

Abstract
Reversing cisplatin resistance of lung cancer cell line A549/DDP through recovering cadherin 13 (CDH13) expression by demethylation was investigated in the current study. RT-PCR was used to measure CDH13 expression in lung cancer A549 and A549/DDP cells with or without 5-Aza-CdR intervention. Methylation-specific PCR was used to detect CDH13 methylation. MTT assay and flow cytometry were used to measure the effects of cisplatin on inhibiting cell proliferation, apoptosis, and the reversal of cisplatin resistance. The IC50 value of cisplatin for A549 and A549/DDP cells was 3.278±0.532 and 28.341±1.435 µmol/l, respectively (P<0.05). The cisplatin-resistance index of A549/DDP cells was up to 8.65. After 2.5, 10, or 40 µmol/l 5-Aza-CdR treatment, the apoptotic rates of A549/DDP cells were 9.4±0.86, 18.1±1.42 and 42±2.01%, respectively, which were significantly different to those of the control group (P<0.05). Methylation-specific PCR detected both methylation (M) and unmethylation (U) bands at CDH13 promoter region before 5-Aza-CdR intervention while it only detected an unmethylation band after the treatment with a higher concentration of 5-Aza-CdR, which indicates the transformation to unmethylation state. When 10 µmol/l 5-Aza-CdR was added, the IC50 of cisplatin to A549/DDP cells was 8.472±0.415 µmol/l, and cisplatin resistance was reversed by 3.35-fold. CDH13 methylation is related to the cisplatin resistance of A549/DDP cells. 5-Aza-CdR can inhibit CDH13 methylation and recover CDH13 expression. With the increase in 5-Aza-CdR concentration, the unmethylation state of CDH13 is enhanced, which can strengthen the function of cisplatin inhibiting proliferation and apoptosis in A549/DDP cells.
Keywords: CDH13, methylation, non-small cell lung cancer, cisplatin
Introduction
Recently, several studies have shown that cadherin 13 (CDH13; T-cadherin, H-cadherin) functioned as an anti-oncogene and that its polymorphisms were associated with the development of different cancers (1–3).
CDH13 is a close phylogenetic relative of classical cadherins and shares with them the typical organization and overall sequence similarity of the extracellular domain (4). CDH13 is encoded by a single gene in each vertebrate genome, and is thought to have appeared recently in evolution as a result of duplication of a gene of a classical cadherin (4). Yet, the EC1 subdomain of CDH13 shows replacements of several amino acid residues crucial for homophilic interactions, in particular the Trp2Ile replacement. Therefore, it was suggested that CDH13 may be unable to efficiently form homodimers, which was supported by NMR studies (5). However, recent findings have identified a novel mechanism of homophilic adhesion of CDH13 (and possibly other nonclassical cadherins) via an alternative interface near the EC1-EC2 calcium-binding sites (6). Another unique feature of CDH13 is that it is devoid of a transmembrane domain and is anchored to the exterior surface of the plasma membrane via a glycosyl-phosphatidylinositol (GPI) anchor. Similar to all cadherins, CDH13 is glycosylated on several sites (7,8). Structural and functional aspects of CDH13 have been covered in considerable detail in recent reviews (9,10). The highest CDH13 expression has been reported in the nervous and cardiovascular system (in endothelial and smooth muscle cells). In this respect, it resembles type II cadherins, which are primarily expressed in the nervous system and vasculature (6), although CDH13 is closer to type I classical cadherins in terms of sequence similarity (4).
Classical cadherins are often located at and contribute to the formation of intercellular junctions. Loss of intercellular adhesion facilitates cell motility and growth. By contrast, CDH13 is not strongly associated with the areas of cell-cell contacts in confluent cell cultures, and shows globular punctate distribution (9). In migrating cells, it is located primarily at the leading edge (9). Recently, its nuclear and centrosomal location has been reported in endothelial cells (11). In HEK 293A cells, CDH13 was detected in the centrosomes but not in the nucleus (11). Nuclear localization of cleaved intracellular domains of some other members of the cadherin superfamily, such as protocadherins a (12), c (13) and Fat1 (14,15), as well as classical E-cadherin (16), N-cadherin (17) has been reported. Cleaved intracellular domains may act as transcription factors or modulate gene expression (18). However, CDH13 has no corresponding intracellular domain and presents in the nucleus as a full-length molecule (11); therefore, functional parallels with other cadherins may not be relevant in this case.
The CDH13 gene is a new member of the cadherin superfamily, which was isolated recently and has been mapped to 16q24 (19). Cadherins are transmembrane glycoproteins expressed on the epithelial cell surface that mediate intercellular Ca2+-dependent adhesion, which is important for maintaining normal tissue structure. Abnormalities in the CDH13 gene have been identified in human malignancies (20,21). Moreover, an association between the abnormal expression of CDH13 and its promoter methylation in lung cancer has been demonstrated (22–24). Recent studies have reported that CDH13 functioned as an anti-oncogene in lung (1), breast (25), ovarian (3), bladder (26), esophageal (27) and gastric cancer (28).
CDH13 promoter methylation plays a key role in cancer development by promoting the inactivation of tumor suppressor genes, activation of oncogenes, and increase in chromosomal instability (29). This study investigated the mechanism between CDH13 promoter methylation and the drug resistance of lung cancer cells during chemotherapy and aimed to clarify whether CDH13 can serve as a molecular marker for predicting the efficacy of cisplatin treatment during adjuvant chemotherapy.
Materials and methods
Materials
A549, a human lung adenocarcinoma cell line (obtained from the American Type Culture Collection and preserved by the Respiratory Department of the Second People's Hospital of Guangdong); A549/DDP, a drug-resistant cell line of lung adenocarcinoma (purchased from the Cell Resource Center of Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai, China); cisplatin (Qilu Pharmaceutical Co., Ltd., Jinan, China); 5-Aza-CdR (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany); the Methylcode™ Bisulfite Conversion kit and the total RNA isolation reagent TRIzol (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA); the reverse transcription kit (Qiagen GmbH, Hilden, Germany). Cells were divided into 7 groups to measure CDH13 mRNA expression level. Group 1 was A549 cells with no 5-Aza-CdR treatment, and groups 2–7 were the A549/DDP cells with different concentration (0, 0.5, 1, 5, 10, and 20 µmol/l) of 5-Aza-CdR treated in 48 h. The study was approved by the Ethics Committee of Guangdong Second Provincial General Hospital (Guangzhou, China).
Methods
Measurement of CDH13 mRNA expression level by transcription-polymerase chain reaction (RT-PCR)
According to the principles of PCR primer design, CDH13 and GAPDH primers were designed. GAPDH served as the positive control for RT-PCR. PCR primers were produced by the Beijing Genomics Institute (Beijing, China). CDH13 primers were F5′-AGTGTTCCATATCAATCAATCAGCCAG-3′ and R5′-CGAGACCTCATAGCGTAGCTT-3′. GAPDH primers were F5′-GAAAGCCTGCCGGTGACTAA-3′ and R5′-GCCCAATACGACCAAATCAGAG-3′.
The PCR solution (25 µl) contained 12.5 µl 2X PCR Master Mix, 0.5 µl of each primer (25 µmol/l), 1 µl DNA template, and DEPC water. The PCR reaction conditions for CDH13 and GAPDH: 5 µl cDNA, 10 µl SYBR® Premix Ex Taq™ (Tli RNaseH Plus) (2X Conc.) (Takara Bio, Inc., Otsu, Japan), 0.5 µl of each primer, 4 µl dH2O, 95°C 30 sec and 40 cycles of 95°C 3 sec and 60°C 34 sec. Each of 5 µl PCR products was separated in 0.15% agarose gel by electrophoresis for 20 min.
Detection of CDH13 methylation in cell lines
The EZDNA methylation kit (Zymo Research, Orange, CA, USA) was used to perform DNA methylation. A total of 900 µl sterile ultrapure water, 50 µl M-dissolving buffer, and 300 µl M-dilution buffer were added into a CT conversion reagent tube. The solution was mixed by agitation. In each PCR tube, 20 µl DNA was added into 130 µl CT conversion reagent. The PCR tubes were placed into the PCR instrument and kept at 98°C for 10 min and then 64°C for 2.5 h. Then, 600 µl M-binding buffer was added into the purification column which was inserted into a liquid collection tube. The sample was added into the purification column, and the tube was centrifuged at 10,000 × g for 30 sec at 4°C. The liquid in collection tube was discarded and 100 µl M-wash buffer was added into the purification column. The tube was centrifuged at 10,000 × g for 30 sec at 4°C and the liquid in collection tube was discarded. A total of 200 µl M-desulphonation buffer was added into the purification column and left at room temperature for 20 min. The tube was centrifuged at 10,000 × g for 30 sec at 4°C. A total of 200 µl M-wash buffer was added and the tube was centrifuged at 10,000 × g for 30 sec at 4°C, and the step was repeat once. The liquid in collection tube was discarded and 20 µl M-elution buffer was added to elute the DNA. The tube was centrifuged at 10,000 × g for 30 sec at 4°C, and the eluted solution was used for PCR amplification.
Detection of DNA methylation using methylation-specific PCR (MSP)
The primer sequences for detecting CDH13 methylation (the length of amplified fragments was 243 bp) were: F, 5′-TCGCGGGGTTCGTTTTTCGC-3′, R, 5′-GACGTTTTCATTCATACACGCG-3′; and primer sequences for detecting CDH13 unmethylation (the length of amplified fragments was 242 bp) were: F, 5′-TTGTGGGGTTTGTTTTTTGT-3′, R, 5′-AACTTTTCATTCATACACACA-3′.
PCR amplification: in 20 µl PCR solution, the blood genomic DNA of healthy subjects was treated by the methylase SssI and served as the positive control, and that not treated by SssI served as the negative control. Sterile double-distilled water served as the blank control. The amplification conditions were 12 cycles of 94°C 30 sec, 64°C 30 sec, and 72°C 40 sec. PCR product (5 µl) of methylation or unmethylation DNA template was mixed with 1 µl loading buffer and then subjected to gel electrophoresis. The gel was stained by using a nucleic acid silver staining kit (Beijing Dingguo Changsheng Biotechnology Co., Ltd., Beijing, China) according to the manufacturer's protocol.
Methylation of blood genomic DNA from healthy subjects: the reaction system (50 µl) contained 5 µl 10X NBE buffer, 0.25 µl SAM (32 mM), 2 U SssI methylase, 2 µl DNA, and 40.75 µl sterile double-distilled water. It was placed in a 37°C water bath for 3 h and then 65°C water bath for 10 min. Then, 6 µl PCR product was mixed with 2 µl loading buffer and loaded into a 2% agarose gel containing a nucleic acid dye. Electrophoresis was performed at 120 V for 30 min. The gel was observed under a gel imaging system (UVP).
Detecting the reversal of drug resistance in A549/DDP cells by MTT
Concentrations of 0, 1.25, 2.5, 5, 7.5, and 10 µmol/l of cisplatin were used to treat A549 cells, and 0, 10, 20, 30, 40 and 50 µmol/l cisplatin was used to treat A549/DDP cells.
Following treatment with a non-toxic dose of 5-Aza-CdR (10 µmol/l) for 48 h, A549/DDP cells were treated with cisplatin (the same concentrations as above) for 48 h. For the control group, an equal volume of DMSO was added. Each group had 3 repeats. After 48 h, 20 µl MTT (5 mg/ml) was added to each well. After 4 h of culture, the culture medium was centrifuged at 2,000 × g for 10 min at 4°C. The supernatant was discarded, and 150 µl DMSO was added into each well. After MTT was completely dissolved, OD 490 nm was measured in a microplate reader and cell viability was calculated. Inhibition rate (%) was calculated as: (A492 of control cells - A492 of intervened cells)/A492 of control cells ×100%. Reversal index of drug resistance = IC50 of the drug-resistant cells only treated with cisplatin/IC50 of the drug-resistant cells treated with both cisplatin and 5-Aza-CdR. Drug resistance index = IC50 of drug-resistant cells/IC50 of parental cells.
Morphological changes of cisplatin-treated apoptotic A549/DDP cells before and after 5-Aza-CdR intervention detected by Hoechst 33258
The cells in the logarithmic growth phase were inoculated into 6-well cell culture plates (3×105/well). The cells were then divided into four groups: i) A549/DDP group; ii) A549/DDP + 5-Aza-CdR (10 µmol/l; 48 h) group; iii) A549/DDP + DDP (20 µmol/l) group; iv) A549/DDP + 5-Aza-CdR (10 µmol/l; 48 h) + DDP (20 µmol/l) group. The cells were cultured in 2 ml medium/well at 37°C and 5% CO2 for 48 h. The culture medium was discarded, and 0.5 ml 4% paraformaldehyde solution was added to fix the cells for 10 min. The cells were washed twice with PBS for 3 min each time. The cells were treated with 1 ml Hoechst 33258 staining solution (10 µg/ml) for 5 min. The staining solution was discarded, and the cells were rinsed 3 times with PBS for 5 min each time. One drop of anti-fluorescence quenching liquid was added onto the coverslip which was then observed under a fluorescence microscope (IX70, Olympus Corporation, Tokyo, Japan).
Measurement of cisplatin-induced A549/DDP cell apoptosis before and after 5-Aza-CdR intervention by flow cytometry. Cells in the logarithmic growth phase were cultured at 37°C and 5% CO2 for 48 h, and 1 ml cell suspension (2.5–5×105/ml) was transferred into a 5 ml centrifuge tube which was centrifuged at 1,000 × g at 4°C for 5 min. The supernatant was discarded and the precipitate was rinsed twice with PBS. The precipitate was dissolved with 200 µl binding buffer, and the solution was treated with 10 µl FITC-labeled Annexin-V and 5 µl PI in the dark for 15 min. After adding 300 µl binding buffer, the solution was measured by flow cytometry. At the same time, three control groups were conducted respectively as follows: i) unstained cells; ii) the cells stained only by AnnexinV-FITC; iii) the cells stained only by PI. The concentrations of cisplatin treatment for A549/DDP cells were 0, 2.5, 10 and 40 µmol/l respectively, for 48 h. After 48 h of 5-Aza-CdR (10 µmol/l) and A549/DDP, the concentration of cisplatin treatment for A549/DDP cells and the time were the same as above.
Statistical analysis
Data were analyzed using SPSS 16.0 software (SPSS, Inc., Chicago, IL, USA). The quantitative data following a normal distribution are presented by mean ± standard deviation. Analysis of variance and post hoc test (Least Significant Difference) was used to compare more than two groups of data following a normal distribution. The t-test was used to compare the data between two groups. P<0.05 was considered to indicate a statistically significant difference. IC50 was calculated using linear regression.
Results
CDH13 mRNA expression levels in A549 cells and A549/DDP cells
CDH13 mRNA expression levels in A549 cells and A549/DDP cells were measured by RT-PCR
The OD value of GAPDH served as the internal reference. The relative CDH13 mRNA expression level was represented by the ratio of CDH13 OD value to GAPDH OD value. The PCR products of A549 cells and A549/DDP cells showed a CDH13 band with the size of 243 bp (Table I; Fig. 1). The CDH13 mRNA expression level in A549 cells was significantly higher than that of A549/DDP concentration. CDH13 mRNA expression level was upregulated, indicating that at a certain range, the relative expression of CDH13 mRNA increased with the increase of 5-Aza-CdR concentration. The CDH13 mRNA expression level in A549/DDP cells with 10.0 µmol/l or 20.0 µmol/l 5-Aza-CdR treatment showed no significant difference (P>0.05), indicating that 10.0 µmol/l 5-Aza-CdR treatment can already induce a high expression level of CDH13 mRNA in A549/DDP cells.
Table I.
Relative CDH13 mRNA expression levels in A549 and A549/DDP cells treated with different concentrations of 5-Aza-CdR (mean ± standard deviation).
| No. | Relative CDH13 mRNA expression levels |
|---|---|
| 1 | 0.8395±0.0045a |
| 2 | 0.2797±0.0041b |
| 3 | 0.3421±0.0037b |
| 4 | 0.4735±0.0016b |
| 5 | 0.6383±0.0051b |
| 6 | 0.8381±0.0034b |
| 7 | 0.8393±0.0056 |
P<0.05, comparison with the group 2.
P<0.05, six groups were compared pairwise. Group 1 was A549 cells with no 5-Aza-CdR treatment, groups 2–7 were the A549/DDP cells with different concentration (0, 0.5, 1, 5, 10, and 20 µmol/l) of 5-Aza-CdR treated in 48 h. CDH13, cadherin 13.
Figure 1.

The RT-PCR results of A549 and A549/DDP cells. Lane 1 was the RT-PCR results of CDH13 mRNA in A549 cells; lanes 2–7 were the RT-PCR results of CDH13 mRNA in A549/DDP cells treated with 0, 0.5, 1, 5, 10, and 20 µmol/l 5-Aza-CdR, respectively. CDH13, cadherin 13.
CDH13 promoter methylation in A549 and A549/DDP cells before 5-Aza-CdR intervention and that in A549/DDP cells after 5-Aza-CdR intervention
MSP was used to measure CDH13 methylation level in A549 and A549/DDP cells. The results showed that A549 cells only had an unmethylation band (U) of CDH13 (Figs. 2 and 3). Drug-resistant A549/DDP cells had both methylation (M) and unmethylation (U) bands of CDH13, which suggests the partial methylation of CDH13. When treated with a non-toxic dose of 5-Aza-CdR (10 µmol/l), A549/DDP cells showed an unmethylation band. The average OD values before and after 5-Aza-CdR treatment were calculated by the gel electrophoresis software QuantityOne and showed a statistically significant difference (P<0.05).
Figure 2.

Electrophoresis results of MSP products. MSP, methylation-specific PCR.
Figure 3.

MSP method for detecting the average OD values of the methylation status of the CDH13 gene. Group 1, A549/DDP cells treated with 40 µmol/l 5-Aza-CdR; group 2, A549/DDP cells treated with 20 µmol/l 5-Aza-CdR; group 3, A549/DDP cells. M, methylation band; U, unmethylation band. CDH13, cadherin 13; MSP, methylation-specific PCR. *P<0.05, compared with methylation band of Group 1.
Results of reversal of drug resistance of A549/DDP before and after 5-Aza-CdR intervention detected by MTT
MTT results showed that the IC50 value of A549 cells with 48 h cisplatin treatment was 3.278±0.532 µmol/l, while that of A549/DDP cells was 28.341±1.435 µmol/l (8.65-fold) (Fig. 4). It demonstrated that cisplatin could inhibit A549/DDP cell proliferation. However, A549/DDP cells showed a stronger cisplatin tolerance than that of A549 cells. After 48 h of 10 µmol/l 5-Aza-CdR treatment, the IC50 value of A549/DDP cells was 8.472±0.415 µmol/l, and the reversal index of cisplatin resistance was 3.35. These results indicated that 10 µmol/l 5-Aza-CdR treatment can increase the cisplatin sensitivity of A549/DDP cells. However, A549/DDP cells still showed a significantly lower cisplatin sensitivity than that of A549 cells (P<0.05).
Figure 4.
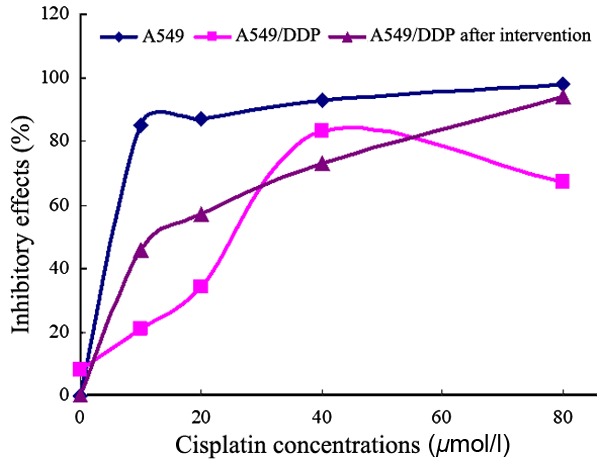
The inhibitory effects of different concentrations of cisplatin on A549 and A549/DDP cells
Morphological changes of cell apoptosis detected by Hoechst 33258
The results showed that in cell apoptosis, the uptake of Hoechst 33258 by cell membrane was increased (Fig. 5). Since the chromosomes were in a highly condensed state, their binding with Hoechst 33258 was strong, and the stained chromosomes showed a strong blue fluorescence. The chromosomes of normal cells only showed a weak fluorescence, and the dead cells were not stained. After 48 h of 10 µmol/l 5-Aza-CdR intervention, A549/DDP cells showed normal (round) morphologies and weak blue fluorescence, and there was few apoptotic cells. After cisplatin treatment, part of the A549/DDP cells showed apoptosis. Their nuclei and cytoplasm had dense fluorescent signals with different sizes and irregular or plum blossom-like shape. Apoptotic bodies could be observed. After 5-Aza-CdR intervention, apoptotic A549/DDP cells were significantly increased.
Figure 5.

Cell apoptosis detected by Hoechst 33258. (A) A549/DDP cells with no treatment. (B) A549/DDP cells treated with 10 µmol/l 5-Aza-CdR. (C) A549/DDP cells treated with 20 µmol/l cisplatin for 48 h. (D) A549/DDP cells treated with 10 µmol/l 5-Aza-CdR and then 20 µmol/l cisplatin for 48 h.
Cell apoptosis detected by flow cytometry
Flow cytometry results showed that A549/DDP cells had a low apoptotic ratio of 0.8±0.10% after 10 µmol/l 5-Aza-CdR intervention, which indicates there is no cell apoptosis caused by 10 µmol/l 5-Aza-CdR intervention after 48 h. The apoptotic ratios of A549/DDP cells after 2.5, 10, or 40 µmol/l cisplatin and 10 µmol/l 5-Aza-CdR treatments were 9.40±0.86, 18.10±1.42, and 42.00±2.01%, respectively (Fig. 6; Table II), which were significantly higher than that of the A549/DDP cells only treated with 5-Aza-CdR (P<0.05).
Figure 6.
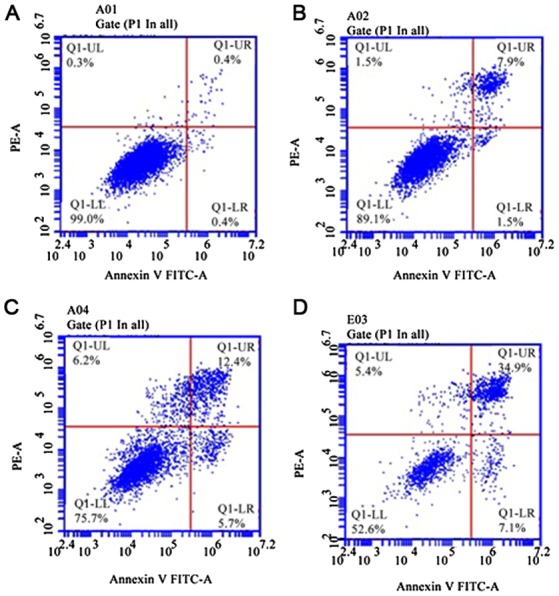
The effects of 5-Aza-CdR and different concentrations of cisplatin on A549/DDP cell apoptosis. A549/DDP cells treated with 0, 2.5, 10 or 40 µmol/l cisplatin. (A) A549/DDP cells without treatment of cisplatin; (B) A549/DDP cells treated with 2.5 µmol/l cisplatin; (C) A549/DDP cells treated with 10 µmol/l cisplatin; (D) A549/DDP cells treated with 40 µmol/l cisplatin.
Table II.
Comparison of apoptotic ratios of cisplatin-treated A549/DDP cells before and after 5-Aza-CdR intervention.
| Apoptotic ratios/% (mean ± standard deviation) | ||||
|---|---|---|---|---|
| 0 µmol/l | 2.5 µmol/l | 10 µmol/l | 40 µmol/l | |
| A549/DDP cells after 5-Aza-CdR intervention | 0.80±0.10 | 9.40±0.86 | 18.10±1.42 | 42.00±2.01 |
Discussion
General chemotherapy is one of the major means of treating non-small cell lung cancer (NSCLC) patients. Platinum drugs are a widely used class of chemotherapy drugs and have good treatment efficacy. Drug resistance in cancer cells is one the most common causes of failure in tumor chemotherapy. CDH13 is closely related to the occurrence and development of cancers. Previous studies found that CDH13 expression is downregulated in bladder, breast, cervical, and colon cancers, which is closely related to CDH13 methylation (30–34).
DNA methylation is a covalent modification to cytosine at the CpG sites of gene promoter region and exon 1. Abnormal methylation of CpG at promoter region can lead to silencing of gene transcription, e.g., the extremely low or no expression of many important genes, such as tumor suppressor genes, cells cycle regulatory genes, apoptosis-related genes (35,36), which promotes the formation of tumor cells. However, DNA methylation is reversible, which provides a theoretical basis and new direction for reversal of tumor drug resistance. The drugs targeting this reversible process can be developed to enhance the drug sensitivity of tumor cells during chemotherapy. 5-Aza-CdR is a DNA methyltransferase inhibitor. Previous findings showed that non-toxic low doses of 5-Aza-CdR can reverse drug resistance in pancreatic cancer, neuroblastoma, ovarian cancer, and colon cancer (37). 5-Aza-CdR was first approved by the U.S. to be applied in chemotherapy of blood system tumors. It is also a first-line drug for chemotherapy of acute nonlymphocytic leukemia in patients older than 65 years. Currently, it has also been used for treating solid tumors, such as ovarian cancer (38–42).
In this study, we used the cisplatin-resistant NSCLC cell line A549/DDP and its parental cell line A549 (as the control group). We discovered that CDH13 mRNA was expressed in both cell lines, but its expression level in A549 cells was significantly higher than that of the cisplatin-resistant A549/DDP cells, which indicates that the cisplatin resistance in lung cancer cells may be related to CDH13 silencing and protein expression deficiency. MSP results showed that 5-Aza-CdR could convert the partial methylation state of CDH13 to unmethylation state in A549/DDP cells and induce CDH13 expression in cisplatin-resistant A549/DDP cells. These results suggest that 5-Aza-CdR can reverse the cisplatin resistance in lung cancer cells.
We further studied the inhibition of A549/DDP cell proliferation and A549/DDP cell apoptosis after the reversal of CDH13 methylation by 5-Aza-CdR treatment. We measured the reversal index of cisplatin resistance in A549/DDP cells after non-toxic low-dose 5-Aza-CdR treatment and analyzed the correlation between CDH13 methylation and cisplatin resistance in lung cancer cells. After 10 µmol/l 5-Aza-CdR intervention, the IC50 value of A549/DDP cells to cisplatin was significantly decreased, which was positively correlated with cell apoptosis and the inhibition of cell proliferation. With the increase in 5-Aza-CdR concentration, the cisplatin sensitivity in A549/DDP cells was significantly enhanced, which indicates that the cisplatin resistance in lung cancer cells may be closely related to methylation of CDH13 promoter region. Although the cisplatin resistance in A549/DDP cells after 5-Aza-CdR intervention was reversed by 3.41-fold, it still had a significant difference with that of the parental A549 cells, which indicates the involvement of other genes in chemotherapy resistance. These drug-resistant genes may induce the drug resistance in lung cancer cells through different targets and mechanisms.
To compare the differentially expressed proteins between A549 cells and DDP (cisplatin)-resistant A549/DDP cells, A549 cells were exposed to DDP for developing the DDP- resistant A549/DDP cells. Differentially expressed proteins in the A549 and A549/DDP cells were separated and identified by proteomics approach. A part of the differentially expressed proteins were validated by RT-PCR, western blot analysis and immunocytochemistry. The functions of these proteins were further explored. There were 8 differentially expressed proteins including POTE, FH (fumarate hydratase), PDE (phosphodiesterase), AKR1C1 (aldo-keto reductase family 1 member Cl), DDH2 (dihydrodiol dehydrogenase 2), S100A10, prefoldin subunit 2, and karyopherin β2-Ran GppNHp nuclear transport complex between A549 and A549/DDP cells, and the difference in expression was >5-fold. These proteins were related with cellular metabolism, apoptosis, cell proliferation, detoxification and signal transduction (43–45).
CDH13, a member of the cadherin family, has been considered as a tumor suppressor, and the introduction of CDH13 in human tumor cells can reduce their invasive potential. In addition, CDH13 can induce the reversion of morphology from an invasive to a normal cell-like type. In a previous study, downregulated CDH13 protein expression was found in bladder TCC (26). However, the effects and mechanisms of downregulated CDH13 expression on bladder TCC invasion need to be further elucidated. The role of CDH13 in cancer invasion is not fully understood. In the current study, we assessed the role of CDH13 in bladder TCC invasiveness by silencing its expression using siRNA and assessed its effects on in vitro invasiveness, migration, and adhesion. We examined the CDH13 expression in CDH13 siRNA-transfected cells, blank control cells and negative control cells using qRT-PCR and western blotting. CDH13 expression was significantly decreased in CDH13 siRNA-treated cells compared with blank and negative controls at the mRNA and protein level (46).
DNA methylation is a DNA modification method. It regulates gene expression mainly through the methylation of cytosine in CpG sequence but does not change the DNA sequence. CpG island is a dispersed or highly aggregated sequence in DNA. Mutation or methylation of CpG island in the promoter region of tumor and suppressor gene CDH13 will affect CDH13 transcription, reduce CDH13 expression, and thus promote tumor cell proliferation and induce lung cancer development (21,47,48). When CDH13 promoter region was methylated, the ability of CDH13 to recognize DNA damage was weakened, and DNA continued to be replicated, which resulted in the formation of drug resistance. This theory was consistent with the finding in our study that CDH13 methylation was associated with cisplatin resistance in lung cancer cells. However, whether the underlying mechanisms were the same still needs further study.
Early discovery is critical for cancer treatment. During the early stage of tumorigenesis, abnormal DNA methylation occurs and can be detected, which provides a basis for early diagnosis of cancer. Since DNA methylation is a reversible gene-modification process, demethylation treatment before tumor generation can restore gene expression and achieve tumor prevention and treatment. In this study, our results showed that methylation of CDH13 promoter region in drug-resistant A549/DDP cells is an important cause of the reduction of CDH13 transcription. Inhibition of CDH13 methylation can restore CDH13 expression, reverse tumor cell biologic activity and change drug resistance phenotype in NSCLC cells. Therefore, this study provided a new target for NSCLC chemotherapy in the future.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.
Authors' contributions
YW, LZ and JW conceived and designed the study. YW, LZ, JY, BL and JW were responsible for the collection and analysis of the experimental data. YW and LZ interpreted the data and drafted the manuscript. JY and JW revised the manuscript critically for important intellectual content. All authors read and approved the final study.
Ethics approval and consent to participate
The study was approved by the Ethics Committee of Guangdong Second Provincial General Hospital (Guangzhou, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Drilon A, Sugita H, Sima CS, Zauderer M, Rudin CM, Kris MG, Rusch VW, Azzoli CG. A prospective study of tumor suppressor gene methylation as a prognostic biomarker in surgically resected stage I to IIIA non-small-cell lung cancers. J Thorac Oncol. 2014;9:1272–1277. doi: 10.1097/JTO.0000000000000256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morisaki H, Yamanaka I, Iwai N, Miyamoto Y, Kokubo Y, Okamura T, Okayama A, Morisaki T. CDH13 gene coding T-cadherin influences variations in plasma adiponectin levels in the Japanese population. Hum Mutat. 2012;33:402–410. doi: 10.1002/humu.21652. [DOI] [PubMed] [Google Scholar]
- 3.Chung CM, Lin TH, Chen JW, Leu HB, Yang HC, Ho HY, Ting CT, Sheu SH, Tsai WC, Chen JH, et al. A genome-wide association study reveals a quantitative trait locus of adiponectin on CDH13 that predicts cardiometabolic outcomes. Diabetes. 2011;60:2417–2423. doi: 10.2337/db10-1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hulpiau P, van Roy F. Molecular evolution of the cadherin superfamily. Int J Biochem Cell Biol. 2009;41:349–369. doi: 10.1016/j.biocel.2008.09.027. [DOI] [PubMed] [Google Scholar]
- 5.Dames SA, Bang E, Haüssinger D, Ahrens T, Engel J, Grzesiek S. Insights into the low adhesive capacity of human T-cadherin from the NMR structure of Its N-terminal extracellular domain. J Biol Chem. 2008;283:23485–23495. doi: 10.1074/jbc.M708335200. [DOI] [PubMed] [Google Scholar]
- 6.Ciatto C, Bahna F, Zampieri N, Van Steenhouse HC, Katsamba PS, Ahlsen G, Harrison OJ, Brasch J, Jin X, Posy S, et al. T-cadherin structures reveal a novel adhesive binding mechanism. Nat Struct Mol Biol. 2010;17:339–347. doi: 10.1038/nsmb.1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu T, Qian WJ, Gritsenko MA, Camp DG, II, Monroe ME, Moore RJ, Smith RD. Human plasma N-glycoproteome analysis by immunoaffinity subtraction, hydrazide chemistry, and mass spectrometry. J Proteome Res. 2005;4:2070–2080. doi: 10.1021/pr0502065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen R, Jiang X, Sun D, Han G, Wang F, Ye M, Wang L, Zou H. Glycoproteomics analysis of human liver tissue by combination of multiple enzyme digestion and hydrazide chemistry. J Proteome Res. 2009;8:651–661. doi: 10.1021/pr8008012. [DOI] [PubMed] [Google Scholar]
- 9.Philippova M, Joshi MB, Kyriakakis E, Pfaff D, Erne P, Resink TJ. A guide and guard: The many faces of T-cadherin. Cell Signal. 2009;21:1035–1044. doi: 10.1016/j.cellsig.2009.01.035. [DOI] [PubMed] [Google Scholar]
- 10.Resink TJ, Philippova M, Joshi MB, Kyriakakis E, Erne P. Cadherins and cardiovascular disease. Swiss Med Wkly. 2009;139:122–134. doi: 10.4414/smw.2009.12429. [DOI] [PubMed] [Google Scholar]
- 11.Andreeva AV, Kutuzov MA, Tkachuk VA, Voyno-Yasenetskaya TA. T-cadherin is located in the nucleus and centrosomes in endothelial cells. Am J Physiol Cell Physiol. 2009;297:C1168–C1177. doi: 10.1152/ajpcell.00237.2009. [DOI] [PubMed] [Google Scholar]
- 12.Emond MR, Jontes JD. Inhibition of protocadherin-alpha function results in neuronal death in the developing zebrafish. Dev Biol. 2008;321:175–187. doi: 10.1016/j.ydbio.2008.06.011. [DOI] [PubMed] [Google Scholar]
- 13.Haas IG, Frank M, Véron N, Kemler R. Presenilin-dependent processing and nuclear function of gamma-protocadherins. J Biol Chem. 2005;280:9313–9319. doi: 10.1074/jbc.M412909200. [DOI] [PubMed] [Google Scholar]
- 14.Magg T, Schreiner D, Solis GP, Bade EG, Hofer HW. Processing of the human protocadherin Fat1 and translocation of its cytoplasmic domain to the nucleus. Exp Cell Res. 2005;307:100–108. doi: 10.1016/j.yexcr.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 15.Hou R, Liu L, Anees S, Hiroyasu S, Sibinga NE. The Fat1 cadherin integrates vascular smooth muscle cell growth and migration signals. J Cell Biol. 2006;173:417–429. doi: 10.1083/jcb.200508121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ferber EC, Kajita M, Wadlow A, Tobiansky L, Niessen C, Ariga H, Daniel J, Fujita Y. A role for the cleaved cytoplasmic domain of E-cadherin in the nucleus. J Biol Chem. 2008;283:12691–12700. doi: 10.1074/jbc.M708887200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shoval I, Ludwig A, Kalcheim C. Antagonistic roles of full-length N-cadherin and its soluble BMP cleavage product in neural crest delamination. Development. 2007;134:491–501. doi: 10.1242/dev.02742. [DOI] [PubMed] [Google Scholar]
- 18.Mukherjee S, Tessema M, Wandinger-Ness A. Vesicular trafficking of tyrosine kinase receptors and associated proteins in the regulation of signaling and vascular function. Circ Res. 2006;98:743–756. doi: 10.1161/01.RES.0000214545.99387.e3. [DOI] [PubMed] [Google Scholar]
- 19.Takeuchi T, Ohtsuki Y. Recent progress in T-cadherin (CDH13, H-cadherin) research. Histol Histopathol. 2001;16:1287–1293. doi: 10.14670/HH-16.1287. [DOI] [PubMed] [Google Scholar]
- 20.Sato M, Mori Y, Sakurada A, Fujimura S, Horii A. The H-cadherin (CDH13) gene is inactivated in human lung cancer. Hum Genet. 1998;103:96–101. doi: 10.1007/s004390050790. [DOI] [PubMed] [Google Scholar]
- 21.Zhong YH, Peng H, Cheng HZ, Wang P. Quantitative assessment of the diagnostic role of CDH13 promoter methylation in lung cancer. Asian Pac J Cancer Prev. 2015;16:1139–1143. doi: 10.7314/APJCP.2015.16.3.1139. [DOI] [PubMed] [Google Scholar]
- 22.Kim H, Kwon YM, Kim JS, Lee H, Park JH, Shim YM, Han J, Park J, Kim DH. Tumor-specific methylation in bronchial lavage for the early detection of non-small-cell lung cancer. J Clin Oncol. 2004;22:2363–2370. doi: 10.1200/JCO.2004.10.077. [DOI] [PubMed] [Google Scholar]
- 23.Hanabata T, Tsukuda K, Toyooka S, Yano M, Aoe M, Nagahiro I, Sano Y, Date H, Shimizu N. DNA methylation of multiple genes and clinicopathological relationship of non-small cell lung cancers. Oncol Rep. 2004;12:177–180. [PubMed] [Google Scholar]
- 24.Toyooka KO, Toyooka S, Virmani AK, Sathyanarayana UG, Euhus DM, Gilcrease M, Minna JD, Gazdar AF. Loss of expression and aberrant methylation of the CDH13 (H-cadherin) gene in breast and lung carcinomas. Cancer Res. 2001;61:4556–4560. [PubMed] [Google Scholar]
- 25.Morisaki H, Yamanaka I, Iwai N, Miyamoto Y, Kokubo Y, Okamura T, Okayama A, Morisaki T. CDH13 gene coding T-cadherin influences variations in plasma adiponectin levels in the Japanese population. Hum Mutat. 2012;33:402–410. doi: 10.1002/humu.21652. [DOI] [PubMed] [Google Scholar]
- 26.Chen F, Huang T, Ren Y, Wei J, Lou Z, Wang X, Fan X, Chen Y, Weng G, Yao X. Clinical significance of CDH13 promoter methylation as a biomarker for bladder cancer: A meta-analysis. BMC Urol. 2016;16:52. doi: 10.1186/s12894-016-0171-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guo Q, Wang HB, Li YH, Li HF, Li TT, Zhang WX, Xiang SS, Sun ZQ. Correlations of promoter methylation in WIF-1, RASSF1A, and CDH13 genes with the risk and prognosis of esophageal cancer. Med Sci Monit. 2016;22:2816–2824. doi: 10.12659/MSM.896877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grady WM, Willis J, Guilford PJ, Dunbier AK, Toro TT, Lynch H, Wiesner G, Ferguson K, Eng C, Park JG, et al. Methylation of the CDH1 promoter as the second genetic hit in hereditary diffuse gastric cancer. Nat Genet. 2000;26:16–17. doi: 10.1038/79120. [DOI] [PubMed] [Google Scholar]
- 29.Andreeva AV, Kutuzov MA. Cadherin 13 in cancer. Genes Chromosomes Cancer. 2010;49:775–790. doi: 10.1002/gcc.20787. [DOI] [PubMed] [Google Scholar]
- 30.Lin YL, Xie PG, Ma JG. Aberrant methylation of CDH13 is a potential biomarker for predicting the recurrence and progression of non muscle invasive bladder cancer. Med Sci Monit. 2014;20:1572–1577. doi: 10.12659/MSM.892130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Widschwendter A, Ivarsson L, Blassnig A, Müller HM, Fiegl H, Wiedemair A, Müller-Holzner E, Goebel G, Marth C, Widschwendter M. CDH1 and CDH13 methylation in serum is an independent prognostic marker in cervical cancer patients. Int J Cancer. 2004;109:163–166. doi: 10.1002/ijc.11706. [DOI] [PubMed] [Google Scholar]
- 32.Yang J, Niu H, Huang Y, Yang K. A systematic analysis of the relationship of CDH13 promoter methylation and breast cancer risk and prognosis. PLoS One. 2016;11:e0149185. doi: 10.1371/journal.pone.0149185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hibi K, Kodera Y, Ito K, Akiyama S, Nakao A. Methylation pattern of CDH13 gene in digestive tract cancers. Br J Cancer. 2004;91:1139–1142. doi: 10.1038/sj.bjc.6602095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ren JZ, Huo JR. Correlation between T-cadherin gene expression and aberrant methylation of T-cadherin promoter in human colon carcinoma cells. Med Oncol. 2012;29:915–918. doi: 10.1007/s12032-011-9836-9. [DOI] [PubMed] [Google Scholar]
- 35.Toyooka S, Toyooka KO, Maruyama R, Virmani AK, Girard L, Miyajima K, Harada K, Ariyoshi Y, Takahashi T, Sugio K, et al. DNA methylation profiles of lung tumors. Mol Cancer Ther. 2001;1:61–67. [PubMed] [Google Scholar]
- 36.Birchmeier W, Behrens J. Cadherin expression in carcinomas: Role in the formation of cell junctions and the prevention of invasiveness. Biochim Biophys Acta. 1994;1198:11–26. doi: 10.1016/0304-419x(94)90003-5. [DOI] [PubMed] [Google Scholar]
- 37.Gayet O, Loncle C, Duconseil P, Gilabert M, Lopez MB, Moutardier V, Turrini O, Calvo E, Ewald J, Giovannini M, et al. A subgroup of pancreatic adenocarcinoma is sensitive to the 5-aza-dC DNA methyltransferase inhibitor. Oncotarget. 2015;6:746–754. doi: 10.18632/oncotarget.2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang AS, Doshi KD, Choi SW, Mason JB, Mannari RK, Gharybian V, Luna R, Rashid A, Shen L, Estecio MR, et al. DNA methylation changes after 5-aza-2′-deoxycytidine therapy in patients with leukemia. Cancer Res. 2006;66:5495–5503. doi: 10.1158/0008-5472.CAN-05-2385. [DOI] [PubMed] [Google Scholar]
- 39.Nguyen TT, Mohrbacher AF, Tsai YC, Groffen J, Heisterkamp N, Nichols PW, Yu MC, Lübbert M, Jones PA. Quantitative measure of c-abl and p15 methylation in chronic myelogenous leukemia: Biological implications. Blood. 2000;95:2990–2992. [PubMed] [Google Scholar]
- 40.Blum W, Schwind S, Tarighat SS, Geyer S, Eisfeld AK, Whitman S, Walker A, Klisovic R, Byrd JC, Santhanam R, et al. Clinical and pharmacodynamic activity of bortezomib and decitabine in acute myeloid leukemia. Blood. 2012;119:6025–6031. doi: 10.1182/blood-2012-03-413898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cowan LA, Talwar S, Yang AS. Will DNA methylation inhibitors work in solid tumors? A review of the clinical experience with azacitidine and decitabine in solid tumors. Epigenomics. 2010;2:71–86. doi: 10.2217/epi.09.44. [DOI] [PubMed] [Google Scholar]
- 42.Nicholson LJ, Smith PR, Hiller L, Szlosarek PW, Kimberley C, Sehouli J, Koensgen D, Mustea A, Schmid P, Crook T. Epigenetic silencing of argininosuccinate synthetase confers resistance to platinum-induced cell death but collateral sensitivity to arginine auxotrophy in ovarian cancer. Int J Cancer. 2009;125:1454–1463. doi: 10.1002/ijc.24546. [DOI] [PubMed] [Google Scholar]
- 43.Kuang P, Li XF, Li B, Wang YS, Li JY, Zhou CC. Comparative proteomic analysis of human lung adenocarcinoma A549 and A549/DDP cells. Tumor. 2012;32:170–176. [Google Scholar]
- 44.Qin X, Yu S, Xu X, Shen B, Feng J. Comparative analysis of microRNA expression profiles between A549, A549/DDP and their respective exosomes. Oncotarget. 2017;8:42125–42135. doi: 10.18632/oncotarget.15009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ye LY, Hu S, Xu HE, Xu RR, Kong H, Zeng XN, Xie WP, Wang H. The effect of tetrandrine combined with cisplatin on proliferation and apoptosis of A549/DDP cells and A549 cells. Cancer Cell Int. 2017;17:40. doi: 10.1186/s12935-017-0410-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lin YL, He ZK, Li ZG, Guan TY. Downregulation of CDH13 expression promotes invasiveness of bladder transitional cell carcinoma. Urol Int. 2013;90:225–232. doi: 10.1159/000345054. [DOI] [PubMed] [Google Scholar]
- 47.Kontic M, Stojsic J, Jovanovic D, Bunjevacki V, Ognjanovic S, Kuriger J, Puumala S, Nelson HH. Aberrant promoter methylation of CDH13 and MGMT genes is associated with clinicopathologic characteristics of primary non-small-cell lung carcinoma. Clin Lung Cancer. 2012;13:297–303. doi: 10.1016/j.cllc.2011.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhai X, Li SJ. Methylation of RASSF1A and CDH13 genes in individualized chemotherapy for patients with non-small cell lung cancer. Asian Pac J Cancer Prev. 2014;15:4925–4928. doi: 10.7314/APJCP.2014.15.12.4925. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.