

Abstract
BACKGROUND
Patients with advanced primary and recurrent salivary duct carcinoma (SDC), a rare and lethal malignancy, have limited therapeutic options. Novel small molecule agents aimed at targeting critical signaling associated with SDC tumorigenesis may lead to new therapeutic options in patients with these tumors. The HER2/PI3K axis, an important oncogenic pathway, has been targeted for therapy in several solid tumors. Currently, little is known on the role and clinical implications of alterations of the HER2/PI3K pathway in SDC.
METHODS
We investigated the clinicopathologic, genetic alterations and expression of key members of the HER2/PI3K pathway in 43 primary tumors and conducted in vitro functional and targeted drug response on cell lines derived from salivary epithelial carcinomas.
RESULTS
In primary tumors, loss of PTEN expression was found in 51% (22/43), overexpression of HER2 was observed in 28% (12/43) and PIK3CA mutations were identified in 28% (12/43) of tumors. Phospho-AKT (p-AKT) was highly expressed in most tumors. The majority of tumors (70%) displayed mutually exclusive alterations of PI3K members, while eight (19%) tumors had two or more concurrent abnormalities. In vitro studies demonstrated direct association between PTEN loss and PI3K pathway activation and evidence of response to combined PI3Kα and β and/or pan-PI3K inhibitors.
CONCLUSIONS
Our study reveals frequent PTEN loss and mutually exclusive alterations of key PI3K pathway members in SDCs and demonstrates in vitro evidence for response to pan-PI3K inhibitors. The study provides a framework for biomarker based sub-stratification of SDC patients’ in future targeted therapy.
Keywords: Salivary gland carcinoma, PI3K pathway, Salivary duct carcinoma, Targeted therapy, PTEN loss
Introduction
Salivary duct carcinoma (SDC), a rare and highly aggressive ductal epithelial malignancy of salivary glands, commonly presents at advanced stage in elderly patients of both sexes1. Histologically SDCs, both de novo and arising in pleomorphic adenoma (Ca ex-PA), display malignant epithelial cell proliferation forming glandular, cribriform, micro-papillary and nesting structures2-5. The primary therapy for SDC is complete surgical removal with lymph node dissection and post-operative radio- and/or chemotherapy. Patients with advanced unresectable primary, recurrent and metastatic disease have limited therapeutic options and are empirically treated with taxane-based and/or targeted agents6, 7. SDC also shares with other salivary adenocarcinomas subtypes and ductal mammary carcinoma common morphologic and biologic characteristics8-12 including alterations of the PI3K pathway11-16. In contrast to mammary carcinomas, however, functional and therapeutic studies of the PI3K pathway in SDC and other salivary adenocarcinomas have been limited, due largely to the lack of cell lines and animal models of this phenotype17, 18. We contend that assessing the differential alterations and the functional consequences of key members of the PI3K pathway may lead to better stratification of patients with salivary duct and other forms of adenocarcinomas for targeted therapy.
PI3K/AKT pathway and HER2 signaling play a critical role in regulating vital cellular processes including proliferation, survival and motility. Overexpression of HER2, an upstream receptor tyrosine kinase (RTK), has been linked to the activation of the PI3K pathway in mammary ductal carcinoma and other epithelial malignancies12-18. PI3K consists of a heterodimer of an 85kDa regulatory and a 110kDa catalytic subunits including PIK3CA, PIK3CB, or PIK3CD that can be activated by RTKs, through phosphorylation of phosphatidylinositol-4,5-biphosphate phosphates to phosphatidylinositol-3,4,5-trisphosphate (PIP3). PIP3 recruits phosphoinositide dependent kinase (PDK1) at cell membrane to induce downstream effectors19, 20. The catalytic activity of the PI3K, which is negatively regulated by PTEN, lead to up-regulation of p-AKT and the stimulation of critical cellular functions including cell proliferation, migration, survival and cell differentiation21, 22.
In this study, we aimed to determine 1) the incidence and inter-relationship of key members of the PI3K pathway in patients managed in a single institution, 2) for the first time the functional and therapeutic implications of PI3K alterations in cell lines developed from salivary epithelial carcinomas.
Material and Methods
Primary SDC tumors
A search of the head and neck tumor bank at the University of Texas, M.D. Anderson Cancer Center for patients with salivary duct carcinoma, either de novo or as a Carcinoma Ex PA, treated primarily at our institution identified 43 patients. Patients’ demographic and clinicopathologic information were available at our institution. Of the 43, 25 have been included in previous publication23 (see Supporting Table 1). The study was approved by the MD Anderson Cancer Center Institutional Review Board (IRB) and all patients have provided written informed consent for genomic and molecular analyses.
PIK3CA, PTEN, and AKT1 gene mutation analysis
Genomic DNA was extracted from tumor tissue samples with more than >70% tumor cells using Gentra Puregene tissue kit (QIAGEN, Valencia, CA) according to manufacturer’s instructions. The primer locations for PIK3CA, PTEN, and AKT1 gene mutation analysis were determined in reference to COSMIC database (http://cancer.sanger.ac.uk/cosmic). Genomic DNAs were PCR amplified using KAPA 2G fast (KAPA biosciences). PCR primer sets are presented in Supporting Table 2. All PCR products were purified using Exo-Sup and subsequently performed Sanger sequencing by Applied Biosystems 3730×1 DNA analyzer at Sequencing and Microarray Facility (SMF) of MD Anderson Cancer Center.
Western blot analysis
Cell lines and fresh tumor tissues were lysed using RIPA buffer containing freshly added protease (Roche Applied, 0505648900) and phosphatase (Roche Applied, 04906837001) inhibitor cocktails. Aliquots of 20 mg of protein lysate were loaded into SDS- PAGE gels and transferred to a nitrocellulose membrane. The blot was probed with anti-PTEN (138G6, rabbit, Cell Signaling), anti-phospho-AKT (p-AKT, Ser473, D9E, rabbit, Cell Signaling), anti-HER2 (D8F12, rabbit, Cell Signaling), and anti-ACTB (AC-15, mouse, Sigma-Aldrich) antibodies.
Immunohistochemistry (IHC)
IHC analysis was performed using 4 μm-thick unstained TMA sections on Autostainer Link 48 (Dako) according to the manufacturer’s instructions. Following incubation with the primary antibodies, HER2 (clone e2-4001, mouse, 1:300, Thermo scientific), PTEN (clone 6H2.1, mouse, 1:100, Dako) or p-AKT (Ser473, clone D9E, rabbit, 1:100, Cell Signaling), and then secondary antibody was applied. HER2 was evaluated for the extent and intensity of membranous and cytoplasmic expression as (+3); strong complete in >10%, (+2); moderate membranous in complete staining in >10%, (+1); weak partial staining of tumor cells in >10%, and (0); negative absence of staining. Only strong (score +3) complete membranous staining was used to define tumors as positive for HER210, 14, 15, 24. PTEN expression was scored in a binary fashion as negative (complete loss) and positive (cytoplasmic and/or nuclear staining) in tumor cells. p- AKT cytoplasmic expression was evaluated based on a binary scale where 0/+1 and +2/+3 were considered as low and high expression.
In vitro cell line studies
Three salivary epithelial derived carcinoma cell lines, the A253, RET98123, 25 and MAC26, were used. The A253 is an ATCC cell line developed from salivary epidermoid carcinoma (squamous-like) and may represent mucoepidermoid primary. The RET981 originated from metastatic malignant mixed tumor of the salivary gland25 with androgen receptor expression and represents SDC23. The MAC cell line is developed from a salivary mucinous adenocarcinoma (salivary duct and other adenocarcinomas can also manifest mucinous differentiation)4, 5, 27. The A253 was maintained in DMEM medium (Thermo Fisher Scientific) with 10% FBS and RET981 (Thermo Fisher Scientific) and MAC were maintained RPMI 1640 medium with 10% FBS using standard cell culture techniques. The prostate cancer cell line LNCaP (ATCC) was used as a PTEN loss control and was maintained in 1640 medium with 10% FBS.
For cellular viability assay to monitor drug toxicity, AZD8186 (PI3Kβ/δ inhibitors, Active Biochem), GSK2636771 (PI3Kβ inhibitor, Selleckchem), BYL719 (PI3Kα inhibitor, Selleckchem), and GDC-0941 (PI3Kα/β/γ/δ inhibitor, Selleckchem) were assessed using a 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) assay. Briefly, cells were seeded at 5000 cells/well in 96-well plates and were exposed to drugs after 24 hours seeded (day 0). MTT assay was performed at 0, 1, 2, 4 days after addition of the drugs.
PTEN knockdown study
PTEN shRNAs and non-silencing control in pGIPZ lentiviral vectors that co-express GFP were obtained from the shRNA/ORF Core Facility (MD Anderson Cancer Center). The 293FT cells (Invitrogen) were transfected with shRNA vector, pCMVR8.74 (addgene, #22036) and pMD2.G (addgene, #12259) using jetPRIME reagent (Polyplus transfection) for lentiviral packaging. SDC cell lines were infected with lentivirus supernatant in the presence of 8ug/mL polybrene. Puromycin selection and GFP sorting by Flow Cytometry and Cellular Imaging Core Facility (FCCICF) of MD Anderson Cancer Center were performed to generate the stable shRNA cell lines.
Statistical analysis
Pathologic findings, markers expression and mutational status, were evaluated by Fisher’s exact test based on the number of comparative groups. A p value of 0.05 was considered significant. Overall survival curves were created by the Kaplan–Meier analysis using GraphPad Prism6 software.
Results
Histopathology
Tumors were composed of 31 de novo tumors and 12 were SDC ex-PA (Supporting Table 1). Tumor cells displayed abundant eosinophilic, cytoplasm and central or eccentric large nuclei with checkered chromatin and occasional nuclei. Abnormal mitosis, apoptotic features and pleomorphism are not uncommonly present. In some tumors, mucinous, rhabdoid, oncocytoid cellular features were observed. These features are present in both de novo and salivary duct carcinoma ex-pleomorphic adenoma. As in previous study23, SDC cells manifested strong nuclear staining for androgen receptor in approximately 70% of tumors irrespective of their derivation (Supporting table 1).
HER2/PI3K pathway alterations in SDCs
A screening set of 20 cases with fresh frozen tumor tissues were tested for PIK3CA, PTEN, and AKT1 genes using Sanger sequencing. Five (20%) of the 20 tumors had hot spot mutations of PIK3CA gene. No mutations were found in both PTEN and AKT1 genes. PTEN and p-AKT expressions with variable levels were observed in all 20 SDCs by western blotting (Supporting Figure 1). Based on these findings, mutation analysis of the PIK3CA and IHC analyses of HER2, PTEN, and p-AKT expression were performed on 43 primary SDC tumors (Figure 1A and 1B). The findings are summarized in Figure 1C. Complete loss of PTEN was found in 22/43 (51%) tumors (Figure 1A; SDC-02 and -24, and Figure 1C) and HER2 was highly expressed in 12/43 (28%) tumors (Figure 1A; SDC-02, and Figure 1C). PIK3CA mutations were detected in 12 of the 43 tumors (28%, Figure 1C). Mutations in either exons 10 and 21 hotspots of PIK3CA (Figure 1B) were found in 10 tumors; p.H1047R (c.3140A>G) in 3 SDCs, p.E542K (c.1624G>A) in 5 SDCs, p.E545K (c.1633G>A) in 2 SDCs. In this study, the p.E542K mutation in PIK3CA was most frequent (Figure 1B, right panel) and one tumor each had p.E81K (c.241G>A) and p.S130_R131del mutations (Figure 1B, left panel), respectively (the latter mutation is not cited in COSMIC database). One patient (SDC-10) with p.S130_R131del mutation that had a remarkable response to a combination of temsirolimus and bevacizumab as previously reported by our group28.
Figure 1. HER2/PI3K/pathway alterations in salivary duct carcinomas.
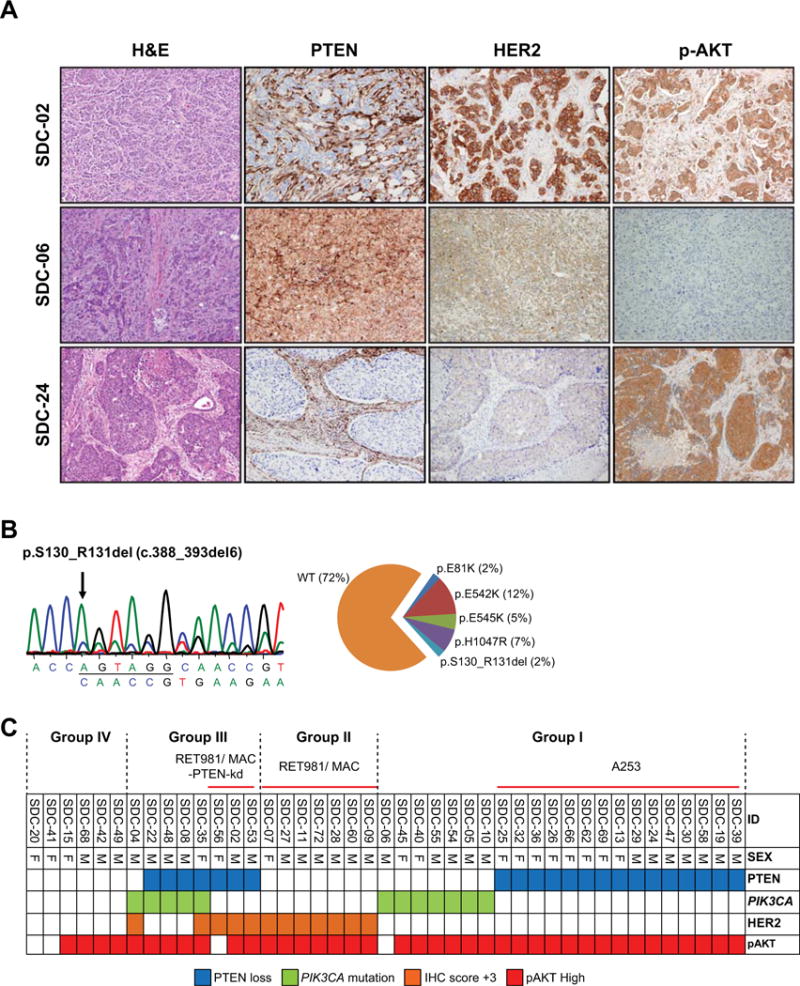
(A) Selected examples of IHC results for PTEN, HER2, and p-AKT in SDC cases. SDC-02 showed PTEN negative (positive in stroma) and high HER2 (+3) with high p-AKT. SDC-06 showed PTEN positive and negative HER2 expression without p-AKT. SDC-24 showed PTEN loss and negative of HER2 with high p-AKT. (B) PIK3CA mutation (p.S130_R131del) in SDC-10. A distribution of PIK3CA mutation in our study showed that p.E542K was most frequent mutation type. (C) A grid plot of alterations profile in PTEN, HER3, and p-AKT expressions and PIK3CA mutation. The numbers above a grit plot indicate the classification of SDC group and applied to our salivary tumor cell lines; Group I (A253): PTEN loss (or PIK3CA mutation)/HER2 negative, Group II (RET981 and MAC): intact PTEN/HER2 positive, Group III (RET981/MAC-PTEN-kd): concomitant alterations with PTEN loss/HER2 positive and/or PIK3CA mutation, and Group IV: no alteration.
Notably, alteration in at least one member of HER2/PI3K pathway was found in 37/43 (86%) and co-alteration of two or more members were found in 8/43 (19%) tumors. The most frequent alteration was the loss of PTEN expression, which was found in more than half of the tumors (22/43, 51%). HER2 overexpression was concurrently found with PIK3CA mutation in only 2/12 (17%) tumors; one of these tumors (SDC-35) also had PTEN loss, PIK3CA mutation, and HER2 overexpression. Consistent with the convergence of PTEN, PIK3CA, and HER2 activations in the PI3K pathway, high expression of p-AKT (Figure 1A, SDC-02 and -24) was observed in 39/43 (91%) tumors. We, therefore, empirically classify SDCs (Figure 1C) into four major groups; Group I: Tumors with single alteration (either PTEN loss or PIK3CA mutation), Group II: Tumors with overexpressing HER2, Group III: Tumors with more than two alterations, and Group IV: Tumor with no alterations.
Clinicopathological correlations and survival status
Table 1 presents the clinicopathologic factors and correlations with alterations in HER2/PI3K pathway members. We found no significant correlation between HER2 overexpression, PTEN loss, and PIK3CA mutation, patients’ outcome, and origin of SDC was found (Table 1, Supporting Figure 2, and Supporting Table 3). Only HER2 overexpression was significantly correlated with lymph node metastasis (p= 0.04) and PTEN loss was correlated with age (p= 0.02).
Table 1.
Incidence and cliniclpathologic correlation of PTEN, HER2, and p-AKT expressions and PIK3CA mutation in salivary ductal carcinomas
| Parameter | PTEN IHC | PIK3CA mutation | HER2 IHC | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Expressed | Lost | P- | Yes | No | P- | High (3+) | Low (0-2) | P- | |
| n (%) | n (%) | Value | n (%) | n (%) | Value | n (%) | n (%) | Value | |
| Age | |||||||||
| >60 | 7 (16) | 16 (37) | 6 (14) | 17 (40) | 4 (9) | 19 (44) | |||
| ≤60 | 14 (33) | 6 (14) | = 0.02 | 6 (14) | 14 (32) | = 1.00 | 8 (19) | 12 (28) | = 0.17 |
| Sex | |||||||||
| Male | 15 (35) | 12 (28) | 9 (21) | 18 (42) | 9 (21) | 18 (42) | |||
| Female | 6 (14) | 10 (23) | = 0.35 | 3 (7) | 13 (30) | = 0.48 | 3 (7) | 13 (30) | = 0.48 |
| Size | |||||||||
| <4 cm | 13 (30) | 10 (23) | 7 (16) | 16 (37) | 4 (9) | 19 (44) | |||
| ≥4 cm | 6 (14) | 9 (21) | = 0.51 | 4 (9) | 11 (26) | = 1.00 | 6 (14) | 9 (21) | = 0.15 |
| Stage | |||||||||
| I-II | 2 (5) | 1 (2) | 2 (5) | 1 (2) | 0 (0) | 3 (7) | |||
| III-IV | 17 (40) | 17 (40) | = 1.00 | 9 (21) | 25 (58) | = 0.21 | 12 (28) | 22 (51) | = 0.54 |
| PNI | |||||||||
| Yes | 15 (35) | 10 (23) | 8 (19) | 17 (40) | 10 (23) | 15 (35) | |||
| No | 3 (7) | 5 (12) | = 0.42 | 3 (7) | 5 (12) | = 1.00 | 2 (5) | 6 (14) | = 0.68 |
| LN met | |||||||||
| Yes | 15 (35) | 17 (40) | 9 (21) | 23 (54) | 12 (28) | 20 (47) | |||
| No | 6 (14) | 4 (9) | = 0.72 | 3 (7) | 7 (16) | = 1.00 | 0 (0) | 10 (23) | = 0.04 |
| Rec/Met | |||||||||
| Yes | 11 (26) | 15 (35) | 6 (14) | 20 (47) | 9 (21) | 17 (40) | |||
| No | 9 (21) | 7 (16) | = 0.53 | 5 (12) | 11 (26) | = 0.72 | 3 (7) | 13 (30) | = 0.32 |
| p-AKT IHC | |||||||||
| positive | 18 (42) | 21 (49) | 11 (26) | 28 (65) | 11 (26) | 28 (65) | |||
| negative | 3 (7) | 1(2) | = 0.35 | 1 (2) | 3 (7) | = 1.00 | 1 (2) | 3 (7) | = 1.00 |
Clinical information is available for age and sex fully and others are limited.
PNI; Perineural invasion, LN met; Lymph node metastasis, Rec/Met; Recurrence/Metastasis.
P-value was calculated by Fisher’s exact test.
PI3K signaling and response in PTEN deficient SDC cell lines
To characterize the HER2/PI3K/PTEN pathway in cell lines derived from salivary ductal epithelial carcinomas, we evaluated expressions of PTEN, HER2, and p-AKT and mutations of PIK3CA and PTEN in A253, RET981, and MAC cell lines. All three cell lines lacked PIK3CA mutation (data not shown). The RET981 and MAC cell lines expressed both PTEN and HER2 proteins (Figure 2A, left panel), while the A253 cell line showed loss of PTEN protein (Figure 2A, left panel) together with PTEN mutation (c.455T>C, Figure 2A right panel also reported in COSMIC database; http://cancer.sanger.ac.uk/cosmic) and low HER2 protein expression. The A253 cell line expressed higher level of p-AKT compared to both MAC and RET981 cells, possibly reflecting the status of PTEN. Based on these findings, A253 cell line was considered to represent SDC tumors with PTEN loss (Figure 1C, Group I), and RET981 and MAC cell lines to represent SDCs with intact PTEN and HER2 overexpression (Figure 1C, Group II).
Figure 2. PTEN status in A253 and RET981 salivary gland tumor cell lines.
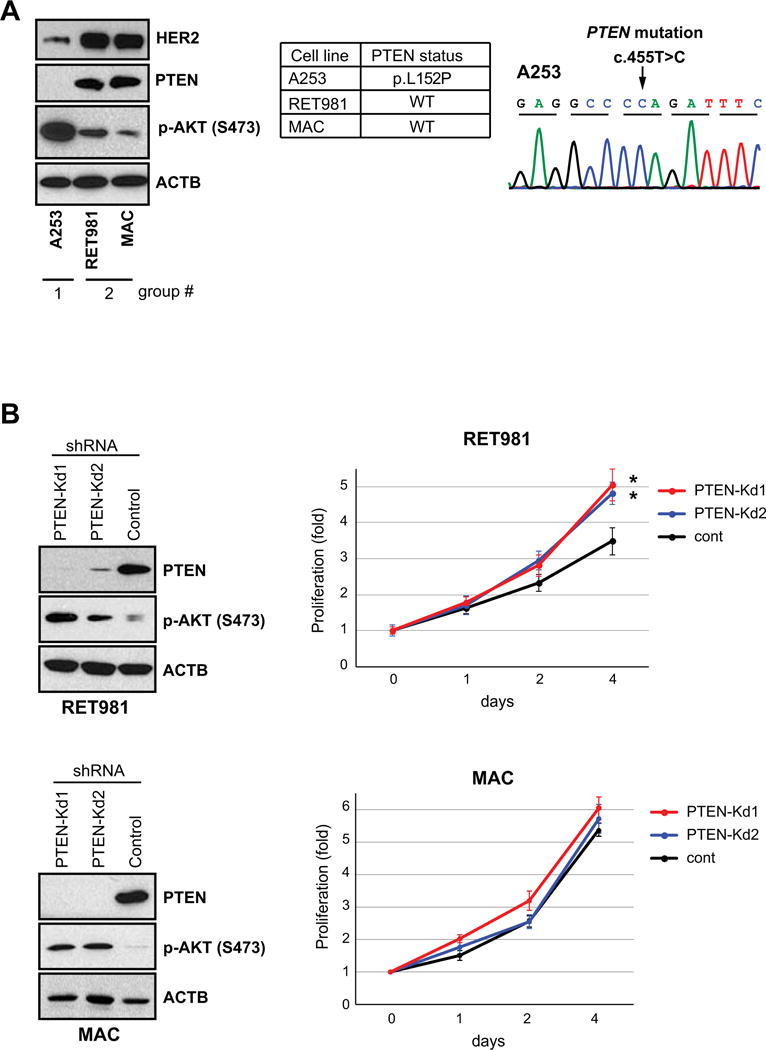
(A) PTEN is expressed in RET981 salivary gland tumor cell line. PTEN loss and its mutation were found in A253 cell line. (B) Left panel, PTEN knockdown (kd) led to increases of p-AKT expression in RET981 cell lines. Right panel, PTEN knockdown (kd) increases cell proliferation as measured by MTT assay and value was normalized to the point of day 0. Error bards represent SD. Asterisks (*) represents significant difference of proliferation activities between PTEN-kd and control lines (p <0.01).
To determine the effect of PTEN loss on the PI3K activation, we stably knocked down PTEN expression (PTEN-kd) in the RET981 and MAC cell lines. Knockdown of PTEN consistently resulted in increase of p-AKT in both PTEN-kd cells (Figure 2B, left panel). Two PTEN-kd RET981 clones showed significant increase of cell proliferation compared to the control (Figure 2B, right panel). In contrast, PTEN-kd MAC lines showed comparatively higher cell proliferation than control. Thus, knockdown of PTEN led to PI3K activation in both RET981 and MAC cells. We, therefore, empirically considered both PTEN-kd of RET981 and MAC cells to mimic tumors with concurrent PTEN loss and HER2 overexpression (Figure 1C, Group III).
Since epithelial malignances with PTEN loss have been associated with PI3Kβ activation29, 30, we investigated the effect of PTEN loss on response to PI3Kβ inhibitors in our cell lines. The effect of AZD8186 (PI3Kβ and PI3Kδ inhibitor) on A253 cell and LNCaP (a prostate cancer cell line with mutation and one allele deletion of PTEN gene) was evaluated. Treatment of LNCaP cells with AZD8186 (0.25 μM) resulted in reduction of cell proliferation (Figure 3A, upper panel) and inhibition of p-AKT expression at 3 hours post-treatment with rebound of p-AKT expression at 12 hours (Figure 3A, lower panel), in agreement with a previous report31. In contrast, treatment with AZD8186 at different doses on the A253 cells lines showed minimal or no effect on cell growth and p-AKT expression (Figure 3B). Similarly, treatment with a different PI3Kβ inhibitor GSK2636771 had no effect on A253 proliferation (Figure 3C). These results indicate that targeting PI3Kβ isoform alone had no effect on the proliferation of A253cell (PTEN loss and low HER2 expression).
Figure 3. Effect of PI3K inhibitors on the proliferation in PTEN deficient (Group I) A253 salivary gland cells.
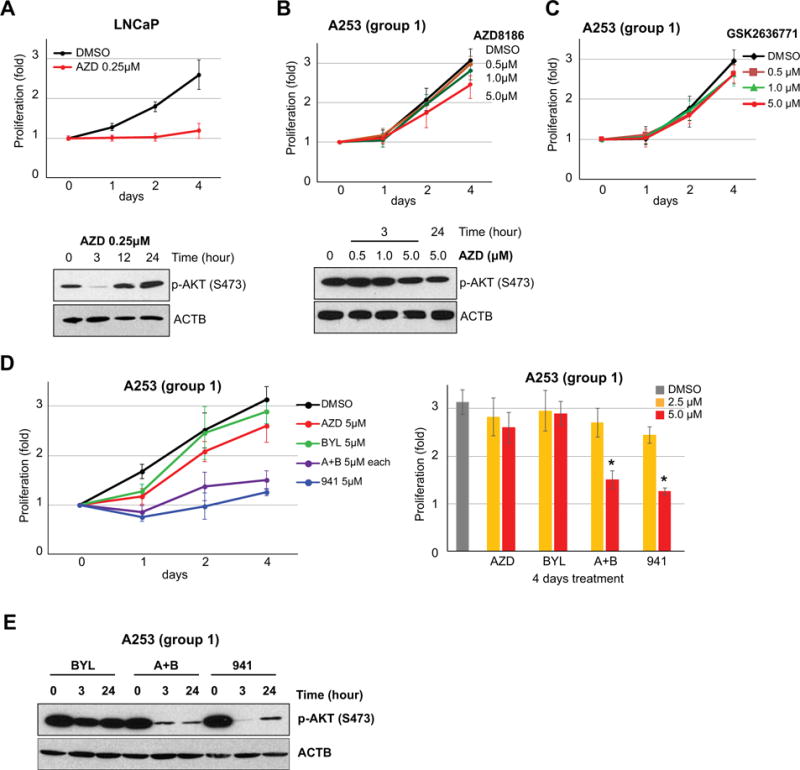
(A) Treatment of LNCaP cells with AZD (0.25μM) showed significant decrease of cell growth. P-AKT was inhibited after 3 hours but rebounded after 12 hours treatment. (B) and (C) High dose (5μM) of PI3Kβ inhibitors (AZD8186 and GSK2636771) had no impact on cell growth and p-AKT expression level of A253 salivary tumor cell. (D) Effects of BYL719 (BYL, 5μM), AZD8186+BYL719 (A+B, 5μM), and GDC941 (941, 5μM) on A253 cell. Left upper panel, A253 cell growth inhibition was repressed by combination treatment of PI3Kα and PI3Kβ inhibitors (A+B, 5μM each) and pan-PI3K inhibitor (941, 5 μM). Left lower panel, comparison of dosage differences (2.5 μM and 5 μM at 4 days treatment) against AZD, BYL, A+B, and 941 on A253 cells. Asterisk (*) indicates that A253 was impacted on cell proliferation by 5 μM of A+B and 941 treatment. (E) P-AKT level after 3h and 24h of treatments. Both combination and pan-PI3K inhibitors maintained inhibition of p-AKT level after 24 h. Error bars in all graph related to cell proliferation indicate SD.
To assess the role of PI3Kα isoform activation on cell proliferation, we treated A253 cell with BYL719 (PI3Kα inhibitor). BYL719 at 5 μM showed no changes in proliferation or p-AKT protein level in A253 cell line (Figure 3D and 3E). To determine whether inhibition of both PI3Kα and PI3Kβ isoforms affect cell kinetics, we treated A253 cells with combination of PI3Kα (BYL719) and PI3Kβ (AZD8186) and to pan-PI3K (GDC-0941) inhibitors. Combined PI3Kα and -β and pan-PI3K treatment at 2.5 μM showed no demonstrable changes in A253 cell proliferation (Figure 3D, right panel). However, treatment with 5 μM dose resulted in significant reduction of proliferation and p-AKT expression in comparison to DMSO control (Figure 3D and 3E). The results suggest that patients with SDC tumors with loss of PTEN and low HER2 expression (Group I) may respond to combined PI3Kα and PI3Kβ inhibitors or pan-PI3K inhibitor.
To investigate the association of HER2 overexpression and intact PTEN (Group II) in response to PI3K inhibitors, we treated HER2 expressing cell lines (RET981 and MAC) with PI3Kα and PI3Kβ inhibitors either alone or in combination and with a pan-PI3K inhibitor. We observed variable responses to individual and combined PI3K inhibitors between both lines. The RET981 cell line showed partial suppression of proliferation at 2.5 μM and complete suppression at 5 μM PI3Kα or PI3Kβ inhibitors alone or in combination or to pan-PI3K inhibitor (Figure 4A). In contrast, MAC cells showed only partial reduction of cell proliferation at 5 μM with pan-PI3K inhibitor or combined PI3Kα and PI3Kβ inhibitors but not to PI3Kα or PI3Kβ inhibitors alone (Figure 4B). These findings suggest that SDC tumors with intact PTEN and HER2 overexpression (Group II) could respond to combined PI3Kα and PI3Kβ inhibitors or pan-PI3K inhibitor.
Figure 4. Effect of PI3K inhibitors on the proliferation in HER2 positive (Group II) salivary gland cells.
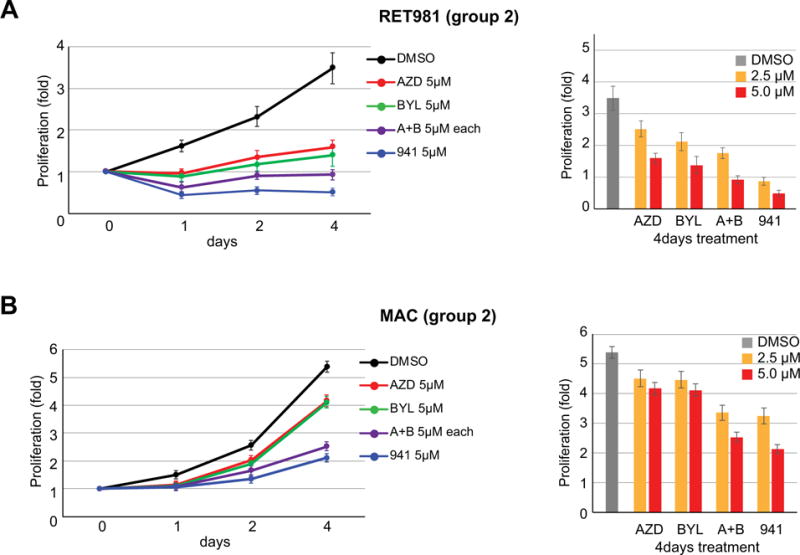
Effects of BYL719 (BYL, 5μM), AZD8186+BYL719 (A+B, 5μM), and GDC941 (941, 5μM) on RET981 (A) and MAC (B) cells. Differential responses to both lines were observed, where RET981 cells was suppressed completely by 5 μM of both PI3Kα and PI3Kβ alone. In contrast, MAC cells demonstrated inhibition of cell proliferation at 5 μM doses of pan-inhibitor or combined PI3Kα and PI3Kβ but not by either PI3Kα or PI3Kβ alone.
To assess whether PTEN loss in HER2 overexpression cells influence response to PI3K inhibitors (Figure 1C, Group III), we treated PTEN-kd RET981 and MAC cell lines with PI3K inhibitors. Proliferation of PTEN-kd RET981 cells was inhibited by treatment with 5 μM AZD8186 (PI3Kβ) or BYL719 (PI3Kα) alone or in combination, and GDC-0941 (pan-PI3K) inhibitor (Figure 5A) and p-AKT expression were markedly reduced (Figure 5B), similar to those observed in parental RET981 cells (Figure 4A). In contrast, MAC PTEN-kd lines showed no response to PI3Kα and PI3Kβ inhibitors but partial inhibition to combination PI3Kα and PI3Kβ and pan-PI3K inhibitor (Figure 5C). Similarly, p-AKT of MAC PTEN-kd lines was inhibited by combination PI3Kα and PI3Kβ and pan-PI3K inhibitor (Figure 5D). Together, these results suggest that all SDC tumor groups could potentially respond to combined PI3Kα and PI3Kβ inhibitors or pan-PI3K inhibitor.
Figure 5. Growth suppression by combined PI3Kα and PI3Kβ inhibition in PTEN-knockout/HER2 overexpression (Group III) SDC cell lines.
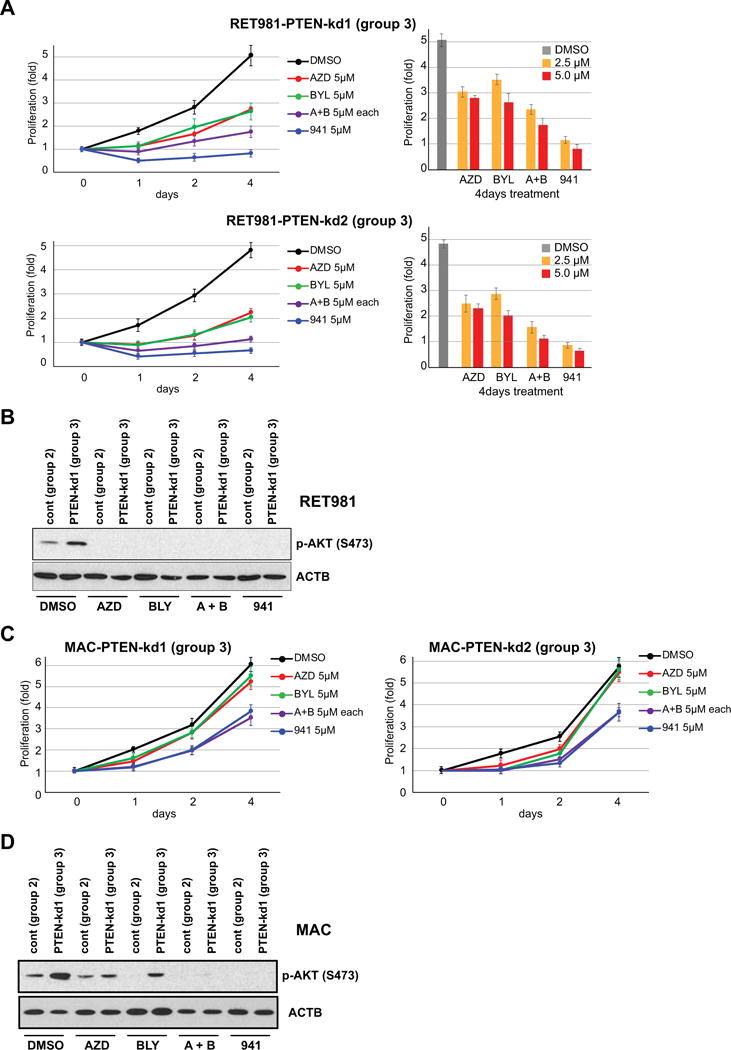
(A) Upper panel, anti-proliferative activity in both PTEN-kd lines was increased by combined treatment (5 μM each) and pan-PI3K (5 μM) inhibitor in PTEN-kd RET981 cells. Lower panel, comparison of dosage differences (2.5 μM and 5 μM at 4 days treatment) against single or combined PI3K inhibitors on PTEN-kd and control RET981 cells. Asterisk (*) indicates that PTEN-kd lines show no changes in cell proliferation at 5 μM of AZD 8186 only compared with other inhibitors. Error bars in all graph related to cell proliferation indicate SD. (B) P-AKT level after 24h of AZD (5μM), BYL (5μM), A+B (5μM), and 941 (5μM) treatments on PTEN-kd of RET981 cells. (C) Both PTEN-kd MAC cell lines indicate more resistance to AZD (5μM), BYL (5μM), A+B (5μM), and 941 (5μM) treatments than control line. (D) P-AKT level after 24h of AZD (5μM), BYL (5μM), A+B (5μM), and 941 (5μM) treatments on PTEN-kd of MAC cells. Of note, p-AKT level retained in control line after single treatment with AZD.
Discussion
Biomarker based stratification of patients for small molecules targeting critical pathways associated with SDC development and progression may lead to sustained response in certain patients32, 33. Our screening of HER2/PI3K pathway revealed frequent and reciprocal alterations of HER2 and key components of the PI3K pathway in majority of SDC patients. We also provide evidence that certain SDC patients with HER2/PI3K pathway abnormalities may benefit from combined PI3Kα and PI3Kβ or pan-PI3K inhibitors. The study also highlight the dominant loss of PTEN expression in SDCs, sustaining previous IHC based findings of salivary and various epithelial malignancies11, 13, 21, 34-37. Together, these data advance the use of IHC as a reliable tool in the determination of PTEN protein loss in SDC irrespective of the genetic, epigenetic and/or post-transcriptional modifications of this gene21, 38.
Consistent with previous studies of SDC and mammary ductal carcinomas, high HER2 expression was found in 25% of SDC either alone or concurrently with different PI3K alterations. The findings sustain that HER2 status may allow the stratification of a subset of patients to either anti-HER2 or PI3K inhibitors based on the presence or absence of other PI3K pathway alterations39-44. Similarly, PIK3CA missense mutations at known hotspots were found in a certain subset of tumors confirming previous studies of SDC11, 45-47 and other carcinomas17, 18, 36, 48-54. Although four tumors (9%) had concurrent PTEN loss and PIK3CA mutation, the significance of these findings cannot be determined due to the lack of PI3K mutation in salivary cell lines. Interestingly, the limited occurrence of both alterations in SDC is at variance with those reported in endometrial carcinoma studies where frequent loss of PTEN and mutation of the PIK3CA were occurred more frequently50. These results underscore a considerable inter-tumor variations and a differential tumor context association of PI3K alterations that may impact their therapeutic stratification.55-58.
Our in vitro findings, for the first time, provide evidence for a direct link between PTEN loss and PI3K activation and a potential response to PI3K inhibitors. The data show that loss of PTEN cell lines was invariably associated with PI3K activation sustaining an important role in SDC tumorigenesis22, 34, 59. Interestingly, salivary cell lines with PTEN loss didn’t respond to PI3Kβ selective inhibitors contrary to reported dependency on PI3K β isoform in PTEN deficient tumors29, 30. However, that response was achieved by combined PI3Kα and PI3Kβ or pan-PI3K inhibitors, suggesting reciprocal relationship between PI3Kα and β activities. Similar findings have recently been reported in PTEN deficient prostate and breast cancer cell lines and were attributed to the functional interactions of PI3Kα and β isoforms31, 60. In contrast to SDCs, head and neck squamous cell carcinoma cells with PTEN loss were commonly resistant to pan-PI3K inhibitor, indicating tumor context differences61. Contrary to reports of the involvement of the HRAS gene mutation in one third of SDC45-47, 62, we found no mutation of the HRAS in this cohort (Supporting Table 5). This, together with the lack of cell lines with mutations of this gene, suggests a minimal functional role of this gene in SDC tumorigenesis in this study. Of note, marked clinical response has been achieved in one of the SDC patients in this cohort with tumor with PIK3CA mutation to combined temsirolimus and bevacizumab treatment28.
Collectively, our findings demonstrate common occurrence of PI3K alterations in SDC and a potential for sub-stratification of patients based on PI3K markers status for targeted PI3K therapy59. Together with cell lines findings, the data suggest that patients can be stratified based on alterations of the PI3K pathway can empirically be categorized into 1) patients with single alteration (Group I) would be stratified for pan-PI3K inhibitor, 2) patients with HER2 only (Group II) to Herceptin or pan-PI3K inhibitor 3) patients with more than two alterations (Group III) to combined anti-HER2 and pan-PI3K therapy or higher dose, 4) patients with no alterations (Group IV) for conventional therapy. In this study, only small number of tumors lacked p-AKT expression, however, the significance and the underlying cause for the loss of this marker. We also observed no significant association between PI3K alterations and patient outcome, likely due to the underlying aggressive nature of this disease. These findings are concordant with previous studies and highlights the short-term survival of these patients regardless of conventional therapy used8, 28, 63, 64.
In summary, our study demonstrates the activation of the of HER2/PI3K alterations in the majority of SDC patients and provides evidence for potential response to combined PI3Kα and PI3Kβ or pan-PI3K targeted therapy in patients with SDC.
Supplementary Material
Acknowledgments
The authors thank Yan Cai, Deborah A. Rodriguez and Cynthia F. Steward for material retrieval and follow-up information.
Grant Support: The study is supported in part by the NIH National Institute of Dental and Craniofacial Research and the NIH Office of Rare Diseases Research grant number U01DE019765, the SGTB (Salivary Gland Tumor Biorepository, HHSN268200900039C 04), the Head and Neck SPORE Program grant number P50 CA097007, The Kenneth D. Muller professorship, The Center for Genetics and NCI CA-16672 grant.
Footnotes
Author Contributions:
Pierre Saintigny: Conceptualization, data curation, formal analysis, investigation, methodology, writing - original draft, and writing - review and editing. Yoshitsugu Mitani: Conceptualization, data curation, formal analysis, investigation, methodology, validation, writing - original draft, and writing - review and editing. Kristen B Pytynia: Resources, and writing - review and editing. Renata Ferratorro: Resources, and writing - review and editing. Dianna B. Roberts: Formal analysis, and writing - review and editing. Randal S. Weber: Resources, and writing - review and editing. Merrill S. Kies: Resources, and writing - review and editing. Sankar N. Maity: Conceptualization, supervision, writing - original draft, and writing - review and editing. Sue-Hwa Lin: Conceptualization, supervision, project administration, writing - original draft, and writing - review and editing. Adel K. El-Naggar: Conceptualization, supervision, funding acquisition, project administration, writing - original draft, and writing - review and editing.
Conflict of Interest Disclosures: The authors of this manuscript have no conflict of interest to disclose.
References
- 1.Wee DT, Thomas AA, Bradley PJ. Salivary duct carcinoma: what is already known, and can we improve survival? J Laryngol Otol. 2012;126(Suppl 2):S2–7. doi: 10.1017/S0022215112000412. [DOI] [PubMed] [Google Scholar]
- 2.Murrah VA, Batsakis JG. Salivary duct carcinoma. Ann Otol Rhinol Laryngol. 1994;103:244–247. doi: 10.1177/000348949410300315. [DOI] [PubMed] [Google Scholar]
- 3.Barnes L, Rao U, Krause J, Contis L, Schwartz A, Scalamogna P. Salivary duct carcinoma. Part I. A clinicopathologic evaluation and DNA image analysis of 13 cases with review of the literature. Oral Surg Oral Med Oral Pathol. 1994;78:64–73. doi: 10.1016/0030-4220(94)90119-8. [DOI] [PubMed] [Google Scholar]
- 4.Simpson RH, Prasad AR, Lewis JE, Skalova A, David L. Mucin-rich variant of salivary duct carcinoma: a clinicopathologic and immunohistochemical study of four cases. Am J Surg Pathol. 2003;27:1070–1079. doi: 10.1097/00000478-200308000-00004. [DOI] [PubMed] [Google Scholar]
- 5.Yakirevich E, Sabo E, Klorin G, et al. Primary mucin-producing tumours of the salivary glands: a clinicopathological and morphometric study. Histopathology. 2010;57:395–409. doi: 10.1111/j.1365-2559.2010.03639.x. [DOI] [PubMed] [Google Scholar]
- 6.Laurie SA, Licitra L. Systemic therapy in the palliative management of advanced salivary gland cancers. J Clin Oncol. 2006;24:2673–2678. doi: 10.1200/JCO.2005.05.3025. [DOI] [PubMed] [Google Scholar]
- 7.Speight PM, Barrett AW. Salivary gland tumours. Oral Dis. 2002;8:229–240. doi: 10.1034/j.1601-0825.2002.02870.x. [DOI] [PubMed] [Google Scholar]
- 8.Glisson B, Colevas AD, Haddad R, et al. HER2 expression in salivary gland carcinomas: Dependence on histological subtype. Clinical Cancer Research. 2004;10:944–946. doi: 10.1158/1078-0432.ccr-03-0253. [DOI] [PubMed] [Google Scholar]
- 9.Jaehne M, Roeser K, Jaekel T, Schepers JD, Albert N, Loning T. Clinical and immunohistologic typing of salivary duct carcinoma - A report of 50 cases. Cancer. 2005;103:2526–2533. doi: 10.1002/cncr.21116. [DOI] [PubMed] [Google Scholar]
- 10.Williams MD, Roberts D, Blumenschein GR, Jr, et al. Differential expression of hormonal and growth factor receptors in salivary duct carcinomas: biologic significance and potential role in therapeutic stratification of patients. Am J Surg Pathol. 2007;31:1645–1652. doi: 10.1097/PAS.0b013e3180caa099. [DOI] [PubMed] [Google Scholar]
- 11.Griffith CC, Seethala RR, Luvison A, Miller M, Chiosea SI. PIK3CA mutations and PTEN loss in salivary duct carcinomas. Am J Surg Pathol. 2013;37:1201–1207. doi: 10.1097/PAS.0b013e3182880d5a. [DOI] [PubMed] [Google Scholar]
- 12.Nardi V, Sadow PM, Juric D, et al. Detection of novel actionable genetic changes in salivary duct carcinoma helps direct patient treatment. Clinical Cancer Research. 2013;19:480–490. doi: 10.1158/1078-0432.CCR-12-1842. [DOI] [PubMed] [Google Scholar]
- 13.Ettl T, Baader K, Stiegler C, et al. Loss of PTEN is associated with elevated EGFR and HER2 expression and worse prognosis in salivary gland cancer. Br J Cancer. 2012;106:719–726. doi: 10.1038/bjc.2011.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Suzuki S, Dobashi Y, Minato H, Tajiri R, Yoshizaki T, Ooi A. EGFR and HER2-Akt-mTOR signaling pathways are activated in subgroups of salivary gland carcinomas. Virchows Arch. 2012;461:271–282. doi: 10.1007/s00428-012-1282-3. [DOI] [PubMed] [Google Scholar]
- 15.Williams MD, Roberts DB, Kies MS, Mao L, Weber RS, El-Naggar AK. Genetic and expression analysis of HER-2 and EGFR genes in salivary duct carcinoma: empirical and therapeutic significance. Clinical Cancer Research. 2010;16:2266–2274. doi: 10.1158/1078-0432.CCR-09-0238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Locati LD, Perrone F, Losa M, et al. Treatment relevant target immunophenotyping of 139 salivary gland carcinomas (SGCs) Oral Oncol. 2009;45:986–990. doi: 10.1016/j.oraloncology.2009.05.635. [DOI] [PubMed] [Google Scholar]
- 17.Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008;68:6084–6091. doi: 10.1158/0008-5472.CAN-07-6854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Faivre S, Kroemer G, Raymond E. Current development of mTOR inhibitors as anticancer agents. Nat Rev Drug Discov. 2006;5:671–688. doi: 10.1038/nrd2062. [DOI] [PubMed] [Google Scholar]
- 20.Massacesi C, di Tomaso E, Fretault N, Hirawat S. Challenges in the clinical development of PI3K inhibitors. Ann N Y Acad Sci. 2013;1280:19–23. doi: 10.1111/nyas.12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol. 2012;13:283–296. doi: 10.1038/nrm3330. [DOI] [PubMed] [Google Scholar]
- 22.Thorpe LM, Yuzugullu H, Zhao JJ. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer. 2015;15:7–24. doi: 10.1038/nrc3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mitani Y, Rao PH, Maity SN, et al. Alterations associated with androgen receptor gene activation in salivary duct carcinoma of both sexes: potential therapeutic ramifications. Clinical Cancer Research. 2014;20:6570–6581. doi: 10.1158/1078-0432.CCR-14-1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vidal L, Tsao MS, Pond GR, et al. Fluorescence in situ hybridization gene amplification analysis of EGFR and HER2 in patients with malignant salivary gland tumors treated with lapatinib. Head Neck. 2009;31:1006–1012. doi: 10.1002/hed.21052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rao PH, Murty VV, Louie DC, Chaganti RS. Nonsyntenic amplification of MYC with CDK4 and MDM2 in a malignant mixed tumor of salivary gland. Cancer Genet Cytogenet. 1998;105:160–163. doi: 10.1016/s0165-4608(98)00013-2. [DOI] [PubMed] [Google Scholar]
- 26.Panaccione A, Zhang Y, Mi Y, et al. Chromosomal abnormalities and molecular landscape of metastasizing mucinous salivary adenocarcinoma. Oral Oncol. 2017;66:38–45. doi: 10.1016/j.oraloncology.2016.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Terasaki M, Terasaki Y, Wakamatsu K, et al. A mucin-rich variant of salivary duct carcinoma with a prominent mucinous component, a tumor that mimics mucinous adenocarcinoma. Oral Surg Oral Med Oral Pathol Oral Radiol. 2013;116:e210–214. doi: 10.1016/j.oooo.2013.01.002. [DOI] [PubMed] [Google Scholar]
- 28.Piha-Paul SA, Cohen PR, Kurzrock R. Salivary duct carcinoma: targeting the phosphatidylinositol 3-kinase pathway by blocking mammalian target of rapamycin with temsirolimus. J Clin Oncol. 2011;29:e727–730. doi: 10.1200/JCO.2011.36.2095. [DOI] [PubMed] [Google Scholar]
- 29.Jia S, Liu Z, Zhang S, et al. Essential roles of PI(3)K-p110beta in cell growth, metabolism and tumorigenesis. Nature. 2008;454:776–779. doi: 10.1038/nature07091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wee S, Wiederschain D, Maira SM, et al. PTEN-deficient cancers depend on PIK3CB. Proc Natl Acad Sci U S A. 2008;105:13057–13062. doi: 10.1073/pnas.0802655105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schwartz S, Wongvipat J, Trigwell CB, et al. Feedback suppression of PI3Kalpha signaling in PTEN-mutated tumors is relieved by selective inhibition of PI3Kbeta. Cancer Cell. 2015;27:109–122. doi: 10.1016/j.ccell.2014.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kelloff GJ, Sigman CC. Cancer biomarkers: selecting the right drug for the right patient. Nat Rev Drug Discov. 2012;11:201–214. doi: 10.1038/nrd3651. [DOI] [PubMed] [Google Scholar]
- 33.Sawyers CL. The cancer biomarker problem. Nature. 2008;452:548–552. doi: 10.1038/nature06913. [DOI] [PubMed] [Google Scholar]
- 34.Chalhoub N, Baker SJ. PTEN and the PI3-kinase pathway in cancer. Annu Rev Pathol. 2009;4:127–150. doi: 10.1146/annurev.pathol.4.110807.092311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hollander MC, Blumenthal GM, Dennis PA. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat Rev Cancer. 2011;11:289–301. doi: 10.1038/nrc3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27:5497–5510. doi: 10.1038/onc.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang S, Yu D. PI(3)king apart PTEN’s role in cancer. Clinical Cancer Research. 2010;16:4325–4330. doi: 10.1158/1078-0432.CCR-09-2990. [DOI] [PubMed] [Google Scholar]
- 38.Worby CA, Dixon JE. Pten. Annu Rev Biochem. 2014;83:641–669. doi: 10.1146/annurev-biochem-082411-113907. [DOI] [PubMed] [Google Scholar]
- 39.Berns K, Horlings HM, Hennessy BT, et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 2007;12:395–402. doi: 10.1016/j.ccr.2007.08.030. [DOI] [PubMed] [Google Scholar]
- 40.Chandarlapaty S, Sakr RA, Giri D, et al. Frequent mutational activation of the PI3K-AKT pathway in trastuzumab-resistant breast cancer. Clinical Cancer Research. 2012;18:6784–6791. doi: 10.1158/1078-0432.CCR-12-1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cizkova M, Dujaric ME, Lehmann-Che J, et al. Outcome impact of PIK3CA mutations in HER2-positive breast cancer patients treated with trastuzumab. Br J Cancer. 2013;108:1807–1809. doi: 10.1038/bjc.2013.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gajria D, Chandarlapaty S. HER2-amplified breast cancer: mechanisms of trastuzumab resistance and novel targeted therapies. Expert Rev Anticancer Ther. 2011;11:263–275. doi: 10.1586/era.10.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nagata Y, Lan KH, Zhou X, et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004;6:117–127. doi: 10.1016/j.ccr.2004.06.022. [DOI] [PubMed] [Google Scholar]
- 44.Loibl S, von Minckwitz G, Schneeweiss A, et al. PIK3CA mutations are associated with lower rates of pathologic complete response to anti-human epidermal growth factor receptor 2 (her2) therapy in primary HER2-overexpressing breast cancer. J Clin Oncol. 2014;32:3212–3220. doi: 10.1200/JCO.2014.55.7876. [DOI] [PubMed] [Google Scholar]
- 45.Dalin MG, Desrichard A, Katabi N, et al. Comprehensive molecular characterization of salivary duct carcinoma reveals actionable targets and similarity to apocrine breast cancer. Clinical Cancer Research. 2016 doi: 10.1158/1078-0432.CCR-16-0637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang K, Russell JS, McDermott JD, et al. Profiling of 149 Salivary Duct Carcinomas, Carcinoma Ex Pleomorphic Adenomas, and Adenocarcinomas, Not Otherwise Specified Reveals Actionable Genomic Alterations. Clinical Cancer Research. 2016;22:6061–6068. doi: 10.1158/1078-0432.CCR-15-2568. [DOI] [PubMed] [Google Scholar]
- 47.Chiosea SI, Thompson LD, Weinreb I, et al. Subsets of salivary duct carcinoma defined by morphologic evidence of pleomorphic adenoma, PLAG1 or HMGA2 rearrangements, and common genetic alterations. Cancer. 2016;122:3136–3144. doi: 10.1002/cncr.30179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lui VW, Hedberg ML, Li H, et al. Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer Discov. 2013;3:761–769. doi: 10.1158/2159-8290.CD-13-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Massion PP, Taflan PM, Shyr Y, et al. Early involvement of the phosphatidylinositol 3-kinase/Akt pathway in lung cancer progression. Am J Respir Crit Care Med. 2004;170:1088–1094. doi: 10.1164/rccm.200404-487OC. [DOI] [PubMed] [Google Scholar]
- 50.Oda K, Stokoe D, Taketani Y, McCormick F. High frequency of coexistent mutations of PIK3CA and PTEN genes in endometrial carcinoma. Cancer Res. 2005;65:10669–10673. doi: 10.1158/0008-5472.CAN-05-2620. [DOI] [PubMed] [Google Scholar]
- 51.Samuels Y, Wang Z, Bardelli A, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 52.Taylor BS, Schultz N, Hieronymus H, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vander Broek R, Mohan S, Eytan DF, Chen Z, Van Waes C. The PI3K/Akt/mTOR axis in head and neck cancer: functions, aberrations, cross-talk, and therapies. Oral Dis. 2015;21:815–825. doi: 10.1111/odi.12206. [DOI] [PubMed] [Google Scholar]
- 54.Zhang J, Roberts TM, Shivdasani RA. Targeting PI3K Signaling as a Therapeutic Approach for Colorectal Cancer. Gastroenterology. 2011;141:50–61. doi: 10.1053/j.gastro.2011.05.010. [DOI] [PubMed] [Google Scholar]
- 55.Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lawrence MS, Stojanov P, Polak P, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu B, Mitani Y, Rao X, et al. Spatio-Temporal Genomic Heterogeneity, Phylogeny, and Metastatic Evolution in Salivary Adenoid Cystic Carcinoma. JNCI: Journal of the National Cancer Institute. 2017;109:djx033–djx033. doi: 10.1093/jnci/djx033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bissell MJ, Labarge MA. Context, tissue plasticity, and cancer: are tumor stem cells also regulated by the microenvironment? Cancer Cell. 2005;7:17–23. doi: 10.1016/j.ccr.2004.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Janku F, Hong DS, Fu S, et al. Assessing PIK3CA and PTEN in early-phase trials with PI3K/AKT/mTOR inhibitors. Cell Rep. 2014;6:377–387. doi: 10.1016/j.celrep.2013.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cescon DW, Gorrini C, Mak TW. Breaking up is hard to do: PI3K isoforms on the rebound. Cancer Cell. 2015;27:5–7. doi: 10.1016/j.ccell.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 61.Mazumdar T, Byers LA, Ng PK, et al. A comprehensive evaluation of biomarkers predictive of response to PI3K inhibitors and of resistance mechanisms in head and neck squamous cell carcinoma. Mol Cancer Ther. 2014;13:2738–2750. doi: 10.1158/1535-7163.MCT-13-1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Khoo TK, Yu B, Smith JA, et al. Somatic mutations in salivary duct carcinoma and potential therapeutic targets. Oncotarget. 2017;8:75893–75903. doi: 10.18632/oncotarget.18173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Haddad R, Colevas AD, Krane JF, et al. Herceptin in patients with advanced or metastatic salivary gland carcinomas. A phase II study. Oral Oncol. 2003;39:724–727. doi: 10.1016/s1368-8375(03)00097-6. [DOI] [PubMed] [Google Scholar]
- 64.Limaye SA, Posner MR, Krane JF, et al. Trastuzumab for the treatment of salivary duct carcinoma. Oncologist. 2013;18:294–300. doi: 10.1634/theoncologist.2012-0369. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.