

Significance
Bacteria, especially actinomycetes, produce the majority of our clinically useful small-molecule antibiotics. Genomic analyses of antibiotic-producing strains indicate that earlier discovery efforts revealed only a fraction of the likely antibiotic candidates. In an effort to uncover these previously missed candidates, we developed an approach that utilizes the ability of microbial communities to produce antibiotics that are not produced by any single member in isolation. Successful communities were established and deconvoluted to identify both producers and inducers of antibiotic activity. One inducer–producer pair made amycomicin, a potent and specific antibiotic against Staphylococcus aureus, an important human pathogen. Amycomicin targets fatty acid biosynthesis and exhibits in vivo efficacy against antibiotic-resistant skin infections in a mouse model.
Keywords: interspecies interactions, antibiotic, actinomycete, Staphylococcus aureus, fatty acid biosynthesis
Abstract
The rapid emergence of antibiotic-resistant pathogenic bacteria has accelerated the search for new antibiotics. Many clinically used antibacterials were discovered through culturing a single microbial species under nutrient-rich conditions, but in the environment, bacteria constantly encounter poor nutrient conditions and interact with neighboring microbial species. In an effort to recapitulate this environment, we generated a nine-strain actinomycete community and used 16S rDNA sequencing to deconvolute the stochastic production of antimicrobial activity that was not observed from any of the axenic cultures. We subsequently simplified the community to just two strains and identified Amycolatopsis sp. AA4 as the producing strain and Streptomyces coelicolor M145 as an inducing strain. Bioassay-guided isolation identified amycomicin (AMY), a highly modified fatty acid containing an epoxide isonitrile warhead as a potent and specific inhibitor of Staphylococcus aureus. Amycomicin targets an essential enzyme (FabH) in fatty acid biosynthesis and reduces S. aureus infection in a mouse skin-infection model. The discovery of AMY demonstrates the utility of screening complex communities against specific targets to discover small-molecule antibiotics.
Researchers have responded to the increasing incidence of antibiotic-resistant bacterial infections with attempts to discover new small-molecule antibiotics (1, 2). Many of these attempts apply new techniques to traditional sources, including the environmental bacteria that have provided the suite of antibiotics currently being threatened by resistance (3). This return to traditional sources began with the realization that earlier explorations had captured only a small fraction of the biosynthetic potential seen in bacterial genomes (2, 4). These missing molecules, frequently called “cryptic” metabolites, can be inferred by identifying their biosynthetic genes but have not yet been confirmed in the laboratory (5). Previous studies may have missed these molecules because they are produced on an “as-needed” basis and culturing a single strain in rich media may not create the requisite need.
Bacteria sense and respond to the world around them with small molecules, and adding nonkin microbes, environmental, or host factors during culturing can induce production of cryptic metabolites (6–15). We have developed a discovery approach in which metabolite expression is induced through coculturing interactions in a complex community simultaneously linked to a therapeutically relevant target assay, an approach we call a targeted-interaction screen.
For the interacting organisms, we used a collection of nine actinomycete strains. Actinomycetes have been a prolific source of antibiotics, and their social lifestyles suggest that they may produce numerous small molecules in response to neighboring microbes (3, 9, 10, 16). The specific nine strains, described in more detail below, were selected because none of them made any significant amount of antibiotics when grown in isolation under our conditions. In addition, the nine strains had been at least partially characterized in our laboratories, leading to draft genome sequences. A dilute complex medium was used for culturing because previous studies indicated that such conditions are conducive to the growth of coexisting strains as well as interspecific induction of antibiosis (10, 17, 18). For the target, we selected the important human pathogen, Staphylococcus aureus. After an initial screen of the nine-member community, to reduce complexity, we focused on a strong response in which Streptomyces coelicolor M145 produces a signal that causes Amycolatopsis sp. AA4 to produce an antibiotic that potently [minimal inhibitory concentration (MIC) ∼ 30 nM] and specifically kills S. aureus in both in vitro and in vivo assays. Further analysis led to the identity of the antibiotic, which we named amycomicin (AMY). AMY is an unusual fatty-acid–based antibiotic that has been highly functionalized, including incorporation of a ketone, a hydroxyl, an isonitrile-epoxide, and an olefin. We also identified AMY’s biosynthetic gene cluster in AA4, which contains a recently identified isonitrile synthase gene (19), and AMY’s target as FabH, an enzyme in the fatty acid biosynthesis pathway. Finally, AMY is effective in a murine skin-infection model of S. aureus.
Results and Discussion
Interstrain Interactions Induce Production of Antimicrobial Activity.
In actinomycetes, antibiotic production is linked to developmental stage, so we used solid phase culturing in transwell plates, allowing continuous monitoring without disrupting the community (Fig. 1A) (16). A transwell sealed by a permeable membrane was filled with a 0.75% agarose solution. Once the agarose solidified, the transwell was partially submerged into liquid medium within a larger well. Actinomycete spores were then inoculated on the agarose surface. The nutrients present in the liquid medium diffused through the membrane and the agarose plug, allowing the bacteria to grow. As the bacterial community proliferated, secreted metabolites diffused through the agarose into the liquid medium. The conditioned liquid medium was periodically tested for antibiotic activity and/or exchanged to provide fresh nutrients.
Fig. 1.

Antimicrobial activity induced by interspecies interactions in actinomycete communities. (A) Microbial strains or communities were inoculated onto an agarose plug that was supported by a permeable membrane within a transwell. The conditioned liquid medium was periodically sampled for antibiotic (see SI Appendix for details). (B) Individual strains do not produce strong antimicrobial activity against S. aureus. Amycolatopsis sp. AA4 (AA4), Streptomyces sp. Mg1 (Mg1), S. sviceus ATCC 29083 (29083), Kutzneria sp. 744 (744), Streptomyces pristinaespiralis ATCC 25486 (25486), Streptomyces sp. E14 (E14), Micromonospora sp. M42 (M42), S. coelicolor A3 (2) M145 (M145), Streptomyces hygroscopicus ATCC 53653 (53653). Genome accession numbers are listed in SI Appendix. (C) A community of all nine strains (mix) sometimes produced and sometimes did not produce antimicrobial activity. Ampicillin (amp) and fresh medium (medium) were used as positive and negative controls, respectively. (D) Relative abundance of each strain in the microbial communities, as determined by 16S rDNA sequencing, at the time of inoculation (I) and at the time when antibiotic was produced in communities that produced relatively high (H), medium (M), low (L) or no activity (N). (E) Zone of inhibition produced by a bipartite community of AA4 and M145 (AA4 + M145). Other combinations tested (AA4 + E14...) did not produce inhibitory activity. A sample from the mix was placed in the middle as a control. (F and G) Zones of growth inhibition produced by medium that was sequentially conditioned by AA4 and then M145 (F) or by M145 and then AA4 (G). (Scale bars: 10 mm.)
To capture multiple interactions and avoid detection of known antibiotics, we began with nine strains of actinomycetes that did not produce significant antimicrobial activity when grown in isolation under tested conditions (Fig. 1B). Spores were inoculated onto the 200-μL agarose plugs of the transwell system singly or as a mix of all nine species. One milliliter of dilute complex medium (SI Appendix) was added to the bottom wells. To assess secreted antimicrobial activity, 10-µL samples were periodically removed from the bottom well and spotted onto agar plates freshly seeded with a lawn of S. aureus (Fig. 1A).
Old medium was replaced with fresh medium 3 wk after inoculation, and 1–2 d after that we sometimes detected the production of a strong antimicrobial activity against S. aureus. While none of the individual strains produced strong antibiosis (Fig. 1B), cultures that started with approximately equal numbers of all nine strains occasionally produced a strong activity (Fig. 1C). This variability was attributed to the stochasticity of complex microbial communities (20, 21).
We used 16S rDNA sequencing to quantify each strain’s relative abundance at the time of antibiotic production (Fig. 1D). At the time antimicrobial activity was detected, the communities were dominated by three of the original nine strains: Amycolatopsis sp. AA4 (henceforth AA4), S. coelicolor A3(2)/M145 (henceforth M145), and Streptomyces sviceus ATCC 29083. Most importantly, the proportion of AA4 present correlated loosely with activity. For example, the community producing the most activity (column H in Fig. 1D) contained 22% AA4, while a community with no detectable activity (column N in Fig. 1D) contained only 0.11% AA4. Ultimately, we found robust antibiotic productions in cocultures of AA4 and M145 (Fig. 1E).
To determine the inducer and the producer, we cultured the strains independently in transwells, collected the conditioned medium from each isolated culture, and used that medium to grow the other strain. Addition of M145-conditioned medium induced robust antibiotic production by AA4 (Fig. 1G), but addition of AA4-conditioned medium to M145 did not induce antibiotic production (Fig. 1F). These results suggested that a M145-derived compound(s) stimulates the production of an antimicrobial agent by AA4.
Since antibiotic biosynthesis in actinomycetes is intricately linked to carbohydrate metabolism we measured the carbohydrate content of M145-conditioned medium, AA4-conditioned medium, and fresh medium (22). Glucose was the predominant sugar in fresh medium (SI Appendix, Fig. S2A). Conditioning of the medium by M145 reduced the amount of glucose and increased the concentration of galactose (SI Appendix, Fig. S2A). Adding galactose, as well as N-acetyl-glucosamine (GlcNAc), glucosamine, xylose, or arabinose, robustly induced antibiotic production in AA4 in the absence of M145 (SI Appendix, Fig. S2B). This agrees with previous studies that found that GlcNAc and xylose induce antibiotic synthesis in actinomycetes through gene regulation (23, 24). Possible sources of galactose in the M145-conditioned medium are teichulosonic acid and polydiglycosylphosphate polysaccharide components of M145, accounting for up to one-third of its cell-wall dry mass (25, 26). Importantly, different actinomycete species contain distinct cell-associated saccharide mixtures, a feature that has been used to classify them (27).
Purification and Absolute Structure Determination of AMY.
Bioassay-guided isolation of the crude extract from large-scale fermentation of AA4 on solid agar media containing GlcNAc led to the purification of AMY. AMY has the molecular formula C19H29NO5, based on HR-q-ToFMS. Both the 1H and 13C NMR spectra suggested that AMY is a highly functionalized fatty acid with carbon chemical shifts indicating a carboxylic acid (δC 174.7), a ketone (δC 210.5), an olefin (δC 123.7 and 132.7), and three oxygenated sp3 carbons (δC 65.9, 67.2, and 67.6).
Analysis of AMY’s 2D NMR spectra (HSQC, H2BC, COSY, HMBC, and ROESY) revealed a relationship to two previously reported metabolites, YM-47515 and aerocyanidin (28, 29). Both contain a highly unstable epoxide isonitrile functionality at the end of the fatty acid chain. Further analysis of AMY’s 13C NMR spectra revealed a small peak at 162.0 ppm (isonitrile carbon) that had no correlations in any of the 2D NMR spectra. Additionally, infrared spectra on AMY consisted of peaks at 3,350, 2,133, and 1,704/1,651 cm−1, which indicate a hydroxyl/carboxylic acid, a triple bond, and two carbonyl groups, respectively. Thus, analysis of the NMR dataset and comparison with both YM-47515 and aerocyanidin led to the planar structural assignment of AMY as shown in Fig. 2A, a highly modified fatty acid containing an epoxy-isonitrile warhead. AMY inhibits growth of Staphylococcaceae at 30 nM and does not inhibit other tested microbes (SI Appendix, Fig. S9).
Fig. 2.

Molecular structure and biosynthetic gene cluster of AMY. (A) Molecular structures of AMY (1) and epoxyketone AMY (2). (B) Gene conservation between biosynthetic gene clusters of isonitrile lipopetide from M. marinum (mma), AMY from AA4 (amc), and aerocyanidin from Chromobacterium sp. ATCC 53434 (aec). Percentages indicate amino acid sequence identity. (C–E) Halo (or lack thereof) produced within a lawn of S. aureus by 10 μL of filtered supernatant from wild-type ATCC 53434 (C) or ΔaecE (D) containing an empty expression plasmid and from ΔaecE containing a plasmid expressing aecE (E). (Scale bars: 10 mm.)
An epoxy-isonitrile functional group is quite labile; therefore, its decomposition in basic media was used as part of our structural analysis, and it likely forms a key feature in AMY’s mechanism of action. Relative stereochemistry of AMY was assigned to be syn by a key ROESY correlation between H14 and H16. The relative C13 hydroxyl stereochemistry was determined by converting AMY to epoxyketone-AMY with inversion at C14 (30), which was subsequently assigned as a trans-epoxide because of the measured 2-Hz coupling between H13 and H14 (Fig. 2A, Bottom). Conversion of AMY to epoxyketone-AMY releases a molecule of hydrogen cyanide and leaves behind an epoxyketone (Fig. 2A, Bottom), which is a familiar component of naturally occurring biologically active molecules (31). The absolute stereochemical assignment of epoxyketone-AMY was assigned by comparing its CD Cotton effect and specific optical rotation to known epoxy-ketone–containing metabolites (32–34).
Identification of a Putative Amycomicin Biosynthetic Gene Cluster.
The isonitrile moiety led to the identification of AMY’s putative biosynthetic gene cluster. The isonitrile function is uncommon in natural products, but two different genes have been identified in bacteria that are able to install this moiety. The first isonitrile synthase (IsnA) to be identified utilizes ribose as a source of the isonitrile carbon, which is attached to an existing amine (35, 36). AA4 has no IsnA homologs. In the second pathway, an isonitrile moiety is created on an existing acyl chain via the action of four proteins, which were recently discovered to install the isonitrile functionality in a lipopeptide produced by Mycobacterium marinum (19). We found homologs of mmaB-E clustered within the AA4 genome (Fig. 2B). We named this gene cluster amc. In addition to genes predicted to synthesize isonitrile (amcA, -B, -E, and -H), this cluster also contains genes encoding putative polyketide synthases (amcF, -G) and two cytochrome P450s (amcC, -D), which could be responsible for the biosynthesis of the AMY’s polyketide backbone and epoxide, respectively.
As we were unable to generate mutations in the genome of AA4, we sequenced the genome of Chromobacterium sp. ATCC 53434, which produces aerocyanidin, another member of the epoxide isonitrile antibiotic family (29). In the genome of ATCC 53434, we found a gene cluster (which we named aec) that is very similar to amc (Fig. 2B). Mutation of ATCC 53434’s putative acyl-ACP-ligase gene (ΔaecE) abolished production of aerocyanidin, and the mutant’s supernatant did not inhibit S. aureus. Production of aerocyanidin was restored by supplementation of aecE in trans (Fig. 2 C–E and SI Appendix, Fig. S5). Given the clear structural similarities between aerocyanidin and AMY, linking of aerocyanidin biosynthesis to the cluster provides strong evidence that the similar gene cluster within the AA4 genome is responsible for biosynthesis of AMY.
Fatty Acids Protect S. aureus Against AMY.
Serendipitously, we found that another AA4 metabolite protects S. aureus from AMY. We sometimes observed that AA4-conditioned medium led to a faint zone of growth within the zone of inhibition produced by AMY (Fig. 3A), which suggested that AA4 could produce a factor that served as an “antidote” to AMY. We purified this factor from AA4-conditioned medium using high-performance liquid chromatography tracking anti-AMY activity. This strategy identified a compound that allowed S. aureus growth in AMY’s presence (Fig. 3 B and C). Mass spectrometry (MS), NMR analysis, and gas chromatography MS identified the compound as palmitoleic acid, a 16-carbon straight-chain unsaturated fatty acid (SCUFA) (SI Appendix, Fig. S6). Commercially available palmitoleic acid as well oleic acid, an 18-carbon fatty acid, allowed growth of S. aureus in the presence of AMY (Fig. 3 D and E).
Fig. 3.

Reversal of AMY activity by fatty acids. (A) Small zone of growth within a zone of growth inhibition produced by a batch of AA4-conditioned medium. (B–F) Halos (or lack thereof) produced by (B) 100 ng AMY, (C) AMY + “anti-AMY,” (D) AMY + palmitoleic acid, (E) AMY + oleic acid, or (F) AMY + 12-MTA. (G and H) Results of adding AMY + 12-MTA to a lawn of wild-type S. aureus USA300 (G) and to a lawn of USA300 fakA transposon mutant (H). (Scale bars: 10 mm.)
S. aureus does not synthesize unsaturated fatty acids such as oleic acid or palmitoleic acid, but it can incorporate them from the medium (37, 38). Instead, S. aureus synthesizes branched chain fatty acids (BCFAs) that function similarly to SCUFAs (39). The most abundant BCFA produced by S. aureus is 12-methyltetradecanoic acid (12-MTA, anteiso-15:0). We found that addition of 12-MTA also inhibits AMY’s activity (Fig. 3F). Addition of increasing amounts of either 12-MTA or oleic acid protected S. aureus from AMY in a dose-dependent manner up to a concentration where the fatty acids became toxic (SI Appendix, Fig. S7 A and B). In contrast, straight-chain saturated fatty acids (SCSFA) do not protect S. aureus against AMY (SI Appendix, Fig. S7 C and D).
Incorporation of external fatty acids into the membrane of S. aureus requires fatty acid kinase A (FakA) (37). A S. aureus mutant with a transposon insertion in the gene encoding FakA (fakA) was susceptible to AMY even in the presence of 12-MTA (Fig. 3 G and H). We noted frequent emergence of AMY-resistant mutants in the fakA mutant background in the presence of 12-MTA. These mutants reverted to a wild-type phenotype where they gained the ability to utilize 12-MTA to counteract AMY but were still susceptible to AMY in the absence of added fatty acids (SI Appendix, Fig. S6 E and F). These observations pointed to the possibility that AMY targets fatty acid biosynthesis. Interestingly, a number of fatty acid biosynthesis genes are located adjacent to amc (SI Appendix, Fig. S11).
AMY Alters Cell Morphology of S. aureus.
To investigate the effect of AMY on cell morphology, we utilized transmission electron microscopy on ultrathin-sectioned S. aureus. Compared with untreated cells, bacteria treated with AMY displayed gross alterations. Specifically, the cells contained thickened cell walls and a severely shrunken cytoplasmic compartment (Fig. 4 A and B). Similar phenotypes were observed in cells treated with the fatty acid biosynthesis inhibitors platensimycin and irgasan (Fig. 4 C and D) (40, 41). These results indicate that AMY alters cell morphology in a manner similar to known fatty acid biosynthesis inhibitors.
Fig. 4.

Morphological changes in S. aureus induced by treatment with AMY. Transmission electron micrographs of sectioned S. aureus cells that were (A) not treated with antibiotics or treated with (B) AMY, (C) platensimycin, or (D) irgasan. (Scale bar: 100 nm.)
FabH Overexpression Increases Resistance to AMY.
It seemed likely that AMY targets an enzyme involved in fatty acid biosynthesis; therefore, we manipulated expression levels of fatty acid biosynthetic enzymes, an approach that has previously been used to identify antibiotic targets (42–45). As a control, we cloned the 3-oxoacyl-[acyl-carrier-protein] synthase 2 gene (fabF) into a plasmid under a constitutive promoter; tested its effect on the susceptibility of S. aureus to platensimycin, a well-characterized FabF inhibitor (40); and observed a fourfold increase in resistance to platensimycin (SI Appendix, Fig. S8). AMY has an MIC of ∼30 nM (∼10 ng/mL) against S. aureus grown at 37 °C at pH 7.0. In assays against strains overexpressing individual Fab proteins, overexpression of 3-oxoacyl-[acyl-carrier-protein] synthase 3 (FabH) displayed a fourfold increase in the MIC against AMY while overexpression of other Fab proteins had no effect (Fig. 5A).
Fig. 5.
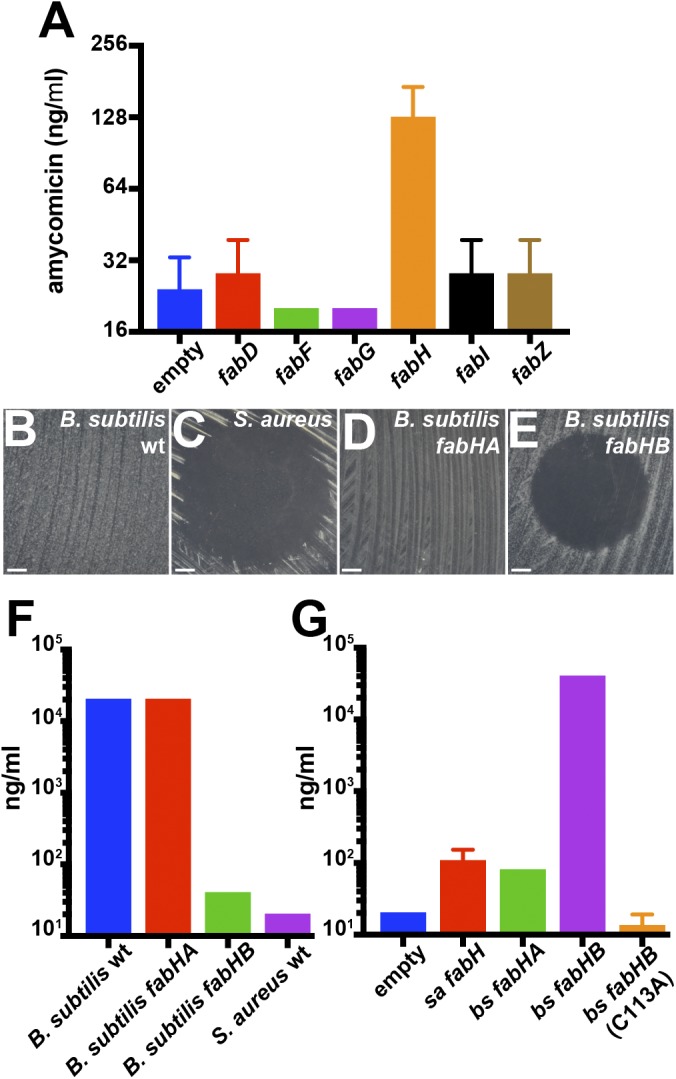
Protection against AMY inhibition in S. aureus by FabH overexpression. (A) Minimal inhibitory concentrations of AMY on strains of S. aureus carrying a plasmid overexpressing individual S. aureus fatty acid biosynthesis genes. (B–E) Effect of adding 100 ng AMY to lawns of (B) wild-type B. subtilis, (C) wild-type S. aureus, (D) B. subtilis fabHA, and (E) B. subtilis fabHB (F). (Scale bars: 10 mm.) Minimal inhibitory concentrations of AMY against the indicated strains of B. subtilis and S. aureus (G). Minimal inhibitory concentrations of AMY S. aureus strains harboring the indicated plasmids, which led to the overexpression of the native S. aureus FabH or the B. subtilis FabHA or FabHB. Error bars represent SDs.
Bacillus subtilis FabHB Confers Resistance Against AMY.
Members of the Staphylococcacceae family are uniquely susceptible to AMY (SI Appendix, Fig. S9). We employed B. subtilis that is resistant to AMY (Fig. 5 B and C) to understand this unique susceptibility. B. subtilis encodes two homologs of FabH, and thus single-knockout mutants are viable (46, 47). FabHA is more closely related to S. aureus FabH (58% identity, 76% similarity) than FabHB (42% identity, 59% similarity) (47). B. subtilis fabHA is resistant to AMY, but B. subtilis fabHB is susceptible (Fig. 5 D and E). The MIC measurements indicated a nearly three orders of magnitude decrease in resistance to AMY of B. subtilis fabHB knockout compared with wild-type B. subtilis or B. subtilis fabHA knockout (Fig. 5F). Furthermore, expressing fabHB from B. subtilis in S. aureus resulted in a 500-fold increase in the MIC of AMY, whereas expression of fabHA did not (Fig. 5G). Expression of a catalytically inactive C113A mutant of FabHB did not increase resistance of S. aureus to AMY. Collectively, these results identify S. aureus FabH as AMY’s target. The exact mechanism of FabH inhibition by AMY is being investigated. It is important to note, in regard to our original goal of developing a targeted interaction screen, that a previous high-throughput screen identified all natural product inhibitors of FabH known to date, but did not discover AMY (48).
AMY Alters Fatty Acid Composition of S. aureus.
FabH’s preference for branched-chain or straight chain acyl-CoA substrates determines the membrane BCFA/SCSFA ratio (46). S. aureus growing in exponential phase was treated with a range of concentrations of AMY. In untreated cells, 12-MTA (anteiso-15:0) was the most abundant fatty acid (SI Appendix, Table S3). S. aureus cells treated with AMY displayed an altered BCFA/SCFA ratio with a decrease of a total fraction of all BCFAs (from 77 to 46%) and 12-MTA specifically (from 37 to 22%) (SI Appendix, Fig. S7G and Table S3). Consistent with this observation, thiolactomycin, a known inhibitor of FabH, also reduces BCFA content in Streptomyces collinus (49). These results are also consistent with the observation that addition of BCFAs reverses the activity of AMY (Fig. 3).
AMY Protects Against S. aureus Infection in a Mouse Skin Model of Infection.
To explore the potential of AMY as a therapeutic agent, we used a superficial mouse skin-infection model (50). Methicillin-resistant S. aureus USA300 containing a modified lux operon allowed us to monitor the progression of infection in live animals (51). An infection was untreated for 24 h, and the mice were separated into three groups based on the treatment to be received. Bioluminescence was measured to ensure that the mice in the three groups had similar burdens of infection (SI Appendix, Fig. S10A). Mice in the first group were left untreated (negative control), mice in the second group were treated with a topical formulation of 2% mupirocin, and mice in the third group were treated with a 0.001% solution of AMY once a day for 3 d. The mice treated with AMY for 3 d showed a marked decrease in luminescence compared with untreated mice (Fig. 6). The decrease was similar to that observed in infection sites treated with mupirocin. The decrease was confirmed by the reduction in number of colony-forming units isolated from the infection sites (SI Appendix, Fig. S10B). These data demonstrate that AMY has in vivo activity in a mouse model of S. aureus skin infections. While AMY’s potency and specificity against S. aureus are highly desirable, its lability makes it an unlikely candidate to advance along a therapeutic pipeline. Although AMY resulted in decreased infection (Fig. 6), in our initial study it acted more slowly than mupirocin (SI Appendix, Fig. S10 C and D). While mupirocin treatment resulted in a significant decrease in infection 1 d after treatment, AMY did not have an effect until day 2. However, mupirocin used as a control in this experiment was applied in the form of a pharmaceutical-grade ointment at a 2% concentration, a concentration much higher than the AMY concentration (0.001%) we used. Throughout the experiment the mice were handled according to the Vanderbilt University Institutional Animal Care and Use Committee regulations.
Fig. 6.

Murine S. aureus skin infections and treatment. (A) Bioluminescence expressed as radiance units (RU) detected in the infected sites 4 d postinfection and after three daily doses of the indicated treatment. Horizontal bars indicate averages and SDs of log10-transformed values. Welch’s t test was used to determine the P values. *P < 0.05. (B) Representative images of bioluminescence emanating from infected sites after three doses of the indicated treatment.
Conclusion.
In an effort to discover new antibiotics, we designed and tested an approach coupling the ability of bacterial cocultures to elicit the expression of cryptic metabolites with phenotypic screening against an important human pathogen. The transwell assay that we describe allows for the growth of a complex actinomycete community on solid medium coupled to frequent testing of the liquid phase for antibiotic activity without disrupting the community. The system also allows for altering the growth conditions by, for example, providing fresh nutrients to the bacterial community. The screen exploits the stochastic populations produced by identical but complex starting sets and the ability of rapid 16S rDNA sequencing to deconvolute and identify productive populations. While this initial targeted interaction approach to discovering new antibiotics was successful, it is important to note a paradox that was troubling from the outset: the logic of the interaction screen is based on taking advantage of existing environmental signals. AA4 is a soil actinomycete and as such is not likely to encounter S. aureus, a common inhabitant of humans. AMY does not inhibit M145, B. subtilis, or any other soil microbe that we tested it against. While we do not understand the ecological role of AMY, our results are encouraging for future applications of target-based interaction screening as the starting community need not reflect inhabitants of the target’s community. Assuming robust readout, similar larger-scale screens can theoretically be applied to any cultivable multispecies community of high complexity, such as host-associated microbiota. This methodology can be further improved by applying techniques such as RNA-Seq and untargeted mass spectrometry to identify bioactive molecules and interactions leading to their production.
Supplementary Material
Acknowledgments
We thank members of the R.K. and J.C. laboratories for valuable advice. We also thank the Harvard Medical School Center for Macromolecular Interactions, the Institute of Chemistry and Cell Biology (ICCB)-Longwood Screening Facility, Small Molecule Mass Spectrometry Core Facility at Harvard University, and the East Quad NMR Facility for analytical services. This work was supported by NIH Grants GM058213 and R21AI117025 (to R.K.), R01AT009874 (to J.C.), and R01AI073843 (to E.P.S.). S.K. was supported by the Intramural Research Program of the National Human Genome Research Institute, NIH. This work utilized the computational resources of the NIH High-Performance Computing Biowulf cluster (https://hpc.nih.gov). Carbohydrate analysis was supported by the Chemical Sciences, Geosciences, and Biosciences Division, Office of Basic Energy Sciences, US Department of Energy Grant DE-FG02-93ER20097.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The sequences reported in this paper have been deposited in the GenBank database (accession nos. CP024894–CP024896 and CP025429).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1807613115/-/DCSupplemental.
References
- 1.Demain AL, Sanchez S. Microbial drug discovery: 80 years of progress. J Antibiot (Tokyo) 2009;62:5–16. doi: 10.1038/ja.2008.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Silver LL. Challenges of antibacterial discovery. Clin Microbiol Rev. 2011;24:71–109. doi: 10.1128/CMR.00030-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barka EA, et al. Taxonomy, physiology, and natural products of Actinobacteria. Microbiol Mol Biol Rev. 2015;80:1–43. doi: 10.1128/MMBR.00019-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nett M, Ikeda H, Moore BS. Genomic basis for natural product biosynthetic diversity in the actinomycetes. Nat Prod Rep. 2009;26:1362–1384. doi: 10.1039/b817069j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rutledge PJ, Challis GL. Discovery of microbial natural products by activation of silent biosynthetic gene clusters. Nat Rev Microbiol. 2015;13:509–523. doi: 10.1038/nrmicro3496. [DOI] [PubMed] [Google Scholar]
- 6.Straight PD, Kolter R. Interspecies chemical communication in bacterial development. Annu Rev Microbiol. 2009;63:99–118. doi: 10.1146/annurev.micro.091208.073248. [DOI] [PubMed] [Google Scholar]
- 7.Romero D, Traxler MF, López D, Kolter R. Antibiotics as signal molecules. Chem Rev. 2011;111:5492–5505. doi: 10.1021/cr2000509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Traxler MF, Kolter R. Natural products in soil microbe interactions and evolution. Nat Prod Rep. 2015;32:956–970. doi: 10.1039/c5np00013k. [DOI] [PubMed] [Google Scholar]
- 9.Traxler MF, Watrous JD, Alexandrov T, Dorrestein PC, Kolter R. Interspecies interactions stimulate diversification of the Streptomyces coelicolor secreted metabolome. MBio. 2013;4:e00459-13. doi: 10.1128/mBio.00459-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abrudan MI, et al. Socially mediated induction and suppression of antibiosis during bacterial coexistence. Proc Natl Acad Sci USA. 2015;112:11054–11059. doi: 10.1073/pnas.1504076112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Netzker T, et al. Microbial communication leading to the activation of silent fungal secondary metabolite gene clusters. Front Microbiol. 2015;6:299. doi: 10.3389/fmicb.2015.00299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ueda K, Beppu T. Antibiotics in microbial coculture. J Antibiot (Tokyo) 2017;70:361–365. doi: 10.1038/ja.2016.127. [DOI] [PubMed] [Google Scholar]
- 13.Schroeckh V, et al. Intimate bacterial-fungal interaction triggers biosynthesis of archetypal polyketides in Aspergillus nidulans. Proc Natl Acad Sci USA. 2009;106:14558–14563. doi: 10.1073/pnas.0901870106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bertrand S, et al. Metabolite induction via microorganism co-culture: A potential way to enhance chemical diversity for drug discovery. Biotechnol Adv. 2014;32:1180–1204. doi: 10.1016/j.biotechadv.2014.03.001. [DOI] [PubMed] [Google Scholar]
- 15.Chodkowski JL, Shade A. A synthetic community system for probing microbial interactions driven by exometabolites. mSystems. 2017;2:e00129-17. doi: 10.1128/mSystems.00129-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hopwood DA. 2007. Streptomyces in Nature and Medicine: The Antibiotic Makers (Oxford Univ Press, New York), 250 pp.
- 17.Song W, Kim M, Tripathi BM, Kim H, Adams JM. Predictable communities of soil bacteria in relation to nutrient concentration and successional stage in a laboratory culture experiment. Environ Microbiol. 2016;18:1740–1753. doi: 10.1111/1462-2920.12879. [DOI] [PubMed] [Google Scholar]
- 18.Rivett DW, et al. Resource-dependent attenuation of species interactions during bacterial succession. ISME J. 2016;10:2259–2268. doi: 10.1038/ismej.2016.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harris NC, et al. Biosynthesis of isonitrile lipopeptides by conserved nonribosomal peptide synthetase gene clusters in Actinobacteria. Proc Natl Acad Sci USA. 2017;114:7025–7030. doi: 10.1073/pnas.1705016114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hao YQ, Zhao XF, Zhang DY. Field experimental evidence that stochastic processes predominate in the initial assembly of bacterial communities. Environ Microbiol. 2016;18:1730–1739. doi: 10.1111/1462-2920.12858. [DOI] [PubMed] [Google Scholar]
- 21.Vega NM, Gore J. Stochastic assembly produces heterogeneous communities in the Caenorhabditis elegans intestine. PLoS Biol. 2017;15:e2000633. doi: 10.1371/journal.pbio.2000633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Urem M, Świątek-Połatyńska MA, Rigali S, van Wezel GP. Intertwining nutrient-sensory networks and the control of antibiotic production in Streptomyces. Mol Microbiol. 2016;102:183–195. doi: 10.1111/mmi.13464. [DOI] [PubMed] [Google Scholar]
- 23.Świątek MA, et al. The ROK family regulator Rok7B7 pleiotropically affects xylose utilization, carbon catabolite repression, and antibiotic production in Streptomyces coelicolor. J Bacteriol. 2013;195:1236–1248. doi: 10.1128/JB.02191-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rigali S, et al. Feast or famine: The global regulator DasR links nutrient stress to antibiotic production by Streptomyces. EMBO Rep. 2008;9:670–675. doi: 10.1038/embor.2008.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shashkov AS, et al. Novel teichulosonic acid from cell wall of Streptomyces coelicolor M145. Carbohydr Res. 2012;359:70–75. doi: 10.1016/j.carres.2012.05.018. [DOI] [PubMed] [Google Scholar]
- 26.Sigle S, Steblau N, Wohlleben W, Muth G. A toolbox to measure changes in the cell wall glycopolymer composition during differentiation of Streptomyces coelicolor A3(2) J Microbiol Methods. 2016;128:52–57. doi: 10.1016/j.mimet.2016.07.002. [DOI] [PubMed] [Google Scholar]
- 27.Lechevalier MP, Lechevalier H. Chemical composition as a criterion in the classification of aerobic actinomycetes. Int J Syst Bacteriol. 1970;20:435–443. [Google Scholar]
- 28.Sugawara T, Tanaka A, Imai H, Nagai K, Suzuki K. YM-47515, a novel isonitrile antibiotic from Micromonospora echinospora subsp. echinospora. J Antibiot (Tokyo) 1997;50:944–948. doi: 10.7164/antibiotics.50.944. [DOI] [PubMed] [Google Scholar]
- 29.Parker WL, et al. Aerocyanidin, a new antibiotic produced by Chromobacterium violaceum. J Antibiot (Tokyo) 1988;41:454–460. doi: 10.7164/antibiotics.41.454. [DOI] [PubMed] [Google Scholar]
- 30.Behrens CHKS, Sharpless KB, Walker FJ. Selective transformation of 2,3-epoxy alcohols and related derivatives. Strategies for nucleophilic attack at carbon-1. J Org Chem. 1985;50:5687–5696. [Google Scholar]
- 31.Kim KB, Crews CM. From epoxomicin to carfilzomib: Chemistry, biology, and medical outcomes. Nat Prod Rep. 2013;30:600–604. doi: 10.1039/c3np20126k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nemoto T, Ohshima T, Yamaguchi K, Shibasaki M. Catalytic asymmetric epoxidation of enones using La-BINOL-triphenylarsine oxide complex: Structural determination of the asymmetric catalyst. J Am Chem Soc. 2001;123:2725–2732. doi: 10.1021/ja004201e. [DOI] [PubMed] [Google Scholar]
- 33.Lu Y, Zheng C, Yang Y, Zhao G, Zou G. Highly enantioselective epoxidation of α,β-unsaturated ketones catalyzed by primary-secondary diamines. Adv Synth Catal. 2011;353:3129–3133. [Google Scholar]
- 34.Watanabe S, Arai T, Sasai H, Bougauchi M, Shibasaki M. The first catalytic enantioselective synthesis of cis-epoxyketones from cis-enones. J Org Chem. 1998;63:8090–8091. [Google Scholar]
- 35.Brady SF, Clardy J. Cloning and heterologous expression of isocyanide biosynthetic genes from environmental DNA. Angew Chem Int Ed Engl. 2005;44:7063–7065. doi: 10.1002/anie.200501941. [DOI] [PubMed] [Google Scholar]
- 36.Brady SF, Clardy J. Systematic investigation of the Escherichia coli metabolome for the biosynthetic origin of an isocyanide carbon atom. Angew Chem Int Ed Engl. 2005;44:7045–7048. doi: 10.1002/anie.200501942. [DOI] [PubMed] [Google Scholar]
- 37.Parsons JB, et al. Identification of a two-component fatty acid kinase responsible for host fatty acid incorporation by Staphylococcus aureus. Proc Natl Acad Sci USA. 2014;111:10532–10537. doi: 10.1073/pnas.1408797111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang YM, Rock CO. Membrane lipid homeostasis in bacteria. Nat Rev Microbiol. 2008;6:222–233. doi: 10.1038/nrmicro1839. [DOI] [PubMed] [Google Scholar]
- 39.Schleifer KH, Kroppenstedt RM. Chemical and molecular classification of staphylococci. Soc Appl Bacteriol Symp Ser. 1990;19:9S–24S. doi: 10.1111/j.1365-2672.1990.tb01794.x. [DOI] [PubMed] [Google Scholar]
- 40.Wang J, et al. Platensimycin is a selective FabF inhibitor with potent antibiotic properties. Nature. 2006;441:358–361. doi: 10.1038/nature04784. [DOI] [PubMed] [Google Scholar]
- 41.McMurry LM, Oethinger M, Levy SB. Triclosan targets lipid synthesis. Nature. 1998;394:531–532. doi: 10.1038/28970. [DOI] [PubMed] [Google Scholar]
- 42.Rine J, Hansen W, Hardeman E, Davis RW. Targeted selection of recombinant clones through gene dosage effects. Proc Natl Acad Sci USA. 1983;80:6750–6754. doi: 10.1073/pnas.80.22.6750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Banerjee A, et al. inhA, a gene encoding a target for isoniazid and ethionamide in Mycobacterium tuberculosis. Science. 1994;263:227–230. doi: 10.1126/science.8284673. [DOI] [PubMed] [Google Scholar]
- 44.Sugden CJ, Roper JR, Williams JG. Engineered gene over-expression as a method of drug target identification. Biochem Biophys Res Commun. 2005;334:555–560. doi: 10.1016/j.bbrc.2005.06.117. [DOI] [PubMed] [Google Scholar]
- 45.Shi W, et al. Pyrazinamide inhibits trans-translation in Mycobacterium tuberculosis. Science. 2011;333:1630–1632. doi: 10.1126/science.1208813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Choi KH, Heath RJ, Rock CO. Beta-ketoacyl-acyl carrier protein synthase III (FabH) is a determining factor in branched-chain fatty acid biosynthesis. J Bacteriol. 2000;182:365–370. doi: 10.1128/jb.182.2.365-370.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kingston AW, Subramanian C, Rock CO, Helmann JD. A σW-dependent stress response in Bacillus subtilis that reduces membrane fluidity. Mol Microbiol. 2011;81:69–79. doi: 10.1111/j.1365-2958.2011.07679.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Young K, et al. Discovery of FabH/FabF inhibitors from natural products. Antimicrob Agents Chemother. 2006;50:519–526. doi: 10.1128/AAC.50.2.519-526.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wallace KK, Lobo S, Han L, McArthur HA, Reynolds KA. In vivo and in vitro effects of thiolactomycin on fatty acid biosynthesis in Streptomyces collinus. J Bacteriol. 1997;179:3884–3891. doi: 10.1128/jb.179.12.3884-3891.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kugelberg E, et al. Establishment of a superficial skin infection model in mice by using Staphylococcus aureus and Streptococcus pyogenes. Antimicrob Agents Chemother. 2005;49:3435–3441. doi: 10.1128/AAC.49.8.3435-3441.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Plaut RD, Mocca CP, Prabhakara R, Merkel TJ, Stibitz S. Stably luminescent Staphylococcus aureus clinical strains for use in bioluminescent imaging. PLoS One. 2013;8:e59232. doi: 10.1371/journal.pone.0059232. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.