

Abstract
Rationale:
Activation of monocytes/macrophages by hyperlipidemia associated with diabetes and obesity contributes to the development of atherosclerosis. PKCδ expression and activity in monocytes were increased by hyperlipidemia and diabetes with unknown consequences to atherosclerosis.
Objective:
To investigate the effect of PKCδ activation in macrophages on the severity of atherosclerosis.
Methods and Results:
PKCδ expression and activity were increased in Zucker diabetic rats. Mice with selective deletion of PKCδ in macrophages were generated by breeding PKCδ flox/flox mice with LyzM-Cre and ApoE−/− mice (MPKCδKO/ApoE−/− mice) and studied in atherogenic (AD) and very high fat diet (HFD). Mice fed AD and HFD exhibited hyperlipidemia, but only HFD fed mice had insulin resistance and mild diabetes. Surprisingly, MPKCδKO/ApoE−/− mice exhibited accelerated aortic atherosclerotic lesions by 2-fold vs. ApoE−/− mice on AD or HFD. Splenomegaly was observed in MPKCδKO/ApoE−/− mice on AD and HFD, but not on regular chow. Both the AD or HFD increased macrophage numbers in aortic plaques and spleen by 1.7 and 2-fold, respectively, in MPKCδKO/ApoE−/− vs. ApoE−/− mice due to decreased apoptosis (62%) and increased proliferation (1.9 fold), and not due to uptake, with parallel increased expressions of inflammatory cytokines. Mechanisms for the increased macrophages in MPKCδKO/ApoE−/− were associated with elevated phosphorylation levels of pro-survival cell signaling proteins, Akt and FoxO3a, with reduction of pro-apoptotic protein Bim associated with PKCδ induced inhibition of P85/PI3K.
Conclusion:
Accelerated development of atherosclerosis induced by insulin resistance and hyperlipidemia may be partially limited by PKCδ isoform activation in the monocytes, which decreased its number and inflammatory responses in the arterial wall.
Keywords: Atherosclerosis, Macrophages, protein kinase C delta, apoptosis, cell proliferation, Animal Models of Human Disease, Atherosclerosis, Diabetes, Type 2
INTRODUCTION
Dyslipidemia accompanying systemic metabolic diseases, such as metabolic syndrome and type 2 diabetes, is a major risk factor for the development of atherosclerosis. The accumulation of cholesterol and its oxidized products in the arterial wall stimulates an inflammatory process that recruits monocyte into the vascular wall where they differentiate into macrophages and become foam cells to form atherosclerotic plaques1. Hyperlipidemia can also induce monocytosis that can enhance the uptake and accumulation of monocytes into the vascular wall2. Macrophage turnover in the atherosclerotic plaque can also be important since a high turnover rate within four weeks has been reported3. The removal of macrophages from atherosclerotic plaque is mainly through apoptosis since their ability to egress is significantly reduced after differentiation into foam cells4. Studies from atherosclerotic regression models support that macrophage apoptosis is the major pathway for macrophage removal from the plaque5. However, recent studies suggested that macrophages in the arterial wall/plaque can proliferate3. In diabetes, multiple studies have shown that hyperglycemia and lipids modified by glycation (glyLDL), oxidation (OxLDL), and acetylation (AcLDL) can activate monocytes and enhance their uptake into the arterial wall6, 7. Our laboratory has been characterizing PKC activation and its effects on cells involved in the atherosclerotic processes in the presence of hyperglycemia, dyslipidemia, and oxidative stress8–12. Many reports have shown that in diabetic and insulin resistant states, glucose and lipids, such as OxLDL, can activate PKC isoforms, especially the β isoform, to accelerate many pathological processes including atherosclerosis11–15. In the present study, we characterize the ability of OxLDL and TNFα to activate PKCδ isoform, and its effects on PKC isoform on monocyte/macrophage homeostasis in the vascular wall in the presence of obesity-induced insulin resistance and hypercholesterolemia. PKCδ isoform activation was studied since its expression and activity were found to be selectively increased in the monocytes by these conditions.
METHODS
Animals.
PKCδ flox/flox mice were generated, as previously described40. All protocols for animal use and euthanasia were reviewed and approved by the Animal Care Committee of the Joslin Diabetes Center, in accordance with NIH guidelines following the standards established by the Animal Welfare Acts and by the documents entitled “Principles for Use of Animals” and “Guide for the Care and Use of Laboratory Animals.”
Statistical analysis.
Comparisons of the two groups were made using a paired or unpaired t-test as appropriate. Comparison among more than two groups was performed by a one-way ANOVA or two-way ANOVA followed by the post hoc analysis with a paired or unpaired t-test to evaluate statistical significance between the two groups. Statistical significance was defined as p<0.05. In text and graphs, data were presented as the mean ± standard error.
RESULTS
Characterization of PKCδ activation in monocytes/macrophages of rodent models with diet-induced hyperlipidemia and diabetes.
PKC activities and the expressions of its isoforms were studied in PBMC isolated from ZDF rats that exhibit hyperlipidemia and hyperglycemia (Supplemental Table I) and in lean non-diabetic control rats (ZL). Total in situ PKC activities were increased by 2-fold in PBMC from ZDF compared to ZL rats (p<0.05) (Figure 1A). Expressions of PKC isoforms in PBMC showed that PKCδ mRNA were significantly increased by 2.9±0.6-fold in ZDF compared to ZL control rats (Figure 1B), but levels of PKCα or β isoforms were not changed (Figure 1B). PKC isoforms were also studied in circulating monocytes labeled with CD11b+Ly6G-, neutrophils with Ly6G+, B cells with CD19+, and T cells with CD3+. These cells, co-stained with anti- PKCδ antibodies, indicated that the expression of PKCδ in monocytes was 3.5, 4, and 7 folds higher than in neutrophils, B, and T cells, respectively (Figure 1C). Compared to ZL rats, PKCδ expression in the circulating monocytes and T cells were increased by 40% and 57%, respectively in ZDF rats, but were not different in the circulating neutrophils and B cells. PKCδ protein expression was evaluated in spleen macrophages, which increased significantly by 50% in ApoE−/− mice on 16 weeks of HFD compared with normal chow (NC) ( p<0.05, Fig 1D). Potential causes for the increased expressions of PKCδ in monocytes/macrophages due to hyperlipidemia and diabetes were studied using cultured BM macrophages initially with OxLDL and LPS, which increased PKCδ mRNA by 2.1±0.1- and 2.2±0.2-fold, respectively, and protein levels by 2.4±0.2- and 2.8±0.2-fold, respectively (Figures 1E-G). Further analysis was performed with inflammatory cytokines that are reported to be elevated in diabetes, including TNF-α, IL-6, or Ac-LDL which was used as a surrogate for modified LDL such as glycated or oxidized LDL, or fatty acids such as palmitic acid, oleic acid, or arachidonic acid on PKCδ expression. The results showed that TNFα and Ac-LDL increased gene expressions of PKCδ in the macrophages by 2 fold, whereas IL6 and fatty acids were ineffective (Supplemental Fig I). No differences were observed when macrophages were incubated with glucose levels of 5.6mM or 25mM. PKC activities, measured by the translocation of PKCδ proteins from cytosol to cell membrane, were increased after stimulation with either OxLDL or LPS for 8 hours, but not during the first 10 minutes. For positive control, the addition of PMA for 10 minutes increased the translocation of PKCδ to cell membrane by 5 fold (Supplemental Fig II).
Figure 1. Regulation of PKCδ isoform in monocytes/macrophages by hyperlipidemia, oxLDL and LPS.
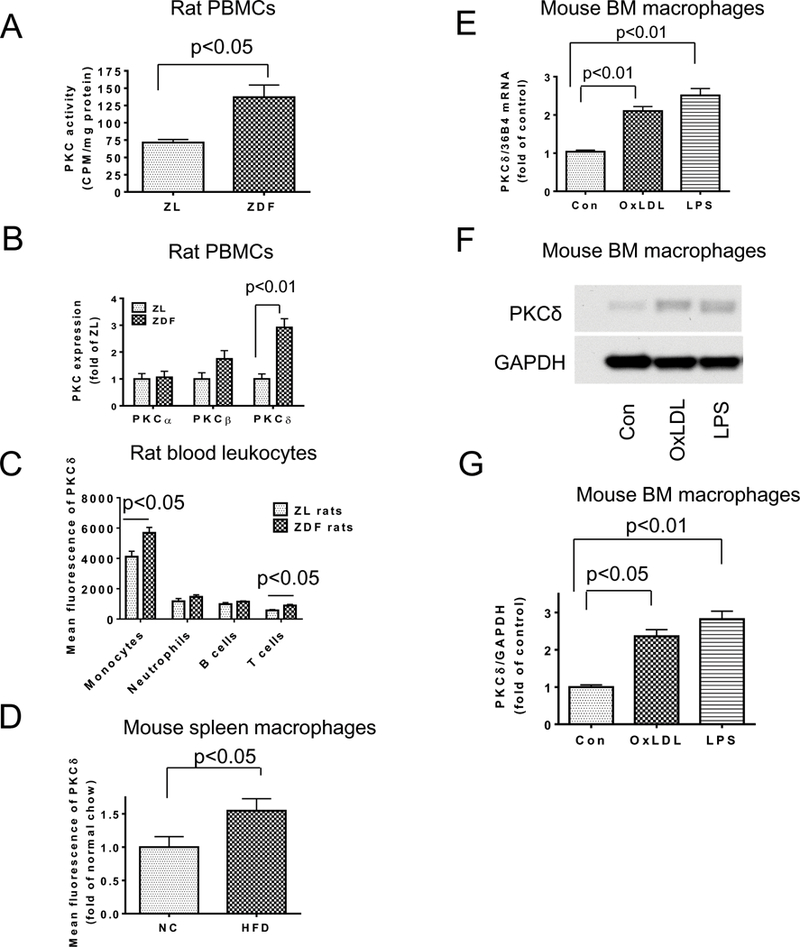
A. PKC activity in PBMC was measured by in situ PKC assay (n=6 per group). B. Expression of PKCα, β and δ in PBMCs from ZDF rats or ZL control rats was determined by qRT-PCR (n=4 per group). C. PKCδ expression in blood monocytes, neutrophils, B cells and T cells of ZDF rats or control rats was determined by flow cytometry (n=4 per group). D. PKCδ expression in spleen macrophages from mice fed with NC or HFD. Mean fluorescence of PKCδ in spleen macrophages was determined by flow cytometry (n=5 per group). E. BM macrophages were stimulated with oxLDL (50 ug/ml) or LPS (100 ng/ml) for 4 h and PKCδ gene expression was measured by qRT-PCR and normalized by 36B4. Control (Con), n=10; oxLDL, n=10; LPS n=6). (F-G). PKCδ protein expression in BM macrophages stimulated with oxLDL (50 ug/ml) or LPS (100 ng/ml) for 8 h was determined by Western blotting and normalized with GAPDH (n=4 per group).
Characterization of MPKCδKO/ApoE−/− mice.
To determine the physiological significance of PKCδ activation, macrophage specific deletion of PKCδ mice was made by breeding PKCδ flox/flox mice with lysozyme M Cre mice and then ApoE−/− mice to obtain MPKCδKO/ApoE−/− mice. As shown by qRT-PCR and Western blotting analysis (Supplemental Figure IIIA-B), PKCδ expression was deficient in cultured BM macrophages from MPKCδKO/ApoE−/− mice. Specificity of the PKCδ knockout was supported by the finding that protein expressions of PKCδ were not different in circulating B and T cells between MPKCδKO/ApoE−/− mice vs. ApoE−/− mice as measured by flow cytometry, although the expression of PKCδ was decreased in neutrophils (Supplemental Figures IIIC).
The Effect of PKCδ deletion on macrophage expression of inflammatory cytokines and functions.
Activation of PKC by PMA in BM macrophages from ApoE−/− mice significantly increased the expression of pro-inflammatory cytokines IL1B, MCP-1, TNFα, and anti-inflammatory cytokines, IL1 receptor antagonist (IL1RA) and arginase (Supplemental Figure IV). However, in macrophages from MPKCδKO/ApoE−/− mice, the effect of PMA on the expression of both pro- and anti-inflammatory cytokines was reduced dramatically (Supplemental Figure IV). In contrast, the addition of LPS and OxLDL, which increased the expression of IL1B, MCP-1, and TNFα production, did not result in differences in BM macrophages between ApoE−/− and MPKCδKO/ApoE−/− mice (Supplemental Figure V). Macrophage migration was evaluated by Transwell assay using vehicle, MCP-1, PMA, or PMA+MCP-1. Supp. Fig. VI showed that macrophages from MPKCδKO/ApoE−/− mice migrated faster (2 fold, p <0.05) than WT macrophages at basal level. MCP-1 increased macrophage migration in WT macrophages, which was inhibited by PKC activator PMA. Macrophages from MPKCδKO/ApoE−/− mice did not respond to PMA to inhibit the effects of MCP-1.
Macrophage phagocytosis, studied by the uptake of FITC-conjugated IgG coated latex beads, did not exhibit changes between macrophages derived from ApoE−/− or MPKCδKO/ApoE−/− mice, either at basal level or after PMA treatment. Similarly, monocyte differentiation to macrophages, characterized by using GM-CSF and assessing the presence of CD11b, F4/80 and CD3616, were not different between WT and MPKCδKO/ApoE−/− mice (Supplemental Figure 6D). Monocyte/macrophage differentiation was further determined in vivo. Thioglycollate was injected intraperitoneally. After 3 days, the expression of CD11b, F4/80, and CD36 were assessed by flow cytometry in circulatory monocytes (PI-CD45+CD115+) and peritoneal macrophages (PI-CD45+CD115+) and were significantly increased when circulatory monocytes differentiated into peritoneal macrophages with the injection of thioglycollate, but their expression again was not different between ApoE−/− mice and MPKCδKO/ApoE−/− mice, either in circulating monocytes or in peritoneal macrophages (Supplemental Figure VIE).
Effect of PKCδ deletion on atherosclerosis in mice that were fed an atherogenic diet (AD), and that had hyperlipidemia but no diabetes or insulin resistance.
Non diabetic and non-insulin-resistant MPKCδKO/ApoE−/− mice and ApoE−/− mice were studied after being fed an AD, which induced hypercholesterolemia. The body weight of MPKCδKO/ApoE−/− mice was slightly higher compared to ApoE−/− mice after 12 weeks of AD (Supplemental Figure VIIA). Mean blood pressure, fasting insulin, and IPGTT were similar between the two groups (Supplemental Figure VIIB-D). Plasma cholesterol and triglycerides were similar after 8 weeks of AD, but cholesterol (1142.3±25.4mg/dl vs. 1394.9±90.9mg/dl, p<0.05) and triglycerides (80.6±11.6 mg/dl vs. 140.3±19.6 mg/dl, p<0.05) were lower in MPKCδKO/ApoE−/− mice after 12 weeks of AD (Supplemental Figure VIIIA). FPLC showed VLDL cholesterol levels were lower in MPKCδKO/ApoE−/− after 8 and 12 weeks of AD (Supplemental Figures VIIIB-E), but plasma free fatty acid was not different between the two groups of mice. However, circulating numbers of monocytes, B and T cells were decreased in MPKCδKO/ApoE−/− compared to ApoE−/− mice after 12 weeks of AD (Supplemental Figure IX).
After 8 or 12 weeks of AD, the extent of atherosclerotic plaque area, as measured by en face staining with Sudan IV, increased by 2.1 (p<0.01) or 1.8 (p<0.05) fold, respectively, in the aorta of MPKCδKO/ApoE−/− mice compared to ApoE−/− mice (Figures 2A-B). However, the amount of atherosclerosis was similar in aortic roots (data not shown), but they were elevated by 3.3-fold in the abdominal aorta of MPKCδKO/ApoE−/− mice vs. ApoE−/− mice (6.0±1.1% vs. 1.8±0.3%, p<0.01). The macrophage (PI-CD45+CD11b+Ly6G- cells) in the aorta of MPKCδKO/ApoE−/− mice was increased by 1.7-fold in MPKCδKO/ApoE−/− mice compared to ApoE−/− mice (14109±2569 cells/aorta vs. 8447±1130 cells/aorta, p<0.05) (Figure 2C). The complexity of the atherosclerotic plaques, assessed by macrophage content (MAC2 staining), increased by 1.7-fold in the aortic lesions from MPKCδKO/ApoE−/− mice vs. ApoE−/− mice (52.7±5.6% vs. 30.5±5.4%, p<0.05) (Figures 2D-E). The number of smooth muscle cells (SMC) in the plaques, showing a positive staining to α-SMC actin, decreased by 31% in MPKCδKO/ApoE−/− mice vs. ApoE−/− mice (p<0.05) (Supplemental Figure X A-B), but the collagen contents and areas of necrosis in the plaques did not differ (Supplemental Figure X C-H).
Figure 2. Atherosclerosis and macrophages in the aorta of MPKCδKO/ApoE−/− and ApoE−/− mice on atherogenic diet (AD).
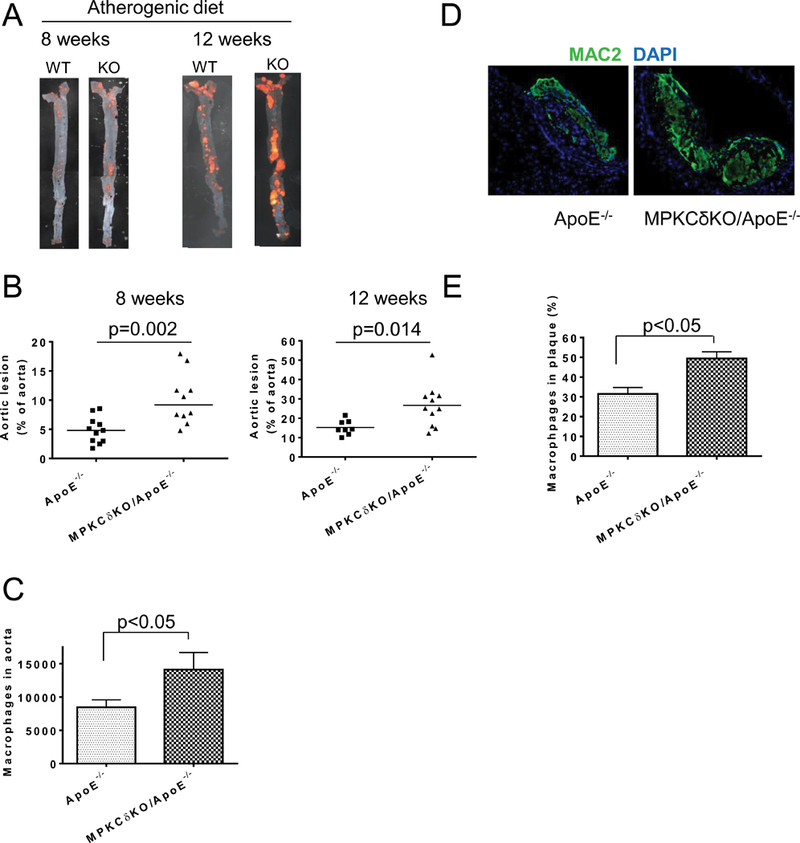
A. Representative en face Sudan IV staining of aorta. B. Quantitative analysis of atherosclerotic lesions. Left, AD at 8 weeks (ApoE−/−, n=11; MPKCδKO/ApoE−/−, n=10) and right AD at 12 weeks (ApoE−/−, n=8; MPKCδKO/ApoE−/−, n=11). C. Macrophage numbers in the aorta of mice fed with AD for 12 weeks. Aorta was digested into single cell and PI-CD45+CD11b+Ly6G- macrophages were determined by flow cytometry (ApoE−/−, n=14; MPKCδKO/ApoE−/−, n=11). (D-E). Immunostaining of the abdominal aorta of ApoE−/− mice or MPKCδKO/ApoE−/− mice with MAC2 on AD for 12 weeks. D. Representative images. E. Quantitative analysis (n=5 for each group).
Macrophage apoptosis and proliferation in the aorta.
The paradoxical findings of decreased levels of circulating monocytes and elevated levels of macrophage content in the aortic wall suggested that turnover of macrophage could be changed in MPKCδKO/ApoE−/− mice. Macrophage apoptosis in atherosclerotic plaques, determined by TUNEL and MAC2 staining, was significantly lower in the aortic plaques of MPKCδKO/ApoE−/− mice compared with ApoE−/− mice (0.37±0.10% vs. 1.17±0.26%, p<0.05) (Figure 3A). Proliferation of macrophage assessed by double staining of Ki67and MAC2, was increased by 2.7-fold in the aortic plaque of MPKCδKO/ApoE−/− mice compared to ApoE−/− mice (9.6±6.1/plaque vs. 3.5±0.8/plaque, p<0.05) (Figures 3B). The extent of cellular proliferation was confirmed by the number of isolated aortic cells incorporating BrdU. The aorta from MPKCδKO/ApoE−/− mice contained 2.3-fold more BrdU /CD11b positive cells than ApoE−/− mice (p<0.05) (Figure 3C). The increase in BrdU+ macrophages in the aortic wall could also be due to the recruitment of circulating BrdU+ monocytes. However, pertussis toxin, an inhibitor of monocyte recruitment17, reduced monocyte recruitment into the peritoneal cavity by 70%, but did not reduce the elevation of BrdU+ macrophages in the aorta of MPKCδKO/ApoE−/− mice vs. ApoE−/− mice (Figure 3C).
Figure 3. Assessment of apoptosis and proliferation in the aorta on AD.
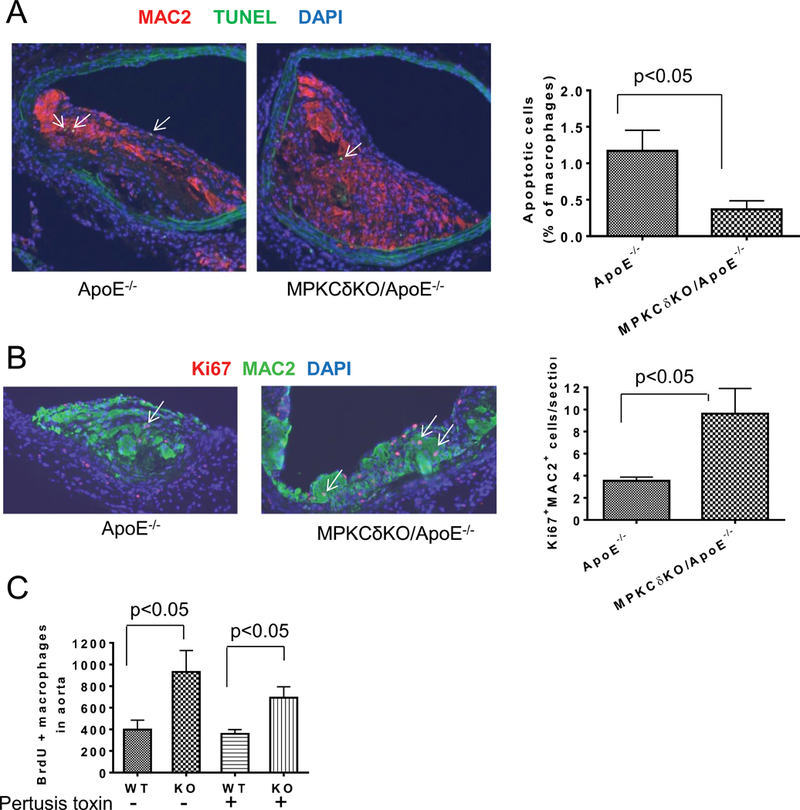
A. Double staining of the abdominal aorta of mice with MAC2 antibody and TUNEL. Left, representative images and right, quantitative analysis (ApoE−/−, n=7; MPKCδKO/ApoE−/− n=8). (B-C). Macrophage proliferation. B, MAC2 and Ki67 double staining in atherosclerotic plaque. Left, representative images. Right, quantification of the number of MAC2 and Ki67 double positive cells in the plaque (ApoE−/−, n=7; MPKCδKO/ApoE−/− n=8). C. BrdU positive macrophages in the aorta. The mice were infused with BrdU with or without pertussis toxin pretreatment for 3 days. Aorta was digested into single cells and BrdU positive macrophages in the aorta were determined by flow cytometry (ApoE−/− without pertussis toxin, n=7; MPKCδKO/ApoE−/− (KO) without pertussis toxin, n=6; ApoE−/− and MPKCδKO/ApoE−/− with pertussis toxin, n=7 per group).
Inflammatory cytokine expression in the aorta and lipid uptake into macrophages.
After 12 weeks of AD, mRNA expressions of F4/80, IL2, CCL5 and CXCL9 were elevated significantly in the aorta of MPKCδKO/ApoE−/− mice by 1.6±0.3, 2.1±0.8, 2.5±0.8, 4.4±3.0-fold, vs. ApoE−/− mice, respectively (Supplemental Figure XI).
PKC activation has been reported to regulate the uptake of LDL in monocytes via CD36, which can be important in the formation of foam cells18, 19. The uptake of AcLDL by peritoneal macrophages, characterized by using Alexa488 labeled AcLDL, did not differ between MPKCδKO/ApoE−/− and ApoE−/− mice (Supplemental Figure XII).
Effect of HFD on ApoE−/− and MPKCδKO/ApoE−/− mice.
The severity of atherosclerosis in MPKCδKO/ApoE−/− mice on HFD was studied since HFD with 60% of calories from fat, unlike AD, induces obesity, insulin resistance, and hyperglycemia in addition to hyperlipidemia20. The body weight of MPKCδKO/ApoE−/− mice and ApoE−/− mice were similar either on NC or HFD for 16 weeks (Supplemental Figure XIIIA), although HFD increased body weight significantly in both types of mice compared to NC. Plasma cholesterol and triglyceride levels were not different between the two groups of mice on NC, but cholesterol (490.0±12.6 mg/dl vs. 577.3±21.9 mg/dl, p<0.01) and triglycerides (89.3±4.4 mg/dl vs. 121.9±11.8 mg/dl, p<0.05) were lower in MPKCδKO/ApoE−/− mice after 16 weeks of HFD (Supplemental Figures XIIIB-C). FPLC showed VLDL cholesterol levels were lower in MPKCδKO/ApoE−/− mice after 16 weeks of HFD compared to ApoE−/− mice (Supplemental Figure XIIID). Blood pressure (Supplemental Figure XIIIE), IPITT, and IPGTT (Supplemental Figure XIV) were similar between the two groups of mice on HFD, but HFD increased blood glucose levels at 30 min to greater than 500mg/dl, indicating mild diabetes.
Circulating monocyte levels were not different on NC, but their levels in MPKCδKO/ApoE−/− mice fed with HFD for 4 weeks showed a 55% decrease (p=0.08, Figure 4A) compared to ApoE−/− mice, whereas circulating neutrophils, B cells, and T cells were similar in the two groups of mice on NC or HFD after 4 weeks (Figures 4B-D). After 16 weeks on HFD, circulating monocytes, neutrophils, B cells, and T cells were all significantly decreased in MPKCδKO/ApoE−/− compared to ApoE−/− mice (Figures 4A-D). Because the circulating white blood cells were significantly decreased in MPKCδKO/ApoE−/− mice, we examined the following cells in the BM: hematopoietic stem cells (HSC, Lin-Sca-1+c-Kit+), monocytes (CD45+CD115+), neutrophils (CD45+Ly6G+), B cells (CD45+CD19+), and T cells (CD45+TCRβ+), which were not different in the BM of the two groups of mice fed either NC or HFD for 4 weeks (Figure 4E-F).
Figure 4. Analysis of leukocytes in mice fed with NC or HFD for 4 (4W) or 16 weeks (16W).
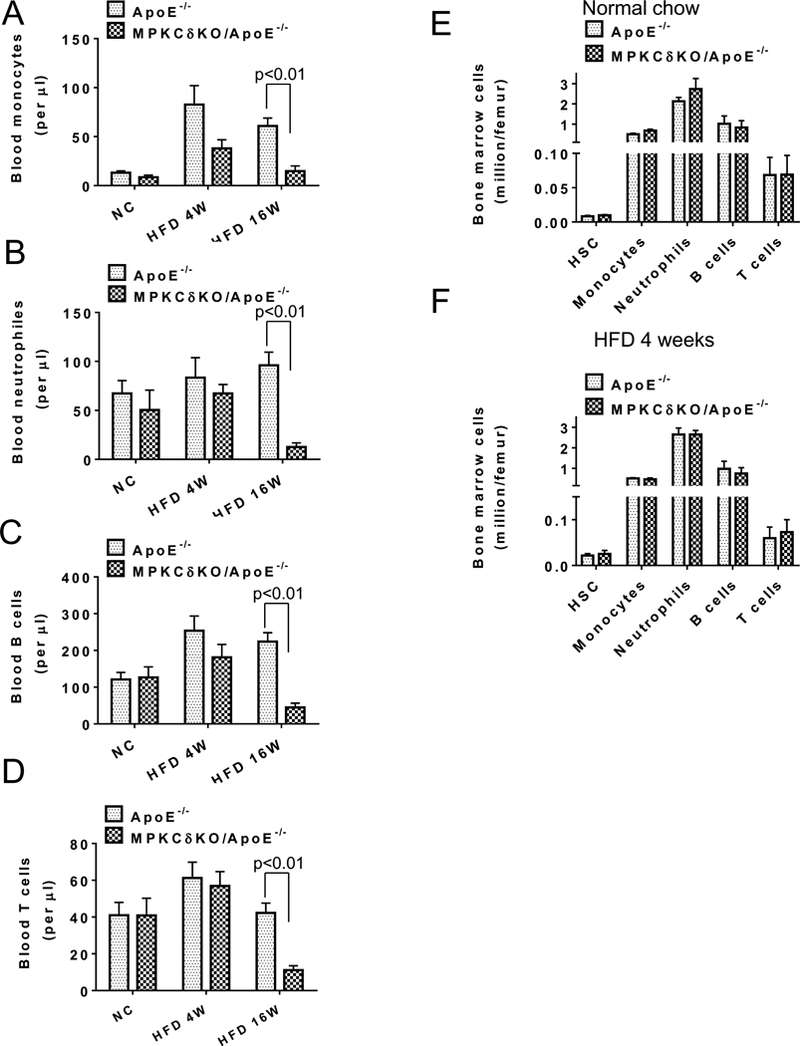
(A-F). Circulating monocytes (A), neutrophils (B), B cells (C), T cells (D) were determined by flow cytometry. (ApoE−/− NC, n=6; MPKCδKO/ApoE−/− NC, n=5; ApoE−/− HFD 4W, n=7; MPKCδKO/ApoE−/− HFD 4W, n= 7; ApoE−/− HFD 16W, n=7; MPKCδKO/ApoE−/− HFD 16W, n=8). E. Bone marrow cells in mice under normal chow (n=3–6 per group). F. Bone marrow cells in mice under HFD for 4 weeks. (n=3–6 per group).
Severity of atherosclerotic lesions and macrophage contents on HFD.
After 16 weeks of HFD, even with decreased levels of plasma cholesterol, the extent of the atherosclerotic plaque area increased by 1.6-fold in the aorta of glucose intolerant MPKCδKO/ApoE−/− mice compared to ApoE−/− mice (5.62±0.64% vs. 3.57±0.35%, p<0.01) (Figure 5A-C). The collagen content also increased in the plaque of MPKCδKO/ApoE−/− mice, but the difference was not significant (Figures 5B and 5D). The content of macrophages, estimated in the plaque of HFD MPKCδKO/ApoE−/− mice by MAC2 staining, was 48% greater than HFD ApoE−/− mice (p<0.05, Figures 5E-F).
Figure 5. Measurements of atherosclerotic lesions and macrophage contents in mice fed on HFD for 16 weeks.
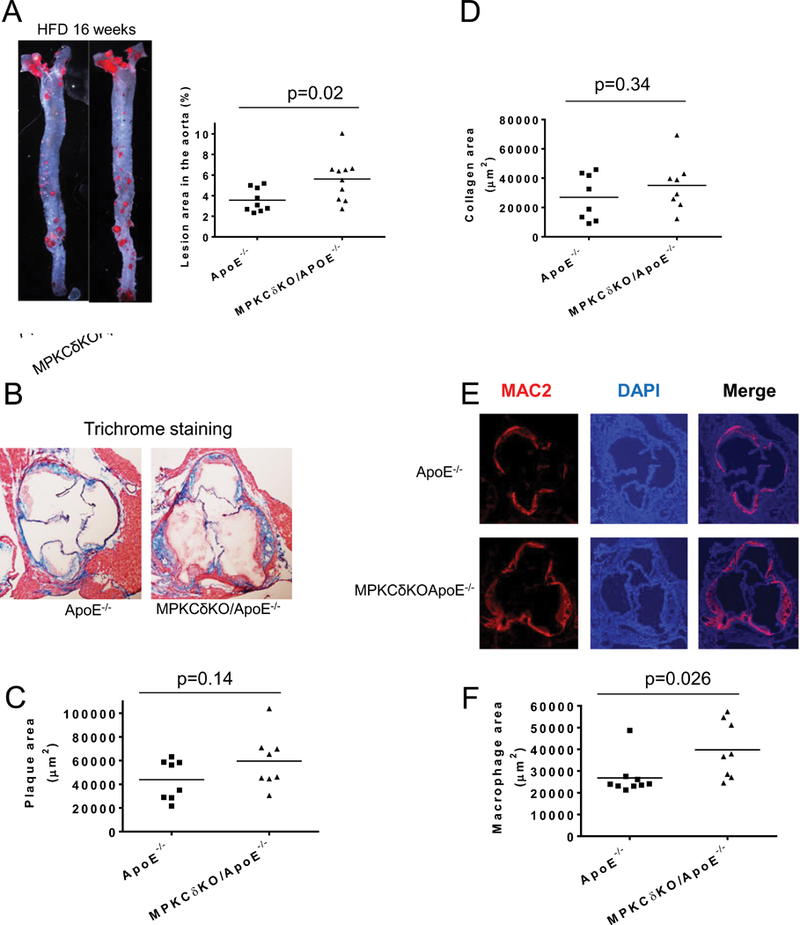
A. Representative en face Sudan IV staining of aorta (left) and quantitative analysis of the ratio of atherosclerotic lesions to the total aortic area (right) (ApoE−/−, n=9; MPKCδKO/ApoE−/−, n=10). (B-D). Trichrome staining with representative images (B) and quantitative analysis of plaque area (C). D. Collagen content as shown (ApoE−/−, n=8; MPKCδKO/ApoE−/−, n=8). (E-F). Macrophage staining. Aorta roots were stained with anti-MAC2 antibody. (E), representative images and (F), quantitative analysis (ApoE−/−, n=9; MPKCδKO/ApoE−/− n=8).
Analysis of macrophage apoptosis in the aortic plaques on HFD.
PKCδ activation is known to induce apoptosis in multiple cell types21–24. Therefore, macrophage apoptosis was compared in the aortic plaque of MPKCδKO/ApoE−/− and ApoE−/− mice, by MAC2 and TUNEL double staining. The results showed that the number of macrophages exhibiting apoptosis was decreased by 42% in MPKCδKO/ApoE−/− compared to ApoE−/− mice on HFD (p<0.05, Figure 6A). The proliferation of macrophages, as determined by MAC2 and Ki67 double staining, increased by 2.6-fold in the aortic plaque of MPKCδKO/ApoE−/− mice compared to ApoE−/− mice on HFD (12.9±2.5/section vs. 5.1±1.1/section, p<0.05) (Figures 6B). The differences in apoptosis in macrophages in cultured BM macrophages derived from MPKCδKO/ApoE−/− vs. ApoE−/− mice were also studied. By withdrawing growth factor or OxLDL, plus a low dose of thapsigargin, an inducer of ER stress and a method to induce macrophage apoptosis25, BM macrophages from MPKCδKO/ApoE−/− mice exhibited approximately 50% less apoptosis than those from ApoE−/− mice (p<0.05; Figure 6C).
Figure 6. Macrophage apoptosis and proliferation in the aorta and BM macrophages of mice fed with HFD for 16 weeks.
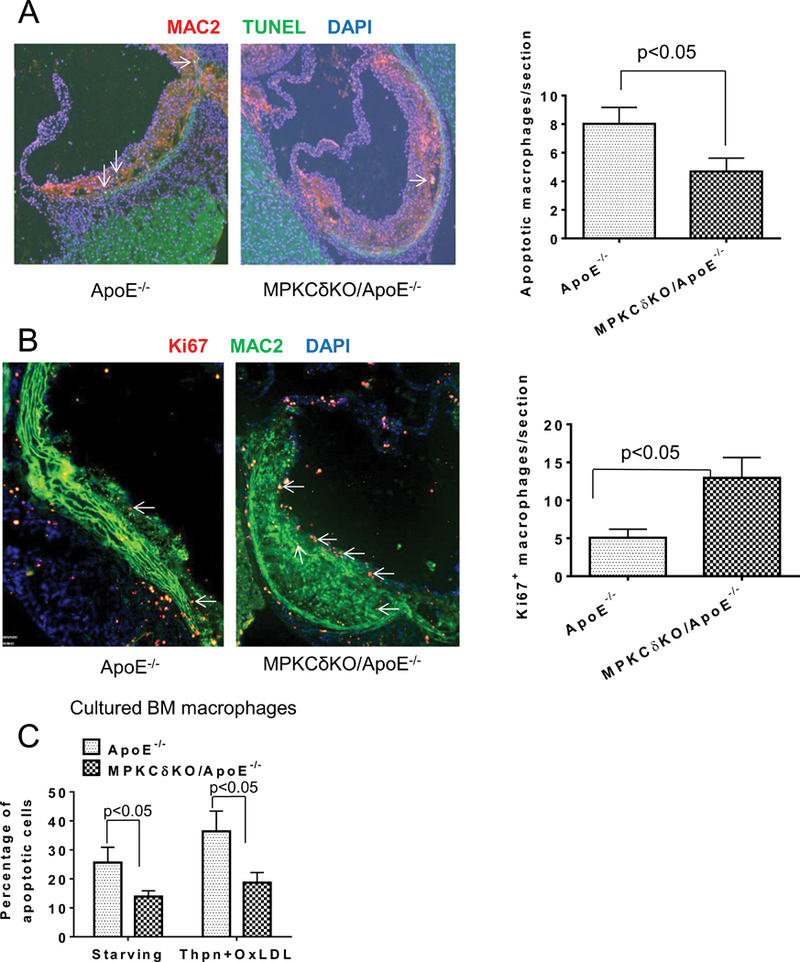
Macrophage apoptosis(A) and proliferation(B) in the aortic root were determined by MAC2 and TUNEL double staining. Left, representative images and right, quantitative analysis. (ApoE−/−, n=9; MPKCδKOApoE−/−, n=9). C. Apoptosis in cultured BM macrophages induced by withdrawing growth factor or treatment with thapsigargin (Thpn, 0.25 uM) plus oxLDL (50 ug/ml). Apoptotic cells (Annexin V and PI double positive cells) were determined by flow cytometry (n=3 per group).
Characterization of plaque stability.
Plaque stability was evaluated by the necrotic area, the extent of smooth muscle cells, fibrosis cap, and collagen. The area of necrosis was not different in the plaques between ApoE−/− and MPKCδKO/ApoE−/− mice (Supplemental Figure XE, H and Supplemental Figure XVC). Collagen content (Figure 5D, Supplemental Figure XD, G) and fibrosis cap (Supplemental Figure XVB) were also comparable between ApoE−/− and MPKCδKO/ApoE−/− mice. The percentage of smooth muscle cells was decreased in the plaque of MPKCδKO/ApoE−/− mice (Supplemental Figure XA) compared to ApoE−/− mice (p < 0.05). The result of increased macrophage content and decreased vascular smooth muscle cells in the plaque suggested that the atherosclerotic plaque in the aorta of MPKCδKO/ApoE−/− mice could be less stable than those in ApoE−/− mice.
Characterization of Akt phosphorylation and its signaling in macrophages.
PKC activation can inhibit Akt phosphorylation (pAkt) and its signaling of several growth factors to increase apoptosis10, 23. Basal pAkt levels were 5.3-fold higher in MPKCδKO/ApoE−/− (Figure 7A) compared to ApoE−/− mice. Activation of PKC with PMA significantly inhibited pAkt (by 78%) in ApoE−/− mice, but in PKCδ deficient macrophages, pAkt levels remained higher than cells from ApoE−/− mice after PMA stimulation (p<0.01). Incubation of BM macrophages with thapsigargin and OxLDL for 3 hours inhibited pAkt by 63% in control macrophages, but their effect on pAkt in MPKCδKO/ApoE−/− mice was reduced only by 12% (p<0.05) (Figure 7B). Thapsigargin dramatically inhibited pAkt and its effects were further enhanced by OxLDL in ApoE−/− macrophages (Figure 7C). This data was consistent with a previous report that OxLDL can enhance low dose thapsigargin induced macrophages apoptosis25. In contrast, OxLDL didn’t enhance thapsigargin’s inhibition effects on pAkt in PKCδ deficient macrophages (Figure 7C). The pAkt levels in the splenic macrophages, determined by flow cytometry, showed that pAkt levels were increased by 52% in macrophages from MPKCδKO/ApoE−/− mice compared to ApoE−/− mice (Figure 7D).
Figure 7. Akt and P85 phosphorylation in cultured BM macrophages.
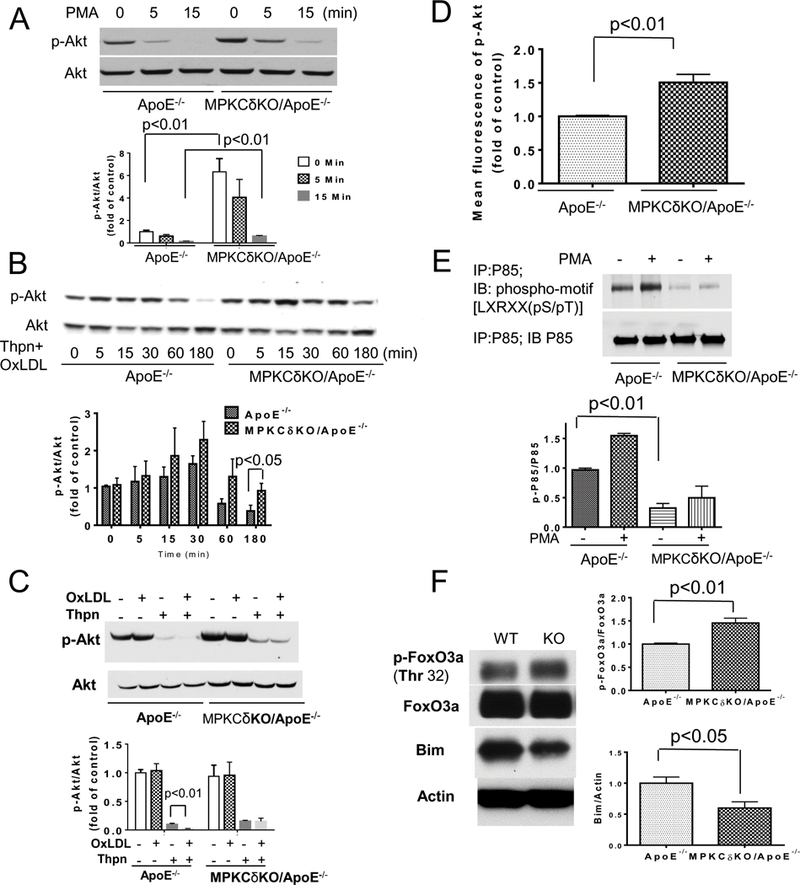
A. p-Akt at basal level (starved with DMEM containing 1% FBS and 2% conditioned L929 medium for 16 h) and after PKC activation induced by 100 nM PMA was determined at indicated times (n=4 per group).(B-C). P-Akt after Thpn (0.25 uM) plus OxLDL (50 ug/ml) stimulation in macrophages cultured in growth medium (DMEM with 10% FBS and 20% conditioned L929 medium ) for indicated times (B, n=5–7 per group) or after OxLDL (50 ug/ml), or Thpn (0.25 uM), or OxLDL plus Thpn stimulation for 3 hours (C, n=6 per group). D. pAkt in isolated spleen macrophages which were measured by flow cytometry (ApoE−/−, n=7; MPKCδKO/ApoE−/−, n=6). E. P85 phosphorylation was determined by antibody which recognizes PKC phosphorylation sites (n=3 per group). F. FoxO3a phosphorylation and Bim expression in macrophages starved with DMEM containing 1% FBS and 2% condition L929 medium for 16 h (n=8 per group).
Levels of Akt in the macrophage cell line Raw264.7 was reduced (by 92%) by lentivirus containing Akt shRNA, compared to control shRNA, which paralleled increases in cellular apoptosis, as determined by Annexin V and PI staining by 35%. Compared with cells grown in medium containing serum, withdrawing growth factor increased apoptosis by 266% in macrophages from ApoE−/− mice in contrast to 46% inhibition in macrophages from MPKCδKO/ApoE−/− mice. Inhibition of Akt by wortmannin, a specific inhibitor of PI3 kinase, further increased macrophage apoptosis by 400% in both type of cells, indicating that inhibition of Akt activation will induce apoptosis in macrophages (Supplemental Figure XVIC).
The effect of deleting PKCδ on the phosphorylation of P85, a subunit of PI3Kinase, was studied since the PKC-induced phosphorylation of P85 has been reported to inhibit the activation of PI3Kinase9, 26. PKC induced phosphorylations of P85/PI3K of macrophages between ApoE−/− and PKCδ deficient mice were identified by anti-phospho-motif antibody, which recognized PKC phosphorylation sites on P85/PI3Kinase30. PMA increased P85 phosphorylation by 1.6-fold in ApoE−/− macrophages (Figure 7E). In contrast, PMA-induced P85 phosphorylation increased by less than 10% (Figure 7E) in MPKCδKO/ApoE−/− mice.
Effects of PKCδ to inhibit Akt activation through ser/thre phosphorylation of IRS2 was also studied since macrophages mainly expressed IRS227. We have previously identified that Ser343 on IRS2, could be phosphorylated by PKC activation to inhibit insulin-induced pAkt in endothelial cells8, 28. In the macrophages, the phosphorylation of Ser343 on IRS2 was increased by PKC activation using PMA in WT macrophages, but was not observed in PKCδ deficient macrophages (Supplemental Figure XVII).
One downstream action of pAkt is to inhibit transcriptional activities of FoxO by its phosphorylation levels29. Parallel with increased pAkt, FoxO3a phosphorylation at threonine 32 was increased by 42% in PKCδ deficient macrophages compared to ApoE−/− mice (Figure 7F, p<0.01). Bim, a BH3-only member of BCl2, is a downstream target of FoxO3a activation30. Consistent with increased phosphorylation of FoxO3a, Bim expression was decreased by 41% in BM macrophages from MPKCδKO/ApoE−/− compared to ApoE−/− mice (p<0.05, Figure 7F). Similarly, we confirmed the reduction of Bim expression in circulating monocytes (CD45+CD11b+Ly6G-) positive to anti-Bim antibody, which decreased by 50% in monocytes from MPKCδKO/ApoE−/− compared to ApoE−/− mice (Supplemental Figure XVIII). Further, PKCδ was overexpressed by 6.8-fold in Raw 264.7 cells by lentivirus containing PKCδ full length plasmid. In parallel with PKCδ overexpression, the expression of Bim was increased by 3.8-fold, whereas the level of pAkt and p-FoxO3a was decreased by 53% and 39%, respectively (p<0.05, Supplemental Figure XIX). Overexpression of PKCδ also inhibited cell proliferation by 72% and increased cell apoptosis by 67% (p<0.05, Supplemental Figure XIX). When the expression of PKCδ in the macrophages of MPKCδKO/ApoE−/− mice was re-constituted by infection with lentivirus containing full length plasmid of PKCδ, which increased its expression by 2 folds, Akt and FoxO3a phosphorylation levels were significantly decreased after treatment with thapsigargin plus OxLDL (Supplemental Figure XXI).
Effect of AD or HFD diet on monocyte expansion in the spleen.
Spleen to body ratio by weight was studied in MPKCδKO/ApoE−/− mice due to the splenomegaly that was noted when these mice were fed chronically with either AD or HFD diet (Figure 8A). Interestingly, no differences in spleen size between MPKCδKO/ApoE−/− mice and ApoE−/− mice were observed while they were fed with NC (Figure 8B). However, after 8 and 12 weeks of AD, the spleen body ratios were higher in MPKCδKO/ApoE−/− mice than that in ApoE−/− mice by 1.6 and 2.5-fold, respectively (p<0.05) (Figures 8A-B). HE staining showed that the red pulp of spleen from MPKCδKO/ApoE−/− mice were greatly expanded, and immunofluorescence staining for F4/80 demonstrated that macrophages content in the red pulp was significantly more than ApoE−/− mice (Supplemental Figure XXII), which was confirmed by a 2-fold increase of F4/80 positive cells in the spleen, per flow cytometry, in MPKCδKO/ApoE−/− mice compared to ApoE−/− mice (Figure 8C). However, on HFD, the weight of spleen of MPKCδKO/ApoE−/− mice was the same as ApoE−/− mice after 4 weeks, and was 1.7 fold greater only after 16 weeks in MPKCδKO/ApoE−/− vs. ApoE−/− mice (Supplemental Figure XXIIIA). Spleen cellular content showed that the macrophage (CD45+CD19-TCRβ-F4/80+) numbers were not different when fed with NC, but the macrophage numbers in MPKCδKO/ApoE−/− mice were elevated by 2-fold compared to ApoE−/− mice when fed HFD for 16 weeks (Supplemental Figure XXIIIB, p<0.05).
Figure 8. Analysis of splenomegaly and its macrophages on AD.
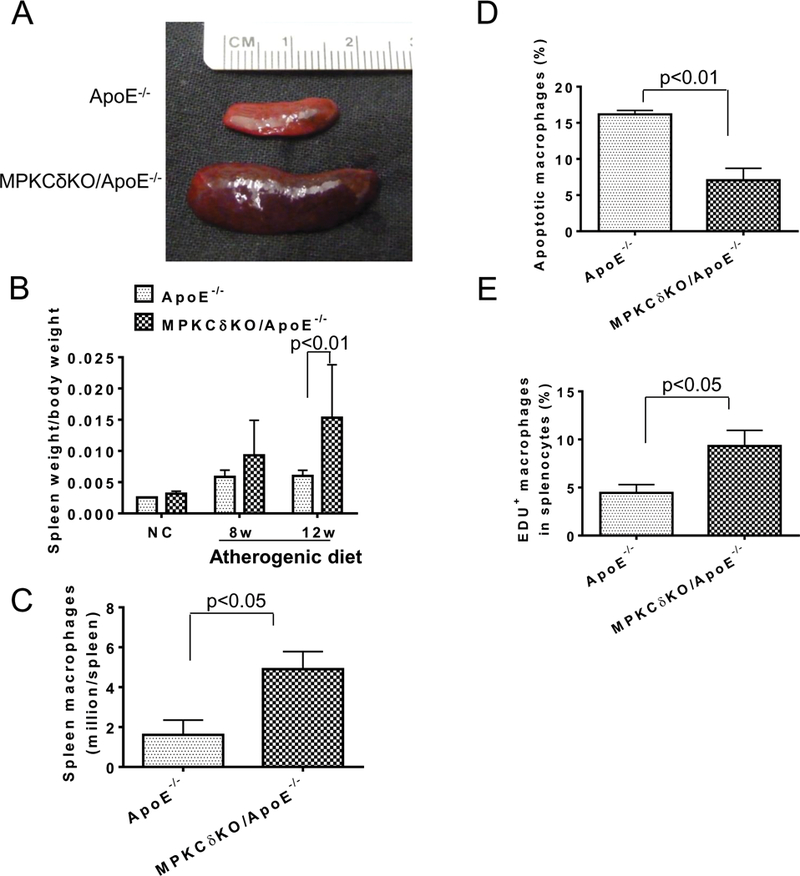
(A-B). Splenomegaly on AD. A. Representative image of spleen from mice fed with AD for 12 weeks. B. Quantification of spleen/body weight ratio (NC, n=3; AD 8 weeks, ApoE−/−, n=5; MPKCδKO/ApoE−/−, n=7; AD12 weeks, ApoE−/−, n=13; MPKCδKO/ApoE−/−, n=11). C. Splenic macrophages after AD for 12 weeks. Macrophages (PI-CD45+F4/80+) in the spleen were determined by flow cytometry (ApoE−/−, n=3; MPKCδKO/ApoE−/−, n=4). D. Apoptotic macrophages (CD45+F4/80+Annexin V+PI+) in the spleen after AD for 12 weeks were determined by flow cytometry (ApoE−/−, n=3; MPKCδKO/ApoE−/−, n=4). E. EDU incorporation into splenic macrophages. EDU and F4/80 double positive cells in the spleen of mice fed with AD for 12 weeks were determined by flow cytometry after 18 h of EDU injection (n=5 per group).
Analysis of macrophage proliferation and apoptosis in the spleen.
Apoptosis in the splenic macrophages was determined by labeling with antibodies to CD45, F4/80, annexin V and PI, which showed that the apoptosis was decreased by 57% in cells from MPKCδKO/ApoE−/− mice compared to ApoE−/− mice (Figures 8D, p<0.01). The extent of macrophage proliferation, assessed by Ki67 incorporation, was markedly increased in the spleen of MPKCδKO/ApoE−/− mice compared to ApoE−/− mice (Supplemental Figure XXIVA). Further, the percentage of cells that incorporated EDU and positive for F4/80+ increased by 2.1-fold in MPKCδKO/ApoE−/− compared to ApoE−/− mice (9.3±1.5% vs. 4.4±0.8%, p<0.05) (Figure 8E). Lastly, the cell cycle of isolated splenocytes in the G0/G1 phase was decreased (75.93±3.59% vs. 86.00±1.85%, p<0.05) and in the S phase were increased (13.4±1.75 vs. 6.34±3.15%, p<0.05) in MPKCδKO/ApoE−/− mice compared to ApoE−/− mice (Supplemental Figure XXIVB). The extent of apoptosis of splenic macrophages in mice fed with HFD, determined by Annexin V and PI double staining, was decreased by 63% in MPKCδKO/ApoE−/− vs. ApoE−/− mice (Supplemental Figure XXV, p<0.05).
DISCUSSION
Our initial observation showed that in ZDF rats, a model of hyperlipidemia, obesity, diabetes, and insulin resistance, PKC, especially the δ isoform in the monocytes, was activated and overexpressed. This is consistent with previous reports that several PKC isoforms α, β, and δ are preferentially activated in monocytes and vascular cells by diabetes and insulin resistance. Overexpression of PKCβ in macrophages exhibited an increase in FBS-stimulated proliferation by 80% (Supplemental Figure XX). In contrast, PKCδ activation, induced by lipids, exhibited pro-apoptotic actions in monocytes/macrophages, which reduced inflammation and severity of atherosclerosis in insulin resistant or hyperlipidemic states.
The elevation of PKCδ isoform expression in PBMC of diabetic and hyperlipidemic rodents is likely due to increased lipid levels or inflammatory cytokines since elevated glucose levels were ineffective. The ability of altered lipids (OxLDL, AcLDL) or cytokines (TNFα) to increase the expression of PKCδ is consistent with previous reports that oxidative stress or inflammation can increase its expression via activation of SP1 promoter 5’ regions of PKCδ gene31. The surprising results are that PKCδ deletion targeted to the macrophages increased the severity of atherosclerosis in MPKCδKO/ApoE−/− mice, which occurred even in the presence of lower total circulating monocytes, cholesterol, and triglycerides levels in MPKCδKO/ApoE−/− mice. Initially, we assumed the activation of PKCδ in the monocyte could contribute to the enhancement of atherosclerosis caused by insulin resistance in diabetes, possibly due to PKCδ activation by angiotensin. The specificity of the effect is mainly attributed in the monocyte since no changes in PKCδ expression were observed in the T and B cells of PKCδ KO/ApoE−/− mice. However, the contribution of neutrophils to the extent of atherosclerosis will need to be clarified since PKCδ expression was decreased in the neutrophils of MPKCδKO/ApoE−/− mice.
The pathology of the atherosclerotic plaques in MPKCδKO/ApoE−/− mice indicated that the increased severity of atherosclerosis was related to elevated numbers of macrophages and decreased smooth muscle cells and collagen content. These changes suggest that the plaque may be less stable than those in ApoE−/− mice. The increased numbers of macrophages in the atherosclerotic plaque was likely responsible for the observed elevated expression of inflammatory cytokines IL2, CCL5, and CXCL9 in the aorta of MPKCδKO/ApoE-/−. Interestingly, results from the in vitro studies showed that PKCδ isoform deletion decreased the productions of both pro- and anti-inflammatory cytokines in response to PKC activators, but was not changed to LPS. However, deletion of PKCδ isoform in monocytes did elevate macrophage migration, which indicates increased macrophage activation. Thus, the elevations of inflammatory cytokines in vivo are likely the results of increased numbers of activated macrophages in the aortic wall.
Surprisingly, these findings suggested that the mechanisms for the increased accumulation of macrophages in the aortic plaque in MPKCδKO/ApoE−/− mice were the result of elevated proliferation and decreased apoptosis, rather than monocytes/macrophages differentiation or monocyte uptake. Most studies in obesity-related or diabetic models of atherosclerosis have shown that elevated levels of circulating monocytes and their uptake are the cause for increased macrophage in the aortic wall32. However, the present study showed that homeostasis of macrophages turnover in the aortic wall in association with hyperlipidemic states could be important for the severity of atherosclerotic plaques. The increases in PKCδ expression and activation induced by oxidized or glycated lipids can enhance apoptosis and inhibit proliferation, thus lowering macrophage numbers and inflammatory cytokine levels. These effects of PKCδ on monocytes/macrophages are consistent with several other studies on its effects on inflammatory and immune cells. For example, activation of PKCδ has been reported to decrease B cell proliferation and associated with splenomegaly and autoimmune diseases33–35,36. Elevation of PKCδ in capillary pericytes enhanced its apoptotic rates and caused retinopathy21. Our results showed that PKCδ deletion or reduction promoted macrophage survival in the plaque and accelerated the development of atherosclerosis. In addition, the stability of the plaque may be decreased in MPKCδKO/ApoE−/− mice vs. ApoE−/− mice with more macrophages and less VSMC. PKCδ activation induced specifically by hyperlipidemia, and not hyperglycemia, probably has a critical role in modulating the progression of early atherosclerotic lesions.
The mechanism by which PKCδ is regulating macrophage proliferation and apoptosis is related to the inhibition of Akt, which is not surprising since activation of Akt is known to be anti-apoptotic and to enhance proliferation in many cells, including macrophages37. Our results supported this mechanism since pAkt was higher in PKCδ deficient macrophages and was inhibited by re-constituting PKCδ in the PKCδ deficient macrophages. The target of PKCδ activation to inhibit pAkt is likely related to the phosphorylation of IRS2/PI3 kinases, reported in endothelial cells8, 9. Previously, we showed that PKCδ can decrease pAkt by phosphorylating ser343 on IRS2, the pre-dominant mediators of insulin receptor’s actions in monocytes, and P85. We showed that phosphorylation of FoxO3a was increased in PKCδ deficient macrophages, which resulted in reduced expression of pro-apoptotic gene Bim. Thus, elevated glycated or oxidized lipids caused increases in PKCδ expression and activation to regulate apoptosis through IRS2/PI3K -Akt-FoxO3a-Bim pathway in macrophages.
The modulating effect of PKCδ on monocyte/macrophage survival and proliferation is not limited to the vascular wall, as shown by splenomegaly in MPKCδKO/ApoE−/− mice fed only on HFD and AD. The splenomegaly in MPKCδKO/ApoE−/− mice is responsible for the decrease in circulating neutrophils, T and B cells since their blood levels were not diminished after 4 weeks on HFD, and no changes were noted in the BM. These findings have emphasized the important contributions of macrophage turnover in the arterial wall on the severity of atherosclerosis. There is substantial evidence to support the idea that macrophages can proliferate in the arterial wall3, 17, 38. The proliferation of macrophages in the atherosclerotic plaques in humans and animals has been reported by several groups3, 17, 38. Our study clearly demonstrated that macrophages can proliferate in the aortic wall with significant elevation of Ki67 and MAC2 double positive cells in the MPKCδKO/ApoE−/− mice vs. ApoE−/− mice.
To exclude the possibility that PKCδ deletion affected hematopoiesis, we examined HSC, monocytes, neutrophils, B cells, and T cells in bone marrow, which did not differ in the two groups of mice under NC or HFD after 4 weeks (Figure 4E-F). This finding suggests that PKCδ deletion targeted to the monocyte/macrophages did not affect the development of monocytes, neutrophils, B cells, and T cells in the bone marrow. This is not surprising since previous reports have shown that whole body knockout of PKCδ resulted in decreases in B cell apoptosis with increased bone marrow and circulating B cell numbers while leaving T cells and other myeloid cells unchanged34. Thus, we believe the specific decreases in B and T cells in circulation in MPKCδKO/ ApoE−/− mice were mainly due to the function of splenomegaly, which is known to increase the uptake and clearance of circulating blood cells, likely due to increased monocytes or macrophages in the red pulp of the spleen to cause increased retention of all cell types, even platelets. In multiple studies have shown the surgical removal of the enlarged spleen will rapidly return the circulatory cells to normal levels39. It is commonly found that bone marrow progenitor cells of these circulatory cells are not abnormal unless the basic pathogenesis of the disease also affected those bone marrow stem cells, such as those involved in autoimmune diseases.
In summary, these findings clarify for the first time that PKCδ activation in monocytes/macrophages induced by hyperlipidemia in insulin resistance and diabetes could be a negative modulator of macrophage numbers in the vascular wall and systemic tissues probably mediated by the inhibition of pAkt. Inhibiting PKCδ to improve insulin resistance and microvascular complications may have contrary effects on the progression of atherosclerosis in the hyperlipidemic state induced by diabetes and insulin resistance.
Supplementary Material
NOVELTY AND SIGNIFICANCE
What Is Known?
Diabetes and insulin resistance associated with obesity can activate monocytes to increase inflammation and accelerate atherosclerosis.
Hyperglycemia and elevated free fatty acids can induce monocyte to macrophage differentiation by increasing lipid synthesis and activating multiple isoforms of protein kinase C (PKC).
Activation PKCα and β isoforms in monocytes by diabetes can contribute to the accelerated atherosclerosis in diabetes. However, the role of PKCδ activation in monocytes on the severity of atherosclerosis has not been determined.
What New Information Does This Article Contribute?
The PKCδ isoform is activated in monocytes/macrophages in rodent models of obesity-induced diabetes and insulin resistance; the PKCδ activation was replicated in cultured macrophages exposed to acetylated-LDL and oxidized-LDL, but not to elevated glucose concentrations.
Deletion of PKCδ specifically in myeloid cells in ApoE−/− mice accelerated atherosclerosis and splenomegaly by inducing proliferation and decreasing apoptosis of the macrophage in the arterial wall and in the spleen.
Activated PKCδ induced by elevated lipids and diabetes can phosphorylate PI3Kinase to inhibit pAKT activation and increase apoptosis of macrophage in the arterial wall.
Abnormal lipids induced by diabetes and insulin resistance can elicit anti-inflammatory responses as well as inflammatory actions in macrophages, which in turn can affect atherosclerosis.
The effect of increased PKCδ expression and activity in monocytes in response to hyperlipidemia and diabetes on atherosclerosis was previously unknown. PKCδ expression and activity were increased in Zucker diabetic rats. Mice with selective deletion of PKCδ in myeloid cells were generated by breeding PKCδ flox/flox mice with LyzM-Cre and ApoE−/− mice (MPKCδKO/ApoE−/− mice), and the mice were fed a very high fat diet (HFD). Mice fed HFD exhibited hyperlipidemia, insulin resistance and mild diabetes. MPKCδKO/ApoE−/− mice exhibited increased aortic atherosclerosis by 2-fold and splenomegaly versus control ApoE−/− mice fed HFD. HFD increased macrophage numbers in aortic plaques and spleens in MPKCδKO/ApoE−/− vs. ApoE−/− mice due to decreased apoptosis (62%) and increased proliferation (1.9 fold), and not due to monocyte recruitment, with parallel increases in expression of inflammatory cytokines. The increased macrophage accumulation in MPKCδKO/ApoE−/− mice was associated with elevated phosphorylation levels of the pro-survival cell signaling proteins, Akt and FoxO3a, and with a reduction of the pro-apoptotic protein Bim. These changes were associated with PKCδ induced inhibition of P85/PI3K. Development of atherosclerosis induced by insulin resistance and hyperlipidemia may be partially limited by PKCδ activation in monocytes.
ACKNOWLEDGMENT
We are grateful for the expert technical assistance provided by Advanced Microscopy Core, Flow Cytometry Core, Mouse Physiology Core, Genomic Core which are supported by NIH grant 5P30DK036836 and S10OD021740. Plasma lipid profiles were measured at the Lipid, Lipoprotein, and Atherosclerosis Core of the Vanderbilt Mouse Metabolic Phenotype Centers with support from NIH grant DK59637. The content of this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
SOURCES OF FUNDING
G.L.K.: NIH grant 5P30DK036836 & NIH R01 DK053105 grant.
Q.L.: ADA mentor fellowship.
Nonstandard Abbreviations and Acronyms:
- PKC
Protein kinase C
- PBMC
Peripheral blood mononuclear cells
- ZDF
Zucker diabetic fatty rats
- qRT PCR
Real time PCR
- NC
Normal chow
- AD
Atherogenic diet
- HFD
High fat diet
- FPLC
Fast protein liquid chromatography
- PI
Propidium iodide
- BM
Bone marrow
- PMA
Phorbol-12-Myristate-13-Acetate
- IPGTT
Intraperitoneal glucose tolerance test
- IPITT
Intraperitoneal insulin tolerance test
- Thpn
Thapsigargin
Footnotes
DISCLOSURES
None.
REFERENCES
- 1.Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell 2011;145:341–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, Pittet MJ. Ly-6chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. The Journal of clinical investigation 2007;117:195–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robbins CS, Hilgendorf I, Weber GF, Theurl I, Iwamoto Y, Figueiredo JL, Gorbatov R, Sukhova GK, Gerhardt LM, Smyth D, Zavitz CC, Shikatani EA, Parsons M, van Rooijen N, Lin HY, Husain M, Libby P, Nahrendorf M, Weissleder R, Swirski FK. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nature medicine 2013;19:1166–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pataki M, Lusztig G, Robenek H. Endocytosis of oxidized ldl and reversibility of migration inhibition in macrophage-derived foam cells in vitro. A mechanism for atherosclerosis regression? Arteriosclerosis and thrombosis : a journal of vascular biology / American Heart Association 1992;12:936–944 [DOI] [PubMed] [Google Scholar]
- 5.Potteaux S, Gautier EL, Hutchison SB, van Rooijen N, Rader DJ, Thomas MJ, Sorci-Thomas MG, Randolph GJ. Suppressed monocyte recruitment drives macrophage removal from atherosclerotic plaques of apoe−/− mice during disease regression. The Journal of clinical investigation 2011;121:2025–2036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoogeveen RC, Ballantyne CM, Bang H, Heiss G, Duncan BB, Folsom AR, Pankow JS. Circulating oxidised low-density lipoprotein and intercellular adhesion molecule-1 and risk of type 2 diabetes mellitus: The atherosclerosis risk in communities study. Diabetologia 2007;50:36–42 [DOI] [PubMed] [Google Scholar]
- 7.Gonen B, Baenziger J, Schonfeld G, Jacobson D, Farrar P. Nonenzymatic glycosylation of low density lipoproteins in vitro. Effects on cell-interactive properties. Diabetes 1981;30:875–878 [DOI] [PubMed] [Google Scholar]
- 8.Park K, Li Q, Rask-Madsen C, Mima A, Mizutani K, Winnay J, Maeda Y, D’Aquino K, White MF, Feener EP, King GL. Serine phosphorylation sites on irs2 activated by angiotensin ii and protein kinase c to induce selective insulin resistance in endothelial cells. Molecular and cellular biology 2013;33:3227–3241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maeno Y, Li Q, Park K, Rask-Madsen C, Gao B, Matsumoto M, Liu Y, Wu IH, White MF, Feener EP, King GL. Inhibition of insulin signaling in endothelial cells by protein kinase c-induced phosphorylation of p85 subunit of phosphatidylinositol 3-kinase (pi3k). The Journal of biological chemistry 2012;287:4518–4530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Naruse K, Rask-Madsen C, Takahara N, Ha SW, Suzuma K, Way KJ, Jacobs JR, Clermont AC, Ueki K, Ohshiro Y, Zhang J, Goldfine AB, King GL. Activation of vascular protein kinase c-beta inhibits akt-dependent endothelial nitric oxide synthase function in obesity-associated insulin resistance. Diabetes 2006;55:691–698 [DOI] [PubMed] [Google Scholar]
- 11.Igarashi M, Wakasaki H, Takahara N, Ishii H, Jiang ZY, Yamauchi T, Kuboki K, Meier M, Rhodes CJ, King GL. Glucose or diabetes activates p38 mitogen-activated protein kinase via different pathways. J Clin Invest 1999;103:185–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Inoguchi T, Battan R, Handler E, Sportsman JR, Heath W, King GL. Preferential elevation of protein kinase c isoform beta ii and diacylglycerol levels in the aorta and heart of diabetic rats: Differential reversibility to glycemic control by islet cell transplantation. Proceedings of the National Academy of Sciences of the United States of America 1992;89:11059–11063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ceolotto G, Gallo A, Miola M, Sartori M, Trevisan R, Del Prato S, Semplicini A, Avogaro A. Protein kinase c activity is acutely regulated by plasma glucose concentration in human monocytes in vivo. Diabetes 1999;48:1316–1322 [DOI] [PubMed] [Google Scholar]
- 14.Sugiyama S, Kugiyama K, Ohgushi M, Fujimoto K, Yasue H. Lysophosphatidylcholine in oxidized low-density lipoprotein increases endothelial susceptibility to polymorphonuclear leukocyte-induced endothelial dysfunction in porcine coronary arteries. Role of protein kinase c. Circulation research 1994;74:565–575 [DOI] [PubMed] [Google Scholar]
- 15.Matsumura T, Sakai M, Kobori S, Biwa T, Takemura T, Matsuda H, Hakamata H, Horiuchi S, Shichiri M. Two intracellular signaling pathways for activation of protein kinase c are involved in oxidized low-density lipoprotein-induced macrophage growth. Arteriosclerosis, thrombosis, and vascular biology 1997;17:3013–3020 [DOI] [PubMed] [Google Scholar]
- 16.Jakubzick CV, Randolph GJ, Henson PM. Monocyte differentiation and antigen-presenting functions. Nature reviews. Immunology 2017;17:349–362 [DOI] [PubMed] [Google Scholar]
- 17.Zhu SN, Chen M, Jongstra-Bilen J, Cybulsky MI. Gm-csf regulates intimal cell proliferation in nascent atherosclerotic lesions. The Journal of experimental medicine 2009;206:2141–2149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feng J, Han J, Pearce SF, Silverstein RL, Gotto AM Jr., Hajjar DP, Nicholson AC. Induction of cd36 expression by oxidized ldl and il-4 by a common signaling pathway dependent on protein kinase c and ppar-gamma. Journal of lipid research 2000;41:688–696 [PubMed] [Google Scholar]
- 19.Lin CS, Lin FY, Ho LJ, Tsai CS, Cheng SM, Wu WL, Huang CY, Lian CH, Yang SP, Lai JH. Pkcdelta signalling regulates sr-a and cd36 expression and foam cell formation. Cardiovascular research 2012;95:346–355 [DOI] [PubMed] [Google Scholar]
- 20.Park K, Mima A, Li Q, Rask-Madsen C, He P, Mizutani K, Katagiri S, Maeda Y, Wu IH, Khamaisi M, Preil SR, Maddaloni E, Sorensen D, Rasmussen LM, Huang PL, King GL. Insulin decreases atherosclerosis by inducing endothelin receptor b expression. JCI insight 2016;1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Geraldes P, Hiraoka-Yamamoto J, Matsumoto M, Clermont A, Leitges M, Marette A, Aiello LP, Kern TS, King GL. Activation of pkc-delta and shp-1 by hyperglycemia causes vascular cell apoptosis and diabetic retinopathy. Nature medicine 2009;15:1298–1306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leitges M, Mayr M, Braun U, Mayr U, Li C, Pfister G, Ghaffari-Tabrizi N, Baier G, Hu Y, Xu Q. Exacerbated vein graft arteriosclerosis in protein kinase cdelta-null mice. J Clin Invest 2001;108:1505–1512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li L, Sampat K, Hu N, Zakari J, Yuspa SH. Protein kinase c negatively regulates akt activity and modifies uvc-induced apoptosis in mouse keratinocytes. The Journal of biological chemistry 2006;281:3237–3243 [DOI] [PubMed] [Google Scholar]
- 24.Limnander A, Depeille P, Freedman TS, Liou J, Leitges M, Kurosaki T, Roose JP, Weiss A. Stim1, pkc-delta and rasgrp set a threshold for proapoptotic erk signaling during b cell development. Nature immunology 2011;12:425–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seimon TA, Nadolski MJ, Liao X, Magallon J, Nguyen M, Feric NT, Koschinsky ML, Harkewicz R, Witztum JL, Tsimikas S, Golenbock D, Moore KJ, Tabas I. Atherogenic lipids and lipoproteins trigger cd36-tlr2-dependent apoptosis in macrophages undergoing endoplasmic reticulum stress. Cell metabolism 2010;12:467–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee JY, Chiu YH, Asara J, Cantley LC. Inhibition of pi3k binding to activators by serine phosphorylation of pi3k regulatory subunit p85alpha src homology-2 domains. Proc Natl Acad Sci U S A 2011;108:14157–14162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Welham MJ, Bone H, Levings M, Learmonth L, Wang LM, Leslie KB, Pierce JH, Schrader JW. Insulin receptor substrate-2 is the major 170-kda protein phosphorylated on tyrosine in response to cytokines in murine lymphohemopoietic cells. The Journal of biological chemistry 1997;272:1377–1381 [DOI] [PubMed] [Google Scholar]
- 28.Yu C, Chen Y, Cline GW, Zhang D, Zong H, Wang Y, Bergeron R, Kim JK, Cushman SW, Cooney GJ, Atcheson B, White MF, Kraegen EW, Shulman GI. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (irs-1)-associated phosphatidylinositol 3-kinase activity in muscle. The Journal of biological chemistry 2002;277:50230–50236 [DOI] [PubMed] [Google Scholar]
- 29.Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell 1999;96:857–868 [DOI] [PubMed] [Google Scholar]
- 30.Gilley J, Coffer PJ, Ham J. Foxo transcription factors directly activate bim gene expression and promote apoptosis in sympathetic neurons. J Cell Biol 2003;162:613–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jin H, Kanthasamy A, Anantharam V, Rana A, Kanthasamy AG. Transcriptional regulation of pro-apoptotic protein kinase cdelta: Implications for oxidative stress-induced neuronal cell death. The Journal of biological chemistry 2011;286:19840–19859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bornfeldt KE. Uncomplicating the macrovascular complications of diabetes: The 2014 edwin bierman award lecture. Diabetes 2015;64:2689–2697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuehn HS, Niemela JE, Rangel-Santos A, Zhang M, Pittaluga S, Stoddard JL, Hussey AA, Evbuomwan MO, Priel DA, Kuhns DB, Park CL, Fleisher TA, Uzel G, Oliveira JB. Loss-of-function of the protein kinase c delta (pkcdelta) causes a b-cell lymphoproliferative syndrome in humans. Blood 2013;121:3117–3125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miyamoto A, Nakayama K, Imaki H, Hirose S, Jiang Y, Abe M, Tsukiyama T, Nagahama H, Ohno S, Hatakeyama S, Nakayama KI. Increased proliferation of b cells and auto-immunity in mice lacking protein kinase cdelta. Nature 2002;416:865–869 [DOI] [PubMed] [Google Scholar]
- 35.Salzer E, Santos-Valente E, Klaver S, Ban SA, Emminger W, Prengemann NK, Garncarz W, Mullauer L, Kain R, Boztug H, Heitger A, Arbeiter K, Eitelberger F, Seidel MG, Holter W, Pollak A, Pickl WF, Forster-Waldl E, Boztug K. B-cell deficiency and severe autoimmunity caused by deficiency of protein kinase c delta. Blood 2013;121:3112–3116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mecklenbrauker I, Saijo K, Zheng NY, Leitges M, Tarakhovsky A. Protein kinase cdelta controls self-antigen-induced b-cell tolerance. Nature 2002;416:860–865 [DOI] [PubMed] [Google Scholar]
- 37.Seimon TA, Wang Y, Han S, Senokuchi T, Schrijvers DM, Kuriakose G, Tall AR, Tabas IA. Macrophage deficiency of p38alpha mapk promotes apoptosis and plaque necrosis in advanced atherosclerotic lesions in mice. The Journal of clinical investigation 2009;119:886–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lamharzi N, Renard CB, Kramer F, Pennathur S, Heinecke JW, Chait A, Bornfeldt KE. Hyperlipidemia in concert with hyperglycemia stimulates the proliferation of macrophages in atherosclerotic lesions: Potential role of glucose-oxidized ldl. Diabetes 2004;53:3217–3225 [DOI] [PubMed] [Google Scholar]
- 39.Lv Y, Lau WY, Li Y, Deng J, Han X, Gong X, Liu N, Wu H. Hypersplenism: History and current status. Experimental and therapeutic medicine 2016;12:2377–2382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bezy O, Tran TT, Pihlajamaki J, Suzuki R, Emanuelli B, Winnay J, Mori MA, Haas J, Biddinger SB, Leitges M, Goldfine AB, Patti ME, King GL, Kahn CR. Pkcdelta regulates hepatic insulin sensitivity and hepatosteatosis in mice and humans. J Clin Invest 2011;121:2504–2517 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.