microbe. 2009;6:343–353. doi: 10.1016/j.chom.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mege JL, Mehraj V, Capo C. Macrophage polarization and bacterial infections. Curr Opin Infect Dis. 2011;24:230–234. doi: 10.1097/QCO.0b013e328344b73e. [DOI] [PubMed] [Google Scholar]
- 5.Italiani P, Boraschi D. From monocytes to M1/M2 macrophages: phenotypical vs. functional differentiation. Front Immunol. 2014;5:514. doi: 10.3389/fimmu.2014.00514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ridley DS. Histological classification and the immunological spectrum of leprosy. Bull World Health Organ. 1974;51:451–465. [PMC free article] [PubMed] [Google Scholar]
- 7.Joosten SA, van Meijgaarden KE, Savage ND, de Boer T, Triebel F, van der Wal A, de Heer E, Klein MR, Geluk A, Ottenhoff TH. Identification of a human CD8+ regulatory T cell subset that mediates suppression through the chemokine CC chemokine ligand 4. Proc Natl Acad Sci U S A. 2007;104:8029–8034. doi: 10.1073/pnas.0702257104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bobosha K, Wilson L, van Meijgaarden KE, Bekele Y, Zewdie M, van der Ploeg-van Schip JJ, Abebe M, Hussein J, Khadge S, Neupane KD, Hagge DA, Jordanova ES, Aseffa A, Ottenhoff TH, Geluk A. T-cell regulation in lepromatous leprosy. PLoS Negl Trop Dis. 2014;8:e2773. doi: 10.1371/journal.pntd.0002773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Palermo ML, Pagliari C, Trindade MA, Yamashitafuji TM, Duarte AJ, Cacere CR, Benard G. Increased expression of regulatory T cells and down-regulatory molecules in lepromatous leprosy. Am J Trop Med Hyg. 2012;86:878–883. doi: 10.4269/ajtmh.2012.12-0088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Modlin RL. The innate immune response in leprosy. Curr Opin Immunol. 2010;22:48–54. doi: 10.1016/j.coi.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dockrell HM, Young SK, Britton K, Brennan PJ, Rivoire B, Waters MF, Lucas SB, Shahid F, Dojki M, Chiang TJ, Ehsan Q, McAdam KP, Hussain R. Induction of Th1 cytokine responses by mycobacterial antigens in leprosy. Infect Immun. 1996;64:4385–4389. doi: 10.1128/iai.64.10.4385-4389.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crotzer VL, Blum JS. Autophagy and its role in MHC-mediated antigen presentation. J Immunol. 2009;182:3335–3341. doi: 10.4049/jimmunol.0803458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choi AJ, Ryter SW. Autophagy in inflammatory diseases. Int J Cell Biol. 2011;2011:732798. doi: 10.1155/2011/732798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barker RN, Erwig LP, Hill KS, Devine A, Pearce WP, Rees AJ. Antigen presentation by macrophages is enhanced by the uptake of necrotic, but not apoptotic, cells. Clin Exp Immunol. 2002;127:220–225. doi: 10.1046/j.1365-2249.2002.01774.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gan H, Lee J, Ren F, Chen M, Kornfeld H, Remold HG. Mycobacterium tuberculosis blocks crosslinking of annexin-1 and apoptotic envelope formation on infected macrophages to maintain virulence. Nat Immunol. 2008;9:1189–1197. doi: 10.1038/ni.1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cooney R, Baker J, Brain O, Danis B, Pichulik T, Allan P, Ferguson DJ, Campbell BJ, Jewell D, Simmons A. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med. 2010;16:90–97. doi: 10.1038/nm.2069. [DOI] [PubMed] [Google Scholar]
- 17.Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhaes JG, Yuan L, Soares F, Chea E, Le Bourhis L, Boneca IG, Allaoui A, Jones NL, Nunez G, Girardin SE, Philpott DJ. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol. 2010;11:55–62. doi: 10.1038/ni.1823. [DOI] [PubMed] [Google Scholar]
- 18.Netea MG, Kullberg BJ, van der Meer JW. Genomewide association study of leprosy. N Engl J Med. 2010;362:1447. author reply 1447-1448. [PubMed] [Google Scholar]
- 19.Chauhan S, Mandell MA, Deretic V. Mechanism of action of the tuberculosis and Crohn disease risk factor IRGM in autophagy. Autophagy. 2016;12:429–431. doi: 10.1080/15548627.2015.1084457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang D, Chen J, Shi C, Jing Z, Song N. Autophagy gene polymorphism is associated with susceptibility to leprosy by affecting inflammatory cytokines. Inflammation. 2014;37:593–598. doi: 10.1007/s10753-013-9773-1. [DOI] [PubMed] [Google Scholar]
- 21.Lahiri R, Randhawa B, Krahenbuhl JL. Infection of mouse macrophages with viable Mycobacterium leprae does not induce apoptosis. J Infect Dis. 2010;201:1736–1742. doi: 10.1086/652499. [DOI] [PubMed] [Google Scholar]
- 22.Truman RW, Krahenbuhl JL. Viable M. Leprae as a research reagent. Int J Lepr Other Mycobact Dis. 2001;69:1–12. [PubMed] [Google Scholar]
- 23.Shepard CC, McRae DH. A method for counting acid-fast bacteria. Int J Lepr Other Mycobact Dis. 1968;36:78–82. [PubMed] [Google Scholar]
- 24.Lahiri R, Randhawa B, Krahenbuhl J. Application of a viability-staining method for Mycobacterium leprae derived from the athymic (nu/nu) mouse foot pad. J Med Microbiol. 2005;54:235–242. doi: 10.1099/jmm.0.45700-0. [DOI] [PubMed] [Google Scholar]
- 25.Petkova DS, Viret C, Faure M. IRGM in autophagy and viral infections. Front Immunol. 2012;3:426. doi: 10.3389/fimmu.2012.00426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.King KY, Lew JD, Ha NP, Lin JS, Ma X, Graviss EA, Goodell MA. Polymorphic allele of human IRGM1 is associated with susceptibility to tuberculosis in African Americans. PLoS One. 2011;6:e16317. doi: 10.1371/journal.pone.0016317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Che N, Li S, Gao T, Zhang Z, Han Y, Zhang X, Sun Y, Liu Y, Sun Z, Zhang J, Ren W, Tian M, Li Y, Li W, Cheng J, Li C. Identification of a novel IRGM promoter single nucleotide polymorphism associated with tuberculosis. Clin Chim Acta. 2010;411:1645–1649. doi: 10.1016/j.cca.2010.06.009. [DOI] [PubMed] [Google Scholar]
- 28.Schaible UE, Winau F, Sieling PA, Fischer K, Collins HL, Hagens K, Modlin RL, Brinkmann V, Kaufmann SH. Apoptosis facilitates antigen presentation to T lymphocytes through MHC-I and CD1 in tuberculosis. Nat Med. 2003;9:1039–1046. doi: 10.1038/nm906. [DOI] [PubMed] [Google Scholar]
- 29.Miksa M, Komura H, Wu R, Shah KG, Wang P. A novel method to determine the engulfment of apoptotic cells by macrophages using pHrodo succinimidyl ester. J Immunol Methods. 2009;342:71–77. doi: 10.1016/j.jim.2008.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cruz D, Watson AD, Miller CS, Montoya D, Ochoa MT, Sieling PA, Gutierrez MA, Navab M, Reddy ST, Witztum JL, Fogelman AM, Rea TH, Eisenberg D, Berliner J, Modlin RL. Hostderived oxidized phospholipids and HDL regulate innate immunity in human leprosy. J Clin Invest. 2008;118:2917–2928. doi: 10.1172/JCI34189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Keane J, Remold HG, Kornfeld H. Virulent Mycobacterium tuberculosis strains evade apoptosis of infected alveolar macrophages. J Immunol. 2000;164:2016–2020. doi: 10.4049/jimmunol.164.4.2016. [DOI] [PubMed] [Google Scholar]
- 32.Kremer L, Estaquier J, Brandt E, Ameisen JC, Locht C. Mycobacterium bovis Bacillus Calmette Guerin infection prevents apoptosis of resting human monocytes. Eur J Immunol. 1997;27:2450–2456. doi: 10.1002/eji.1830270945. [DOI] [PubMed] [Google Scholar]
- 33.Velmurugan K, Chen B, Miller JL, Azogue S, Gurses S, Hsu T, Glickman M, Jacobs WR Jr, Porcelli SA, Briken V. Mycobacterium tuberculosis nuoG is a virulence gene that inhibits apoptosis of infected host cells. PLoS Pathog. 2007;3:e110. doi: 10.1371/journal.ppat.0030110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Q, Liu S, Tang Y, Liu Q, Yao Y. MPT64 protein from Mycobacterium tuberculosis inhibits apoptosis of macrophages through NFkB-miRNA21-Bcl-2 pathway. PLoS One. 2014;9:e100949. doi: 10.1371/journal.pone.0100949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harris J, Master SS, De Haro SA, Delgado M, Roberts EA, Hope JC, Keane J, Deretic V. Th1-Th2 polarisation and autophagy in the control of intracellular mycobacteria by macrophages. Vet Immunol Immunopathol. 2009;128:37–43. doi: 10.1016/j.vetimm.2008.10.293. [DOI] [PMC free article] [PubMed] [Google Scholar]