

Abstract
Intercellular communication is critical for integrating complex signals in multicellular eukaryotes. Vascular endothelial cells and T lymphocytes closely interact during the recirculation and trans-endothelial migration of T cells. In addition to direct cell–cell contact, we show that T cell derived extracellular vesicles can interact with endothelial cells and modulate their cellular functions. Thrombospondin-1 and its receptor CD47 are expressed on exosomes/ectosomes derived from T cells, and these extracellular vesicles are internalized and modulate signaling in both T cells and endothelial cells. Extracellular vesicles released from cells expressing or lacking CD47 differentially regulate activation of T cells induced by engaging the T cell receptor. Similarly, T cell-derived extracellular vesicles modulate endothelial cell responses to vascular endothelial growth factor and tube formation in a CD47-dependent manner. Uptake of T cell derived extracellular vesicles by recipient endothelial cells globally alters gene expression in a CD47-dependent manner. CD47 also regulates the mRNA content of extracellular vesicles in a manner consistent with some of the resulting alterations in target endothelial cell gene expression. Therefore, the thrombospondin-1 receptor CD47 directly or indirectly regulates intercellular communication mediated by the transfer of extracellular vesicles between vascular cells.
Keywords: Thrombospondin-1, CD47, Extracellular vesicles, Intercellular communication
1. Introduction
Intercellular communication is an essential process for the development and function of multicellular organisms. Cell to cell communications can be mediated via cell surface receptors that recognize secreted soluble ligands or directly via formation of gap junctions and tunneling micro-nanotubules that allow coupling and transport of metabolites among cells (Abounit and Zurzolo, 2012; Grimmelikhuijzen and Hauser, 2012). Recent studies have identified an additional mechanism of intercellular communication mediated by release of extracellular vesicles (EVs) from cells. Depending on the mechanism of release, these vesicles are referred to as microparticles, ectosomes (right-side-out), exosomes (right-side or inside-out), or shedding vesicles (Holme et al., 1994; Hess et al., 1999; Gasser et al., 2003; Cocucci and Meldolesi, 2011). Electron micrographs often show a cup shaped morphology and sizes between 40 and 120 nm. The presence of EVs has been reported in simple eukaryotes such as Dictyostelium discoideum (Lavialle et al., 2009) and Trypanosoma cruzi (Bayer-Santos et al., 2013), plants (Regente et al., 2012), invertebrates such as Drosophila melanogaster (Korkut et al., 2013) and Caenorhabditis elegans (Liegeois et al., 2006), and higher vertebrates. The broad presence of EVs in eukaryotes suggests that intercellular communication mediated by these vesicles must be of fundamental importance and evolutionary conserved.
Although the release of apoptotic bodies from cells undergoing programmed cell death and EVs from healthy live cells has been known for long time, their perceived role was long limited to the removal of unwanted or damaged cell contents. Exosomes have been isolated in vitro from cultured cells and in vivo from body fluids including saliva (Ogawa et al., 2011), urine (Pisitkun et al., 2004), seminal fluid (Stridsberg et al., 1996), blood (Caby et al., 2005), breast milk (Admyre et al., 2007), plasma, amniotic fluid (Asea et al., 2008), malignant ascites (Andre et al., 2002), cerebrospinal fluid (Street et al., 2012), bile (Masyuk et al., 2010), and synovial fluid (Skriner et al., 2006).
The protein content of exosomes has been analyzed by flow cytometry and proteomic methods. Highly purified exosomes are devoid of serum proteins and most protein components of intracellular compartments. As a consequence of their origin from the plasma membrane, exosomes express proteins that mediate antigen presentation (MHC-I, MHC-II), cell adhesion (integrins), cell structure and motility (actins, tubulin, myosin, etc.), stress regulators (e.g. heat shock proteins 70 and 90), metabolic enzymes (β-enolase, peroxidases, pyruvate kinase) proteins of the ESCRT machinery, signaling cascade proteins (kinases), tetraspanins (CD9, CD63, CD81, CD82), proteins involved in transcription and protein synthesis (histones, ribosomal proteins, ubiquitin), and proteins involved in trafficking and membrane fusion (Rabs, annexins) (Lakkaraju and Rodriguez-Boulan, 2008). In addition to proteins, exosomes contain a specific subset of mRNAs and microRNAs that can regulate gene expression in recipient cells (Mittelbrunn et al., 2011; Vickers et al., 2011; Boon and Vickers, 2013). Exosomes are also enriched in elements of lipid rafts including cholesterol, sphingomyelin and ceramide. Inverted vesicles also exhibit phosphatidylserine (Thery et al., 2009). Bioactive lipids such as prostaglandins are also sorted into exosomes (Subra et al., 2010).
The interaction of exosomes with target cells can follow two alternative routes: endocytosis of the whole vesicles or fusion with the plasma membrane. Surface molecules such as integrins, tetraspanins and phosphatidylserine in exosomes can form complexes with cell surface molecules and participate in the attachment of exosomes, as studied in dendritic cells (Ostrowski et al., 2010). In these cells, the whole exosome is internalized and sorted into recycling endosomes and then through late endosomes/lysosomes (Morelli et al., 2004). Studies to date have identified roles of exosomes in essential processes such as development, angiogenesis, and inflammation in cancer and tumor metastasis (Peinado et al., 2012), and in the transmission of infectious agents including prions and viruses (Fevrier et al., 2004; Lenassi et al., 2010).
CD47 is a cell surface receptor that interacts laterally with VEGFR2 and integrins in endothelial cells (Brown and Frazier, 2001; Kaur et al., 2010). CD47 can be cleaved via proteolysis from the surface of endothelial and smooth muscle cells and is important for SHP2-dependent insulin growth factor signaling (Maile et al., 2009). CD47 shed into conditioned medium by endothelial cells, smooth muscle cells, and T cells has heparan sulfate modification (Kaur et al., 2011). CD47 was identi-fied as a protein on ectosomes released from platelets (Sadallah et al., 2011). Proteomic analysis of exosomes from mesenchymal stem cells also showed the presence of CD47 (Kim et al., 2012). Although the presence of CD47 on EVs released from several cell types is clear, its function on EVs is unknown.
Intercellular communication between endothelial cells and circulating T cells is critical for T cell homing and immune surveillance (Knolle, 2006). Based on the important role CD47 plays as a signaling receptor for the matricellular protein thrombospondin-1 in both T cells and endothelial cells, we have examined the functional role of CD47 in the activities of EVs released by T cells. Here we report that CD47 is constitutively present on EVs produced by Jurkat T cells. We further demonstrate that T cell-derived EVs can alter endothelial gene expression and physiological processes such as cell proliferation and tube formation in CD47-dependent manner.
2. Results
2.1. Jurkat T cell conditioned medium modulates T cell activation in a CD47-dependent manner
Our previous studies have demonstrated that CD47 is released into conditioned medium by Jurkat T cells (Kaur et al., 2011). This could be soluble CD47 produced by proteolytic cleavage of its IgV domain (Maile et al., 2009) or intact CD47 associated with EVs. In order to identify a potential functional role of this released CD47 in T cell activation, we transferred conditioned media (CM) between wild type Jurkat and CD47-deficient JinB8 T cell cultures. We plated the treated cells onto anti-CD3 coated plates to induce activation and compared TCR signaling in the presence or absence of the CD47 ligand TSP1. To assess early TCR activation we quantified mRNA of the T cell activation marker CD69. As we reported previously, anti-CD3 induced CD69 mRNA expression at 6 h, and TSP1 significantly inhibited this induction in Jurkat T cells. In contrast, anti-CD3 induction of CD69 mRNA in the CD47-deficient JinB8 clone was not inhibited by TSP1, rather as reported previously, TSP1 had a positive effect on CD69 mRNA induction by anti-CD3 compared to anti-CD3 alone (Fig. 1A, C). Remarkably, transfer of conditioned medium from JinB8 onto Jurkat cells resulted in a profile characteristic of the CD47-deficient cells. TSP1 did not inhibit but instead enhanced induction of CD69 mRNA expression by anti-CD3 (Fig. 1B). Conversely, transfer of Jurkat conditioned medium onto JinB8 cells increased CD69 mRNA induction by anti-CD3 and restored sensitivity to inhibition by TSP1 (Fig. 1D).
Fig. 1.
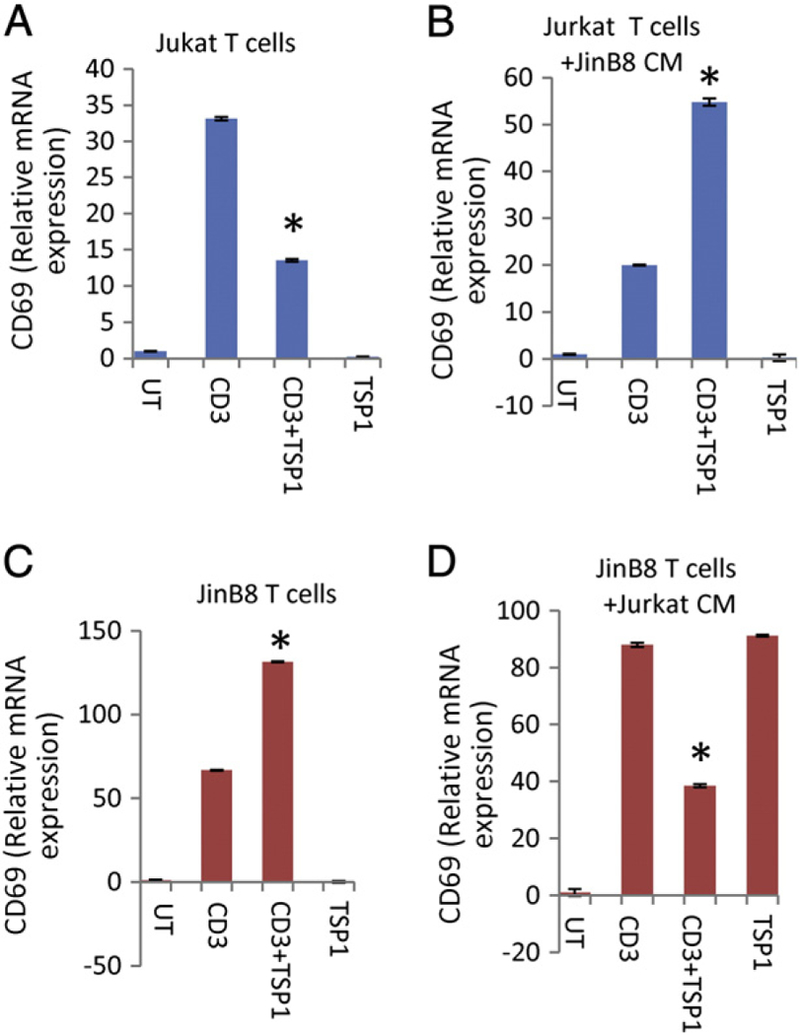
Effects of TSP1 on activation of WT and CD47-deficient T cells are reversed by exposure to heterologous conditioned media. (A and C) Jurkat and CD47-deficient (JinB8) T cells were plated on anti-CD3 coated wells in the presence or absence of TSP1 (1 μg/ml, B and D) in the presence of the indicated cell conditioned media for 6 h. The relative expression of CD69 mRNA was measured using real-time PCR using B2M (A) and HPRT1 (B, C, D) as internal controls.
2.2. Jurkat T cell EV properties and re-uptake by WT and CD47-deficient Jurkat T cells
Because CD47 has been shown to be present on platelet ectosomes (Sadallah et al., 2011) and as a proteolytically cleaved fragment in conditioned media (Maile et al., 2009), we considered whether the transfer of CD47 to the CD47-deficient cells restores their sensitivity to modulation of TCR signaling by TSP1. Alternatively, this effect of CD47 could be indirect, and indirect effects of CD47 are obviously necessary to account for the ability of conditioned medium from CD47-deficient cells to confer resistance of WT Jurkat cells to TSP1 inhibition. To assess the properties and function of EVs released by Jurkat and JinB8 T cells, EVs were isolated from Jurkat and JinB8 cell conditioned medium using a commercial exosome isolation kit. We refer to these preparations as EVs because they may contain both exosomes and ectosomes. EVs exhibited size and morphology characteristic of exosomes when examined using transmission electron microscopy (Fig. 2A, B). Exosomes typically have electron-dense cores due to their high content of miRNAs and other macromolecules. No significant morphological differences were seen between EVs from WT and CD47-deficient cells, but the yield of EVs was consistently higher from JinB8 cells based on the packed volume and confirmed by flow analysis (4400 versus 10,500/10 μl from WT and CD47-deficient cells, respectively).
Fig. 2.
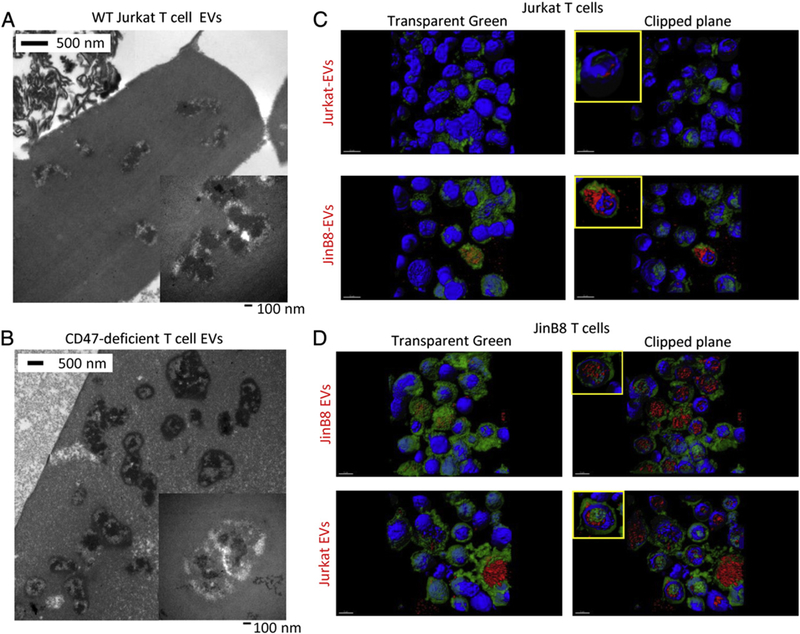
EV morphology and T cell uptake of T cell derived EVs. (A, B) Electron micrographs of EVs derived from Jurkat and JinB8 T cells. Insets at higher magnification with 100 nm scale bars.(C) Uptake of Jurkat and JinB8 EVs (red) by Jurkat cells (green). (D). Uptake of Jurkat and JInB8 EVs by JinB8 T cells.
A recent study reported that T cells do not efficiently take up their own exosomes (Mittelbrunn et al., 2011), but T cell exosomes were more efficiently taken up by antigen presenting cells. Using red fluorescently labeled EVs, we confirmed that Jurkat cell EVs are not efficiently taken up by green labeled WT Jurkat T cells after 6 h incubation (Fig. 2C upper panels). Notably, CD47-deficient Jurkat cell EVs showed better uptake into WT Jurkat cells (Fig. 2C lower panels). However, EVs from both WT and CD47-deficient Jurkat cells were efficiently taken up into CD47-deficient JinB8 cells (Fig. 2D). Treatment of Jurkat and JinB8 cells with TSP1 did not alter uptake of EVs as assessed by confocal analysis (data not shown). Optical sectioning confirmed that the EVs were internalized by the recipient cells (Fig. 2C, D clipped plane images). Analogous to the known role of CD47 to inhibit phagocytic cell uptake (reviewed in (Murata et al., 2014)), these data suggest that CD47 expression on EVs and recipient cells limits their uptake.
To ensure that EVs isolated from conditioned medium were not contaminated with CD47 or other proteins derived from serum EVs, we analyzed EVs that were purified from FCS, human AB serum, and commercial exosome-depleted serum (exo-FBS) using the Exo-quick kit. Although exo-FBS is depleted of CD63- and CD9-positive exosomes, CD47 expression was readily detectable by flow cytometry on EVs isolated from exo-FBS (Fig. 3A). Because the CD47 antibody used recognizes the extracellular domain of CD47 the positive EVs are right-side out. CD47 expression was also observed in EVs purified from FCS but not in EVs purified from human AB serum (Fig. 3B, C). The presence of proteoglycan and lower molecular mass isoforms of CD47 on EVs isolated from Exo-FBS and FCS was further confirmed using western blot analysis (Fig. 3D). CD47 immunoprecipitated from Jurkat T cells was used as a positive control. These data suggest that CD47 is present in a different population of EVs than exosomes containing CD63.
Fig. 3.
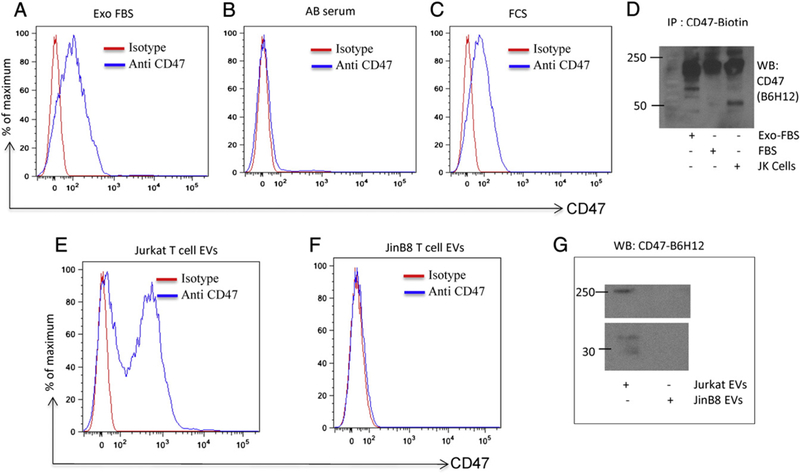
CD47 is present on serum and cell-derived EVs. (A, B and C) CD47-FITC staining of EVs derived from exo-FBS, FCS and Human AB serum analyzed via flow. (D) Immunoprecipitation/Western blot analysis of CD47 on EVs derived from Exo-FBS and FBS. Jurkat cell lysate was used as a positive control. (E and F) CD47-FITC staining of EVs derived from cell supernatants of Jurkat and JinB8 cells via flow. (G) Western blot analysis of CD47 expression by EVs derived from Jurkat and JinB8 T cells.
Because human AB serum contained the least amount of CD47 on EVs, we used this serum to culture Jurkat and JinB8 T cells to characterize their EVs. EVs purified from Jurkat and JinB8 cell conditioned media were analyzed by flow cytometry using human CD47 and isotype control antibodies. CD47 was prominently expressed in EVs released from Jurkat cells but not by EVs from JinB8 T cells as expected (Fig. 3E,F). Expression of proteoglycan and non-proteoglycan isoforms of CD47 on isolated EVs from Jurkat but not JinB8 cells was confirmed by western blotting using the CD47 antibody B6H12 (Fig. 3G).
2.3. EVs recapitulate the CD47-dependent effects of T cell conditioned medium on T cell activation
Incubation of Jurkat T cells with JinB8 EVs did not prevent anti-CD3 induction of CD69 mRNA, but the sensitivity of these cells to inhibition of that induction by TSP1 was lost (Fig. 4A). Conversely, incubation of CD47-deficient JinB8 cells with EVs from WT Jurkat cells restored their sensitivity to inhibition by TSP1 of CD69 mRNA induction (Fig. 4C). Control experiments performed using the EV-depleted Jurkat and JinB8 conditioned media demonstrated that the ability to modulate TSP1 responsiveness is specific to the respective EVs (Fig. 4B, D). TSP1 strongly inhibited anti-CD3-dependent induction of CD69 mRNA in Jurkat cells treated with JinB8 exo-depleted medium (Fig. 4B). Depleted medium from WT Jurkat cells suppressed anti-CD3 induction of CD69, and TSP1 alone enhanced CD69 mRNA expression significantly in these cells, which is consistent with the positive effects of TSP1 alone in Fig. 1D. The TSP1 receptor that mediates this T cell activating activity has not been identified. In control experiments Jurkat T cells that were activated with anti-CD3 in the presence of autologous Jurkat EVs remained sensitive to TSP1 inhibition of CD69 mRNA induction (Fig. S1A). This confirms the specificity of JinB8 EVs to convert the phenotype of WT Jurkat cells to the CD47-null phenotype. Conversely, JinB8 cells incubated with JinB8 EVs were unresponsive to anti-CD3 activation at 6 h, and addition of TSP1 in the presence of anti-CD3 increased mRNA level of CD69 (Fig. S1B). Some previous studies have reported impaired activation of JinB8 T cells that can be overcome by co-stimulation with anti-CD28 (Ticchioni et al., 1997) (Reinhold et al., 1997). Therefore, we activated JinB8 cells using anti-CD3 + anti-CD28 and found that induction of CD69 mRNA remained insensitive to TSP1 inhibition (Fig. S1C).
Fig. 4.
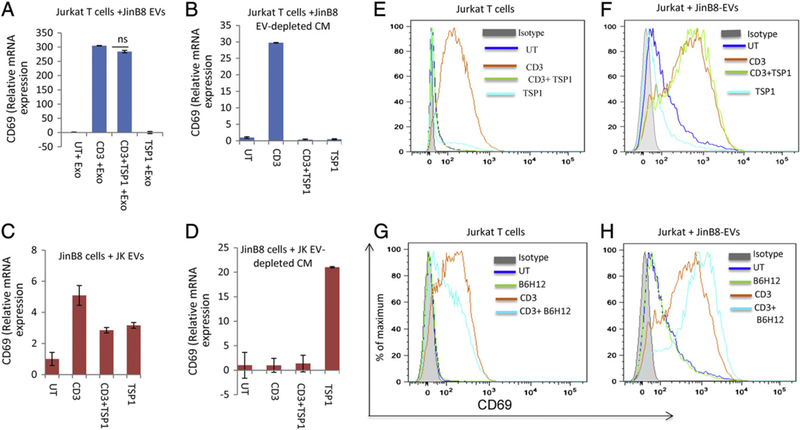
(A, C) JinB8 T cell-derived EVs were added to Jurkat T cells, or Jurkat T cell-derived EVs were added to JinB8 cells and plated on anti-CD3 coated plates in the presence or absence of TSP1 (1 μg/ml) for 6 h. The relative expression of CD69 mRNA was measured using real-time PCR. (B and D) JinB8 T cell exosome-depleted conditioned medium was added to Jurkat T cells, or Jurkat T cell exosome-depleted conditioned medium was added to JinB8 cells and plated on anti-CD3 coated plates in the presence or absence of TSP1 (1 μg/ml) for 6 h. The relative expression of CD69 mRNA was measured using real-time PCR using HPRT1 as control. The relative expression of CD69 protein expression was measured using flow cytometry (E and F). Similarly, Jurkat cells in the presence and absence of JinB8-derived EVs were plated onto anti-CD3 coated wells. The cells were treated with the CD47 antibody B6H12 (1 μg/ml) for 6 h in the presence or absence of EVs. Expression of CD69 was analyzed via flow and is presented with the respective isotype control (G and H).
To further confirm the modulation of Jurkat T cell activation by EVs isolated from CD47-deficient T cells, CD69 protein expression was measured using flow cytometry analysis (Fig. 4E–H). The ability of TSP1 to inhibit induction of cell surface CD69 expression by anti-CD3 (Fig. 4E) was lost in Jurkat T cells incubated with JinB8 EVs (Fig. 4F). Treatment with JinB8 EVs similarly blocked the ability of CD47 antibody B6H12 to inhibit CD69 induction (compare Fig. 4G, H). Therefore, components transferred to Jurkat T cells by JinB8 EVs abolish inhibitory signaling by two independent CD47 ligands. When JinB8 cells were activated on immobilized anti-CD3 for 6 or 26 h in the presence of Jurkat T cell EVs the number of CD69+ cells only increased 9–12%, and signifi-cant inhibition by TSP1 was not observed under these conditions (Fig. S2A). However, the addition of anti-CD28 co-stimulation for 24 h in the presence of Jurkat T cell EVs increased the number of CD69+ cells induced to 36%, and this activation was not significantly attenuated by TSP1 (Fig. S2B). Although we did not see a significant decrease in CD69 protein expression in the presence of EVs, the decrease in CD69 mRNA confirms that exposure to CD47+ EVs can restore sensitivity of the CD47-deficient Jurkat T cell clone to TSP1 inhibition. Many reports have established that mRNA regulation can be uncoupled from protein expression (Glickman and Ciechanover, 2002; Arava et al., 2003; Greenbaum et al., 2003; Khositseth et al., 2011; Grimmelikhuijzen and Hauser, 2012; Taylor et al., 2013).
Although Jurkat EVs express CD47, are internalized by recipient JinB8 cells, and alter CD69 induction, treatment with Jurkat T cell EVs did not confer detectable cell surface CD47 protein expression in recipient JinB8 cells (Fig. S3A). This suggests that CD47 protein is not efficiently transferred from EVs to the plasma membrane of recipient T cells, but low amounts may be transferred and could be sufficient to restore inhibition by TSP1. However, transfer of CD47 cannot explain why CD47-deficient EVs could make Jurkat cells insensitive to TSP1. One potential indirect mechanism is by down-regulating expression of CD47 or downstream signaling components through which CD47 regulates T cell activation. Anti-CD3 activation increased CD47 mRNA expression in WT Jurkat cells, and TSP1 decreased this CD47 induction (Fig. S3B). Notably, treatment of Jurkat cells with JinB8 EVs abolished induction of CD47 mRNA expression following treatment with anti-CD3 (Fig. S3C). As shown above for CD69 induction, exo-depleted medium from JinB8 cells did not prevent the induction of CD47 expression in Jurkat cells by anti-CD3 or the inhibition of this response by TSP1 (Fig. S3D).
Anti-CD3 stimulation is known to induce cell surface TSP1 expression on quiescent T cells (Li et al., 2006). Flow cytometry analysis indicated similar levels of TSP1 on the surface of EVs from WT and CD47-deficient Jurkat cells (Fig. S4A, B). In contrast, TSP1 was barely detectable on the surface of unstimulated Jurkat and JinB8 cells, and anti-CD3 stimulation increased TSP1 surface expression more on WT than on CD47-deficient T cells (Fig. S4C, D). TSP1 treatment in the presence of anti-CD3 reduced the TCR induction of surface TSP1 expression in Jurkat but not significantly in JinB8 cells (Fig. S4C, D). Some of the reduction of TCR-induced cell surface TSP1 by exogenous TSP1 may result from decreased TSP1 biosynthesis because anti-CD3 strongly increased TSP1 mRNA in Jurkat T cells, and exogenous TSP1 inhibited this induction (Fig. S4E). Therefore, TSP1 is present on EVs released by Jurkat T cell independent of CD47 expression, but CD47 signaling can modulate cell surface TSP1 levels on T cells in a CD47-dependent manner.
2.4. Uptake of T cell EVs by endothelial cells is CD47-independent
T lymphocytes regularly come into contact with endothelial cells during circulation and trans-endothelial migration (Choi et al., 2004). To examine the uptake of T cell EVs by human umbilical vein endothelial cells (HUVEC), the cells were co-cultured with green fluorescent labeled Jurkat or JinB8 EVs for 6 h. The cells were fixed and stained for microtubules and analyzed with confocal microscopy (Fig. S5). In contrast to the CD47-dependence of uptake of T cell EVs by Jurkat cells, EVs from WT and CD47-deficient Jurkat cells showed similar uptake by HUVEC. Surface and clipped plane rendering demonstrated that most of the EVs were internalized to a perinuclear location.
2.5. CD47-dependent and -independent regulation of HUVEC gene expression by T cell-derived EVs
HUVEC were cultured with Jurkat or JinB8 T cell-derived EVs for three days. Whole genome expression profiles were generated using total RNA. The global microarray analysis showed broad changes in gene expression of recipient HUVEC treated with Jurkat and JinB8 T cell-derived EVs. A total of 165 HUVEC genes showed significant regulation (>1.5-fold, p < 0.05) by Jurkat cell EVs that was CD47-independent, of which 53 were down-regulated by EVs, and 112 were up-regulated (Table S1). A larger number of HUVEC genes showed responses to T cell EVs that were CD47 dependent (Fig. 5A and Table S2). A total of 281 genes showed CD47-dependent regulation, of which 126 were significantly elevated by EVs from cells expressing CD47 and 155 were down-regulated. Interestingly only 80 of these represent named protein-coding genes, and the remainder represents microRNAs and other non-coding RNAs of unknown function.
Fig. 5.
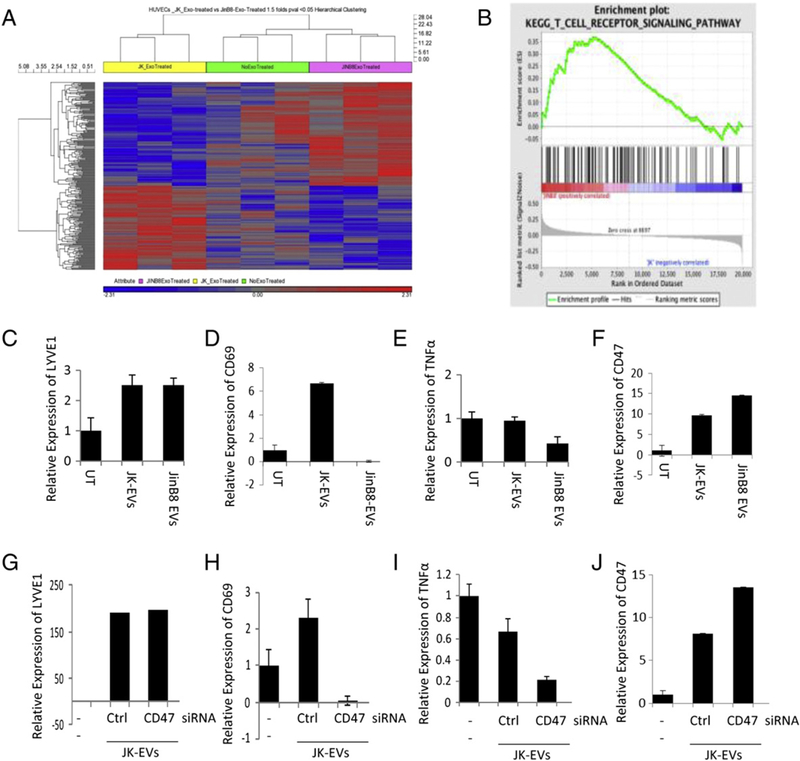
CD47-dependent modulation of endothelial cell gene expression by T cell EVs. (A) HUVEC were treated with Jurkat or JinB8 T cell-derived EVs for 3 days. Hierarchical clustering is presented of HUVEC genes that showed significant differential responses to Jurkat versus JinB8 EVs on microarray analysis. Expression data is in Table S2. (B) Geneset enrichment analysis of T cell signaling genes from microarray data after treatment of HUVEC with Jurkat and JinB8 T cell derived EVs for 3 days. (C–F) Validation of selected HUVEC gene expression regulation by Jurkat EVs using real time PCR. (G–J) Confirmation of CD47-dependent gene expression regulation by EVs in HUVEC treated with EVs derived from Jurkat T cells treated with CD47-siRNA or control si-RNAs. HPRT1 was used as the internal control.
A gene enrichment analysis showed an unexpected increase in T cell receptor signaling gene expression in HUVEC treated with T cell derived EVs (Fig. 5B and Fig. S6A). Angiogenesis-related genes were also modulated by the EVs, and among these integrin α4, which is known to associate with CD47 (Barazi et al., 2002), and the lymphangiogenesis marker LYVE1 were among the most significantly regulated HUVEC genes. Other top EV-dependent HUVEC genes included netrin-4, which is a pro-angiogenic factor in zebrafish development and during cerebral ischemia responses (Hoang et al., 2009; Lambert et al., 2012).
The top ranked gene that was differentially regulated by the presence of CD47 in the EVs was ubiquilin-4. Other ubiquilins were previously identified in a yeast two hybrid screen as cytoplasmic binding partners of CD47 (Wu et al., 1999). Ubiquilins are also regulators of G-protein coupled receptors (N’Diaye et al., 2008), as is the second most significant gene arrestin-β2 (Kopra et al., 2013). CD47 is a known regulator of heterotrimeric G proteins in various cell types (Brown, 2001), suggesting that introduction exosome-derived CD47 into HUVEC results in feedback regulation of its downstream signaling pathways.
The microarray findings were confirmed by analysis of selected gene expression in independent experiments using HUVEC treated with Jurkat or JinB8 EVs for 3 days. Induction of LYVE1 mRNA in HUVEC by WT and CD47-deficient T cell EVs was confirmed by real time PCR (Fig. 5C). Silencing of CD47 by siRNA in Jurkat cells (Fig. S6B, C) did not affect the activity of EVs released by these cells to induce LYVE1 (Fig. 5G). Selective CD47-dependent induction of the T cell activation marker CD69 in HUVEC was validated (Fig. 5D). Treatment of HUVEC with Jurkat EVs increased expression of CD69, whereas treatment of HUVEC with JinB8 EVs decreased CD69 expression as compared to untreated HUVEC. The role of CD47 in induction by Jurkat EVs was confirmed by siRNA silencing, which eliminated the activity of the resulting EVs to induce CD69 in HUVEC (Fig. 5H). Conversely, EVs from CD47-deficient Jurkat cells selectively down-regulated TNFα mRNA in HUVEC (Fig. 5E), and siRNA silencing of CD47 in WT Jurkat cells recapitulated this down-regulation (Fig. 5I).
Exposure of HUVEC to WT or CD47-deficient Jurkat EVs also increased CD47 expression (Fig. 5F). HUVEC treated with JinB8 EVs showed higher CD47 mRNA expression, and this was consistent with the higher induction of CD47 expression in HUVEC exposed to EVs from Jurkat cells where CD47 was silenced (Fig. 5J).
2.6. Jurkat T cell CD47 expression regulates the RNA content of EVs
Expression of T cell activation genes and LYVE1 in treated HUVEC was unexpected and suggested that Jurkat EVs may transfer T cell mRNAs for these genes or noncoding RNAs that can induce these genes in the recipient HUVEC. To assess the former possibility, we performed microarray analyses of EVs derived from Jurkat and JinB8 T cells (Table S3). Relatively few mRNAs were detected in the EVs. Notably, LYVE1 mRNA was present and may account for the increased expression of this gene in treated HUVEC. The top ranked mRNAs that were higher in JinB8 exosomes were mostly of mitochondrial origin including mitochondrially encoded cytochrome c oxidase-III and subunits 1, 2, 4 and 5 of the mitochondrial complex I NADH dehydrogenase, which is consistent with the known negative regulation of mitochondrial biogenesis by CD47 in other cell types (Frazier et al., 2011). Transfer of leukemia cell miRNAs to endothelial cells via exosomes is known to regulate endothelial gene expression (Umezu et al., 2013), suggesting that induction of T cell activation genes in HUVEC may be caused by noncoding RNAs in Jurkat exosomes. Several of the detected micro-RNAs were at significantly different levels, which based on previous reports (Hulsmans and Holvoet, 2013; Matsumoto et al., 2013; Umezu et al., 2013), could account mRNA changes that were observed in HUVEC treated with these EVs. The microarray chips and protocol used were not optimal for assessing miRNAs, so these preliminary results will require further study. However, the present data indicate that some effects of Jurkat T cell EVs on HUVEC gene expression are indirect and mediated by transfer of their RNA content.
2.7. CD47-dependent and -independent effects of T cell EVs on endothelial cells
Some of the changes in HUVEC gene expression suggested that T cell EVs could alter their angiogenic phenotype. In vitro tube formation in Matrigel mimics early aspects of vasculogenesis and angiogenesis. As expected, VEGF treatment of control HUVEC increased the number of rings (p = 0.006) but did not significantly increase tube length (Fig. 6A, B and Fig. S7 top panels). Treatment with Jurkat EVs did not significantly change basal tube formation, but the number of rings induced by VEGF decreased (p = 0.01) while the tube length increased (p = 0.03) (Fig. 6A, B and Fig. S7 middle panels). Treatment with JinB8 EVs tended to increase the basal number of rings and tube length, but stimulation by VEGF was lost (Fig. 6A, B and Fig. S7 bottom panels). This indicates that CD47 delivered via exosomes can modulate a functional response of endothelial cells to VEGF.
Fig. 6.
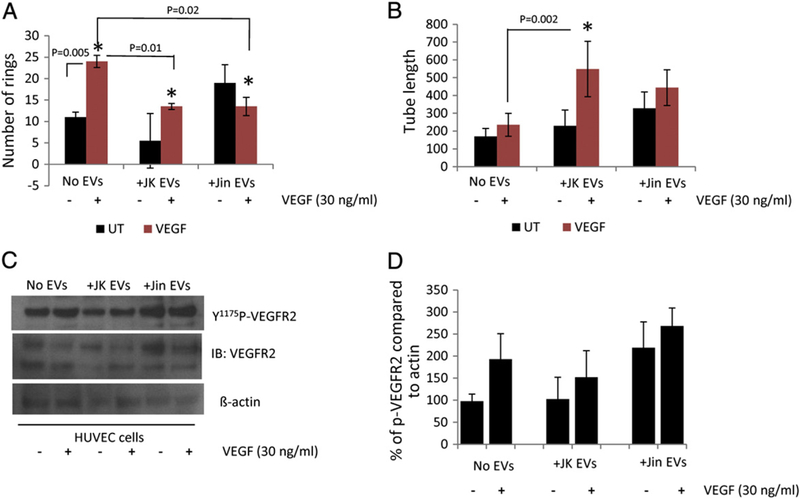
CD47-dependent modulation of endothelial cell angiogenic responses by T cell EVs. (A and B) Quantitative analysis of ring number and tube length for HUVEC cells treated with Jurkat or JinB8 T cell derived EVs for 16 h. Significance values were calculated using t-tests for UT vs VEGF and ANOVA two way analysis with replication was used for VEGF (no EVs), VEGF + JK-EV and VEGF + JinB8-EV analyses. (C) Representative western blot analysis of VEGFR2 Y1175 phosphorylation in HUVEC treated with 30 ng/ml VEGF in the presence or absence of T cell-derived EVs. (D) Quantification of VEGFR2 Y1175 phosphorylation from two independent experiments compared to actin controls.
2.8. T cell exosome-induced VEGF phosphorylation is CD47 dependent
VEGFR2 phosphorylation at Y1175 is critical for VEGF-induced downstream signal transduction (Takahashi et al., 2001; Sakurai et al., 2005). To determine the effect of HUVEC, Jurkat, and JinB8 EVs on VEGFR2 phosphorylation, HUVEC were co-cultured with freshly purified EVs for 6 h and then were stimulated with VEGF (30 ng/ml) for 5 min, and western blotting was performed using VEGFR2 Y1175 antibody. Treatment of HUVEC with EVs derived from Jurkat T cells (JK EVs) in the presence of VEGF did not further increase phosphorylation as compared HUVEC (-EVs). On the other hand, treatment of HUVEC with JinB8 T cell-derived EVs increased basal VEGFR2 Y1175 phosphorylation (Jin EVs) but did not significantly enhance VEGF-induced VEGFR2 Y1175 phosphorylation (Fig. 6C and D). These results are consistent with our previous findings that CD47 null endothelial cells exhibit increased VEGFR2 phosphorylation (Kaur et al., 2010). These findings demonstrate that T cell EVs can regulate endothelial cell VEGF signaling in a CD47-dependent manner, although further studies will be required to distinguish direct effects of CD47 transfer from CD47-dependent transfer of modulatory RNAs.
3. Discussion
TSP1 signaling through CD47 inhibits pro-angiogenic signaling in endothelial cells and TCR signaling to inhibit T cell activation (Li et al., 2001; Isenberg et al., 2006; Kaur et al., 2010; Kaur et al., 2011). These functions have been presumed to be mediated by CD47 resident in the plasma membrane of the respective cells. However, several studies have identified CD47 as a component of exosomes or ectosomes, and we now show that EVs derived from T cells can be internalized by endothelial cells and modulate signaling and gene expression in the target cells in a CD47-dependent manner. Because altering CD47 expression in the source T cells modulates the RNA and protein content of EVs that T cells release, we cannot distinguish direct effects of CD47 in EVs on target cell behavior from altered target cell responses caused by RNA and other proteins that are delivered by these EVs. Our flow cytometry data establishes that CD47 is present on EVs released by Jurkat T cells, and its extracellular ligand binding domain is exposed in at least a subset of these EVs, presumably ectosomes (Rechavi et al., 2009). We identified changes in target cell gene expression that depend on the presence of CD47 but presumably involve its regulation of the incorporation of RNAs from the source T cells into exosomes that then deliver these RNAs into the recipient endothelial cells. Thus, CD47 in a source cell may directly and indirectly control the biological activities of EVs that are released.
TSP1 inhibits anti-CD3 induced expression of the early activation marker CD69 in T cells (Li et al., 2001). A CD47 binding peptide inhibited this early TCR activation, suggesting that TSP1 inhibits CD69 induction through CD47. Although TSP1 does not inhibit TCR-stimulated CD69 expression in CD47-null murine T cells and human JinB8 T cells (Kaur et al., 2011), exposure of JinB8 cells to conditioned medium from CD47-expressing Jurkat cells remarkably restores inhibitory signaling by TSP1. Conversely, conditioned medium from the CD47-deficient cells decreased the sensitivity of Jurkat T cells to TSP1. This result could be replicated by exposing Jurkat T cells to JinB8-derived EVs but not exosome depleted conditioned medium. These findings suggest that CD47 transferred to CD47-deficient target cells via EV trafficking can regulate TSP1 signaling in target T cells. However, the ability of CD47-deficient EVs to modulate signaling in recipient T cells that express CD47 also indicates that components of EVs other than CD47 can alter responses of target cells.
Ectosomes released by platelets can modulate the function of dendritic and macrophage cells. CD47 is also present on platelet ectosomes (Sadallah et al., 2011). Proteomic analysis of mesenchymal stem cells also revealed the presence of CD47 on exosomes (Kim et al., 2012). Our results from cell supernatants and serum further confirm that CD47 is present on EVs.
Further studies are required to define which components of EVs are required to modulate specific functions of target cells. Transfer of functional signaling receptors to target cells via EVs is becoming well documented (Rechavi et al., 2009). Notably, counter-receptors for the inhibitory receptor CTLA-4 can be transferred from antigen-presenting cells to T cells by this process (Qureshi et al., 2011). Our data indicates that the presence of CD47 on EVs is not necessary for their uptake into endothelial cells, but we could not detect acquisition of CD47 in the plasma membrane of target CD47-deficient T cells. Exosomes released by immune cells can also modulate target cells by transferring mRNAs and noncoding RNAs (Umezu et al., 2013), and our expression studies demonstrated significant differences in RNA expression in endothelial cells exposed to CD47-positive versus CD47-negative EVs, including some that coincide with observed alterations in the targeted HUVEC gene expression.
T cell derived exosomes are known to be enriched in non-coding RNA and miRNAs (Mittelbrunn et al., 2011). Noncoding RNAs in the T cell EVs may play a major role in altering HUVEC gene expression via CD47-dependent and -independent mechanisms. Major CD47-independent targets include angiogenesis, lymphangiogenesis and T cell activation genes. Further studies are required to define CD47-dependent miRNAs in T cell exosomes and their biological functions.
Cross talk between endothelial cells and T lymphocytes in normal physiological as well as pathological conditions has been well established (Vora et al., 1997; Choi et al., 2004). Our data demonstrate that T cell derived EVs can alter endothelial VEGF signaling, tube formation, and gene expression. Interestingly, JinB8 EVs enhanced basal VEGFR2 phosphorylation, suggesting that CD47 could indirectly modulate VEGF–VEGFR2 signaling on target endothelial cells via EV trafficking. This may be important in cancers, where high expression of CD47 on tumor cells is a negative prognostic factor (Willingham et al., 2012). Our work suggests that CD47 transferred to the tumor vasculature from tumor cells could also modulate tumor angiogenesis.
4. Experimental procedures
4.1. Cells and reagents
HUVEC were purchased from (Lonza) and were maintained using EGM2 medium (Lonza). HUVEC were used between passages 4 and 5. Jurkat T cells (E6.1, ATCC) and the corresponding CD47-deficient Jurkat mutant JinB8 were provided by Dr. Eric Brown, Genentech (Reinhold et al., 1999) and were maintained at 2.0 × 105 cells per ml in RPMI 1640 medium (Life Technologies) supplemented with 10% FBS, glutamine, penicillin, and streptomycin. Jurkat cells were maintained for a maximum of 4 weeks for experiments. Human anti-CD3 and anti-CD47-FITC (BD Biosciences), anti-CD69 (R&D systems), anti-actin, anti-tubulin, anti-VEGFR2, anti-VEGFR2 Y1175 (Cell Signaling), anti-CD47 (B6H12, Abcam) and azide-free functional grade anti-CD47 (e-Biosciences) were purchased from the indicated vendors.
4.2. Isolation of exosomes from serum and cell supernatants
A 250 μl volume of fetal bovine serum fetal calf serum (FCS), Exo-FBS™ Exosome-depleted FBS (SBI, System Bioscience) and human serum AB was used. The exosomes were isolated using exoQuick kit according to manufacturer’s instructions (SBI, System Bioscience). The purified exosomes were washed with PBS and analyzed for flow cytometry analysis.
Before exosome isolation, the cells were cultured using low human AB serum in their respective media for at least one passage. The T cells were cultured in 75 cm3 flasks (Corning) and conditioned medium from ~40 × 106 cells (~100 ml) was collected. The conditioned medium was further centrifuged for 10 min at 1530 ×g to remove cell debris. The supernatants were transferred to new 50 ml tubes. The supernatants were concentrated using centrifugal filter device YM-10 (Centricon). The final volume of supernatant was ~1 ml. The concentrated super-natants (~ 250 μl) from the respective cells were used to purify exosomes. The exosomes were isolated using the exoQuick kit (SBI System Biosciences) according to the manufacturer’s instructions. The supernatant was completely removed via centrifugation 2 times for 30 min at 1500 ×g. The white exosome pellet was diluted into 1 ml of ultra-pure water (~10 × 106 cells). Purified exosomes were analyzed using CD47 antibody via flow cytometry and western blots. For exosome treatment experiments only 10–20 μl of purified exosomes (equivalent to that released by ~250–500,000 cells) were added from 1 ml of stock. The numbers of EVs were quantified using SPHERo AccuCount Particles from Spherotech.
4.3. Western blots
HUVEC were incubated with Jurkat and JinB8 T cell-derived exosomes using EGM2 media. After 6 h, the cells were treated with VEGF165 for 5 min at 37 °C. The cells were washed with cold PBS containing phosphatase inhibitors (Active Motif). The cells were scraped, and cell lysates were made using RIPA buffer (Active Motif). The cell lysates were centrifuged at 17,949 ×g for 10 min at 4 °C. The supernatants were collected into new Eppendorf tubes. Lysate samples containing equal amounts of protein were boiled at 95 °C for 5 min. SDS-PAGE was performed using Bis–tris 10% acrylamide gels with MES running buffer (Life technologies). Primary P-VEGFR2 Y1175 and VEGFR2 1:1000 and secondary horseradish peroxidase-conjugated anti-mouse IgG antibodies 1:5000 (Amersham) were used for western blots. For total loading control, the western blots were re-probed using actin antibodies. Quantitative analysis of P-VEGFR2 normalized to actin controls was performed using ImageJ.
4.4. Immunoprecipitation
Supernatants from Jurkat and JinB8 cells were centrifuged for 10 min at 1530 ×g rpm to remove cell debris. The supernatants were further concentrated using centrifugal filter devices from Centricon. Jurkat, JinB8, FCS and exo-FBS derived exosomes were purified and lysates were made using RIPA buffer. The immunoprecipitation of Jurkat cell lysate and isolated exosomes was performed using anti-CD47 antibody. The immunoprecipitation was performed as previously described (Kaur et al., 2010). The CD47 immunoprecipitation was analyzed using SDA-PAGE (Bis− tris 4–12% gels). Primary antibodies were used at 1:1000 and secondary HRP-conjugated antibodies at 1:5000.
4.5. Exosome uptake
Similarly, Jurkat- and JinB8-derived EV uptake was measured by using confocal microscopy with Zeiss 780. The EVs were labeled with CSFE (Life technologies). The purified Jurkat or JinB8 T cell derived EVs were co-cultured for 6 h and fixed with 4% paraformaldehyde for 15 min. the cells were blocked using 5% BSA for 30 min at room temperature. The cells were stained using anti-tubulin antibody at 1:200 for 1 h at room temperature. The cells were washed 3 times with PBS-Tween 20. The secondary Alexa Fluor® 568 (1:500 dilutions) was incubated for 1 h at RT. The cells were washed and mounted using Vectashield DAPI (Vector Laboratories).
The Jurkat and JinB8 T cells were labeled with PKH67 (green) and their exosomes were labeled with PKH26 (red) according to the manufacturer’s instructions. Jurkat and JinB8 T cells were co-cultured with their own EVs or with EVs from the opposite cells. After 6 h, the cells were harvested, centrifuged and plated on L-polylysine coated plates using fresh medium. Live cell confocal microscopy using Zeiss 780 was performed at 37 °C with 5% CO2 during imaging.
Confocal images were sequentially acquired with Zeiss ZEN 2011 software on a Zeiss LSM 780 Confocal system (Carl Zeiss Inc., Thornwood, NY) with a Zeiss Observer Z1 inverted microscope and a 30 mW Diode laser tuned to 405 nm, a 25 mW Argon visible laser tuned to 488 nm and a 20 mW DPSS laser tuned to 561 nm. A 63× Plan-Apochromat 1.4 NA oil immersion objective was used and digital images were 512 × 512 pixels with 0.190 μm pixel size. Emission signals after sequential excitation of DAPI, CSFE, and PKH26 by the 405 nm, 488 nm or 561 nm laser lines were collected with a BP 411–482 nm filter, BP 491–553 nm filter, and BP 553–738 nm filter respectively, using individual photomultipliers. Z-stacks consisted of 9–22 slices at 0.5 intervals and these stacks were examined with Bitplane Imaris software (v6.0; Zurich, Switzerland) for surface rendering. In some cases, a cutting plane was used to expose internal surface, or the outer surface was made semi-transparent.
4.6. Endothelial tube formation assay
HUVEC were cultured in EGM2 medium using exo-FBS for one passage in 75 cm2 flasks. 96-well plates were coated with BD Matrigel overnight at 4 °C. The tube formation assay was performed according to the manufacturer’s instructions using triplicates for each treatment. Briefly, 15,000 HUVEC were placed on Matrigel along with 10 μl of Jurkat or JinB8 EVs. After 6–16 h, images were captured, and tube ring formation was counted manually. The tube length and the area of tube were calculated using Image J program after 16 h of incubation.
4.7. T cell activation assay
The surface of 12-well plates was coated with anti-CD3 (1 μg/ml) or in combination with anti-CD28 (3 μg/ml) overnight at 4 °C. Jurkat and JinB8 T cells were cultured overnight using 75 cm2 flasks with RPMI 1640 containing 5% human AB serum. The cells were centrifuged, and supernatants were used for exosome analysis. The Jurkat and JinB8 cells (100,000 cells per well) were plated for 6 h. The Jurkat T cell derived EVs were plated on JinB8 cells or vice versa for 6 h. The cells were then plated in the presence or absence of immobilized anti-CD3. Where indicated these cells were treated with TSP1 (1 μg/ml) and anti-CD47 B6H12 for 6 h. Expression of the early TCR marker CD69 was used to analyze T cell activation either via flow cytometry analysis or via quantitative RT-PCR. Total RNA was prepared using TriZol (Roche). cDNA was made using the Maxima kit from Thermo Scientific, and real time PCR was performed using CD69, TNFα, CD47, TSP1, LYVE1 and β2 microglobulin (B2M) or HPRT1 primers to determine normalized mRNA expression. PCR primers for human were identified using Fast PCR on-line software and are listed in Table S4. Real time PCR was performed using SYBR Green (Roche) on an MJ Research Opticon I instrument (Bio-Rad) with the following amplification program: 95 °C for 15 min, followed by 40 cycles of 95 °C for 15 s, 58 °C for 20 s, 72 °C for 25 s, and 72 °C for 1 min. Melting curves were performed for each product from 30 to 95 °C, reading every 0.5 °C with a 6-s dwell time. Fold change in mRNA expression was calculated by normalizing to B2M or HPRT1 mRNA levels.
4.8. Flow cytometry analysis
The Jurkat and JinB8 cells and exosomes purified from serum and supernatants of Jurkat and JinB8 cells were harvested by centrifugation. For analysis of cell surface proteins, cells were stained with fluorophoreconjugated anti-CD69 and anti-CD47 in separate tubes for 30 min at room temperature in FACS buffer (HBSS containing 1% FBS) and washed twice with the same buffer. Fluorescence minus one controls and isotype control antibodies were used to validate flow cytometric results. EVs isolated from different cell types were stained overnight with Flk-1 (A-3) mouse monoclonal IgG1 specific for TSP1 antibody F18 1G8 (IgG1) (Calzada et al., 2008) followed by goat anti mouse IgG1-FITC (Santa Cruz Biotechnology) for 30 min at 4 °C. Flow cytometric acquisition was performed on a LSR II cytometer with FACSDiva software (BD Biosciences). Flow cytometry data were analyzed by using FlowJO software (Tree Star, San Carlos, CA).
4.9. Exosome RNA extraction and array analysis
Exosome pellets were purified from Jurkat and JinB8 T cell conditioned medium using the Exo Quick kit TC (SBI Biosciences). The pellets were dissolved into 100 μl of 1 × PBS. Exosome RNA was extracted using a total RNA kit from Qiagen (MD, USA). Where indicated, CD47 expression in the source Jurkat T cells was reduced by siRNA knockdown. A siRNA targeting CD47 was identified using siRNA Target Finder algorithm (Applied Biosystems) and expressed using the Silencer siRNA Construction kit (Applied Biosystems). The target sequences were 5′-AAG ATG GAT AAG AGT GAT GCT CCT GTC TC-3′ and 5′-AAA GCA TCA CTC TTA TCC ATC CCT GTC TC-3′. A scrambled version of sequence was used as control. Transfections (n = 3) were performed using an Amexa Kit (Lonza) according to the manufacturer’s conditions using Human AB serum RPM1 media. After 24 h, conditioned media from three replicates were pooled. The knockdown of CD47-siRNA in Jurkat T cells was confirmed by real time PCR. The exosomes were isolated using Exo-quick kit as described earlier.
RNA samples were prepared according to Affymetrix protocols (Affymetrix, Inc.). RNA quality and quantity were ensured using the Bioanalyzer (Agilent, Inc.) and NanoDrop (Thermo Scientific, Inc.), respectively. Per RNA labeling, 200 ng of total RNA was used in conjunction with the Affymetrix recommended protocol for the GeneChip 1.0 ST chips.
The hybridization cocktail containing the fragmented and labeled cDNAs was hybridized to The Affymetrix Human Genome ST 1.0 GeneChip. The chips were washed and stained by the Affymetrix Fluidics Station using the standard format and protocols as described by Affymetrix. The probe arrays were stained with streptavidin phycoerythrin solution (Molecular Probes, Carlsbad, CA) and enhanced by using an antibody solution containing 0.5 mg/mL of biotinylated anti-streptavidin (Vector Laboratories, Burlingame, CA). An Affymetrix Gene Chip Scanner 3000 was used to scan the probe arrays. Gene expression intensities were calculated using Affymetrix AGCC software. Partek Genomic Suite was used to RMA (Robust Multichip Analysis) normalize, summarize, log transform the data, and run ANOVA analysis and Hierarchical clustering.
4.10. Statistical analysis
The t-test was used for analyzing the number of ring formation and ANOVA with Two Way Factor for tube length data. ANOVA with Two Way Factor with replicates was used for all real time PCR data. A p-value less than 0.05 was considered significant.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.matbio.2014.05.007.
Supplementary Material
Acknowledgments
We thank Dr. Elise Kohn for sharing PKH26 and PKH67 dyes and Susan Garfield and Lim Langston for their help for confocal microscopy. We thank Dr. Michael L Pendrak for designing primers and making the CD47 siRNA construct. This work was supported by the Intramural Research Programs of the Center for Cancer Research, National Cancer Institute (ZIA SC 009172), National Institute of Allergy and Infectious Diseases, and National Human Genome Research Institute.
Abbreviations:
- EVs
extracellular vesicles
- HUVEC
human umbilical vein endothelial cells
- TSP1
thrombospondin-1
- TCR
T cell antigen receptor
- VEGFR2
vascular endothelial growth factor receptor-2
References
- Abounit S, Zurzolo C, 2012. Wiring through tunneling nanotubes — from electrical signals to organelle transfer. J. Cell Sci 125, 1089–1098. [DOI] [PubMed] [Google Scholar]
- Admyre C, Johansson SM, Qazi KR, Filen JJ, Lahesmaa R, Norman M, Neve EP, Scheynius A, Gabrielsson S, 2007. Exosomes with immune modulatory features are present in human breast milk. J. Immunol 179, 1969–1978. [DOI] [PubMed] [Google Scholar]
- Andre F, Schartz NE, Movassagh M, Flament C, Pautier P, Morice P, Pomel C, Lhomme C, Escudier B, Le Chevalier T, Tursz T, Amigorena S, Raposo G, Angevin E, Zitvogel L, 2002. Malignant effusions and immunogenic tumour-derived exosomes. Lancet 360, 295–305. [DOI] [PubMed] [Google Scholar]
- Arava Y, Wang Y, Storey JD, Liu CL, Brown PO, Herschlag D, 2003. Genome-wide analysis of mRNA translation profiles in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U. S. A 100, 3889–3894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asea A, Jean-Pierre C, Kaur P, Rao P, Linhares IM, Skupski D, Witkin SS, 2008. Heat shock protein-containing exosomes in mid-trimester amniotic fluids. J. Reprod. Immunol 79, 12–17. [DOI] [PubMed] [Google Scholar]
- Barazi HO, Li Z, Cashel JA, Krutzsch HC, Annis DS, Mosher DF, Roberts DD, 2002. Regulation of integrin function by CD47 ligands. Differential effects on alpha vbeta 3 and alpha 4beta1 integrin-mediated adhesion. J Biol Chem 277, 42859–42866. [DOI] [PubMed] [Google Scholar]
- Bayer-Santos E, Aguilar-Bonavides C, Rodrigues SP, Cordero EM, Marques AF, Varela-Ramirez A, Choi H, Yoshida N, da Silveira JF, Almeida IC, 2013. Proteomic analysis of Trypanosoma cruzi secretome: characterization of two populations of extracellular vesicles and soluble proteins. J. Proteome Res 12, 883–897. [DOI] [PubMed] [Google Scholar]
- Boon RA, Vickers KC, 2013. Intercellular transport of microRNAs. Arterioscler. Thromb. Vasc. Biol 33, 186–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown E, 2001. Integrin-associated protein (CD47): an unusual activator of G protein signaling. J Clin Invest 107, 1499–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown EJ, Frazier WA, 2001. Integrin-associated protein (CD47) and its ligands. Trends Cell Biol 11, 130–135. [DOI] [PubMed] [Google Scholar]
- Caby MP, Lankar D, Vincendeau-Scherrer C, Raposo G, Bonnerot C, 2005. Exosomal-like vesicles are present in human blood plasma. Int. Immunol 17, 879–887. [DOI] [PubMed] [Google Scholar]
- Calzada MJ, Kuznetsova SA, Sipes JM, Rodrigues RG, Cashel JA, Annis DS, Mosher DF, Roberts DD, 2008. Calcium indirectly regulates immunochemical reactivity and functional activities of the N-domain of thrombospondin-1. Matrix Biol 27, 339–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J, Enis DR, Koh KP, Shiao SL, Pober JS, 2004. T lymphocyte–endothelial cell interactions. Annu. Rev. Immunol 22, 683–709. [DOI] [PubMed] [Google Scholar]
- Cocucci E, Meldolesi J, 2011. Ectosomes. Curr. Biol 21, R940–R941. [DOI] [PubMed] [Google Scholar]
- Fevrier B, Vilette D, Archer F, Loew D, Faigle W, Vidal M, Laude H, Raposo G, 2004. Cells release prions in association with exosomes. Proc. Natl. Acad. Sci. U. S. A 101, 9683–9688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazier EP, Isenberg JS, Shiva S, Zhao L, Schlesinger P, Dimitry J, Abu-Asab MS, Tsokos M, Roberts DD, Frazier WA, 2011. Age-dependent regulation of skeletal muscle mitochondria by the thrombospondin-1 receptor CD47. Matrix Biol 30, 154–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasser O, Hess C, Miot S, Deon C, Sanchez JC, Schifferli JA, 2003. Characterisation and properties of ectosomes released by human polymorphonuclear neutrophils. Exp. Cell Res 285, 243–257. [DOI] [PubMed] [Google Scholar]
- Glickman MH, Ciechanover A, 2002. The ubiquitin–proteasome proteolytic pathway: destruction for the sake of construction. Physiol. Rev 82, 373–428. [DOI] [PubMed] [Google Scholar]
- Greenbaum D, Colangelo C, Williams K, Gerstein M, 2003. Comparing protein abundance and mRNA expression levels on a genomic scale. Genome Biol 4, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimmelikhuijzen CJ, Hauser F, 2012. Mini-review: the evolution of neuropeptide signaling. Regul. Pept 177, S6–S9 (Suppl.). [DOI] [PubMed] [Google Scholar]
- Hess C, Sadallah S, Hefti A, Landmann R, Schifferli JA, 1999. Ectosomes released by human neutrophils are specialized functional units. J. Immunol 163, 4564–4573. [PubMed] [Google Scholar]
- Hulsmans M, Holvoet P, 2013. MicroRNA-containing microvesicles regulating inflammation in association with atherosclerotic disease. Cardiovasc Res 100, 7–18. [DOI] [PubMed] [Google Scholar]
- Hoang S, Liauw J, Choi M, Choi M, Guzman RG, Steinberg GK, 2009. Netrin-4 enhances angiogenesis and neurologic outcome after cerebral ischemia. J Cereb Blood Flow Metab 29, 385–397. [DOI] [PubMed] [Google Scholar]
- Holme PA, Solum NO, Brosstad F, Roger M, Abdelnoor M, 1994. Demonstration of platelet-derived microvesicles in blood from patients with activated coagulation and fibrinolysis using a filtration technique and western blotting. Thromb. Haemost 72, 666–671. [PubMed] [Google Scholar]
- Isenberg JS, Ridnour LA, Dimitry J, Frazier WA, Wink DA, Roberts DD, 2006. CD47 is necessary for inhibition of nitric oxide-stimulated vascular cell responses by thrombospondin-1. J. Biol. Chem 281, 26069–26080. [DOI] [PubMed] [Google Scholar]
- Kaur S, Martin-Manso G, Pendrak ML, Garfield SH, Isenberg JS, Roberts DD, 2010. Thrombospondin-1 inhibits VEGF receptor-2 signaling by disrupting its association with CD47. J. Biol. Chem 285, 38923–38932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur S, Kuznetsova SA, Pendrak ML, Sipes JM, Romeo MJ, Li Z, Zhang L, Roberts DD, 2011. Heparan sulfate modification of the transmembrane receptor CD47 is necessary for inhibition of T cell receptor signaling by thrombospondin-1. J. Biol. Chem 286, 14991–15002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khositseth S, Pisitkun T, Slentz DH, Wang G, Hoffert JD, Knepper MA, Yu MJ, 2011. Quantitative protein and mRNA profiling shows selective post-transcriptional control of protein expression by vasopressin in kidney cells. Mol. Cell. Proteomics 10 (M110), 004036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HS, Choi DY, Yun SJ, Choi SM, Kang JW, Jung JW, Hwang D, Kim KP, Kim DW, 2012. Proteomic analysis of microvesicles derived from human mesenchymal stem cells. J. Proteome Res 11, 839–849. [DOI] [PubMed] [Google Scholar]
- Knolle PA, 2006. Cognate interaction between endothelial cells and T cells. Results Probl. Cell Differ 43, 151–173. [DOI] [PubMed] [Google Scholar]
- Kopra K, Kainulainen M, Mikkonen P, Rozwandowicz-Jansen A, Hanninen P, Harma H, 2013. Multiparametric homogeneous method for identification of ligand binding to G protein-coupled receptors: receptor-ligand binding and beta-arrestin assay. Anal Chem 85, 2276–2281. [DOI] [PubMed] [Google Scholar]
- Korkut C, Li Y, Koles K, Brewer C, Ashley J, Yoshihara M, Budnik V, 2013. Regulation of postsynaptic retrograde signaling by presynaptic exosome release. Neuron 77, 1039–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakkaraju A, Rodriguez-Boulan E, 2008. Itinerant exosomes: emerging roles in cell and tissue polarity. Trends Cell Biol 18, 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert E, Coissieux MM, Laudet V, Mehlen P, 2012. Netrin-4 acts as a pro-angiogenic factor during zebrafish development. J Biol Chem 287, 3987–3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavialle F, Deshayes S, Gonnet F, Larquet E, Kruglik SG, Boisset N, Daniel R, Alfsen A, Tatischeff I, 2009. Nanovesicles released by Dictyostelium cells: a potential carrier for drug delivery. Int. J. Pharm 380, 206–215. [DOI] [PubMed] [Google Scholar]
- Lenassi M, Cagney G, Liao M, Vaupotic T, Bartholomeeusen K, Cheng Y, Krogan NJ, Plemenitas A, Peterlin BM, 2010. HIV Nef is secreted in exosomes and triggers apoptosis in bystander CD4+ T cells. Traffic 11, 110–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, He L, Wilson K, Roberts D, 2001. Thrombospondin-1 inhibits TCR-mediated T lymphocyte early activation. J. Immunol 166, 2427–2436. [DOI] [PubMed] [Google Scholar]
- Li SS, Liu Z, Uzunel M, Sundqvist KG, 2006. Endogenous thrombospondin-1 is a cell-surface ligand for regulation of integrin-dependent T-lymphocyte adhesion. Blood 108, 3112–3120. [DOI] [PubMed] [Google Scholar]
- Liegeois S, Benedetto A, Garnier JM, Schwab Y, Labouesse M, 2006. The V0-ATPase mediates apical secretion of exosomes containing Hedgehog-related proteins in Caenorhabditis elegans. J. Cell Biol 173, 949–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maile LA, Allen LB, Veluvolu U, Capps BE, Busby WH, Rowland M, Clemmons DR, 2009. Identification of compounds that inhibit IGF-I signaling in hyperglycemia. Exp. Diabetes Res 2009, 267107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masyuk AI, Huang BQ, Ward CJ, Gradilone SA, Banales JM, Masyuk TV, Radtke B, Splinter PL, LaRusso NF, 2010. Biliary exosomes influence cholangiocyte regulatory mechanisms and proliferation through interaction with primary cilia. Am. J. Physiol. Gastrointest. Liver Physiol 299, G990–G999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto S, Sakata Y, Suna S, Nakatani D, Usami M, Hara M, Kitamura T, Hamasaki T, Nanto S, Kawahara Y, Komuro I, 2013. Circulating p53-responsive microRNAs are predictive indicators of heart failure after acute myocardial infarction. Circ Res 113, 322–326. [DOI] [PubMed] [Google Scholar]
- Mittelbrunn M, Gutierrez-Vazquez C, Villarroya-Beltri C, Gonzalez S, Sanchez-Cabo F, Gonzalez MA, Bernad A, Sanchez-Madrid F, 2011. Unidirectional transfer of microRNA-loaded exosomes from T cells to antigen-presenting cells. Nat. Commun 2, 282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morelli AE, Larregina AT, Shufesky WJ, Sullivan ML, Stolz DB, Papworth GD, Zahorchak AF, Logar AJ, Wang Z, Watkins SC, Falo LD Jr., Thomson AW, 2004. Endocytosis, intracellular sorting, and processing of exosomes by dendritic cells. Blood 104, 3257–3266. [DOI] [PubMed] [Google Scholar]
- Murata Y, Kotani T, Ohnishi H, Matozaki T, 2014. The CD47-SIRPalpha signalling system: its physiological roles and therapeutic application. J. Biochem 155, 335–344. [DOI] [PubMed] [Google Scholar]
- N’Diaye EN, Hanyaloglu AC, Kajihara KK, Puthenveedu MA, Wu P, von Zastrow M, Brown EJ, 2008. The ubiquitin-like protein PLIC-2 is a negative regulator of G protein-coupled receptor endocytosis. Mol Biol Cell 19, 1252–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa Y, Miura Y, Harazono A, Kanai-Azuma M, Akimoto Y, Kawakami H, Yamaguchi T, Toda T, Endo T, Tsubuki M, Yanoshita R, 2011. Proteomic analysis of two types of exosomes in human whole saliva. Biol. Pharm. Bull 34, 13–23. [DOI] [PubMed] [Google Scholar]
- Ostrowski M, Carmo NB, Krumeich S, Fanget I, Raposo G, Savina A, Moita CF, Schauer K, Hume AN, Freitas RP, Goud B, Benaroch P, Hacohen N, Fukuda M, Desnos C, Seabra MC, Darchen F, Amigorena S, Moita LF, Thery C, 2010. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat. Cell Biol 12 (19–30), 11–13 (sup). [DOI] [PubMed] [Google Scholar]
- Peinado H, Aleckovic M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G, Hergueta-Redondo M, Williams C, Garcia-Santos G, Ghajar C, Nitadori-Hoshino A, Hoffman C, Badal K, Garcia BA, Callahan MK, Yuan J, Martins VR, Skog J, Kaplan RN, Brady MS, Wolchok JD, Chapman PB, Kang Y, Bromberg J, Lyden D, 2012. Melanoma exosomes educate bone marrow progenitor cells toward a prometastatic phenotype through MET. Nat. Med 18, 883–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisitkun T, Shen RF, Knepper MA, 2004. Identification and proteomic profiling of exosomes in human urine. Proc. Natl. Acad. Sci. U. S. A 101, 13368–13373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, Baker J, Jeffery LE, Kaur S, Briggs Z, Hou TZ, Futter CE, Anderson G, Walker LS, Sansom DM, 2011. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science 332, 600–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rechavi O, Goldstein I, Kloog Y, 2009. Intercellular exchange of proteins: the immune cell habit of sharing. FEBS Lett 583, 1792–1799. [DOI] [PubMed] [Google Scholar]
- Regente M, Pinedo M, Elizalde M, de la Canal L, 2012. Apoplastic exosome-like vesicles: a new way of protein secretion in plants? Plant Signal. Behav 7, 544–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhold MI, Lindberg FP, Kersh GJ, Allen PM, Brown EJ, 1997. Costimulation of T cell activation by integrin-associated protein (CD47) is an adhesion-dependent, CD28-independent signaling pathway. J. Exp. Med 185, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhold MI, Green JM, Lindberg FP, Ticchioni M, Brown EJ, 1999. Cell spreading distinguishes the mechanism of augmentation of T cell activation by integrin-associated protein/CD47 and CD28. Int. Immunol 11, 707–718. [DOI] [PubMed] [Google Scholar]
- Sadallah S, Eken C, Martin PJ, Schifferli JA, 2011. Microparticles (ectosomes) shed by stored human platelets downregulate macrophages and modify the development of dendritic cells. J. Immunol 186, 6543–6552. [DOI] [PubMed] [Google Scholar]
- Sakurai Y, Ohgimoto K, Kataoka Y, Yoshida N, Shibuya M, 2005. Essential role of Flk-1 (VEGF receptor 2) tyrosine residue 1173 in vasculogenesis in mice. Proc. Natl. Acad. Sci. U. S. A 102, 1076–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skriner K, Adolph K, Jungblut PR, Burmester GR, 2006. Association of citrullinated proteins with synovial exosomes. Arthritis Rheum 54, 3809–3814. [DOI] [PubMed] [Google Scholar]
- Street JM, Barran PE, Mackay CL, Weidt S, Balmforth C, Walsh TS, Chalmers RT, Webb DJ, Dear JW, 2012. Identification and proteomic profiling of exosomes in human cerebrospinal fluid. J. Transl. Med 10, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stridsberg M, Fabiani R, Lukinius A, Ronquist G, 1996. Prostasomes are neuroendocrine-like vesicles in human semen. Prostate 29, 287–295. [DOI] [PubMed] [Google Scholar]
- Subra C, Grand D, Laulagnier K, Stella A, Lambeau G, Paillasse M, De Medina P, Monsarrat B, Perret B, Silvente-Poirot S, Poirot M, Record M, 2010. Exosomes account for vesicle-mediated transcellular transport of activatable phospholipases and prostaglandins. J. Lipid Res 51, 2105–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Yamaguchi S, Chida K, Shibuya M, 2001. A single autophosphorylation site on KDR/Flk-1 is essential for VEGF-A-dependent activation of PLC-gamma and DNA synthesis in vascular endothelial cells. EMBO J 20, 2768–2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor RC, Webb Robertson BJ, Markillie LM, Serres MH, Linggi BE, Aldrich JT, Hill EA, Romine MF, Lipton MS, Wiley HS, 2013. Changes in translational efficiency is a dominant regulatory mechanism in the environmental response of bacteria. Integr. Biol. (Camb.) 5, 1393–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thery C, Ostrowski M, Segura E, 2009. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol 9, 581–593. [DOI] [PubMed] [Google Scholar]
- Ticchioni M, Deckert M, Mary F, Bernard G, Brown EJ, Bernard A, 1997. Integrin-associated protein (CD47) is a comitogenic molecule on CD3-activated human T cells. J. Immunol 158, 677–684. [PubMed] [Google Scholar]
- Umezu T, Ohyashiki K, Kuroda M, Ohyashiki JH, 2013. Leukemia cell to endothelial cell communication via exosomal miRNAs. Oncogene 32, 2747–2755. [DOI] [PubMed] [Google Scholar]
- Vickers KC, Palmisano BT, Shoucri BM, Shamburek RD, Remaley AT, 2011. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat. Cell Biol 13, 423–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vora AJ, Kidd D, Miller DH, Perkin GD, Hughes RA, Ellis BA, Dumonde DC, Brown KA, 1997. Lymphocyte–endothelial cell interactions in multiple sclerosis: disease specificity and relationship to circulating tumour necrosis factor-alpha and soluble adhesion molecules. Mult. Scler 3, 171–179. [DOI] [PubMed] [Google Scholar]
- Willingham SB, Volkmer JP, Gentles AJ, Sahoo D, Dalerba P, Mitra SS, Wang J, Contreras-Trujillo H, Martin R, Cohen JD, Lovelace P, Scheeren FA, Chao MP, Weiskopf K, Tang C, Volkmer AK, Naik TJ, Storm TA, Mosley AR, Edris B, Schmid SM, Sun CK, Chua MS, Murillo O, Rajendran P, Cha AC, Chin RK, Kim D, Adorno M, Raveh T, Tseng D, Jaiswal S, Enger PO, Steinberg GK, Li G, So SK, Majeti R, Harsh GR, van de Rijn M, Teng NN, Sunwoo JB, Alizadeh AA, Clarke MF, Weissman IL, 2012. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. U. S. A 109, 6662–6667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu AL, Wang J, Zheleznyak A, Brown EJ, 1999. Ubiquitin-related proteins regulate interaction of vimentin intermediate filaments with the plasma membrane. Mol Cell 4, 619–625. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.