

Abstract
Background:
Recent studies show links between metabolic syndrome and Alzheimer’s disease (AD) neuropathology. Understanding the link between vascular-related health conditions and dementia will help target at risk populations and inform clinical strategies for early detection and prevention of AD.
Objective:
To determine whether metabolic syndrome is associated with global cerebral amyloid-β (Aβ) positivity and longitudinal Aβ accumulation.
Methods:
Prospective study of 165 participants who underwent (11)C-Pittsburgh compound B (PiB) PET neuroimaging to measure Aβ, from June 2005 to May 2016. Metabolic syndrome was defined using the revised Third Adults Treatment Panel of the National Cholesterol Education Program criteria. Participants were classified as PiB+/−. Linear mixed effects models assessed the relationships between baseline metabolic syndrome and PiB status and regional Aβ change over time.
Results:
A total of 165 cognitively normal participants of the Baltimore Longitudinal Study of Aging (BLSA) Neuroimaging substudy, aged 55–92 years (mean baseline age = 76.4 years, 85 participants were male), received an average of 2.5 PET-PiB scans over an average interval of 2.6 (3.08 SD) years between first and last visits. Metabolic syndrome was not associated with baseline PiB positivity or concurrent regional Aβ. Metabolic syndrome was associated with increased rates of Aβ accumulation in superior parietal and precuneus regions over time in the PiB+ group. Elevated fasting glucose and blood pressure showed individual associations with accelerated Aβ accumulation.
Conclusion:
Metabolic syndrome was associated with accelerated Aβ accumulation in PiB+ individuals and may be an important factor in the progression of AD pathology.
Keywords: Alzheimer’s disease, amyloid PET, brain, cholesterol, dementia, diabetes, hypertension, metabolic syndrome, vascular
INTRODUCTION
Alzheimer’s disease (AD), the most common cause of dementia, affects one in nine people aged 65 and older [1], and the prevalence of this disease will continue to escalate as the population ages. Identification of etiological factors that modify the neuropathological trajectory and clinical manifestation of this disease has assumed increasing importance. Cumulative evidence suggests that vascular risk factors may play a role in AD-related pathology [2] and the risk of clinical symptoms of dementia is increased in individuals with infarcts or pre-existing stroke often associated with small vessel disease or microangiopathy [3–5]. Understanding the link between vascular-related health conditions and dementia will help target at risk populations and inform clinical strategies for early detection and prevention of AD.
Metabolic syndrome (MetS) represents the collective burden of five frequently co-occurring vascular risk factors and affects nearly 50% of adults over 60 years of age. A diagnosis of metabolic syndrome requires the presence of three or more health conditions related to body mass, impaired glucose tolerance, dyslipidemia, and hypertension, all of which increase risk for heart disease and other cardiovascular health problems. Several epidemiological studies have linked MetS to increased risk for cognitive impairment [6], vascular dementia [7], and AD [8]. Additionally, it has been suggested that metabolic syndrome as a whole may be a better predictor of AD pathology than each of its individual component conditions [9]. However, without investigation of neuropathological burden, these epidemiological studies provide only indirect evidence of these associations and are limited in their inferences.
The progression of neuropathology in AD is marked by abnormal accumulation of amyloid and tau proteins in the brain, with cerebral amyloid-β (Aβ) deposition thought to begin many years before the onset of clinical symptoms [10, 11]. A growing body of literature suggests that the vascular risk factors comprising MetS are associated with Aβ accumulation. Specifically, amyloid deposition has been associated with obesity [12], diabetes [13], cholesterol levels [14, 15], and hypertension [16, 17]. Though multiple review articles highlight the disruption in Aβ metabolism as a potential link between the development of MetS and AD [18, 19], the neuropathological association between MetS and cerebral Aβ has not been fully explored.
The present study examines the relationship between MetS and cerebral fibrillar Aβ, measured using positron emission tomography (PET) imaging and the radiotracer (11)C-Pittsburgh compound-B (PiB). Our first aim was to determine whether MetS diagnosis is associated with the presence of Aβ in the brain, indicated by PiB positivity. Second, we investigated whether the presence of MetS at baseline was associated with regional baseline Aβ levels and with subsequent longitudinal rates of change in brain regions recognized as areas of early amyloid accumulation. Based on previous findings, we hypothesized that individuals with MetS would exhibit increased concurrent amyloid burden or accelerated amyloid accumulation over time.
METHODS
Participants
We analyzed data from 165 participants enrolled in the neuroimaging substudy of the Baltimore Longitudinal Study of Aging (BLSA-NI) collected from June 2005 to May 2016 [20,21]. All participants received concurrent measures of waist circumference, fasting glucose, triglyceride level, high-density lipoprotein (HDL) cholesterol, and blood pressure at each BLSA visit. Individuals with clinical diagnoses of mild cognitive impairment (MCI), impaired not-MCI, or dementia at the beginning of the study (n = 7) were excluded from the analyses. Individuals who received an MCI, impaired not-MCI, or dementia diagnosis during the study were censored at the year of onset (n = 9). Cognitive impairment was determined by consensus case conference diagnosis using Diagnostic and Statistical Manual of Mental Disorders Third Edition, Revised (DSM-III-R) (1987) criteria for dementia, and the National Institute of Neurological and Communication Disorders and Stroke-Alzheimer’s Disease and Related Disorders Association criteria [22], using clinical data and a battery of neuropsychological tests that assess memory (California Verbal Learning Test [23] and Benton Visual Retention Test [24]), word knowledge (Primary Mental Abilities Vocabulary [25]), attention (Digits Forward [26] and the Trail Making Test-A [27]), executive function (Digits Backward [26], Trail Making Test-B [27], and Verbal Fluency [28]), and visuospatial abilities (Card Rotations Test [29]) [30]. Diagnosis of MCI followed the Petersen criteria [31]. At neuroimaging study enrollment, participants were free of central nervous system disease (such as epilepsy and stroke), psychiatric disorders, severe cardiac disease (myocardial infarction, coronary artery disease requiring angioplasty or bypass surgery), and metastatic cancer. Participant characteristics are presented in Table 1. Baseline was defined as initial amyloid assessment with a concurrent evaluation for MetS.
Table 1.
Participant demographics
| Characteristic | Total Sample |
PiB + Subset |
||
|---|---|---|---|---|
| MetS − | MetS + | MetS − | MetS + | |
| Total number of individuals | 165 | 51 | ||
| by MetS Group, n | 81 | 84 | 24 | 27 |
| Baseline age in years, mean (SD) | 77.3 (9.05) | 75.5 (6.67) | 81.8 (6.43) | 78.3 (6.55) |
| Range | 55.7–92.4 | 59.3–88.5 | 67.3–92.4 | 65.4–88.5 |
| Male, n (%) | 38 (46.9) | 47 (56.0) | 12 (50.0) | 18 (66.7) |
| Education, mean (SD) | 17.2 (2.05) | 16.8 (2.28) | 16.6 (1.76) | 16.5 (2.57) |
| APOE ε4 status, n (%) | 17 (21.0) | 20 (23.8) | 8 (33.3) | 9 (33.3) |
| Total number of PET-PiB scans, n | 409 | 125 | ||
| by MetS Group, n | 196 | 213 | 59 | 66 |
| PET-PiB scans per subject, n | 2.4 | 2.5 | 2.5 | 2.4 |
| Years between first and last scan, mean (SD) | 2.4 (2.96) | 2.8 (3.15) | 2.2 (2.35) | 2.3 (2.98) |
| Mean cortical DVR at last visit (SD) | 1.07 (0.16) | 1.09 (0.18) | 1.26 (0.15) | 1.31 (0.17) |
| PiB+ at first visit, n (%) | 21 (25.9) | 25 (29.8) | ||
| PiB+ at last visit, n (%) | 24 (29.6) | 27 (32.1) | ||
| Baseline Medication | ||||
| Hypertension, n (%) | 9 (11.1) | 45 (53.6) | 1 (4.2) | 10 (37.0) |
| Diabetes, n (%) | 2 (2.5) | 9 (10.7) | 0 (0) | 4 (14.8) |
| Dyslipidemia, n (%) | 20 (24.7) | 70 (83.3) | 1 (4.2) | 21 (77.8) |
DVR, distribution volume ratio; MetS, Metabolic Syndrome; PiB, carbon 11-labeled Pittsburgh Compound B; SD, standard deviation. APOE ε4 positive status required at least one ε4 allele. Baseline is defined as initial amyloid assessment with a concurrent evaluation for MetS.
Imaging acquisition and preprocessing
Cerebral amyloid was measured with PET-PiB acquired over 70 min. Distribution volume ratios (DVR) reflecting Aβ levels were calculated using the cerebellar gray matter reference region and simplified reference tissue model [32] using an in-house implementation in MATLAB (MATLAB and Statistics Toolbox Release 2016a, The Mathworks, Inc., Natick, Massachusetts, USA). DVR images were spatially normalized using co-registered T1-weighted MRI scans to a study-specific template in MNI space using SyN diffeomorphic registration (ANTs version 2.1.0, http://stnava.github.io/ANTs/) [33]. Anatomical labels were obtained from the study-specific template’s FreeSurfer (version 5.1, http://surfer.nmr.mgh.harvard.edu) segmentation [34]. Mean cortical DVR (mcDVR) was calculated by averaging across cortical regions (excluding sensorimotor strip, medial occipital and medial temporal areas). Participants were separated into PiB−/+ groups based on mcDVR at the first PET-PiB assessment (baseline) visit using a Gaussian mixture model threshold of 1.066 computed using the mixtools library version 1.1.0 [35] in R version 3.4.1 (http://www.r-project.org) (See Supplementary Figure 1 for Gaussian mixture model). Image processing in further detail is described elsewhere [36].
Selection of brain regions
To investigate associations between MetS and regional amyloid burden, we selected cortical and subcortical gray matter regions that were determined to be early amyloid accumulators based on quantitative and qualitative visual assessments of our prior longitudinal analysis results [36]. Regions were based on the Desikan-Killiany atlas [34], and those identified as early amyloid accumulators were the caudate and putamen, and the following gyri: superior frontal; rostral and caudal middle frontal; pars opercularis, triangularis, and orbitalis; lateral and medial orbitofrontal; frontal pole; paracentral lobule; superior and inferior parietal; supramarginal; precuneus; superior, middle, and inferior temporal; banks of the superior temporal sulcus; rostral and caudal anterior cingulate; posterior cingulate; and isthmus cingulate.
Metabolic syndrome diagnosis
MetS was defined using the Third Adults Treatment Panel of the National Cholesterol Education Program (NCEPATPIII) criteria, revised by the American Heart Association and National Heart, Lung, and Blood Institute (AHA/NHLBI) [37]. MetS scores were generated based on the presence of 5 conditions (Table 2). The presence or absence of each condition was indicated by a binary score (0 or 1), which were then summed to produce a MetS score for each participant, ranging from 0 to 5. A score of 3 or more indicated a diagnosis of MetS. Baseline MetS was defined as the score calculated at the initial amyloid (PET-PiB) assessment.
Table 2.
Diagnostic Criteria for Metabolic Syndrome, defined according to the National Cholesterol Education Program/Adult Treatment Panel III (NCEP/ATPIII) revised AHA/NHLBIa
| Measureb | Criteria |
|---|---|
| Elevated waist circumference | >40 inches in males >35 inches in females |
| Elevated fasting glucose | ≥100 mg/dl, or Rxc |
| Elevated triglyceride level | ≥150 mg/dl, or Rx |
| Reduced HDL cholesterol | <40 mg/dl in males, <50 mg/dl in females, or Rx |
| Elevated blood pressure | ≥ 130 mm Hg systolic blood pressure, or ≥85 mm Hg diastolic blood pressure, or Rx |
AHA/NHLBI, criteria revised from the report of the National Heart, Lung, and Blood Institute/American Heart Association Conference.
Three or more of the 5 conditions constitute a diagnosis of metabolic syndrome.
Rx, drug treatment for condition.
Statistical analyses
The analyses were performed in three stages. First, the relationship between MetS diagnosis and concurrent PiB positivity, i.e., the presence of amyloid in the brain, was examined in all participants. The significance of the association was determined using a Fisher’s exact test. To assess whether the prevalence of PiB positivity differed by age within the MetS groups, we computed the prevalence of PiB positivity by MetS group within two age bins: ≤77.3 and >77.3, based on the median age of our sample at baseline. We used logistic regression to assess the interaction between age group and MetS on PiB positivity.
Second, we used linear mixed effects models to examine the associations between baseline MetS diagnosis and both concurrent level of amyloid accumulation (DVR) and change in amyloid accumulation over time between PiB+ and PiB− groups. The models included sex and baseline MetS diagnosis as categorical predictors and baseline age (age at initial PET-PiB, mean-centered) and interval (time in years from first PET-PiB) as continuous predictors of mcDVR and regional DVR measures. Interval × baseline MetS × baseline PiB group, as well as the 2-way interactions (baseline MetS × interval, baseline PiB group × interval, baseline MetS × baseline PiB group), were included in the models. Subject-level random effects were specified to account for individual differences in intercept. Separate models were conducted for mcDVR and for regions of early amyloid accumulation, as outlined above. The false discovery rate (FDR) was used to correct for multiple comparisons [38]. Post-hoc contrasts were conducted to compare the effect of MetS on regional DVR rates of change between PiB + and PiB− groups.
Third, we sought to determine whether the individual components of MetS contributed to the association between MetS diagnosis and rate of amyloid accumulation in the PiB+ group. We conducted further analyses substituting for MetS diagnosis each of the five MetS components as categorical binary variables in separate linear mixed effects models. Additionally, we conducted a sensitivity analysis to determine whether treatment for diabetes, dyslipidemia, or hypertension affected the longitudinal relationship between MetS diagnosis and amyloid accumulation. We formed three binary variables to indicate baseline medication treatment for the three respective health conditions, and we included them in the model as categorical covariates. All linear mixed effects models were fitted using the nlme library version 3.1 in R version 3.4.1 (http://www.r- [project.org]).
RESULTS
Participant characteristics
A total of 165 BLSA-NI participants (mean baseline age = 76.4 years, 85 participants were male) received an average of 2.5 PET-PiB scans over an average interval of 2.6 (3.08 SD) years between first and last visits (Table 1). Within the total sample and the PiB+ subset, no characteristics detailed in Table 1 differed significantly between MetS+ and MetS− groups. APOE4 status, defined by the presence of at least one ε4 allele, did not differ between MetS groups.
Metabolic syndrome and PiB positivity
To determine whether PiB positivity differed by MetS diagnosis, we conducted Fisher’s exact test. The analysis revealed no association between baseline MetS diagnosis and PiB positivity (p = 0.605). Additionally, logistic regression revealed that age group, assigned by median split, did not change the association between MetS and PiB positivity.
Metabolic syndrome and regional DVR
The linear mixed models of the relationships between MetS diagnosis at baseline and concurrent measures of mcDVR and regional DVR showed no significant cross-sectional associations (Supplementary Tables 1 and 2). Consistent with our previous reports [39], we found significant increases in Aβ accumulation over time in the PiB+ but not PiB− group (PiB group × interval interaction, p <0.001; Supplementary Table 1). We found no significant overall effect of MetS group on change in amyloid accumulation over time. However, the MetS × PiB group × interval interaction was significant in some regions of early amyloid accumulation, indicating distinct longitudinal relationships between baseline MetS diagnosis and rates of change over time in regional DVR that varied by PiB group (Supplementary Table 2). Covarying for APOE4 status did not change the results.
To determine the patterns of longitudinal associations between baseline MetS and regional DVR rates of change specific to each PiB group, post hoc contrasts were conducted (Table 3 and Fig. 1). In the PiB− group, there was no effect of baseline MetS on rates of change in DVR in any region of early accumulation. In contrast, in the PiB+ group, post hoc contrasts revealed that MetS was associated with accelerated Aβ accumulation in the bilateral superior parietal and left precuneus regions after FDR correction (Fig. 2). The trajectories of amyloid accumulation were similar in several other regions including the right precuneus in the MetS positive group, but these relationships did not survive correction for multiple comparisons (Table 3).
Table 3.
Post hoc contrasts demonstrating the distinct longitudinal effects by PiB group of baseline metabolic syndrome (MetS) diagnosis on distribution volume ratio (DVR) rate of change in regions of early amyloid accumulation
| Baseline MetS × Baseline PiB group × Interval | PiB − group: Baseline MetS × Interval | PiB + group: Baseline MetS × Interval | ||||||
|---|---|---|---|---|---|---|---|---|
| Region | p (FDR-corrected) | Estimate | SE | p | Estimate | SE | p | p (FDR-corrected) |
| L Superior Parietal | 0.001 | 0.000 | 0.001 | 0.807 | 0.013 | 0.003 | <0.001 | <0.001 |
| L Precuneus | 0.002 | −0.002 | 0.002 | 0.350 | 0.015 | 0.004 | <0.001 | 0.002 |
| R Superior Parietal | 0.058 | −0.001 | 0.002 | 0.697 | 0.010 | 0.003 | 0.002 | 0.035 |
| R Precuneus | 0.079 | −0.003 | 0.002 | 0.173 | 0.009 | 0.004 | 0.021 | 0.123 |
| R Middle Frontal | 0.079 | −0.001 | 0.002 | 0.515 | 0.009 | 0.003 | 0.008 | 0.101 |
| R Middle Temporal | 0.112 | −0.001 | 0.002 | 0.544 | 0.009 | 0.004 | 0.014 | 0.123 |
| R Paracentral Lobule | 0.164 | −0.001 | 0.002 | 0.792 | 0.010 | 0.004 | 0.017 | 0.123 |
| R Posterior Cingulate | 0.289 | 0.000 | 0.002 | 0.923 | 0.010 | 0.004 | 0.031 | 0.163 |
Fig. 1.
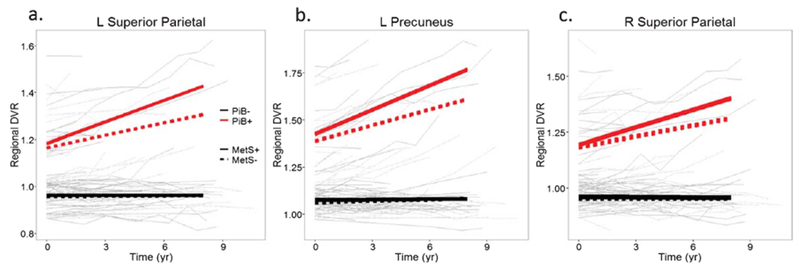
Observed values of PET-PiB regional DVR over time stratified by metabolic syndrome (MetS) diagnosis and PiB group in (a) left superior parietal, (b) left precuneus, and (c) right superior parietal. Within-individual changes are shown by connecting lines. PiB negative individuals are shown in black and PiB positive in red. Dashed lines represent MetS negative individuals. Bold lines depict predicted DVR trajectories by age, MetS group, and PiB group.
Fig. 2.
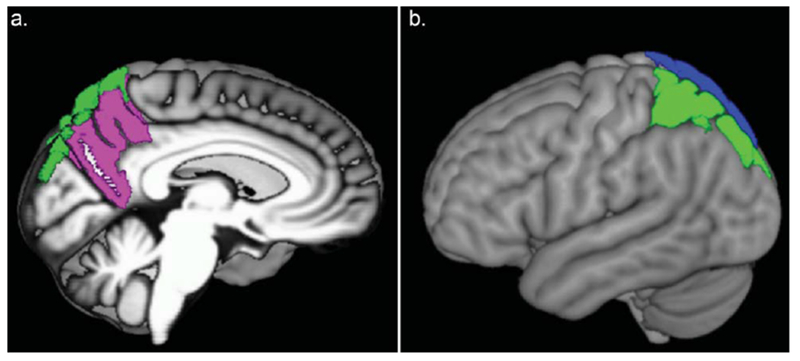
Freesurfer labels of the brain regions in which rate of distribution volume ratio (DVR) change is associated with baseline metabolic syndrome diagnosis in PiB+ individuals. a) Left mid-sagittal view; L precuneus (pink); L superior parietal (green). b) Left lateral view; L superior parietal (green) and R superior parietal (blue).
Metabolic syndrome components and regional DVR rate of change
To test the possibility that individual components of MetS were driving the effects of MetS on rates of Aβ accumulation, we conducted further analyses in the PiB+ group testing each MetS component in separate linear mixed effects models (Table 4). We found that two MetS components, impaired fasting glucose and elevated blood pressure, were individually associated with accelerated amyloid accumulation in the left superior parietal region. Fasting glucose was also associated with accelerated Aβ deposition in the middle frontal region. No associations between waist circumference, triglyceride level, or HDL level and rates of Aβ accumulation were observed. Controlling for diabetes, dyslipidemia, and hypertension treatment did not change the results.
Table 4.
Effects of elevated baseline fasting glucose (FG) and blood pressure (BP) on longitudinal change in regional DVR in regions of early amyloid accumulation
| 4.1. FG linear mixed effects model longitudinal results | ||||
| Baseline FG × Interval |
||||
| Fasting Glucose Regions | Estimate | SE | p | p (FDR-corrected) |
| L Superior Parietal | 0.014 | 0.004 | <0.001 | 0.020 |
| R Middle Frontal | 0.013 | 0.004 | 0.002 | 0.036 |
| 4.2. Rates of change in DVR/year by FG group computed based on linear mixed effects model results | ||||
| Rate of change (DVR/year) |
||||
| Fasting Glucose Regions | Total Sample | FG − | FG + | |
| L Superior Parietal | 0.023 | 0.016 | 0.030 | |
| R Middle Frontal | 0.022 | 0.016 | 0.029 | |
| 4.3. BP linear mixed effects model longitudinal results | ||||
| Baseline BP × Interval |
||||
| Blood Pressure Regions | Estimate | SE | p | p (FDR-corrected) |
| L Superior Parietal | 0.014 | 0.004 | <0.001 | 0.007 |
| 4.4. Rates of change in DVR/year by BP group computed based on linear mixed effects model results | ||||
| Rate of change (DVR/year) |
||||
| Blood Pressure Region | Total Sample | BP − | BP + | |
| L Superior Parietal | 0.024 | 0.017 | 0.031 | |
DISCUSSION
In the present study, we investigated the relationships between MetS diagnosis and amyloid deposition in brain regions known to accumulate amyloid during the preclinical phase of AD. We found that a diagnosis of MetS was not associated with overall PiB positivity, or with group differences in cortical and regional Aβ levels at baseline. However, a MetS diagnosis was associated with greater Aβ accumulation over time in selective regions in nondemented PiB+ individuals, suggesting that MetS may accelerate Aβ accumulation in individuals with preexisting cerebral amyloid. Regarding the individual components of MetS, some but not all showed an individual association with accelerated amyloid accumulation in PiB+ individuals.
MetS defines a pattern of multimorbidity that increases risk for subsequent stroke and cardiovascular disease and may be predictive of both vascular dementia and AD. MetS is also a powerful predictor of cerebrovascular morbidity [37], including infarcts and white matter hyperintensities (WMH), which have been linked to increased risk for incident AD [40] and AD-related pathology [41]. Although we did not examine other cerebrovascular factors in the current study, our findings regarding the relationship between MetS and accumulation of cerebral Aβ, an early marker of the pathophysiological cascade leading to AD, help clarify this epidemiological link between MetS and AD. First, we found no association between MetS diagnosis and PiB positivity, indicating that MetS does not increase significantly the initial likelihood of exhibiting detectable amyloid in the brain. We also found no association between MetS and concurrent regional levels of cerebral Aβ at baseline. This finding suggests that MetS may not be an initiating factor of amyloid accumulation, specifically within brain regions that accumulate Aβ early in the AD trajectory. We did find, however, that PiB+ individuals with MetS demonstrate accelerated accumulation of amyloid over time in bilateral superior parietal and left precuneus regions. This longitudinal finding aligns with cumulative evidence regarding the link between MetS, neurovascular damage, and AD, and is consistent with the idea of a “two or multiple hit” process.
The superior parietal lobe and precuneus are sites not only of early Aβ accumulation in the AD trajectory, but also of hypometabolism and decreased cortical thickness [42]. The latter findings are more proximal to the neurodegeneration and synaptic loss underlying AD and are evident in the preclinical stages of the disease at least 5–10 years before clinical symptom onset [43]. The regional specificity of our findings to the parietal lobe accord with a prior report of an association between WMH and increased amyloid deposition in the parieto-occipital regions [41]. WMH, particularly in the temporoparietal region, have been associated both with MetS and incident AD [40] and may be an indirect marker of impaired cortical drainage pathways, which may hinder amyloid clearance resulting in further pathological Aβ accumulation over time. Portet and colleagues noted that this association was strongest in regions closer to the cortical surface and suggested that these brain areas may be more vulnerable to age-related neurovascular damage than subcortical regions which have other means of lymphatic drainage [41, 44]. Alternatively, abnormalities in amyloid production or clearance may promote demyelination and impaired axonal integrity in WMH [45], further advancing ischemic brain injury and Aβ-mediated neurodegenerative cascades.
PiB+ individuals with WMH have also exhibited greater Aβ accumulation in the parietal and precuneus regions than individuals without vascular pathology [46]. The authors posited that, as these regions are supported by the posterior cerebral artery, they may be more susceptible to blood-brain barrier “disruption” due to vascular injury. This study also observed increased left-right asymmetry in parietal Aβ accumulation, which was likely mediated by WMH topography [46] and which provides support for our left-lateralized results. This evidence, consistent with the regional specificity of our findings, suggests that MetS may be associated with accelerated Aβ deposition via a neurodegenerative or neurovascular pathway.
Of the five conditions comprising MetS, we found elevated fasting glucose and hypertension to be predictive of accelerated Aβ accumulation. Specifically, our results revealed an association between elevated fasting glucose and accelerated amyloid deposition in the left superior parietal and right middle frontal gyri. Prior studies of impaired glycemia and cerebral amyloid are mixed. One study of nondemented older adults found a relationship between impaired fasting glucose and higher concurrent cerebral amyloid burden in several default mode network regions, including the superior parietal cortex [47]; however, other cross-sectional studies using PET-PiB have not found such relationships [16]. An autopsy study linked hyperinsulemia and hyperglycemia with neuritic plaques, suggesting that insulin resistance may underlie this association [48]. Studies of insulin resistance and PET amyloid imaging, however, also show conflicting results. Some studies demonstrate a relationship between insulin resistance and amyloid levels [49], whereas others do not [50]. In previous studies from our laboratory, we found no significant association between repeated measures of glucose intolerance, diabetes, or insulin resistance and either postmortem Aβ or in vivo PET-PiB [51], yet regions associated with amyloid and tau pathology show significantly higher tissue glucose concentrations in AD [52]. Thus, the exact relationship between these factors and amyloid accumulation demonstrated by PET imaging deserves further study.
Nevertheless, it is well established that hyperglycemia creates a toxic environment leading to metabolic and vascular injury in the brain [53]. Recent work in mouse models also shows that hyperglycemia leads to increased Aβ production in young animals, and these effects are enhanced in older mice with preexisting Aβ plaque pathology [54]. Our findings regarding accelerated Aβ deposition may be a result not only of hyperglycemia, however, but also the neuromodulatory changes produced by insulin resistance and mediated by compensatory hyperinsulemia [48]. These diabetes-related factors have been shown to play a role in regulating Aβ degradation and amyloid precursor protein [55] and may act together to accelerate preclinical AD pathogenesis.
We also found an association between hypertension and increased rate of amyloid accumulation, specifically in the left superior parietal region. Hypertension has been linked to not only clinical AD [56] but pathological manifestations of AD as well. Hypertension is a known contributor to WMH and arterial stiffness, which have been associated with CSF markers of Aβ [57] and increased cerebral Aβ in nondemented older adults [58], respectively. A study using PET-PiB imaging found that both systolic blood pressure and pulse pressure were correlated with regional DVR, including parietal and precuneus regions [17]. These earlier findings suggest similar regional involvement to our findings, though we did not find significant associations with baseline PET-PiB measures. Vascular defects such as arterial stiffness due to chronic hypertension may degrade microvascular integrity of the brain, impairing perivascular drainage pathways, and thus impeding Aβ clearance and promoting pathological Aβ accumulation [59].
Together, our findings could lend support to several additional hypotheses which point to Aβ metabolism as a possible mechanism underlying the association between metabolic risk factor burden and AD [18, 19, 60]. The pathological accumulation of Aβ, characteristic of AD, may be induced by disruption of several factors, including Aβ production, transport of Aβ across the blood-brain barrier, Aβ oligomerization and Aβ degradation [18]. Though studies often do not find a correlation between cerebral infarcts and amyloid specifically, it is possible that such vascular injury may have an initiating effect among individuals predisposed to developing amyloid. Additionally, neurovascular injury may heighten permeability of the blood-brain barrier, leading to impaired Aβ clearance [61]. Another explanation might involve insulin resistance, characteristic of diabetes, obesity, and MetS; impaired insulin and insulin-like growth factor signaling leads to increased expression of Aβ protein precursor and thus Aβ [60]. In addition, these processes promote oxidative stress, release of inflammatory cytokines, and neuronal dysfunction, further activating Aβ-mediated neurodegenerative cascades [62].
Our study has several limitations. As BLSA participants are regularly screened for health conditions, our sample receives an unusual degree of medical evaluations typically resulting in pharmacologic treatments. Evidence from FDG-PET studies suggests that, even with treatment, individuals with vascular risk factors such as hypertension may exhibit deleterious age-related brain changes [63, 64]. Our results are consistent with these findings, in that adjusting for disease treatment did not change the association between MetS and regional acceleration of Aβ accumulation. As the AHA/NHLBI revised NCEP/ATPIII diagnosis criteria for MetS incorporate treatment for hyperglycemia, dyslipidemia, and hypertension, it is difficult to distinguish between the effect of disease state and the effect of disease treatment on our results. Additionally, we focused on concurrent measures of MetS in an older sample and midlife measures may have revealed different findings. Finally, our sample size was relatively small, and we restricted our analyses to cognitively normal participants. Cognitively healthy PiB positive individuals are heterogeneous, with some individuals likely to develop future cognitive impairment and as many as 30 to 50% remaining cognitively healthy throughout their lifespan [65].
In summary, our results provide novel evidence of the association between MetS and longitudinal change in cerebral Aβ accumulation, an early marker of the AD pathophysiological cascade, in several regions known for early Aβ accumulation. These findings suggest that the pattern of vascular burden captured by MetS diagnosis may accelerate Aβ accumulation in individuals with preexisting cerebral amyloid levels. Two components of MetS diagnosis—hyperglycemia and hypertension—partially accounted for the pattern of association, suggesting that the effect of MetS extends beyond the effects of its individual components. Although the individuals in this study are cognitively normal at present, amyloid positivity in cognitively normal individuals is associated with heterogeneous cognitive outcomes [66, 67]. On the one hand, there are some amyloid positive individuals who demonstrate resilience in the face of the pathological burden and survive to old age without cognitive impairment [30]. On the other hand, there is a group of individuals whose amyloid positivity will be associated with cognitive decline and impairment. A major advance provided by in vivo amyloid PET is that it is now possible to follow the development of amyloid prospectively in longitudinal studies to provide critical information on factors that predict future cognitive outcomes.
Early identification and modulation of MetS targeting health and lifestyle factors, particularly among individuals with cerebral amyloid burden, may be critical to developing preventative strategies to delay progression of the neuropathology underlying AD. Ongoing prospective studies integrating a variety of imaging and CSF markers will further define the temporal sequence of amyloid and tau deposition in relation to the neurodegenerative processes underlying cognitive impairment. Such studies will also allow the development of intervention strategies to determine whether modifiable risk factors for MetS change the pathophysiologic cascade underlying AD and related dementias.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to the BLSA participants and staff for their dedication to these studies and the staff of the Johns Hopkins PET facility, the Wong lab fellows, and staff at Johns Hopkins University and Hospital for their assistance. This research was supported by the Intramural Research Program of the NIH, National Institute on Aging.
Footnotes
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/18-0297r1).
SUPPLEMENTARY MATERIAL
The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/JAD-180297.
REFERENCES
- [1].Alzheimer’s Association (2016) Alzheimer’s disease facts and figures. Alzheimers Dement 12, 459–509. [DOI] [PubMed] [Google Scholar]
- [2].Panza F, Frisardi V, Capurso C, Imbimbo BP, Vendemiale G, Santamato A, D’Onofrio G, Seripa D, Sancarlo D, Pilotto A, Solfrizzi V (2010) Metabolic syndrome and cognitive impairment: Current epidemiology and possible underlying mechanisms. J Alzheimers Dis 21, 691–724. [DOI] [PubMed] [Google Scholar]
- [3].Gamaldo A, Moghekar A, Kilada S, Resnick SM, Zonderman AB, O’Brien R (2006) Effect of a clinical stroke on the risk of dementia in a prospective cohort. Neurology 67, 1363–1369. [DOI] [PubMed] [Google Scholar]
- [4].Schneider JA, Wilson RS, Bienias JL, Evans DA, Bennett DA (2004) Cerebral infarctions and the likelihood of dementia from Alzheimer disease pathology. Neurology 62, 1148–1155. [DOI] [PubMed] [Google Scholar]
- [5].Troncoso JC, Zonderman AB, Resnick SM, Crain B, Pletnikova O, O’Brien RJ (2008) Effect of infarcts on dementia in the Baltimore Longitudinal Study of Aging. Ann Neurol 64, 168–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Exalto LG, van der Flier WM, van Boheemen CJ, Kappelle LJ, Vrenken H, Teunissen C, Koene T, Scheltens P, Biessels GJ (2015) The metabolic syndrome in a memory clinic population: Relation with clinical profile and prognosis. J Neurol Sci 351, 18–23. [DOI] [PubMed] [Google Scholar]
- [7].Raffaitin C, Gin H, Empana J- P, Helmer C, Berr C, Tzourio C, Portet F, Dartigues J- F, Alpérovitch A, Barberger-Gateau P (2009) Metabolic syndrome and risk for incident Alzheimer’s disease or vascular dementia: The Three-City Study. Diabetes Care 32, 169–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Vanhanen M, Koivisto K, Moilanen L, Helkala EL, Hänninen T, Soininen H, Kervinen K, Kesäniemi YA, Laakso M, Kuusisto J (2006) Association of metabolic syndrome with Alzheimer disease: A population-based study. Neurology 67, 843–847. [DOI] [PubMed] [Google Scholar]
- [9].Yaffe K (2007) Metabolic syndrome and cognitive disorders: Is the sum greater than its parts? Alzheimer Dis Assoc Disord 21, 167–171. [DOI] [PubMed] [Google Scholar]
- [10].Hof P, Giannakopoulos P, Bouras C (1996) The neuropathological changes associated with normal brain aging. Histol Histopathol 11, 1075–1088. [PubMed] [Google Scholar]
- [11].Perl D (2010) Neuropathology of Alzheimer’s disease. Mt Sinai J Med 77, 32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Glodzik L, Rusinek H, Kamer A, Pirraglia E, Tsui W, Mosconi L, Li Y, McHugh P, Murray J, Williams S, Osorio RS, Randall C, Butler T, Deshpande A, Vallabhajolusa S, de Leon M (2016) Effects of vascular risk factors, statins, and antihypertensive drugs on PiB deposition in cognitively normal subjects. Alzheimers Dement (Amst) 2, 95–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Yang Y, Song W (2013) Molecular links between Alzheimer’s disease and diabetes mellitus. Neuroscience 250, 140–150. [DOI] [PubMed] [Google Scholar]
- [14].Reed B, Villeneuve S, Mack W, DeCarli C, Chui HC, Jagust W (2014) Associations between serum cholesterol levels and cerebral amyloidosis. JAMA Neurol 71, 195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hughes TM, Lopez OL, Evans RW, Kamboh MI, Williamson JD, Klunk WE, Mathis CA, Price JC, Cohen AD, Snitz BE, Dekosky ST, Kuller LH (2014) Markers of cholesterol transport are associated with amyloid deposition in the brain. Neurobiol Aging 35, 802–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Toledo JB, Toledo E, Weiner MW, Jack CR Jr, Jagust W, Lee VM, Shaw LM, Trojanowski JQ, Alzheimer’s Disease Neuroimaging Initiative (2012) Cardiovascular risk factors, cortisol, and amyloid-beta deposition in Alzheimer’s Disease Neuroimaging Initiative. Alzheimers Dement 8, 483–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Langbaum JB, Chen K, Launer LJ, Fleisher AS, Lee W, Liu X, Protas HD, Reeder SA, Bandy D, Yu M, Caselli RJ, Reiman EM (2012) Blood pressure is associated with higher brain amyloid burden and lower glucose metabolism in healthy late middle-age persons. Neurobiol Aging 33, 827 e811–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Campos-Pena V, Toral-Rios D, Becerril-Perez F, Sanchez-Torres C, Delgado-Namorado Y, Torres-Ossorio E, Franco-Bocanegra D, Carvajal K (2017) Metabolic syndrome as a risk factor for Alzheimer’s disease: Is Abeta a crucial factor in both pathologies? Antioxid Redox Signal 26, 542–560. [DOI] [PubMed] [Google Scholar]
- [19].Frisardi V, Solfrizzi V, Seripa D, Capurso C, Santamato A, Sancarlo D, Vendemiale G, Pilotto A, Panza F (2010) Metabolic-cognitive syndrome: A cross-talk between metabolic syndrome and Alzheimer’s disease. Ageing Res Rev 9, 399–417. [DOI] [PubMed] [Google Scholar]
- [20].Resnick SM, Goldszal AF, Davatzikos C, Golski S, Kraut MA, Metter EJ, Bryan RN, Zonderman AB(2000) One-year age changes in MRI brain volumes in older adults. Cereb Cortex 10, 464–472. [DOI] [PubMed] [Google Scholar]
- [21].Shock NW, Greulich RC, Costa PT Jr, Andres R, Lakatta EG, Arenberg D, Tobin JD (1984) Normal Human Aging: The Baltimore Longitudinal Study on Aging. NIH, US Government Printing Office, Washington, D.C. [Google Scholar]
- [22].McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM (1984) Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 34, 939–944. [DOI] [PubMed] [Google Scholar]
- [23].Delis DC, Kramer JH, Kaplan E, Ober BA(1987) California Verbal Learning Test: Research edition. The Psychological Corporation, New York. [Google Scholar]
- [24].Benton AL (1968) Differential behavioral effects in frontal lobe disease. Neuropsychologia 6, 53–60. [Google Scholar]
- [25].DeFries JC, Vandenberg SG, McClearn GE, Kuse AR, Wilson JR, Ashton GC, Johnson RC (1974) Near identity of cognitive structure in two ethnic groups. Science 183, 338–339. [DOI] [PubMed] [Google Scholar]
- [26].Wechsler D (1981) Wechsler Adult Intelligence Scale–Revised. Psychological Corporation, New York. [Google Scholar]
- [27].Reitan R (1982) Trail Making Test: Manual for Administration and Scoring. Reitan Neuropsychological Laboratory, Tuscon. [Google Scholar]
- [28].Newcombe F (1969) Missile Wounds of the Brain: A Study of Psychological Deficits. Oxford University Press, London. [Google Scholar]
- [29].Wilson JR, De Fries JC, McClearn GE, Vanderberg SG, Johnson RC, Rashad MN (1975) Cognitive abilities: Use of family data as a control to assess sex and age differences in two ethnic groups. Int J Aging Hum Dev 6, 261–276. [DOI] [PubMed] [Google Scholar]
- [30].Driscoll I, Resnick SM, Troncoso JC, An Y, O’Brien R, Zonderman AB (2006) Impact of Alzheimer’s pathology on cognitive trajectories in nondemented elderly. Ann Neurol 60, 688–695. [DOI] [PubMed] [Google Scholar]
- [31].Petersen RC (2004) Mild cognitive impairment as a diagnostic entity. J Intern Med 256, 183–194. [DOI] [PubMed] [Google Scholar]
- [32].Zhou Y, Resnick SM, Ye W, Fan H, Holt DP, Klunk WE, Mathis CA, Dannals R, Wong DF (2007) Using a reference tissue model with spatial constraint to quantify [(11)C]Pittsburgh compound B PET for early diagnosis of Alzheimer’s disease. Neuroimage 36, 298–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Avants BB, Epstein CL, Grossman M, Gee JC (2008) Symmetric diffeomorphic image registration with cross-correlation: Evaluating automated labeling of elderly and neurodegenerative brain. Med Image Anal 12, 26–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Desikan RS, Segonne F, Fischl B, Quinn BT, Dickerson BC, Blacker D, Buckner RL, Dale AM, Maguire RP, Hyman BT, Albert MS, Killiany RJ (2006) An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage 31, 968–980. [DOI] [PubMed] [Google Scholar]
- [35].Benaglia T, Chauveau D, Hunter D, Young D (2009) mixtools: An R package for analyzing finite mixture models. J Stat Softw 32, 1–29. [Google Scholar]
- [36].Bilgel M, An Y, Zhou Y, Wong DF, Prince JL, Ferrucci L, Resnick SM (2016) Individual estimates of age at detectable amyloid onset for risk factor assessment. Alzheimers Dement 12, 373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Eckel RH, Grundy SM, Zimmet PZ (2005) The metabolic syndrome. Lancet 365, 1415–1428. [DOI] [PubMed] [Google Scholar]
- [38].Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol 57, 289–300. [Google Scholar]
- [39].Sojkova J, Goh J, Bilgel M, Landman B, Yang X, Zhou Y, An Y, Beason-Held LL, Kraut MA, Wong DF, Resnick SM (2015)Voxelwise relationships between distribution volume ratio and cerebral blood flow: Implications for analysis of beta-amyloid images. J Nucl Med 56, 1042–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Portet F, Brickman AM, Stern Y, Scarmeas N, Muraskin J, Provenzano FA, Berr C, Bonafe A, Artero S, Ritchie K, Akbaraly TN (2012) Metabolic syndrome and localization of white matter hyperintensities in the elderly population. Alzheimers Dement 8, S88–95. e81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Grimmer T, Faust M, Auer F, Alexopoulos P, Forstl H, Henriksen G, Perneczky R, Sorg C, Yousefi BH, Drzezga A, Kurz A (2012) White matter hyperintensities predict amyloid increase in Alzheimer’s disease. Neurobiol Aging 33, 2766–2773. [DOI] [PubMed] [Google Scholar]
- [42].Benzinger TL, Blazey T, Jack CR Jr, Koeppe RA, Su Y, Xiong C, Raichle ME, Snyder AZ, Ances BM, Bateman RJ, Cairns NJ, Fagan AM, Goate A, Marcus DS, Aisen PS, Christensen JJ, Ercole L, Hornbeck RC, Farrar AM, Aldea P, Jasielec MS, Owen CJ, Xie X, Mayeux R, Brickman A, McDade E, Klunk W, Mathis CA, Ringman J, Thompson PM, Ghetti B, Saykin AJ, Sperling RA, Johnson KA, Salloway S, Correia S, Schofield PR, Masters CL, Rowe C, Villemagne VL, Martins R, Ourselin S, Rossor MN, Fox NC, Cash DM, Weiner MW, Holtzman DM, Buckles VD, Moulder K, Morris JC (2013) Regional variability of imaging biomarkers in autosomal dominant Alzheimer’s disease. Proc Natl Acad Sci U S A 110, E4502–4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Jack CR Jr, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, Petersen RC, Trojanowski JQ (2010) Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol 9, 119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Szentistvanyi I, Patlak CS, Ellis RA, Cserr HF (1984) Drainage of interstitial fluid from different regions of rat brain. Am J Physiol 246, F835–844. [DOI] [PubMed] [Google Scholar]
- [45].Kalheim LF, Bjørnerud A, Fladby T, Vegge K, Selnes P (2016) White matter hyperintensity microstructure in amyloid dysmetabolism. J Cereb Blood Flow Metab 37, 356–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Noh Y, Seo SW, Jeon S, Lee JM, Kim JS, Lee JH, Kim JH, Kim GH, Ye BS, Cho H, Kim HJ, Yoon CW, Choe YS, Lee KH, Weiner MW, Na DL (2016) The role of cerebrovascular disease in amyloid deposition. J Alzheimers Dis 54, 1015–1026. [DOI] [PubMed] [Google Scholar]
- [47].Morris JK, Vidoni ED, Wilkins HM, Archer AE, Burns NC, Karcher RT, Graves RS, Swerdlow RH, Thyfault JP, Burns JM(2016) Impaired fasting glucose is associated with increased regional cerebral amyloid. Neurobiol Aging 44, 138–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Matsuzaki T, Sasaki K, Tanizaki Y, Hata J, Fujimi K, Matsui Y, Sekita A, Suzuki SO, Kanba S, Kiyohara Y, Iwaki T (2010) Insulin resistance is associated with the pathology of Alzheimer disease: The Hisayama Study. Neurology 75, 764–770. [DOI] [PubMed] [Google Scholar]
- [49].Willette AA, Johnson SC, Birdsill AC, Sager MA, Christian B, Baker LD, Craft S, Oh J, Statz E, Hermann BP, Jonaitis EM, Koscik RL, La Rue A, Asthana S, Bendlin BB (2015) Insulin resistance predicts brain amyloid deposition in late middle-aged adults. Alzheimers Dement 11, 504–510 e501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Laws SM, Gaskin S, Woodfield A, Srikanth V, Bruce D, Fraser PE, Porter T, Newsholme P, Wijesekara N, Burnham S, Dore V, Li QX, Maruff P, Masters CL, Rainey-Smith S, Rowe CC, Salvado O, Villemagne VL, Martins RN, Verdile G (2017) Insulin resistance is associated with reductions in specific cognitive domains and increases in CSF tau in cognitively normal adults. Sci Rep 7, 9766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Thambisetty M, JeffreyMetter E, Yang A, Dolan H, Marano C, Zonderman AB, Troncoso JC, Zhou Y, Wong DF, Ferrucci L, Egan J, Resnick SM, O’Brien RJ (2013) Glucose intolerance, insulin resistance, and pathological features of Alzheimer disease in the Baltimore Longitudinal Study of Aging. JAMA Neurol 70, 1167–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].An Y, Varma VR, Varma S, Casanova R, Dammer E, Pletnikova O, Chia CW, Egan JM, Ferrucci L, Troncoso J, Levey AI, Lah J, Seyfried NT, Legido-Quigley C, O’Brien R, Thambisetty M (2018) Evidence for brain glucose dysregulation in Alzheimer’s disease. Alzheimers Dement 14, 318–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Gispen WH, Biessels G- J (2000) Cognition and synaptic plasticity in diabetes mellitus. Trends Neurosci 23, 542–549. [DOI] [PubMed] [Google Scholar]
- [54].Macauley SL, Stanley M, Caesar EE, Yamada SA, Raichle ME, Perez R, Mahan TE, Sutphen CL, Holtzman DM (2015) Hyperglycemia modulates extracellular amyloid-beta concentrations and neuronal activity in vivo. J Clin Invest 125, 2463–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Farris W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch MP, Eckman CB, Tanzi RE, Selkoe DJ, Guenette S (2003) Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci U S A 100, 4162–4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Qiu C, Winblad B, Viitanen M, Fratiglioni L (2003) Pulse pressure and risk of Alzheimer disease in persons aged 75 years and older: A community-based, longitudinal study. Stroke 34, 594–599. [DOI] [PubMed] [Google Scholar]
- [57].Kester MI, Goos JD, Teunissen CE, Benedictus MR, Bouwman FH, Wattjes MP, Barkhof F, Scheltens P, van der Flier WM (2014) Associations between cerebral small-vessel disease and Alzheimer disease pathology as measured by cerebrospinal fluid biomarkers. JAMA Neurol 71, 855–862. [DOI] [PubMed] [Google Scholar]
- [58].Hughes TM, Kuller LH, Barinas-Mitchell EJ, McDade EM, Klunk WE, Cohen AD, Mathis CA, Dekosky ST, Price JC, Lopez OL (2014) Arterial stiffness and beta-amyloid progression in nondemented elderly adults. JAMA Neurol 71, 562–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Weller RO, Subash M, Preston SD, Mazanti I, Carare RO (2008) Perivascular drainage of amyloid-beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer’s disease. Brain Pathol 18, 253–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].de la Monte SM (2012) Contributions of brin insulin resistance and deficiency in amyloid-related neurodegeneration in Alzheimer’s disease. Drugs 72, 49–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Bell RD, Zlokovic BV (2009) Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer’s disease. Acta Neuropathol 118, 103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Altman R, Rutledge JC (2010) The vascular contribution to Alzheimer’s disease. Clin Sci (Lond) 119, 407–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Beason-Held LL, Moghekar A, Zonderman AB, Kraut MA, Resnick SM (2007) Longitudinal changes in cerebral blood flow in the older hypertensive brain. Stroke 38, 1766–1773. [DOI] [PubMed] [Google Scholar]
- [64].Salerno JA, Grady C, Mentis M, Gonzalez-Aviles A, Wagner E, Schapiro MB, Rapoport SI (1995) Brain metabolic function in older men with chronic essential hypertension. J Gerontol A Biol Sci Med Sci 50, M147–154. [DOI] [PubMed] [Google Scholar]
- [65].Sojkova J, Resnick SM(2011) In vivo human amyloid imaging. Curr Alzheimer Res 8, 366–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Resnick SM, Sojkova J (2011) Amyloid imaging and memory change for prediction of cognitive impairment. Alzheimers Res Ther 3, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Resnick SM, Sojkova J, Zhou Y, An Y, Ye W, Holt DP, Dannals RF, Mathis CA, Klunk WE, Ferrucci L, Kraut MA, Wong DF (2010) Longitudinal cognitive decline is associated with fibrillar amyloid-beta measured by [11C]PiB. Neurology 74, 807–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.