

Abstract
Dendritic cell (DC) based immunotherapy is a promising approach for cancer treatment and has been approved in clinical settings for decades. Clinical trials have demonstrated relatively poor therapeutic efficacy. The efficacy of DC immunotherapy is strongly influenced by their ability to migrate to the draining lymph nodes (LNs). Therefore, it is critical to deliver DCs and monitor the in vivo biodistributions of DCs after administration. The purpose of this study is to determine whether a novel injection route of DCs improves DC migration to LNs, tissues, organs and lymphatics. In the present study, a modified method was investigated to acquire DCs from mouse bone marrow. Cultured antibody labeled DCs were analyzed by flow cytometry. India ink was used to visualize mouse abdominal LNs and PKH26 was utilized to label DCs for intraperitoneal (IP) injection, results were evaluated by histology. Our results showed that large amounts of DCs with a relatively high purity were acquired. IP injection of india ink marked the abdominal LNs and PKH26 labeled DCs showed IP was an effective administration route to increase the absorption of viable DCs, and different time points after IP inject showed no significant difference of the migrated DCs. The findings indicated that large amounts of high purity DCs can be acquired through our method and IP injection accelerates DCs migration to abdominal LNs, which can be directly translated to clinical settings, especially for abdominal cancers. This study makes a foundation for future researches of DC-based immunotherapy as a treatment modality against cancer.
Keywords: Dendritic cells, lymph node, migration, abdomen, intraperitoneal administration
Introduction
Dendritic cells (DCs) can present antigens on their surface to other cells of the immune system [1,2] and have been used in this capacity as potent therapeutic vaccines against human cancers [3]. However, immature DCs in a tumor environment may be functionally defective [4-6]. This limitation can be overcome by generating immature DCs from bone marrow cells and activating them with tumor antigens to allow maturation of these DCs with proinflammatory stimuli in vitro [7,8]. Many experimental immune therapies are now based upon the DC immunotherapies of cancer patients with autologous DCs [6,9-11]. DC-based immunotherapy has clinically relevant mechanisms of action with great potential for the systemic treatment of many cancers in clinical settings [12-14]. However, clinical trials have not yet demonstrated positive therapeutic efficacy and clinical response has been limited to a minority of patients [9-11,15]. The effectiveness of immunization with DC-based immunotherapies is strongly influenced by their successful migration to secondary lymphoid organs and tissues where they orchestrate immune response [6,9-11,16].
The administration route and frequency of DC-based vaccine injections along with the number of injected cells are strongly relevant with the viable DCs migration efficacy and subsequently determine the result of the treatment. Currently the standardized methodology of DC-based cancer vaccine application is not established yet [17]. Though ex vivo generated DCs loaded with specific tumor antigens have been proved to be feasible and superior, the administration route remains controversial [10]. Given that substantial population of LNs reside in the abdomen and together with spleen, consisting the most important secondary lymphoid organs [18], it’s theoretically effective that we inject the DCs intraperitoneally (IP), particularly for the abdominal tumors. However, few studies have demonstrated the efficiency of DC-based cancer vaccine employed through this route.
In this study, to determine whether a novel injection route of DCs improves DC migration to LNs, tissues, organs and lymphatics, which may give new insights into the DC-based anticancer regimen and provide approaches that can be directly translated to clinic and improve the outcome.
Materials and methods
The study was approved by institutional animal care and use committee (IACUC) and were strictly performed in compliance with NIH guidelines.
Animals and reagents
The mouse bone marrow derived DCs were prepared as previously described with some modifications [19]. Briefly, C57BL/6 mice (4-6 weeks age; Charles River, Wilmington, MA) were sacrificed with CO2. After immersed in 70% ethanol for 5 mins, tibias and femurs were carefully dissected. All the muscles and tissues attached to the bones were cleaned with sterilized gauze, and the bones were disinfected by immersion in 70% ethanol for 5 mins. Then the bones were flushed in half with fetal bovine serum (FBS) free RMPI 1640 (Gibco, Waltham, MA) in the hood. When the middle of the bones became visually white, the end of the bone was cut and flushed again. The bone marrow cells were collected and red blood cells were lysed. A total of 1×106 cells in 10 mL DC culture medium that containing 10% FBS (Gibco, Waltham, MA), 100 units/mL penicillin, 100 µg/mL streptomycin, 0.25 µg/mL amphotericin b (antibiotic-antimycotic 100x, Gibco, Waltham, MA), 10 ng/mL rm-GM-CSF and 1 ng/mL rm-IL-4 (both from Shenandoah Biotechnology, Warwick, PA) were plated in a petri dish. If a mature state was preferred, a cocktail of IFNγ, TNFα and LPS (all from Shenandoah Biotechnology, Warwick, PA) was added at 7th day. After 8 days’ culture, floating and loosely attached cells were ready to be harvested and tested by flow cytometry.
FACS
The cultured mouse bone marrow derived cells were collected and washed by cold PBS, incubated for 40 mins at 4°C with 2 µg/3×105 cells anti-mouse PerCP-CyTM5.5 CD11c, PerCP-CyTM5.5 CD11b, APC CD40, APC CD86, PE CD80, PE H-2Db, FITC H-2Kb (all from BD Bioscience, San Jose, CA), PE MHC II (Southern Biotech, Birmingham, AL) and appropriate isotype controls. Cells were identified by flow cytometry (BD LSRFortessaTM cell analyzer, San Jose, CA) and the data were analyzed by FlowJo (FlowJo LLC, Ashland, OR).
Dendritic cell labelling
The labeling of DCs was performed according to production manual. Briefly, at the 8th day of culture when the DCs were mature, 2×106 washed cells in 100 µL Diluent C were mixed with 100 µL Diluent C containing 0.4 µL PKH 26 dye for 5 mins (Sigma-Aldrich, St. Louis, MO). After adding 200 µL FBS and 3 times washing, the DCs were labeled with PKH26 (red) and the labeling efficiency was evaluated by fluorescent microscope (Axiovert 40 CFL, Carl Zeiss, Ontario, CA).
Abdominal LNs visualization
Six mice were IP injected with 100 µL 10% india ink (BD, Sparks, MD) and 2×106 PKH26 (red) labeled DCs respectively. After 30 mins, the india ink injected mice were sacrificed and abdomen were opened. India ink stained LNs and lymphatics can be visually seen. Spleen, pancreas, intestine and surrounding tissues were then collected and fixed in formalin for histological verification of india ink staining. As to PKH26 (red) labeled DCs injected mice, the interval before euthanasia is 1 hour. The collected tissues were embedded in OCT compound (Fisher HealthCare, Houston, TX) infused modes that were placed on dry ice, and freezed at -80°C after 2 mins.
Histology
For india ink injected mice, the organs and tissues collected after euthanization were immediately fixed in formalin solution for 72 hours, and then embedded in paraffin. Slices were cut in 4 µm thick and stained with H&E. All the slices were analyzed with optical microscope (EVOS FL Cell Imaging System, Life Technologies, Carlsbad, CA) from low magnification (10x) to high magnification (40x) by the same person. For PKH26 (red) labeled DCs injected mice, the samples that were placed in OCT compound and freezed at -80°C were cut in 5 µm with the microtome portion of the cryostat and picked up on slides. One group of the slides were mounted by cover glasses with ProLong Glod Antifade Reagent with DAPI (Cell Signaling Technology, Danvers, MA). Another group were stained with H&E, and all the slices were observed under optical microscope as well as florescent microscope (Axioimager Z1, Carl Zeiss, Ontario, CA).
Statistics
Total cells amount and PKH26 positive cells amount of the representative figures of spleen slides were count by ImageJ software (NIH, Bethesda, MD). Student’s t test was used to compare the differences between different time points after IP injection of PKH26 labeled DCs. Data were presented as mean ± SD. P<0.05 was considered as statistically significant.
Results
BMDC amount and morphology development
A total amount of 1.0-1.2×107 cells could be acquired from one mouse bone marrow through our method, as diagrammatized in Figure 1, in contrast to flushing in half only with 0.5-0.8×107 cells. Representative photographs of the cultured cells with typical DC characteristics in a dynamic development were shown in Figure 2. After 8 days’ culture, the bone marrow derived cells with initial concentration of 1×105 cells/mL would proliferate to occupy all the 10 cm petri dishes, and we were able to obtain 1-2×108 cells at this point.
Figure 1.
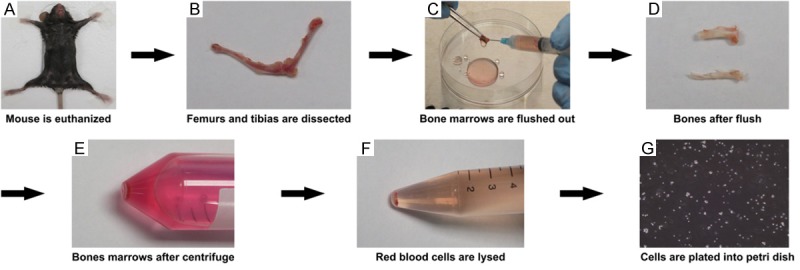
Schematic diagram of BMDC generation. Herein we illustrate the process with objective photograph of mouse BMDC. After sacrificed with CO2 (A), the tibias and femurs are carefully and intactly dissected with surrounding muscles and tissues detached (B). Bones are flushed with medium in half with tips preserved followed by second flush without tips (C), until the bones were flushed white (D). After centrifuge (E) and red blood cells lysed (F), these mouse bone marrow cells are plated into petri dishes at a concentration of 1×105/mL (G), and cultured for 7 days in the presence of GM-CSF and IL-4.
Figure 2.
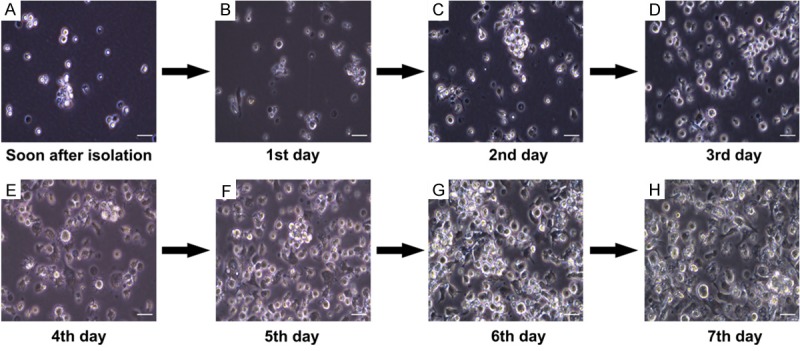
BMDC morphology development. Dynamic morphology changes of the mouse bone marrow cells are recorded everyday under an optical microscope. Representative pictures are shown. Cells were loosely connected and no DC characteristics can be seen in the first two days (A, B). From the second day of culture, cell clusters can be observed (C) and typical DC dendrites grow out in the third day (D). Cells proliferate rapidly (E-G), and at the last day of culture, a large amount with longer dendrites can be acquired (H). Scale bar represents 20 μm.
BMDC purity and maturity
The cultured bone marrow derived cells were collected and tested by flow cytometry. Results suggested a relatively high purity and maturity of DCs with substantial expression of CD11c, CD11b and other DC maturity markers compared to isotype controls (Figure 3A, 3B). Independent repeating experiments of analogous DC markers expression confirmed this method as a stable and repeatable approach to achieve extensive quantity mouse bone marrow derived DCs (Figure 3C).
Figure 3.

Flow cytometry analysis of BMDC. DCs were harvest after 8 days’ culture. CD11c and CD11b were chosen as DC specialty markers, CD40, CD80, CD86, MHC II, H-2Db and H-2Kb were utilized as DC maturity markers (A, B). (C) Average percentage of the chosen markers expression. Error bars represented mean ± SD. Three independent experiments were performed.
Abdominal LNs Visualization by India ink
After 30 mins india ink IP injection, the mouse abdominal LNs and lymphatics could be visually distinguished (Figures 4A, 4B, 5B). The india ink stained organs and tissues were collected for histologic demonstration of the staining efficiency. H&E staining of the slices indicated significant uptake of india ink by abdominal lymph organs (Figures 4C, 4D, 5D). As spleen is the largest lymph organ of the body where large amounts of lymphocytes exist, we compared the macroscopical and histological features of spleen after india ink and equal PBS IP injection, respectively. Results showed considerable stained cells in the spleen (Figure 5).
Figure 4.
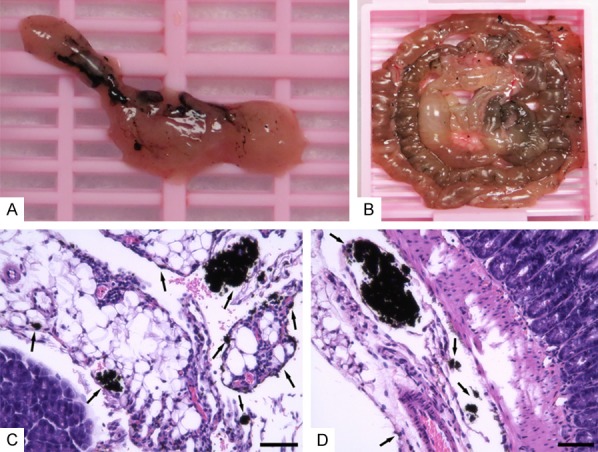
Abdominal LNs visualization by india ink. Mice are injected 100 µL of 10% india ink intraperitoneally and sacrificed after 30 mins. The abdomen is opened and pancreas as well as intestine with visual India ink staining are collected for histology analysis (A, B). Representative pictures of pancreas and intestine slices with H&E staining are shown (C, D). Black arrows indicate LNs and lymphatics stained by india ink. Scale bar represents 40 µm.
Figure 5.
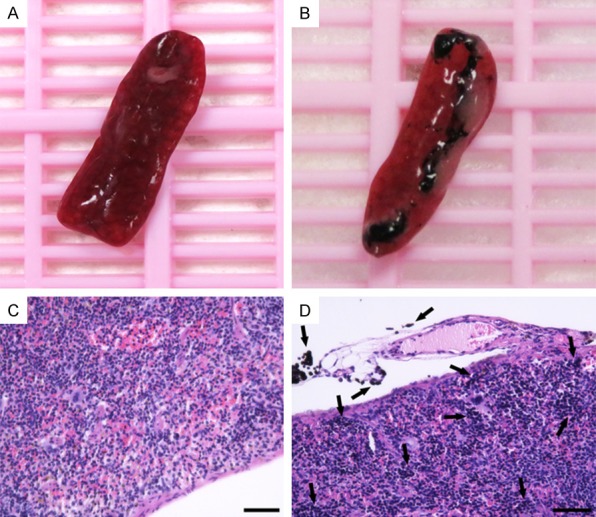
Macroscopical and histological pictures of spleen after india ink injection. Spleens are collected 30 mins after IP injection of 100 µL india ink at 10% concentration or 100 µL saline solution (A, B). Corresponding slices of H&E staining are presented with black arrows indicating LNs and lymphatics staining with india ink (C, D). Scale bar represents 40 µm.
Migration of fluorescence labeled BMDCs to abdominal LNs via IP injection
The PKH26 (red) labeling efficiency of mouse bone marrow derived mature DCs was verified under fluorescent microscope (Figure 6A, 6B). 2×106 labeled cells were injected intraperitoneally and the mice were euthanized 1 hour later. The same organs and tissues to Figures 4 and 5 were collected for frozen sections. After the organ structure were confirmed by H&E staining (Figure 6C-E), fluorescent microscope analysis of the same slides detected the labeled DCs in the LNs, tissues, organs and lymphatics, which was in consistent with india ink injected mice results (Figure 6F-H). The results of spleen were quantified by ImageJ and no significant difference was observed between different time points after IP injection (1 h vs. 6 h, 7.26% vs. 7.73%, P=0.4842; 1 h vs. 12 h, 7.26% vs. 7.59%, P=0.7462; 6 h vs. 12 h, 7.73% vs. 7.59%, P=0.8908) (Figure 6I).
Figure 6.

Fluorescent labeled BMDC in abdominal LNs. The cultured DCs labeled with PKH26 (red) were observed by optical and fluorescent microscope (A, B). Scale bar represents 20 µm. Spleen, pancreas and intestines along with surrounding tissues were collected for frozen sections after 1-hour IP injection of labeled DCs. H&E staining characterized the tissue structure and the labeled DCs were detected in the LNs, tissues, organs and lymphatics under fluorescent microscope (C-H). (C-E) Scale bar represents 40 µm. (F-H) Scale bar represents 20 µm. PKH26 positive DCs in spleen were quantified by ImageJ in different time points after IP injection (I). (#), no statistical difference.
Discussion
Ex vivo isolation of DC precursors from murine bone marrow and following differentiation into DCs has been extensively studied, and it is reported to be feasible and effective [20,21]. However, unified standard is still not reached among diverse exploration of the mouse bone marrow isolation process and cell culture recipe [22-24]. In the present study, a modified method was used to acquire large amounts of DCs from mouse bone marrow. The tips of bones were preserved at first to avoid possible contamination and cut it after first flush to fully wash out the bone marrow. Furthermore, the ratio of rm-GM-CSF and rm-IL-4 combination was investigated for the DC culture medium and found out 10 ng/mL rm-GM-CSF with 1 ng/mL rm-IL-4 to be the preference in our assays. A relatively high DC purity and maturity were founded after 8 days’ culture rather than 6 days, similar as Lutz’s conclusions [25]. These slight modifications enabled us to gain large quantities of highly pure DCs from mouse bone marrow for further research compared to other reports [20,21].
The administration route of DC-based cancer vaccine is directly correlated with consequent antitumor immune response and immune memory [26,27]. Currently, administration routes include subcutaneous injection and intra-LN injection [28,29]. Low dose of DCs can migrate to LNs and organs by subcutaneous injection [28,29]. A cluster of DCs directly delivers to LNs, which affects DC therapeutic function by intra-LN injection [28,29]. Regarding in the anatomic basis of abdomen. It turned out the uptake of IP injected labeled DCs by abdominal LNs, tissues, organs and lymphatics was comparable to india ink IP injection, indicating this administration method may increase the amount of viable DC-based vaccines that are actively absorbed by abdominal lymphoid organs and thus improve outcomes. Our results suggested a more suitable and potential administration route for abdominal cancers.
Previous reports showed that DC-based vaccine might be more valuable and efficient when applied in the early stage of the disease. Current attempts to combine DC-based cancer vaccine with immunostimulatory cytokines, immunomodulating agents, chemotherapy, targeted therapy and immune checkpoint inhibitors have shown clinical benefits [30-33], while the optimal combination regimen remains to be established. However, large amount of highly pure DCs from mouse bone marrow will be needed and IP delivery of DCs will improve DC therapeutic efficacy due to more DCs to abdominal LNs.
In conclusion, we present a modified method to acquire considerable amount of mouse bone marrow derived DCs and testify the efficiency of IP injection of labeled DCs, making a foundation for future researches of DC-based vaccine immunotherapy as a part of comprehensive treatment against cancer, especially for abdominal tumors.
Acknowledgements
This work is supported by the USA National Cancer Institute R01CA209886 and R01CA196967.
Disclosure of conflict of interest
None.
References
- 1.Lion E, Smits EL, Berneman ZN, Van Tendeloo VF. NK cells: key to success of DC-based cancer vaccines? Oncologist. 2012;17:1256–1270. doi: 10.1634/theoncologist.2011-0122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Karthaus N, Torensma R, Tel J. Deciphering the message broadcast by tumor-infiltrating dendritic cells. Am J Pathol. 2012;181:733–742. doi: 10.1016/j.ajpath.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 3.Mocellin S, Mandruzzato S, Bronte V, Lise M, Nitti D. Part I: Vaccines for solid tumours. Lancet Oncol. 2004;5:681–689. doi: 10.1016/S1470-2045(04)01610-9. [DOI] [PubMed] [Google Scholar]
- 4.Avigan D, Rosenblatt J, Kufe D. Dendritic/tumor fusion cells as cancer vaccines. Semin Oncol. 2012;39:287–295. doi: 10.1053/j.seminoncol.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 5.Cintolo JA, Datta J, Mathew SJ, Czerniecki BJ. Dendritic cell-based vaccines: barriers and opportunities. Future Oncol. 2012;8:1273–1299. doi: 10.2217/fon.12.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Melief CJ. Cancer immunotherapy by dendritic cells. Immunity. 2008;29:372–383. doi: 10.1016/j.immuni.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 7.Bapsy PP, Sharan B, Kumar C, Das RP, Rangarajan B, Jain M, Suresh Attili VS, Subramanian S, Aggarwal S, Srivastava M, Vaid A. Openlabel, multi-center, non-randomized, singlearm study to evaluate the safety and efficacy of dendritic cell immunotherapy in patients with refractory solid malignancies, on supportive care. Cytotherapy. 2014;16:234–244. doi: 10.1016/j.jcyt.2013.11.013. [DOI] [PubMed] [Google Scholar]
- 8.Chodon T, Comin-Anduix B, Chmielowski B, Koya RC, Wu Z, Allen-Auerbach MS, Ng C, Avramis E, Seja E, Villanueva A, McCannel TA, Ishiyama A, Czernin J, Radu CG, Wang X, Gjertson DW, Cochran AJ, Cornetta K, Wong DJ, Kaplan-Lefko P, Hamid O, Samlowski W, Cohen PA, Daniels GA, Mukherji B, Yang L, Zack J, Kohn DB, Heath JR, Glaspy JA, Witte ON, Baltimore D, Economou JS, Ribas A. Adoptive transfer of MART-1 T cell receptor transgenic lymphocytes and dendritic cell vaccination in patients with metastatic melanoma. Clin Cancer Res. 2014;14:14. doi: 10.1158/1078-0432.CCR-13-3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oshita C, Takikawa M, Kume A, Miyata H, Ashizawa T, Iizuka A, Kiyohara Y, Yoshikawa S, Tanosaki R, Yamazaki N, Yamamoto A, Takesako K, Yamaguchi K, Akiyama Y. Dendritic cell-based vaccination in metastatic melanoma patients: phase II clinical trial. Oncol Rep. 2012;28:1131–1138. doi: 10.3892/or.2012.1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 2012;12:265–277. doi: 10.1038/nrc3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zahradova L, Mollova K, Ocadlikova D, Kovarova L, Adam Z, Krejci M, Pour L, Krivanova A, Sandecka V, Hajek R. Efficacy and safety of Id-protein-loaded dendritic cell vaccine in patients with multiple myeloma--phase II study results. Neoplasma. 2012;59:440–449. doi: 10.4149/neo_2012_057. [DOI] [PubMed] [Google Scholar]
- 12.Hoos A, Eggermont AM, Janetzki S, Hodi FS, Ibrahim R, Anderson A, Humphrey R, Blumenstein B, Old L, Wolchok J. Improved endpoints for cancer immunotherapy trials. J Natl Cancer Inst. 2010;102:1388–1397. doi: 10.1093/jnci/djq310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aarntzen EH, De Vries IJ, Lesterhuis WJ, Schuurhuis D, Jacobs JF, Bol K, Schreibelt G, Mus R, De Wilt JH, Haanen JB, Schadendorf D, Croockewit A, Blokx WA, Van Rossum MM, Kwok WW, Adema GJ, Punt CJ, Figdor CG. Targeting CD4(+) T-helper cells improves the induction of antitumor responses in dendritic cell-based vaccination. Cancer Res. 2013;73:19–29. doi: 10.1158/0008-5472.CAN-12-1127. [DOI] [PubMed] [Google Scholar]
- 14.Tada F, Abe M, Hirooka M, Ikeda Y, Hiasa Y, Lee Y, Jung NC, Lee WB, Lee HS, Bae YS, Onji M. Phase I/II study of immunotherapy using tumor antigen-pulsed dendritic cells in patients with hepatocellular carcinoma. Int J Oncol. 2012;41:1601–1609. doi: 10.3892/ijo.2012.1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garcia-Marquez MA, Wennhold K, Draube A, von Bergwelt-Baildon M. Results of a Phase II clinical trial with Id-protein-loaded dendritic cell vaccine in multiple myeloma: encouraging or discouraging? Immunotherapy. 2012;4:991–994. doi: 10.2217/imt.12.94. [DOI] [PubMed] [Google Scholar]
- 16.Seyfizadeh N, Muthuswamy R, Mitchell DA, Nierkens S, Seyfizadeh N. Migration of dendritic cells to the lymph nodes and its enhancement to drive anti-tumor responses. Crit Rev Oncol Hematol. 2016;107:100–110. doi: 10.1016/j.critrevonc.2016.09.002. [DOI] [PubMed] [Google Scholar]
- 17.Datta J, Terhune JH, Lowenfeld L, Cintolo JA, Xu S, Roses RE, Czerniecki BJ. Optimizing dendritic cell-based approaches for cancer immunotherapy. Yale J Biol Med. 2014;87:491–518. [PMC free article] [PubMed] [Google Scholar]
- 18.Malik B, Rath G, Goyal AK. Are the anatomical sites for vaccine administration selected judiciously? Int Immunopharmacol. 2014;19:17–26. doi: 10.1016/j.intimp.2013.12.023. [DOI] [PubMed] [Google Scholar]
- 19.Muccioli M, Pate M, Omosebi O, Benencia F. Generation and labeling of murine bone marrow-derived dendritic cells with Qdot nanocrystals for tracking studies. J Vis Exp. 2011 doi: 10.3791/2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bruno L. Differentiation of dendritic cell subsets from mouse bone marrow. Methods Mol Biol. 2007;380:47–57. doi: 10.1007/978-1-59745-395-0_3. [DOI] [PubMed] [Google Scholar]
- 21.Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, Muramatsu S, Steinman RM. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colonystimulating factor. J Exp Med. 1992;176:1693–1702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim SJ, Diamond B. Generation and maturation of bone marrow-derived DCs under serum-free conditions. J Immunol Methods. 2007;323:101–108. doi: 10.1016/j.jim.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yin SY, Wang CY, Yang NS. Interleukin-4 enhances trafficking and functional activities of GM-CSF-stimulated mouse myeloid-derived dendritic cells at late differentiation stage. Exp Cell Res. 2011;317:2210–2221. doi: 10.1016/j.yexcr.2011.06.013. [DOI] [PubMed] [Google Scholar]
- 24.Roney K. Bone marrow-derived dendritic cells. Methods Mol Biol. 2013;1031:71–76. doi: 10.1007/978-1-62703-481-4_9. [DOI] [PubMed] [Google Scholar]
- 25.Lutz MB, Kukutsch N, Ogilvie ALJ, Rossner S, Koch F, Romani N, Schuler G. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods. 1999;223:77–92. doi: 10.1016/s0022-1759(98)00204-x. [DOI] [PubMed] [Google Scholar]
- 26.Mullins DW, Sheasley SL, Ream RM, Bullock TN, Fu YX, Engelhard VH. Route of immunization with peptide-pulsed dendritic cells controls the distribution of memory and effector T cells in lymphoid tissues and determines the pattern of regional tumor control. J Exp Med. 2003;198:1023–1034. doi: 10.1084/jem.20021348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Simon T, Fonteneau JF, Gregoire M. Dendritic cell preparation for immunotherapeutic interventions. Immunotherapy. 2009;1:289–302. doi: 10.2217/1750743X.1.2.289. [DOI] [PubMed] [Google Scholar]
- 28.Galati D, Zanotta S. Hematologic neoplasms: dendritic cells vaccines in motion. Clin Immunol. 2017;183:181–190. doi: 10.1016/j.clim.2017.08.016. [DOI] [PubMed] [Google Scholar]
- 29.Jiang H, Wang Q, Sun X. Lymph node targeting strategies to improve vaccination efficacy. J Control Release. 2017;267:47–56. doi: 10.1016/j.jconrel.2017.08.009. [DOI] [PubMed] [Google Scholar]
- 30.Okada H, Kalinski P, Ueda R, Hoji A, Kohanbash G, Donegan TE, Mintz AH, Engh JA, Bartlett DL, Brown CK, Zeh H, Holtzman MP, Reinhart TA, Whiteside TL, Butterfield LH, Hamilton RL, Potter DM, Pollack IF, Salazar AM, Lieberman FS. Induction of CD8+ T-cell responses against novel glioma-associated antigen peptides and clinical activity by vaccinations with {alpha}-type 1 polarized dendritic cells and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in patients with recurrent malignant glioma. J. Clin. Oncol. 2011;29:330–336. doi: 10.1200/JCO.2010.30.7744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maus MV, June CH. Making Better chimeric antigen receptors for adoptive T-cell therapy. Clin Cancer Res. 2016;22:1875–1884. doi: 10.1158/1078-0432.CCR-15-1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ozao-Choy J, Ma G, Kao J, Wang GX, Meseck M, Sung M, Schwartz M, Divino CM, Pan PY, Chen SH. The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Res. 2009;69:2514–2522. doi: 10.1158/0008-5472.CAN-08-4709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanchez-Paulete AR, Cueto FJ, Martinez-Lopez M, Labiano S, Morales-Kastresana A, Rodriguez-Ruiz ME, Jure-Kunkel M, Azpilikueta A, Aznar MA, Quetglas JI, Sancho D, Melero I. Cancer immunotherapy with immunomodulatory Anti-CD137 and Anti-PD-1 monoclonal antibodies requires BATF3-dependent dendritic cells. Cancer Discov. 2016;6:71–79. doi: 10.1158/2159-8290.CD-15-0510. [DOI] [PMC free article] [PubMed] [Google Scholar]