

Abstract
A 52 year-old male presented with neck pain after undergoing thyroidectomy for a goiter three weeks prior which was complicated by a neck hematoma requiring evacuation. Computed tomography (CT) scan showed a neck hematoma requiring evacuation and he received desmopressin with cessation of bleeding. Coagulation studies were normal. He returned eighteen months later with severe oral mucosal bleeding after a dental procedure and required transfusions with red blood cells, platelets, and fresh frozen plasma (FFP) in addition to desmopressin, Humate-P, aminocaproic acid, and surgical packing. A comprehensive bleeding diathesis workup was normal. He was readmitted six months later due to abdominal pain and distention and found to have massive hepatosplenomegaly on CT. A new coagulopathy workup revealed prolonged INR to 1.5, corrected prothrombin time mixing study, and a low factor VII level (29%), suggesting acquired factor VII deficiency. A transjugular liver biopsy revealed extensive involvement by ALamyloidosis- Kappa type. He then developed a large right retroperitoneal hematoma which required multiple transfusions with FFP, cryoprecipitate, aminocaproic acid, and vitamin K with slight success. Hemorrhage was subsequently stabilized with recombinant factor VIIa administered every four hours which corresponded with correction of factor VII levels and PT and eventual cessation hemorrhage. Acquired factor VII deficiency causing severe coagulopathy was attributed to hepatic amyloidosis ALkappa subtype. We started treatment with bortezomib, dexamethasone, and cyclophosphamide, however, the patient succumbed to uncontrolled hemorrhage. Acquired factor VII deficiency is extremely rare and to our knowledge, this is the only known case of factor VII deficiency secondary to amyloidosis involving the liver.
Key words: Amyloidosis, factor VII deficiency, liver amyloidosis, recombinant factor VIIa
Case Report
A 52 year-old Egyptian male with past medical history significant for a goiter presented to the emergency department (ED) complaining of progressive neck pain, dysphagia, and difficulty breathing for the past seven days. He had undergone a routine thyroidectomy surgery for a goiter three weeks prior to ED presentation and had developed a post-operative hematoma requiring evacuation and placement of a Jackson-Pratt wound drain for 1 week. His past medical and family history were negative for a bleeding diathesis. His physical exam was significant for anterior neck enlargement and neck tenderness. Labs including prothrombin time (PT), international normalized ratio (INR), partial thromboplastin time (PTT), and fibrinogen were all within normal limits. In the ED, computerized tomography (CT) scan showed a neck hematoma. He was admitted and the hematoma required evacuation by the otolaryngology service. He also received desmopressin (DDAVP) with cessation of bleeding and was discharged home.
The patient returned to the ED eighteen months later with persistent severe oral mucosal bleeding after a dental procedure. He was admitted to the intensive care unit (ICU) and required multiple transfusions with red blood cells, platelets and fresh frozen plasma (FFP) in addition to DDAVP, Humate-P®, aminocaproic acid and surgical packing by the oral surgery team which successfully controlled the bleeding. A comprehensive bleeding diathesis workup was initiated which showed normal levels for PTT and Factor II, V,VII, VIII IX, X, XI, XII assays. Other work-up included a normal Von Willebrand Factor antigen and activity levels and normal platelet function assay. PT/INR was noted to be mildly prolonged to 13.9 seconds and 1.3, respectively. A PT mixing study was performed which corrected, indicating a simple factor deficiency. Many of these labs were, however, obtained after transfusion of multiple blood products during the hospitalization for the acute bleeding event. Hence, a definitive diagnosis was not established at the time of discharge. The patient was made aware of his bleeding diathesis and he was educated not to have any elective procedures done without consulting a hematologist and to return to ED in the event of any spontaneous bleeding. The patient was discharged on oral aminocaproic acid taper upon resolution of his bleeding. He was also recommended to follow-up with hematology to complete the work-up as outpatient which he failed to do due to socio-financial issues.
The patient was readmitted six months later due to abdominal pain and distention and was found to have massive hepatosplenomegaly on exam and CT imaging. He had no history of prior liver disease or alcohol abuse. Due to multiple coagulopathy workups that were unreliable in the past due to transfusions of blood products, a new workup was initiated by the hematology team during this admission (Table 1) prior to transfusion of any blood products or factor replacements. The workup was significant for elevated PT and a PT mixing study which corrected, suggesting a simple factor deficiency. Factor levels were obtained which revealed factor VII level that was low at 29% (normal range 40-150%), suggestive of an acquired factor VII deficiency. A transjugular liver biopsy was planned for evaluation of his massive hepatosplenomegaly. Given his high risk of bleeding, recombinant factor VIIa (rFVIIa) (70 mcg/kg) and vitamin K was given prophylactically both before and after the procedure which he tolerated well. We obtained a liver biopsy which was sent to Mayo Clinic (Rochester, Minnesota). Congo red stain confirmed the presence of Congo red-positive amyloid deposits involving the hepatic sinusoids. Liquid chromatography tandem mass spectrometry was performed on peptides from Congo red-positive areas and analysis detected a peptide profile consistent with AL (kappa)- type amyloid deposition. Figure 1 shows extensive involvement by amyloid lightchain (AL) amyloidosis-kappa type. The patient was readmitted 6 days later for bleeding at the transjugular biopsy site and bleeding was controlled with recombinant factor VIIa and vitamin K. The patient was admitted again several days later for worsening abdominal pain and distention. He was found to have a new large right retroperitoneal hematoma with extension along the iliopsoas muscle to right inguinal region (Figure 2) (Figure 3). He was admitted to the ICU and again received multiple transfusions with FFP, cryoprecipitate, aminocaproic acid infusion, and vitamin K supplementation with only slight success. The patient’s retroperitoneal hemorrhage was subsequently stabilized with regular, scheduled doses of rFVIIa (70 mcg/kg) administered every four hours which corresponded well with correction of his factor VII levels and PT/INR. With eventual stabilization in hemoglobin levels and of hemorrhage on serial abdominal CT scans, he was gradually weaned off rFVIIa infusions.
Table 1.
Laboratory tests.
| Test | Value | Normal range |
|---|---|---|
| INR | 1.5 | 0.8-1.2 |
| PTT | 31.2 seconds | 25-30 seconds |
| Factor II | 74% | 40-150% |
| Factor V | 91% | 40-150% |
| Factor VII | 29% | 40-150% |
| Factor VIII | 184% | 50-170% |
| Factor IX | 71% | 40-150% |
| Factor X | 58% | 40-150% |
| Factor XI | 68% | 40-150% |
| Factor XII | 98% | 40-150% |
| Fibrinogen | 327 mg/dL | 190-400 mg/dL |
| Von Willebrand Factor Antigen | 328% | 55-200% |
| Von Willebrand Multimers | Normal |
PT mixing study, 1:1 mix Corrected, simple factor deficiency present
Figure 1.
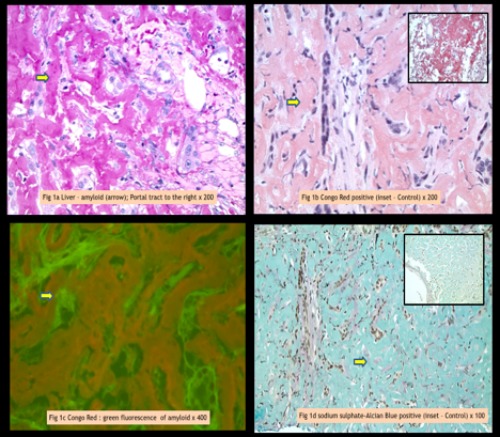
A) Extensive amyloid within liver parenchyma, appearing as amorphous eosinophilic waxy deposits (arrow). Adjacent portal tract is clear H&E stain. B) Brick red staining (arrow) of amyloid deposits using Congo red (Inset shows positive control); C) Amyloid deposits emitting green fluorescence with Congo red stain (arrow); D) Amyloid deposits staining bluish-green (arrow) with Sulfated Alcian Blue (SAB) stain (Inset shows positive control).
Figure 2.
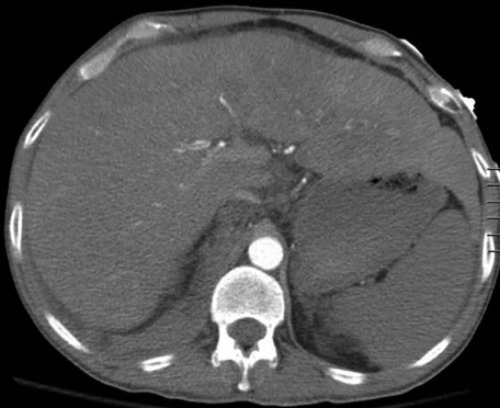
Massive hepatomegaly measuring 27.2 cm in craniocaudal dimension.
Figure 3.
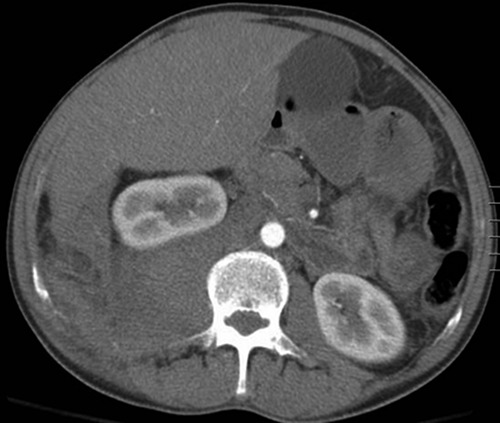
Large right retroperitoneal hematoma displacing right kidney superiorly.
At this time, the patient’s acquired factor VII deficiency was thought to be the cause of his bleeding diathesis which was further attributed to hepatic amyloidosis, AL amyloidosis–kappa subtype. Further workup for amyloidosis included serum protein electrophoresis and urine protein electrophoresis, which did not show an Mspike. Serum free light chain ratio was elevated to 5.65. We offered the patient a bone marrow biopsy, to which he declined given the risk of bleeding. The patient started treatment with bortezomib and dexamethasone for treatment of the underlying amyloidosis and cyclophosphamide was subsequently added and he completed a total of 4 cycles. However, the patient’s clinical condition gradually declined due to progressive liver dysfunction from hepatic amyloidosis and cardiac amyloidosis. Cardiac amyloidosis was diagnosed with cardiac MRI revealing myocardial delayed enhancement suggestive of amyloid infiltration. Echocardiogram showed grade I left ventricular diastolic dysfunction with an ejection fraction of 80% with pro Bnatriuretic peptide level rising up to 2,012 as his disease advanced. With progressive clinical liver dysfunction, liver function tests (ALT and AST) remained within normal limits as INR increased to 3.9 and PTT increased to 43. Renal function remained stable with a creatinine level of 1.3. He developed recurrent esophageal variceal bleeding requiring banding, severe ascites, an episode of spontaneous bacterial peritonitis and multiple episodes of hepatic encephalopathy. He was evaluated for bone marrow transplant and liver transplant but was deemed not to be a candidate due to his advanced amyloidosis, bleeding disorder and poor performance status. His multiple episodes of gastrointestinal (GI) bleeding was controlled each time with rFVIIa in addition to other supportive measures. Unfortunately, he progressed to worsening coagulopathy with elevation of both PTT and PT. Non-conventional therapy with colchicine and prednisone was attempted with a planned 7 day cycle every 6 weeks (of which he completed 1 cycle) towards the end of his life without improvement in his hepatic function or GI bleeding. The patient died at an outside hospital emergency department due to uncontrolled upper GI bleeding. His multiple hospital admissions are summarized in Table 2.
Table 2.
Patient’s multiple hospital admissions.
| 12/2/2013 | Thyroidectomy with postoperative hematoma requiring evacuation and JP drain. |
| 12/19/2013 | Admitted for re-evacuation of neck hematoma, DDAVP administered with cessation of bleeding. |
| 5/2014 | Admitted for severe mucosal bleeding after dental procedure. Required platelets, FFP, DDAVP, Humate-P, and aminocaproic acid. Bleeding diathesis workup inconclusive. |
| 10/2014 | Admitted with abdominal pain and massive hepatosplenomegaly. Coagulopathy workup established factor VII deficiency. Transjugular biopsy of liver obtained with prophylactic rFVIIa administered. Liver biopsy revealed AL-amyloidosis-Kappa type. |
| 11/2014 | Admitted for bleeding at transjugular biopsy site and retroperitoneal hematoma. Hemorrhage was controlled with rFactor VIIa infusions. |
| 12/2015-8/2016 | Received treatment for AL-amyloidosis-Kappa type with bortezomib, dexamethasone, and cyclophosphamide, and later with colchicine and prednisone without improvement in hepatic function. Developed complications from cirrhosis including GI bleeding and SBP. Eventually succumbed to uncontrolled upper GI hemorrhage. |
Discussion and Conclusions
Amyloidosis is a group of protein folding diseases with extracellular deposition of a soluble precursor protein aggregating in the form of insoluble fibrils. There are 4 main types of systemic amyloidosis: AL (light chain), AA (inflammation), ATTR (hereditary and old age), and Aβ2M (dialysis).1 AL amyloidosis is the most common subtype and is caused by a clonal plasma cell dyscrasia and the precursor to AL amyloidosis is either kappa or lambda immunoglobulin free light chains. Sometimes, it is associated with other clonal disorders like multiple myeloma, and rarely non-Hodgkin lymphoma or Waldenstrom macroglobulinemia.2 The most common manifestations of AL amyloidosis involve the kidneys, heart, liver, and peripheral nervous system. Although hepatic involvement can be seen in up to 90 percent of patients with AL amyloidosis, clinical manifestations are usually mild with hepatomegaly and elevated alkaline phosphatase levels and rarely, hyperbilirubinemia.3 The most common presenting symptoms are weight loss and fatigue.2 There are more than 28 different subtypes of amyloidosis and share common structural and chemical characteristics. The accurate identification of amyloidosis subtype is critical since different subtypes of amyloidosis have different etiologies and are associated various clinical manifestations and organ involvement. The diagnosis of amyloidosis requires a tissue biopsy and Congo red stain and electron microscopy are helpful to differentiate between amyloid and other pathologic fibrils. Once amyloidosis is confirmed, immunofluorescence and immunohistochemistry are used to for amyloid typing. The specific treatment of amyloidosis is dependent on the subtype.
In patients with primary AL amyloidosis, coagulopathy is frequently present. Acquired factor X deficiency is a common coagulation factor deficiency noted in AL amyloidosis. It is thought to occur through the adsorption of factor X to amyloid fibrils. Studies have observed that 8.7% of those with AL amyloidosis had factor X levels 50% of normal with 56% of those who had abnormal levels had clinically significant bleeding episodes.4 Patients who were treated with high dose melphalan chemotherapy had an improvement in factor X levels.5 In addition to acquired factor X deficiency, case reports of acquired deficiencies in factors II, V, and IX have also been reported as well as combined factor deficiencies of factors IX and X, and factors V and X.6 Other etiologies of bleeding in AL amyloidosis include hyperfibrinolysis and platelet dysfunction.7
Acquired factor VII deficiency is an extremely rare bleeding disorder and is typically seen in warfarin anticoagulant therapy. Factor VII is also disproportionately lower in chronic liver disease than other Vitamin K dependent factors likely due to its short half-life. Other etiologies of acquired factor VII deficiency described in literature include cases of aplastic anemia, severe sepsis, pleural liposarcoma, stem cell transplant, cirrhosis and in the presence of antiphospholipid antibodies.8,9 Other rare etiologies of acquired factor VII deficiency include cases secondary to administration of penicillins, cephalosporins and diseases such as myeloma, underlying malignancy and usage of anti-thymocyte globulin and IL- 2.10,11 The levels of factor VII in previous cases presenting with severe hemorrhage were between <1% to 38% of normal factor VII levels.11 Additionally, factor VII levels have been measured in the setting of decompensated cirrhosis with mean level of 40%, which is significantly lower than than levels in patients with compensated cirrhosis, obstructive jaundice, and noncirrhotics with alcoholic liver disease. Additionally, of all the vitamin Kdependent factors in cirrhosis, the decline in factor VII levels are more significant that the decline in levels of factors II, IX, X.
To our knowledge, this is the first known case of acquired factor VII deficiency in the setting of AL amyloidosis affecting the liver. The patient initially presented with episodes of delayed post procedure bleeding which gradually increased in severity likely from progressive factor deficiency secondary to worsening liver dysfunction from amyloidosis. Since factor VII has the shortest half-life of the procoagulant factors which is approximately 3-6 hours, its levels were most susceptible to decrease in the setting of liver dysfunction. In this patient, there were possibly other imbalances of hemostasis that were contributory as well. Systemic amyloidosis usually involves the liver and kidneys, however, clinically significant liver dysfunction as noted in this patient is also reported to be very rare.12
In controlling this patient’s recurrent hemorrhage, we found that recombinant factor VIIa (NovoSeven®RT) was the most effective treatment over fresh frozen plasma infusion and aminocaproic acid infusion. There was clinical correlation of controlling the patient’s bleeding with correction of his PT and factor VII assay. Plasma is often not an effective therapy to correct factor VII deficiency due to the low concentration of factor VII in plasma, its short half-life, and problems with fluid overload. rFVIIa is indicated for use in congenital deficiencies of factor VII, acquired hemophilia and patients with acquired inhibitors to hemophilia A and B replacement products13 and other wide variety of uses have been reported in diverse acquired factor VII deficiencies and factor X deficiency.14-18
In our attempt to control hemorrhage with rFVIIa and blood products, we were mindful that treatment of the underlying cause is the definitive solution to correcting the bleeding disorder. Unfortunately, attempts to treat amyloidosis with systemic therapy proved to be unsuccessful. Our patient was not considered to be candidate for other definitive treatment options like autologous stem cell transplant and liver transplant due to his poor functional status. Our case demonstrates a novel etiology for acquired factor VII deficiency that has not been previously described in literature. In future cases of AL amyloidosis with hepatic involvement and bleeding diathesis, acquired factor VII deficiency should be considered as a potential complication and rFVIIa is a useful treatment option in this situation.
References
- 1.Sipe JD, Benson MD, Buxbaum JN, et al. Amyloid fibril proteins and amyloidosis: chemical identification and clinical classification - International Society of Amyloidosis 2016 Nomenclature Guidelines. Amyloid 2016;23:209-13. [DOI] [PubMed] [Google Scholar]
- 2.Hazenberg BP. Amyloidosis: a clinical overview. Rheum Dis Clin North Am 2013;39:323-45. [DOI] [PubMed] [Google Scholar]
- 3.Merlini G, Wechalekar AD, Palladini G. Systemic light chain amyloidosis: an update for treating physicians. Blood 2013;121:5124-30. [DOI] [PubMed] [Google Scholar]
- 4.Choufani EB, Sanchorawala V, Ernst T, et al. Acquired factor X deficiency in patients with amyloid light-chain amyloidosis: incidence, bleeding manifestations, and response to highdose chemotherapy. Blood 2001;97:1885-7. [DOI] [PubMed] [Google Scholar]
- 5.Mahmood S, Blundell J, Drebes A, et al. Utility of factor X concentrate for the treatment of acquired factor X deficiency in systemic light-chain amyloidosis. Blood 2014;123:2899-900. [DOI] [PubMed] [Google Scholar]
- 6.Emori Y, Sakugawa M, Niiya K, et al. Life-threatening bleeding and acquired factor V deficiency associated with primary systemic amyloidosis. Blood Coagul Fibrinolysis 2002;13:555-9. [DOI] [PubMed] [Google Scholar]
- 7.Sucker C, Hetzel GR, Grabensee B, Stockschlaeder M, Scharf RE. Amyloidosis and bleeding: pathophysiology, diagnosis, and therapy. Am J Kidney Dis 2006;47:947-55. [DOI] [PubMed] [Google Scholar]
- 8.Perry DJ. Factor VII deficiency. Br J Haematol 2002;118:689-700. [DOI] [PubMed] [Google Scholar]
- 9.Yokuş O. Acquired factor VII deficiency associated with pneumonia. Amyloid 2011;2:460-2. [Google Scholar]
- 10.Mulliez SM, Devreese KM. Isolated acquired factor VII deficiency: review of the literature. Acta Clin Belg 2016;71:63-70. [DOI] [PubMed] [Google Scholar]
- 11.Girolami A, Santarossa C, Cosi E, et al. Acquired isolated FVII deficiency: an underestimated and potentially important laboratory finding. Clin Appl Thromb Hemost 2016;22:705-11. [DOI] [PubMed] [Google Scholar]
- 12.AL-Saedi M, Faisal B, Yaser D, et al. Liver amyloidosis complicated with liver failure: a case report and review of the literature. J Gastroint Dig Syst 2011;S3. [Google Scholar]
- 13.NovoSeven (Recombinant factor VIIa) [package insert]. 2880 Bagsvaerd, Denmark; Novo Nordisk A/S; Revised 10/13/2006. [Google Scholar]
- 14.Hedner U, Glazer S, Falch J. Recombinant activated factor VII in the treatment of bleeding episodes in patients with inherited and acquired bleeding disorders. Transfus Med Rev 1993;7:78-83. [DOI] [PubMed] [Google Scholar]
- 15.Brenner B, Wiis J. Experience with recombinant-activated factor VII in 30 patients with congenital factor VII deficiency. Hematology 2007;12:55-62. [DOI] [PubMed] [Google Scholar]
- 16.Mullighan CG, Rischbieth A, Duncan EM, Lloyd JV. Acquired isolated factor VII deficiency associated with severe bleeding and successful treatment with recombinant FVIIa (NovoSeven). Blood Coagul Fibrinolysis 2004;15:347-51. [DOI] [PubMed] [Google Scholar]
- 17.Green G, Poller L, Thomson JM, Dymock IW. Factor VII as a marker of hepatocellular synthetic function in liver disease. J Clin Pathol. 1976;29:971-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bernstein DE, Jeffers L, Erhardtsen E, et al. Recombinant factor VIIa corrects prothrombin time in cirrhotic patients: a preliminary study. Gastroenterology 1997;113:1930-7. [DOI] [PubMed] [Google Scholar]