

Abstract
Physiological autophagy plays a crucial role in the regulation of muscle mass and metabolism, while the excessive induction or the inhibition of the autophagic flux contributes to the progression of several diseases. Autophagy can be activated by different stimuli, including cancer, exercise, caloric restriction and denervation. The latter leads to muscle atrophy through the activation of catabolic pathways, i.e. the ubiquitin-proteasome system and autophagy. However, the kinetics of autophagy activation and the upstream molecular pathways in denervated skeletal muscle have not been reported yet. In this study, we characterized the kinetics of autophagic induction, quickly triggered by denervation, and report the Akt/mTOR axis activation. Besides, with the aim to assess the relative contribution of autophagy in neurogenic muscle atrophy, we triggered autophagy with different stimuli along with denervation, and observed that four week-long autophagic induction, by either intermitted fasting or rapamycin treatment, did not significantly affect muscle mass loss. We conclude that: i) autophagy does not play a major role in inducing muscle loss following denervation; ii) nonetheless, autophagy may have a regulatory role in denervation induced muscle atrophy, since it is significantly upregulated as early as eight hours after denervation; iii) Akt/mTOR axis, AMPK and FoxO3a are activated consistently with the progression of muscle atrophy, further highlighting the complexity of the signaling response to the atrophying stimulus deriving from denervation.
Key Words: Denervation, Autophagy, Neurogenic muscle atrophy, Intermittent fasting, Rapamycin
Ethical Publication Statement
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Macroautophagy, or simply autophagy, is a catabolic pathway that leads to the degradation of damaged or misfolded cytoplasmic proteins or organelles, via double-membranes structures, the autophagosomes.1 More than 30 autophagy-related (Atg) genes have been identified in yeast, and many of these are conserved among higher eukaryotes.2 Upon autophagy induction, cytosolic Atg8 / LC3b I, is conjugated to phosphatidylethanolamine, therefore converted to LC3b II (changing its apparent molecular weight from about 16 kDa to about 14 kDa) and incorporated into the autophagic membranes. Another important player in autophagy is p62, a chaperone protein that directs polyubiquitinated proteins to the autophagosomes, by directly binding to LC3b and Gabarapl1 proteins and allowing the degradation of protein aggregates via autophagy.3 The p62 protein is itself incorporated and degraded during autophagy, therefore it is often used as a marker for the progression of the autophagic flux.4 Although autophagy was long considered a non-selective degradation pathway, in the last years autophagy emerged as responsible for specifically removing only selected cellular components. Examples of selective autophagy include aggrephagy, to remove protein aggregates,5 pexophagy, for peroxisomes,6 or ribophagy, to eliminate ribosomes.7 Mitophagy is an essential cellular mechanism controlling mitochondrial quality since its inhibition leads to accumulation of damaged or dysfunctional mitochondria.8 Deficiencies in mitophagy correlate with several pathologies, including neurodegenerative disorders such as Parkinson’s disease.9 Different cellular stimuli trigger autophagy, including cancer, ageing, starvation, exercise, disuse and denervation.10–14 Two kinases, namely Akt and AMP-activated protein kinase (AMPK), modulate autophagy. Akt is a serine/threonine protein kinase that mediates cellular response to insulin, thereby contributing to energy metabolism, inducing protein synthesis and muscle hypertrophy. Consistently, Akt overexpression in adult skeletal muscle counteracts muscle atrophy induced by denervation.15 Indeed, activated phosphorylated Akt blocks atrophy by phosphorylating and thereby inactivating the transcription factor FoxO3a.16 The phosphorylated form of FoxO3a is excluded from the nucleus and therefore unable to activate the muscle-specific E3 ubiquitin ligases atrogin-1 and MuRF1.13 On the contrary, when Akt is dephosphorylated, such as upon starvation, it promotes the inhibition of the mammalian target of rapamycin (mTOR), a potent autophagy suppressor, 17 and the activation of FoxO3a, a major inducer of autophagy- and proteasome-mediated catabolism, by its dephosphorylation and consequent translocation into the nucleus.13 In response to low levels of cellular energy, also AMPK is activated and induces autophagy by modulating the same Akt molecular targets, i.e. mTOR and FoxO3a.18–20 However, while Akt phosphorylates FoxO3a on thr32, ser253 and ser315, AMPK targets Foxo3 on different residues, phosphorylating ser413 and ser588.21 Autophagy has long been regarded as a cellular process leading to muscle wasting and atrophy in several muscular diseases, such as myopathies, dystrophies or neurogenerative disorders.22,23 However, a role for basal, physiological levels of autophagy in the maintenance of muscle mass and homeostasis has been assessed. Indeed, mouse models lacking Atg5 or Atg7 in skeletal muscle, or with compromised autophagy, such as the mutant mice for the histone deacetylase 1 and 2,24 show muscle loss, intracellular protein aggregates, and accumulation of abnormal membranous structures. Interestingly, the absence of Atg7 in skeletal muscle does not preserve muscle mass during catabolic conditions and, surprisingly, exacerbates muscle loss upon denervation.25 Despite the activation of autophagic markers has been reported three days following denervation in skeletal muscle,26 a fine characterization of the kinetics of autophagy activation upon denervation is still missing.
The present study aims to better clarify the kinetics of autophagy activation and the upstream molecular pathways in murine skeletal muscle following denervation, and to assess the contribution of autophagy to neurogenic muscle atrophy in vivo.
Materials and Methods
Animal models and Ethical Approval
Mice were treated in strict accordance with the guidelines of the Institutional Animal Care and Use Committee and to national and European legislation, throughout the experiments. All the experimental protocols were approved by the Charles Darwin Ethics Committee n°5 in Paris and by the Ministry of Research. Adult (2.5-month-old) C57BL/6 female mice were used throughout the experiments.
Denervation. The sciatic nerve of the left limb of anesthetized adult female C57/BL6 mice was cut and a 3 mm piece was excised. The right leg remained innervated and was used as control. Mice were sacrificed at indicated time points for histological and molecular analyses.
Intermittent fasting. In intermittent fasting (IF) experiments, the chow was removed from the cages of 4 mice, for 24 hours, twice a week, for 4 weeks, starting from the day of denervation, to evaluate the effects on both innervated and denervated muscles.
Rapamycin injections. Daily injections of 2 mg/kg rapamycin in physiologic solution containing 1% ethanol (vehicle), or of vehicle, were performed for 4 weeks from the day of denervation, in 4 mice for each condition, to evaluate the effects on both innervated and denervated muscles.
Histological Analyses
Tibialis Anterior (TA) muscles were dissected, embedded in tissue freezing medium (Leica, Wetzlar, Germany) and frozen in liquid nitrogen pre-cooled isopentane. Cryosections (8 μm) were obtained by using a Leica cryostat. Hematoxylin and eosin staining (Sigma-Aldrich) was performed according to the manufacturer’s instructions.
RNA Extraction and Real-time PCR
Total RNA was isolated and purified from 30-50 mg of TA muscles by using Trizol (Invitrogen), following the manufacturer's protocol. One microgram of total RNA was converted to cDNA by using the PrimeScript™ RT reagent Kit (TAKARA). Real-time PCR was performed with the SDS-ABI Prism 7500 (Applied Biosystem), by using the SYBR Green reaction mix (Applied Biosystem). The primer sequences used are reported below:
Atg5 for: GGAGAGAAGAGGAGCCAGGT
Atg5 rev: GCTGGGGGACAATGCTAATA
Atg7 for: GCCTAACACAGATGCTGCAA
Atg7 rev: TGCTCTTAAACCGAGGCTGT
Gaparapl1 for: GTGCCGGTCATCGTGGA
Gabarapl1 rev: TCCTCGTGGTTGTCCTCA
LC3b for: CACTGCTCTGTCTTGTGTAGGTG
LC3b rev: TCGTTGTGCCTTTATTATTAGTGCATC
p62 for: CCCAGTGTCTTGGCATTCTT
p62 rev: AGGGAAAGCAGAGGAAGCTC
GAPDH for: ACCCAGAAGACTGTGGATGG
GAPDH rev: CACATTGGGGGTAGGAACA
Protein Extraction and Western Blot Analyses
TA muscles were dissected, minced, and homogenized in lysis buffer (50 mM Tris-HCl pH 7.4, 1 mM EDTA, 150 Mm NaCl, 1% TRITON) supplemented with protease and phosphatase inhibitors (Sigma-Aldrich). Proteins (30-50 micrograms) were separated by SDS-PAGE and transferred to PVDF membrane (Invitrogen). Unspecific binding was blocked in 5% not fat dry milk in TBST buffer (20 mM Tris HCl pH 7.6, 137 mM NaCl, 0.5% Tween 20), then the membranes were incubated overnight with the primary antibody diluted in 5% BSA (Sigma) in TBST. After washing in TBST, the membranes were incubated with the secondary HRP-conjugate antibodies (Bio-rad 170-6515 or 170-6516) and signals were detected by using ECL chemistry (Advansta). Images were acquired using the ChemiDoc MP imaging system (Bio-Rad). Densitometric analyses were performed by measuring the band intensity for each sample using the Image J software. The following primary antibodies were used: GAPDH (Santa Cruz sc-32233), P-AMPK (Cell Signaling 2535), AMPK (Cell Signaling 2532), P-ACC (Cell Signaling 3661), P-Akt (Cell Signaling 9271), Akt (Cell Signaling 9272), P-FoxO3a (Ser253) (Cell Signaling 9466), Foxo3a (Cell Signaling 2497), P-mTOR (Ser2448) (Cell Signaling 2971), mTOR (Cell Signaling 2983), LC3b (Cell Signaling 2775), p62 (Sigma-Aldrich P0067).
Statistics
Statistical significance was determined using two-tailed Student t-test with a significance level < 0.05 or by using two-way analysis of variance (ANOVA), followed by Tukey’s HSD as a post-hoc test, when more than two conditions needed to be compared. All values are expressed as mean ± standard error of the mean (SEM). VassarStats, a statistical computation website available at http://vassarstats.net/, was used for the statistical analyses.
Results
Autophagy is induced in skeletal muscle one day following denervation
Aiming to investigate the kinetics of denervation-induced autophagy in skeletal muscle, we resected the sciatic nerve of one limb of adult mice and analyzed the target skeletal muscle over time. We used the contralateral muscle as control, since we had previously established that these muscles are unaffected by denervation.27 The expression levels of several autophagic genes were monitored by real-time PCR, following denervation (Fig. 1A). The expression of some autophagy related genes, such as Atg5, LC3b and p62, was up-regulated as soon as eight hours upon denervation and increased over time, reaching statistical significance within eight to twenty-four hours; while the expression of others, such as Atg7 and Gabarapl1 significantly increased only from day three following denervation. Noticeably, LC3b expression showed a different, specific pattern, reaching its maximal induction one day following denervation and then decreasing, while still remaining significantly higher than control muscles (Fig. 1A). To further confirm these findings, we analyzed the lipidation of LC3b and the accumulation of p62 protein at one, three and seven days following denervation (Fig. 1B). Denervation induced a significant accumulation of the lipidated form of LC3b and p62 after 7 days, indicative of effective autophagosome formation upon denervation in skeletal muscle.
Fig 1.
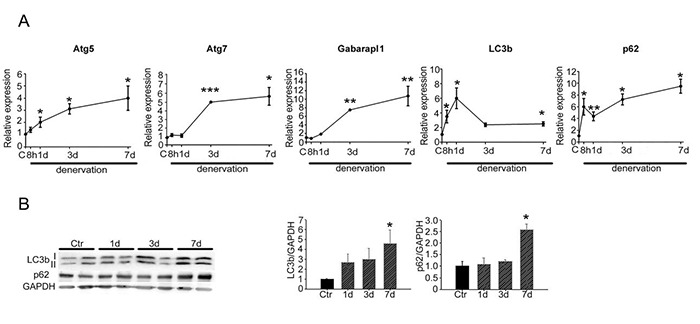
Autophagy is quickly induced in skeletal muscle in response to denervation. (A) Real-time PCR for autophagic markers in skeletal muscle following denervation at indicated time points, relative to control (C) muscles. Values represent mean ± SEM. n=4-6 mice for each condition; *p<0.05; **p<0.001; ***p<0.0001 by Student’s t test vs control muscles. (B) Western blotting analyses and quantification for LC3b and p62 proteins, one, three and seven days following denervation. GAPDH was used as loading control. Values represent mean ± SEM. n=4 mice for each condition; *p<0.05 by Student’s t test vs control muscles.
Autophagy is associated with activation of both Akt- and AMPK- dependent pathways
To elucidate the signaling pathways involved in autophagy, we quantified the expression and the activation of specific up-stream proteins controlling autophagy, one three and seven days following denervation. Increased levels of activated phospho (P)-AMPK, but not total AMPK, were detected seven days following denervation, in respect to control muscles. Consistently, higher levels of phospho-acetyl-CoA Carboxylase, a direct target of AMPK,28 were observed seven days after denervation, compared to control muscles, indicative of AMPK activation (Fig. 2A). Moreover, both P-Akt and total Akt were significantly increased seven days following denervation, as well as P-mTOR and total mTOR, indicative of Akt activation (Fig. 2B). In addition, an important downregulation of P-FoxO3 was detected three days following denervation, indicative of a nuclear localization and activation of this transcription factor (Fig. 2C).
Fig 2.
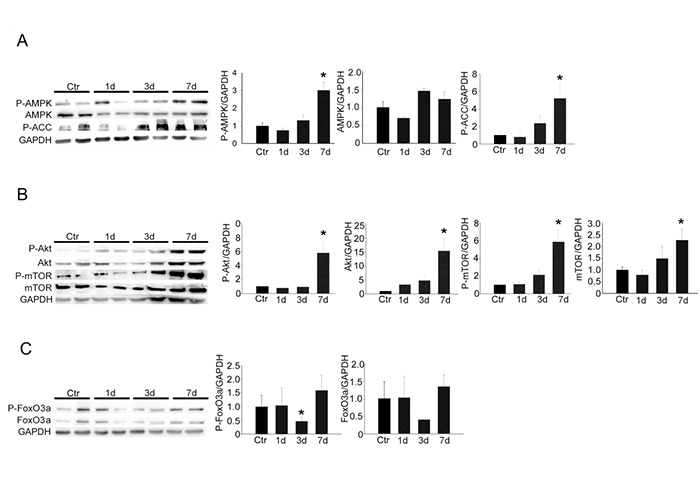
Up-stream activators of the autophagic pathway are activated upon denervation. (A) Western blotting analyses and quantification of P-AMPK, AMPK and P-ACC proteins, at indicated time points following denervation. GAPDH was used as control. Values represent mean ± SEM; n=4 mice for each condition; *p<0.05 by Student’s t test vs control (Ctr) muscles. (B) Western blotting analyses and quantification of Akt/mTOR pathway, at indicated time points following denervation. GAPDH was used as loading control. Values represent mean ± SEM. n=4 mice for each condition; *p<0.05 by Student’s t test vs Ctr muscles. c Western blotting analyses and quantification of P-FoxO3 and FoxO3 proteins, at indicated time points following denervation. GAPDH was used as loading control. Values represent mean ± SEM. n=4 mice for each condition; *p<0.05 by Student’s t test vs Ctr muscles.
Autophagy does not contribute to muscle atrophy following denervation
To investigate the contribution of autophagy to neurogenic muscle atrophy, we triggered autophagy in skeletal muscle from the day of denervation to day 28 following denervation, by using two different approaches: a metabolic stimulus, i.e. intermittent fasting (IF) 14 and daily injections of the known autophagy activator, rapamycin (Rapa).10 We first verified that IF effectively activated autophagy in skeletal muscle following denervation, by confirming the significant accumulation of the lipidated form of LC3b and of p62, by western blot analyses (Fig. 3). Muscle mass evaluation and histological analyses revealed that neither IF, nor Rapa induce muscle atrophy in control muscles, or exacerbated muscle fiber atrophy in denervated muscles, despite increased autophagy for up to four weeks following denervation (Fig. 4A and 4B). Morphometric analysis, i.e. fiber cross-sectional area distribution, confirmed these results. Indeed, while denervation significantly affected myofiber CSA, in the 500-1500, 1000-1500, and >2500 μm2 classes, IF or Rapa did not significantly change myofiber CSA distribution of control or denervated muscles (Fig. 4C and D). These results clearly indicate that, despite the activation of autophagy following denervation, further activation of this pathway does not exacerbate neurogenic muscle atrophy, nor induce muscle atrophy in control muscles.
Fig 3.
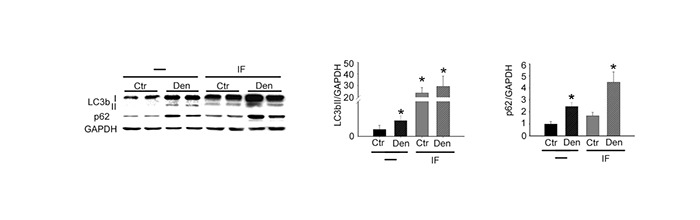
Intermittent fasting activates autophagy in skeletal muscle. Western blotting analyses and quantification of LC3b and p62 proteins in mice subjected (IF) or not (-) to intermittent fasting, seven days following denervation. GAPDH was used as loading control. Values represent mean ± SEM. n=4 mice for each condition; two-way ANOVA revealed an interaction between treatments (F=5.52; df 1; p=0.036 for LC3 and F=6.93; df 1; p=0.025 for p62); *p<0.05 vs untreated Ctr, by Tukey’s HSD test.
Fig 4.
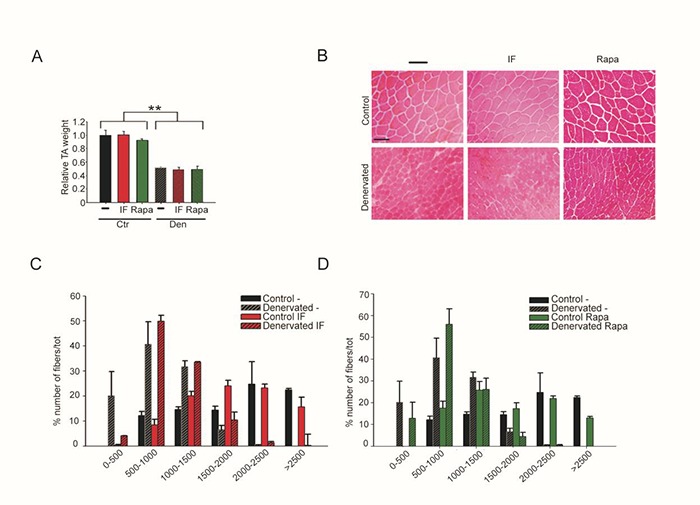
Autophagy does not contribute to muscle atrophy. (A) TA muscle weight of mice subjected to IF or treated with rapamycin (Rapa), or untreated (-), 28 days following denervation, relative to untreated control (Ctr) TA muscles. Values represent mean ± SEM. n=4 mice for each condition; two-way ANOVA revealed an effect of denervation (F=73 df 1; p=0.0001 for IF and F=98; df 1; p=0.0001 for Rapa), without interaction between treatments. (B) Representative pictures of the histology of treated (IF and Rapa) or not (-), control and denervated TA muscles, 28 days following denervation. Scale bar=50 micron. (C) Myofiber cross-sectional area distribution of TA muscles of mice without (-) or after IF treatment, four weeks after denervation. Data are shown as mean ± SEM; n=4 mice per each condition. Two-way ANOVA revealed an effect of denervation in the 500-1500, 1000-1500, and >2500 classes (F=39.61; df 1; p=0.0001; F=30.98; df 1; p=0.0001 and F=39.14; df 1; p=0.0001, respectively) without interaction between treatments. (D) Myofiber cross-sectional area distribution of TA muscles of mice without (-) or after rapamycin (Rapa) treatment, four weeks after denervation. Data are shown as mean ± SEM; n=4 mice per each condition. Two-way ANOVA revealed an effect of denervation in the 500-1500, 1000-1500 and >2500 classes (F=53.95; df 1; p=0.0001; F=14.23; df 1; p=0.0054 and F=91.95; df 1; p=0.0001, respectively) without interaction between treatments.
Discussion
Autophagy is a physiological process necessary to deliver dysfunctional organelles and proteins to lysosomes for degradation. Increased autophagy has long been considered as a harmful cellular process, which contributes to muscle wasting in several muscle diseases, such as myopathies, dystrophies or neurodegenerative disorders.22,23,29 Consistently, chronic activation of autophagy leads to muscle wasting.13,30 However, inhibition of autophagy is detrimental to skeletal muscle mass and architecture, indicating its essential physiological role in preserving muscle homeostasis.25,31 The relevance of autophagy for skeletal muscle homeostasis was also recently demonstrated by our study, showing that the autophagic flux is overloaded in skeletal muscles of colon carcinoma patients and mice and that aerobic exercise or pharmacological treatments that promote autophagy rescue muscle homeostasis in tumor-bearing mice.10 Given the importance for muscle homeostasis of the dynamic balance between autophagy hyper-activation and inhibition, it is important to shed light on the activation of autophagy and the molecular pathways involved in its regulation in pathological settings, such as denervation. To date, several studies characterized skeletal muscle response to denervation,27,32,33 but conflicting results have been published on autophagy induction. Some studies reported that autophagy is triggered seven days after denervation, prevalently targeting mitochondria for their removal,34,35 while according to others autophagy is rather suppressed in denervated muscles.36 None of the studies investigated earlier time points following denervation. Here, for the first time, we reported a significant increase in the expression levels of the autophagic markers LC3b and p62, as soon as eight hours following denervation. Being FoxO3a activated only three days following denervation, either other factors trigger the transcription of these autophagic genes, such as E2F1 or TFEB,37,38 or FoxO factors are transiently activated earlier than one day following denervation. The molecular pathways controlling autophagic induction in skeletal muscle upon denervation have been only partially elucidated and contradictory explanations have been reported. We observed that both the total and phosphorylated forms of Akt and mTOR were higher in denervated muscles after seven days, compared to control muscles, in agreement with what previously reported by others.39,40 Akt is a crucial regulator of muscle growth and hypertrophy, also preventing muscle atrophy.41,42 The activation of the Akt pathway following denervation, therefore, could represent an attempt to counteract muscle atrophy, by promoting muscle growth36 which may explain the persistence of muscle regeneration in long term denervated condition.32 Alternatively, the denervation-induced increase mTOR activity may produce a feedback inhibition of Akt pathway43, or even contributes to increase proteasome levels.44 According to others, however, neurogenic muscle atrophy is independent of Akt and mTOR activation,45 supporting the hypothesis that other signals trigger the ubiquitin-proteasome pathway. Since mTOR inhibits autophagy,17 we propose that mTOR activation has no major effects on autophagy regulation upon denervation, differently from muscle atrophy induced by other stimuli.46 AMPK is an important mediator of skeletal muscle metabolism47 and is one of the kinases known to regulate autophagy, together with Akt, by inhibiting mTOR and by activating FoxO3a.18,20 We found that P-AMPK and its downstream target ACC are accumulated in denervated muscles, one week following denervation, in respect to the control muscles. We reported a decrease in P-FoxO3a, three days after denervation, implying its transcriptional activation at early time points following denervation. Therefore, our data support the model that neither Akt, nor AMPK are responsible for the FoxO3a transcriptional activation at this early time point. Other kinases known to regulate the phosphorylation status of FoxO3a, such as P38 or JUNK,48,49 may be involved in this early response to denervation.
To investigate the contribution of autophagy to muscle trophism, we induced autophagy in skeletal muscle following denervation by using two different approaches: IF and rapamycin. Importantly, the effects of IF on LC3b and p62 accumulation are independent of denervation, and the two stimuli seem to be additive. These data indicate that denervation does not induce a maximal activation of autophagy in skeletal muscle, but additional stimuli can further increase this process. Muscle atrophy was evaluated four weeks following denervation, a time point chosen to evaluate the effects on both innervated and denervated muscles. Muscle mass, histological and morphometrical analyses proved that both treatments did not induce muscle atrophy in control muscles, neither enhanced muscle atrophy in denervated ones, indicating that autophagy does not induce nor exacerbate muscle fiber atrophy following denervation. In agreement with our study, inhibition of autophagy does not preserve skeletal muscle mass following denervation,25 indicating that autophagy activation is not required for denervation-dependent muscle atrophy and rapamycin treatment. Moreover, rapamycin treatment has been already shown to do not exacerbate neurogenic muscle atrophy, or to induce muscle atrophy, after three weeks of denervation.45 Since skeletal muscle denervation induces a shift in fiber metabolism,50 being autophagy involved in the regulation of skeletal muscle metabolism,24,25 we speculate that autophagy activation could mediate the denervation-dependent metabolic changes in skeletal muscle.
In conclusion, we monitored the activation of autophagy since the early phases following denervation, thus contributing new evidence to a model whereby neurogenic muscle atrophy and autophagic induction occurs despite Akt activation. Moreover, increased autophagy does not cause muscle fiber atrophy per se, nor it exacerbates muscle fiber atrophy in response to denervation, highlighting the importance of autophagy in regulating cellular responses other than muscle trophism.
Acknowledgments
This work was supported by FIRB 2012 (RBFR12BUMH) grant from the Italian Ministry of Education, Universities and Research; and by Sapienza research project 2016 (SapMedi2016) and Sapienza research project 2017 (RM11715C78539BD8).
List of acronyms
- Akt
Protein kinase B
- AMPK
AMP-activated protein kinase
- Atg
Autophagy-related genes
- FoxO3a
Forkhead box O3
- Gabarapl1
GABA-A Receptor-Associated Protein-Like 1
- GAPDH
Glyceraldehyde-3-phosphate dehydrogenase
- IF
intermittent fasting
- LC3b
Microtubule-associated proteins 1A/1B Light Chain 3B
- mTOR
mammalian target of rapamycin
- MuRF1
Muscle RING-finger protein-1
Funding Statement
Funding: MIUR - FIRB; Sapienza University of Rome.
Contributor Information
Eva Pigna, Email: eva.pigna@uniroma1.it.
Krizia Sanna, Email: krizia.sanna@live.it.
Dario Coletti, Email: dario.coletti@uniroma1.it.
Zhenlin Li, Email: zhenlin.li@upmc.fr.
Ara Parlakian, Email: ara.parlakian@upmc.fr.
Viviana Moresi, Email: viviana.moresi@uniroma1.it.
References
- 1.Bonaldo P, Sandri M. Cellular and molecular mechanisms of muscle atrophy. Dis Model Mech 2013;6:25-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res 2014;24:24-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shvets E, Fass E, Scherz-Shouval R, Elazar Z. The n-terminus and phe52 residue of lc3 recruit p62/sqstm1 into autophagosomes. J Cell Sci. 2008, 121:2685-95. [DOI] [PubMed] [Google Scholar]
- 4.Klionsky DJ, Klionsky DJ, Abdelmohsen K, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016; 12:1-222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lamark T, Johansen T. Aggrephagy: selective disposal of protein aggregates by macroautophagy. Int J Cell Biol 2012;2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hutchins MU, Veenhuis M, Klionsky DJ. Peroxisome degradation in saccharomyces cerevisiae is dependent on machinery of macroautophagy and the cvt pathway. J Cell Sci 1999;112:4079-87. [DOI] [PubMed] [Google Scholar]
- 7.Kraft C, Deplazes A, Sohrmann M, Peter M. Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the ubp3p/bre5p ubiquitin protease. Nat Cell Biol 2008;10:602. [DOI] [PubMed] [Google Scholar]
- 8.Palacino JJ, Sagi D, Goldberg MS, et al. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J Biol Chem 2004;279:18614-22. [DOI] [PubMed] [Google Scholar]
- 9.Redmann M, Dodson M, Boyer-Guittaut M, et al. Mitophagy mechanisms and role in human diseases. Int J Biochem Cell Biol 2014;53:127-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pigna E, Berardi E, Aulino P, et al. Aerobic exercise and pharmacological treatments counteract cachexia by modulating autophagy in colon cancer. Sci Rep 2016;6:26991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cudkowicz ME, Andres PL, Macdonald SA, et al. Phase 2 study of sodium phenylbutyrate in als. Amyotroph Lateral Scler 2009;10:99-106. [DOI] [PubMed] [Google Scholar]
- 12.Zha Z, Wang J, Wang X, et al. Involvement of pink1/parkin-mediated mitophagy in age-induced cardiomyocyte aging. Int J Cardiol 2017;227:201-08. [DOI] [PubMed] [Google Scholar]
- 13.Mammucari C, Milan G, Romanello V, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab 2007;6:458-71. [DOI] [PubMed] [Google Scholar]
- 14.Mizushima N. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 2003; 15:1101-1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bodine SC, Stitt TN, Gonzalez M, et al. Akt/mtor pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat Cell Biol 2001; 3(11):1014. [DOI] [PubMed] [Google Scholar]
- 16.Sandri M, Sandri C, Gilbert A, et al. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 2004;117:399-412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jung CH, Ro SH, Cao J, et al. MTOR regulation of autophagy. FEBS Lett 2010. 584:1287-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bolster DR, Crozier SJ, Kimball SR, Jefferson LS. AMP-activated protein kinase suppresses protein synthesis in rat skeletal muscle through down-regulated mammalian target of rapamycin (mtor) signaling. J Biol Chem 2002; 277:23977-980. [DOI] [PubMed] [Google Scholar]
- 19.Greer EL, Dowlatshahi D, Banko MR, et al. An ampk-foxo pathway mediates longevity induced by a novel method of dietary restriction in c. elegans. Curr Biol 2007; 17:1646-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Greer EL, Oskoui PR, Banko MR, et al. The energy sensor amp-activated protein kinase directly regulates the mammalian foxo3 transcription factor. J Biol Chem 2007; 282:30107-119. [DOI] [PubMed] [Google Scholar]
- 21.Brunet A, Bonni A, Zigmond MJ, et al. Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell 1999;96:857-868. [DOI] [PubMed] [Google Scholar]
- 22.Penna F, Costamagna D, Pin F, et al. Autophagic degradation contributes to muscle wasting in cancer cachexia. Am J Pathol 2013;182:1367-78. [DOI] [PubMed] [Google Scholar]
- 23.Dobrowolny G, Aucello M, Rizzuto E, et al. Skeletal muscle is a primary target of sod1g93a-mediated toxicity. Cell Metab 2008;8:425-36. [DOI] [PubMed] [Google Scholar]
- 24.Moresi V, Carrer M, Grueter CE, et al. Histone deacetylases 1 and 2 regulate autophagy flux and skeletal muscle homeostasis in mice. Proc Natl Acad Sci 2012;109:1649-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Masiero E, Agatea L, Mammucari C, et al. Autophagy is required to maintain muscle mass. Cell Metab 2009;10:507-515. [DOI] [PubMed] [Google Scholar]
- 26.Milan G, Romanello V, Pescatore F, et al. Regulation of autophagy and the ubiquitin-proteasome system by the foxo transcriptional network during muscle atrophy. Nat Commun 2015;6:6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pigna E, Greco E, Morozzi G, et al. Denervation does not induce muscle atrophy through oxidative stress. Eur J Transl Myol 2017;27:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ha J, Daniel S, Broyles SS, Kim KH. Critical phosphorylation sites for acetyl-coa carboxylase activity. J Biol Chem 1994. 269:22162-168. [PubMed] [Google Scholar]
- 29.Madaro L, Marrocco V, Carnio S, et al. Intracellular signaling in er stress-induced autophagy in skeletal muscle cells. FASEB J 2013;27:1990-2000. [DOI] [PubMed] [Google Scholar]
- 30.Brocca L, Cannavino J, Coletto L, et al. The time course of the adaptations of human muscle proteome to bed rest and the underlying mechanisms. J Physiol 2012; 590:5211-5230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raben N, Schreiner C, Baum R, et al. Suppression of autophagy permits successful enzyme replacement therapy in a lysosomal storage disorder—murine pompe disease. Autophagy 2010;6:1078-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carraro U, Boncompagni S, Gobbo V, et al. Persistent muscle fiber regeneration in long term denervation. past, present, future. Eur J Transl Myol 2015;25:4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kern H, Carraro U. Home-based functional electrical stimulation for long-term denervated human muscle: history, basics, results and perspectives of the vienna rehabilitation strategy. Eur J Transl Myol 2014;24:3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O’Leary MFN, Hood DA. Denervation-induced oxidative stress and autophagy signaling in muscle. Autophagy 2009;5:230-31. [DOI] [PubMed] [Google Scholar]
- 35.Vainshtein A, Desjardins EMA, Armani A, et al. PGC-1α modulates denervation-induced mitophagy in skeletal muscle. Skelet Muscle 2015;5:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Quy PN, Kuma A, Pierres P, Mizushima N. Proteasome-dependent activation of mammalian target of rapamycin complex 1 (mtorc1) is essential for autophagy suppression and muscle remodeling following denervation. J Biol Chem 2013; 288:1125-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Polager S, Ofir M, Ginsberg D. E2F1 regulates autophagy and the transcription of autophagy genes. Oncogene 2008;27:4860. [DOI] [PubMed] [Google Scholar]
- 38.Settembre C, Di Malta C, Polito VA, et al. TFEB links autophagy to lysosomal biogenesis. Science 2011;332:1429-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Machida M, Takeda K, Yokono H, et al. Reduction of ribosome biogenesis with activation of the mtor pathway in denervated atrophic muscle. J Cell Physiol 2012; 227:1569-76. [DOI] [PubMed] [Google Scholar]
- 40.Argadine HM, Mantilla CB, Zhan W-Z, Sieck GC. Intracellular signaling pathways regulating net protein balance following diaphragm muscle denervation. Am J Physiol Cell Physiol 2011;300:C318-C327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bodine SC, Latres E, Baumhueter S, et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001;294:1704-08. [DOI] [PubMed] [Google Scholar]
- 42.Stitt TN, Drujan D, Clarke BA, et al. The igf-1/pi3k/akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting foxo transcription factors. Mol Cell 2004;14:395-403. [DOI] [PubMed] [Google Scholar]
- 43.Tang H, Inoki K, Lee M, et al. MTORC1 promotes denervation-induced muscle atrophy through a mechanism involving the activation of foxo and e3 ubiquitin ligases. Sci Signal 2014;7:ra18-ra18. [DOI] [PubMed] [Google Scholar]
- 44.Zhang Y, Manning BD. MTORC1 signaling activates nrf1 to increase cellular proteasome levels. Cell Cycle 2015;14:2011-2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.MacDonald EM, Andres-Mateos E, Mejias R, et al. Denervation atrophy is independent from akt and mtor activation and is not rescued by myostatin inhibition. Dis Model Mech 2014;7:471-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Malavaki CJ, Sakkas GK, Mitrou GI, et al. Skeletal muscle atrophy: disease-induced mechanisms may mask disuse atrophy. J Muscle Res Cell Motil 2015;36:405-21. [DOI] [PubMed] [Google Scholar]
- 47.Huber K, Petzold J, Rehfeldt C, et al. Muscle energy metabolism: structural and functional features in different types of porcine striated muscles. J Muscle Res Cell Motil 2007;28:249-58. [DOI] [PubMed] [Google Scholar]
- 48.Clavel S, Siffroi-Fernandez S, Coldefy AS, et al. Regulation of the intracellular localization of foxo3a by stress-activated protein kinase signaling pathways in skeletal muscle cells. Mol Cell Biol 2010;30:470-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Diebold I, Petry A, Burger M, et al. NOX4 mediates activation of foxo3a and matrix metalloproteinase-2 expression by urotensin-ii. Mol Biol Cell 2011;22:4424-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Patterson MF, Stephenson GMM, Stephenson DG. Denervation produces different single fiber phenotypes in fast- and slow-twitch hindlimb muscles of the rat. Am J Physiol Physiol 2006;291:C518-28. [DOI] [PubMed] [Google Scholar]