

Abstract
Because of the increase in the number of immunocompromised patients, the incidence of invasive fungal infections is growing, but the treatment efficiency remains unacceptably low. The most potent clinical systemic antifungals (azoles) are the derivatives of two scaffolds: ketoconazole and fluconazole. Being the safest antifungal drugs, they still have shortcomings, mainly because of pharmacokinetics and resistance. Here, we report the successful use of the target fungal enzyme, sterol 14α−demethylase (CYP51), for structure-based design of novel antifungal drug candidates by minor modifications of VNI [(R)-N-(1-(2,4-dichlorophenyl)-2-(1H−imidazol-1-yl)ethyl)-4-(5-phenyl-1,3,4-oxadiazol-2-yl)-benzamide)], an inhibitor of protozoan CYP51 that cures Chagas disease. The synthesis of fungi-oriented VNI derivatives, analysis of their potencies to inhibit CYP51s from two major fungal pathogens (Aspergillus fumigatus and Candida albicans), microsomal stability, effects in fungal cells, and structural characterization of A. fumigatus CYP51 in complexes with the most potent compound are described, offering a new antifungal drug scaffold and outlining directions for its further optimization.
INTRODUCTION
In the last two decades, the mortality and morbidity of invasive fungal infections (IFIs) has greatly increased, and it has been estimated that over 1.6 million infected people die each year. The lack of access to timely diagnosis, coinfections with other diseases (HIV/tuberculosis), use of immunosuppressive therapies (chemotherapy/transplants), some medical devices (central venous catheters), and growing resistance to commercial antifungal drugs significantly contribute to the higher incidence of IFIs. In addition, the socio-economic environment in different countries has a great impact on antifungal treatment (i.e., expensive drugs and commercial availability).1 Of the different fungal species, Candida and Aspergillus genera are responsible for the vast majority of lifethreatening IFIs. Candida albicans remains the most prevalent human pathogen, yet non-albicans Candida species represent a growing global emergency. Highly virulent strains less susceptible to antifungal medicines, for example, C. auris, a multidrug-resistant deadly pathogen, are often present in healthcare facilities.2 Aspergillus is a ubiquitous fungus, transmitted to humans by inhalation of conidia. Approximately 200 000 cases of invasive aspergillosis, mostly in immunocompromised patients, are caused each year by Aspergillus fumigatus, which is also the major species responsible for chronic pulmonary aspergillosis and asthma.3
The therapeutic antifungal drugs available for systemic clinical use act on three different targets: (1) sterol biosynthesis (six azoles and terbinafine); (2) cell wall integrity (amphotericin B and caspofungin); and (3) DNA synthesis (flucytosine) (Figure S1A,C). All of these drugs have clinical limitations,4 and therefore even upon treatment the mortality rates from IFIs often exceed 50%.3,5 More efficient new medicines are needed.
Several attempts to discover new fungal drug targets have been undertaken, and a few compounds with different modes of action are in preclinical studies.6 Whether they will progress into clinical trials remain uncertain. On the other hand, further development of antifungal azoles, inhibitors of fungal sterol 14α−demethylase (CYP51, EC 1.14.13.70), represents a successful area for investigation because azoles, as compared to other classes of current systemic antifungals, clearly display higher potency, better safety profile, and broader spectrum of activity.6,7 Indeed, in 2015 a new azole, the triazole-based isavuconazole, was approved for treatment of IFIs,8 while another triazole (albaconazole) and a tetrazole (VT-1161) are in clinical trials (Figure S1B) [https://clinicaltrials.gov].
Determination of the crystal structures of A. fumigatus9 and C. albicans10 CYP51 in complexes with azole drugs (voriconazole and posaconazole) and drug candidates (VNI, VT-1161, and VT-159811) opened new opportunities for rational, structure-based design of more efficient compounds. The objective of this work was to use the knowledge that we acquired from our structure/function studies to modify our experimental inhibitor of protozoan CYP51, VNI,12,13 in such a way that its inhibitory effect on fungal CYP51 is enhanced. We selected VNI because it is active in vivo against Chagas disease and visceral leishmaniasis, nontoxic, nonmutagenic, with excellent cellular permeability, metabolic stability, and pharmacokinetics (including oral bioavailability and tissue distribution that are substantially more favorable than those of systemic clinical antifungal azoles7). Moreover, although VNI is a relatively weaker inhibitor of fungal CYP51 than the protozoan orthologues, we found that it is more effective in killing A. fumigatus cells than voriconazole, a first line drug against aspergillosis, with the minimum inhibitory concentration (MIC) values being 0.5 versus 0.7 μg/mL, respectively, for VNI and voriconazole.9 Importantly, the VNI scaffold is easy to synthesize and modify.
The rationale for this study was based on the observation that fungal and protozoan CYP51 structures, although displaying high three-dimensional similarity, differ noticeably in the features of substrate entry, which in the fungal CYP51 structures is open much wider (due to the phylum-specific differences between the protozoan and fungal amino acid sequences).10 The hypothesis to test was whether antifungal potency of the VNI scaffold can be enhanced through the formation of additional contacts between the inhibitor molecule and the substrate entrance residues of the fungal CYP51 enzyme.
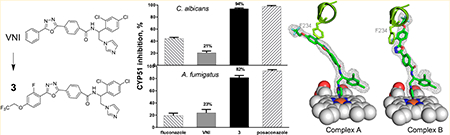
A small in-house library of analogues carrying various substituents in the ortho, meta, and para positions of the distal (5-phenyl) ring of the VNI long arm (Figure 1) has been synthesized and analyzed, with the results serving as the initial proof of concept. Potencies of most of the new compounds to inhibit C. albicans and A. fumigatus CYP51 were increased, some of them approaching the potency of posaconazole, which is presently one of the most powerful inhibitors of fungal CYP51 (example shown in table of content graphic).
Figure 1.

Sites of modifications in the VNI molecule.
RESULTS
Synthesis.
The new in-house library of VNI derivatives was prepared using the general procedures outlined in Scheme 1. The synthesis started by bromination of 1-(2,4-dichlorophen-yl)-ethan-1-one, followed by condensation with the imidazole ring. Subsequently, asymmetric transfer hydrogenation with RuCl(p-cymene)[(S,S)-Ts-DPEN] catalyst was performed to obtain (S)-1-(2,4-dichlorophenyl)-2-(1H−imidazol-1-yl)ethan-1-ol, as described previously.14 The direct conversion of (S)-1-(2,4-dichlorophenyl)-2-(1H−imidazol-1-yl)ethan-1-ol to (R)-1-(2-azido-2-(2,4-dichlorophenyl)ethyl)-1H−imidazole was carried out by dissolving the alcohol in N,N−dimethylformamide (DMF) and adding diphenyl phosphorazidate (DPPA) and 1,5-diazabiciclo[5.4.0]undec-7-ene (DBU) at 0 °C. (R)-1-(2,4-Dichlorophenyl)-2-(1H−imidazol-1-yl)ethan-1-amine was then prepared by reduction of the azide into an amine using LiAlH4.
Scheme 1.

Synthesis of New VNI Derivatives
The final compounds 1–14 were synthesized by coupling between (R)-1-(2,4-dichlorophenyl)-2-(1H−imidazol-1-yl)-ethan-1-amine and 4-(5-phenyl-1,3,4-oxadiazol-2-yl)benzoic acids containing various substituents in the 5-phenyl ring (intermediate c, Scheme 2). The synthesis of intermediate c was performed using three reaction steps. The acylation of the hydrazide with methyl 4-(chlorocarbonyl)benzoate was followed by cyclization of the 1,3,4-oxadiazole in POCl3, and the hydrolysis of ester into carboxylic acid by LiOH H2O.
Scheme 2.

Synthesis of Intermediate c
However, different approaches were used for preparing variously substituted benzohydrazines (intermediate b). Thus, 1b and 6b were available commercially, and the intermediates b for compounds 2, 5, 7, 8, and 14 were synthesized by the addition of the hydrazine monohydrate (N2H4∙H2O) to the commercial available carboxylic acid or ester, as shown in Scheme 3. The synthesis of intermediates b for 3, 4, and 9–13 included preparation of intermediates a. Intermediates a for compounds 3 and 4 were made by addition of the electrophile 2,2,2-trifluoroethyl trifluoromethanesulfonate to the corresponding carboxylic acid. Intermediates a for 9–13 were synthesized using [1,1′-bis(diphenylphosphino)ferrocene]-dichloropalladium(II) (Pd(dppf)Cl2) as a catalyst for Suzuki coupling, and it was performed in a microwave reactor to increase the efficiency (Scheme 3).
Scheme 3.

Synthesis of Benzohydrazinesa
aH2SO4 was not used for hydrazinolysis of 9a–13a.
Evaluation of New Compounds as Inhibitors of Fungal CYP51 Activity.
Even minor modifications in the 5-phenyl ring composition produced remarkably strong effects on the potencies of the new compounds to inhibit enzymatic activity of C. albicans and A.fumigatus CYP51 (Table 1). Thus, quite encouragingly, addition of only twofluorine atoms, in the meta and para positions relative to 1,3,4-oxadiazole (Figure 1) (compound 1, 5-(3,4-difluorophenyl)-1,3,4-oxadiazole), enhanced inhibition of C. albicans CYP51 activity from 20% to 90% and inhibition of A. fumigatus CYP51 activity from 24% to 60% (under the same conditions, the inhibitory effects of posaconazole were 98% and 93%, respectively). When chlorines were substituted for fluorines (atomic radius 0.99 A vs 0.71 A) in the same meta and para positions (compound 5), the inhibitory effect on C. albicans increased slightly (92%), but inhibition of A. fumigatus CYP51 became weaker (55%). Because the larger size of a chlorine atom did not appear to be crucial for inhibition, and introduction of a fluorine has a lower impact on drug lipophilicity (e.g., the log P values of 1 and 5 are 4.88 and 5.68, respectively, Table 1), we mainly used fluorine instead of chlorine substituents in other (non-aromatic) analogues, except for compound 14 (log P 3.83), whose potency toward both fungal enzymes turned out to be about 2-fold lower than that of 5. On the other hand, substitution of a para fluorine with a trifluoromethyl group in 2 (log P 5.65) increased the ability of the compound to inhibit A. fumigatus (68%) without influencing its inhibition of C. albicans CYP51 (89%). Our strongest inhibitor, compound 3, has one fluorine atom in the ortho position and a trifluoroethoxy group in the para position (94% and 82% inhibition of C. albicans and A. fumigatus CYP51 activity, respectively). However, when the same substituents are in two meta positions, compound 4, the potency drops, although the inhibitory effect is still more than twice that of the parent compound VNI. Because all of the modifications above produced inhibitors with higher potencies, thus supporting our hypothesis, a number of bulkier substituents were introduced to decorate the 5-phenyl ring of the VNI arm. A single bromine atom (atomic radius 1.14 Å) in the meta position (compound 6) enhanced the inhibition to 84% (C. albicans) and to 41% (A. fumigatus). When the meta position was substituted with a morpholine ring, however (compound 7), the potency for inhibition of A. fumigatus CYP51 decreased (15% inhibition), and insertion of a morpholine ring into the para position produced an even weaker inhibitor (compound 8, 29% and 3% inhibition toward C. albicans and A. fumigatus CYP51, respectively). While it is possible that the morpholine substitutions are unfavorable as a result of the ring hydrophilicity, because the CYP51 substrate entry, being immersed into the endoplasmic reticulum membrane in vivo, is composed mainly of hydrophobic amino acid residues, a general trend of the meta position being more preferable than the para position for a heterocyclic substituent is apparent (compounds 9, 10, 11, 12, 13, and 15 in Table 1). Among these six analogues, 11 and 15 were most potent toward both fungal enzymes (87/80% and 92/57%, C. albicans/A fumigatus CYP51, respectively).
Table 1.
New VNI Analogues: 5-Phenyl Ring Composition and Inhibition of Fungal CYP51 Activitya
| Cmp | 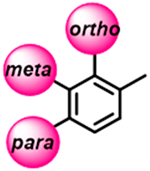 |
CYP51 inhibition, % | Cmp | 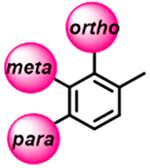 |
CYP51 inhibition, % | ||
|---|---|---|---|---|---|---|---|
| C. albicans | A. fumigatus | C. albicans | A. fumigatus | ||||
| VNI |  |
20 ± 3 | 24 ± 6 | 8 | 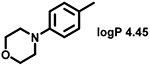 |
29 ± 3 | 3 ± 1 |
| 1 | 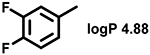 |
90 ± 3 | 58 ± 3 | 9 | 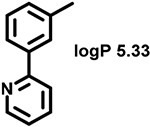 |
77 ± 3 | 34 ± 4 |
| 2 | 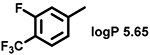 |
89 ± 2 | 65 ± 13 | 10 |  |
66 ± 6 | 23 ± 1 |
| 3 |  |
94 ± 1 | 82 ± 4 | 11 |  |
87 ± 3 | 80 ± 5 |
| 4 |  |
65 ± 4 | 51 ± 5 | 12 | 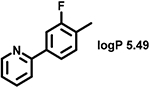 |
61 ± 4 | 26 ± 4 |
| 5 | 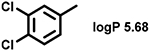 |
92 ± 3 | 55 + 4 | 13 | 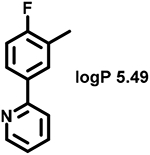 |
83 ± 1 | 24 ± 5 |
| 6 | 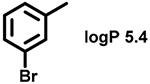 |
84 ± 4 | 41 ± 4 | 14 | 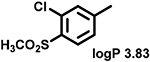 |
53 ± 1 | 32 ± 3 |
| 7 | 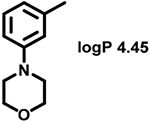 |
58 ± 2 | 15 ± 1 | 15 | 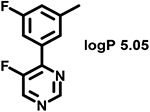 |
92 ± 1 | 57 ± 1 |
A 1-h reaction was used to distinguish the strongest inhibitors, with the potential propensity to act as functionally irreversible (see Experimental Section, Discussion and Conclusions). The P450 concentration was 0.5 μM, and the molar excess of each inhibitor over enzyme was 1.5-fold (0.75 μM).
Evaluation of Selected Compounds as CYP51 Binding Ligands.
Five of the strongest inhibitors of C. albicans and A. fumigatus CYP51 activity were also assayed as binding ligands of the enzymes (Table 2). VNI, posaconazole, and voriconazole were used as controls. Previously, we observed a clear lack of correlation between the apparent spectral binding affinities of some ligands and their potencies to inhibit CYP51 enzymatic activity, particularly in long reactions.14–16 Accordingly, we compared spectral titrations with close structural analogues (e.g., as observed in ref 17). Overall, the data support a relationship between binding and inhibitory potency, particularly for compounds 1, 2, and 3, which have the highest structural similarities to VNI (see Table 1 and Figure S2).
Table 2.
Dissociation Constants Determined by Spectral Titration of C. albicans and A. fumigatus CYP51 with VNI, Compounds 1, 2, 3, 11, and 15, Posaconazole, and Voriconazole
| cmpds | C. albicans Kd, nM | fold enhancement (Kd VNI/Kdna) | A. fumigatus Kd, nM | fold enhancement (Kd VNI/Kdn) |
|---|---|---|---|---|
| VNI | 290 ± 27 | 203 ± 10 | ||
| 1 | 13 ± 1 | 22 | 57 ± 6 | 4 |
| 2 | 13 ± 1 | 22 | 55 ± 3 | 4 |
| 3 | 10 ± 6 | 29 | 20 ± 4 | 10 |
| 11 | 25 ± 5 | 12 | 61 ± 3 | 3 |
| 15 | 95 ± 11 | 3 | 132 ± 2 | 1.5 |
| posaconazole | 81 ± 16 | 3.5 | 131 ± 11 | 1.5 |
| voriconazole | 165 ± 9 | 1.8 | 56 ±4 | 3.6 |
n, compound number.
Estimated Intrinsic and Hepatic Clearance in Human, Rat, and Mouse Microsomes.
Because high metabolic stability is one of the many advantages of VNI as a potential drug,18 we examined the parameters for the new compounds. Moreover, simultaneous analysis in human and animal microsomes is highly informative in allowing for a prediction of the likelihood of acceptable pharmacokinetics in clinical trials. VNI and 13 new compounds with the increased potencies to inhibit both fungal CYP51 enzymes (all except for the morpholine-containing 7 and 8) were tested in human, rat, and mouse liver microsomes (Table 3). The data show that, overall, none of the modifications drastically affected the stability of the new compounds, with all but one (11, human microsomes) displaying low to moderate hepatic clearance. The stability of compounds 1 and 2 in human and mouse systems was even higher than that of VNI. Replacement of the twofluorine atoms with chlorine (5), however, had slightly negative effects in human/mouse but not in rat microsomes. Although the pyridine ring can be metabolically vulnerable,19 compounds 9 and 12, 13 also showed rather good stability, not inferior to compound 15 (5-fluoro-6 pyrimidinyl analogue of 11).
Table 3.
Microsomal Stability: Half-Lives and Estimated Clearance of VNI and New Derivatives in Human, Rat, and Mouse Microsomes
| human | rat | mouse | |||||||
|---|---|---|---|---|---|---|---|---|---|
| cmpds | CLINTa | CLHEP | t1/2 (min)b | CLINT | CLHEP | t1/2 (min) | CLINT | CLHEP | t1/2 (min) |
| VNI | 8.31 | 5.95 | 150 | 26.4 | 19.2 | 106 | 87 | 44 | 62 |
| 1 | 7.53 | 5.54 | 166 | 31 | 21 | 91 | 55 | 34 | 98 |
| 2 | 6.06 | 4.07 | 206 | 36 | 24 | 77 | 60 | 36 | 92 |
| 3 | 16.5 | 9.24 | 76 | 64 | 34 | 44 | 88 | 44 | 62 |
| 4 | 11.3 | 7.33 | 111 | 50.4 | 29 | 56 | 84.7 | 43.6 | 65 |
| 5 | 11.4 | 7.39 | 110 | 70 | 35 | 40 | 107 | 49 | 51 |
| 6 | 9.47 | 6.53 | 132 | 27 | 19 | 106 | 126 | 55 | 43 |
| 9 | 13.1 | 8.06 | 95 | 68 | 35 | 41 | 152 | 56 | 36 |
| 10 | 10.8 | 7.15 | 115 | 94 | 40 | 30 | 95 | 46 | 57 |
| 11 | 96.6 | 17.3 | 13 | 36.0 | 24.0 | 78 | 55.0 | 34 | 99 |
| 12 | 15 | 8.75 | 83 | 51.6 | 29.7 | 54 | 36.5 | 26.0 | 149 |
| 13 | 10.2 | 6.85 | 123 | 49.4 | 29.0 | 57 | 238 | 65 | 23 |
| 14 | 9.31 | 6.45 | 134 | 104 | 42 | 27 | 56 | 34.5 | 97 |
| 15 | 14.9 | 8.37 | 84 | 27.0 | 19.5 | 104 | 104 | 48.3 | 52 |
Intrinsic (CLINT) and hepatic clearance (CLHEP) expressed in mL/min/kg body weight.
In vitro half-life values in minutes.
Cellular Effects in A. fumigatus Strains.
Four compounds (1, 2, 3, and 15) that revealed both the highest potencies to inhibit A. fumigatus CYP51 activity and favorable stability in human microsomes (Figure 2) were further tested in cellular experiments. VNI was used as a control (Table 4). All of the derivatives displayed stronger inhibition of fungal cell growth, and their fungicidal potencies correlated well with the effects on the target enzyme. The lowest MIC values, 2.4- and 2.5-fold lower than the MICs of VNI on average, were observed for compounds 2 and 3, respectively.
Figure 2.

Half-life time of VNI, selected analogues, and a drug verapamil (positive control) in human microsomes. R2 values were all >0.7.
Table 4.
Antifungal Activity of VNI and Derivatives (MIC ± SEM, μg/mL)
| A fumigatus strain | VNI | 1 | p- value | 2 | p- value | 3 | p- value | 15 | p- value |
|---|---|---|---|---|---|---|---|---|---|
| 1022 | 0.48 ± 0.05 | 0.29 ± 0.03 | 0.0001a | 0.19 ± 0.03 | 0.0003a | 0.19 ± 0.03 | 0.0002a | 0.48 ± 0.05 | 1 |
| 32820 | 0.08 ± 0.03 | 0.08 ± 0.03 | 0.0009a | 0.09 ± 0.02 | 0.0027a | 0.06 ± 0.01 | 0.0004a | 0.19 ± 0.03 | 0.21 |
| MYA-3626 | 0.08 ± 0.03 | 0.78 ± 0.03 | 0.0035a | 0.39 ± 0.05 | 0.0001a | 0.39 ± 0.05 | 0.0001a | 0.78 ± 0.04 | 0.0035a |
Statistically significant differences; p−values were analyzed by one-way ANOVA.
X-ray Crystallography.
Compound 3 that displayed the strongest inhibition of both fungal CYP51 enzymes (Table 1) and the most expressed enhancement in the apparent binding affinity (29- and 10-fold in comparison to VNI, C. albicans and A. fumigatus CYP51, respectively, Table 2) was selected for cocrystallization. In addition to the goal of observing inhibitorspecific interactions with the enzyme (i.e., structural basis for the increased inhibitory potency), we examined whether the modifications alter the ligand orientation in the fungal CYP51 substrate binding cavity. While the cocrystals of C. albicans CYP51 with 3 did not diffract well, the A. fumigatus complexes produced crystals that diffracted to ~2.4 Å. Several data sets were collected, the structures were determined, and each of them indicated that upon binding to the enzyme the inhibitor can adopt two different conformations, both of them different from the conformations of VNI in A. fumigatus or in T. brucei CYP51 (Figure 3A). The structure of the best diffracting crystal has been refined and deposited in the Protein Data Bank (PDB ID 6CR2). Table S4 summarizes the diffraction and refinement statistics. The 2Fo − Fc electron density maps of the inhibitor molecules in both conformations contoured at 1.2 σ and shown as gray mesh can be seen in the table of content graphic.
Figure 3.

LFV (compound 3) adopts two alternative conformations in the active site of A.fumigatus CYP51B. (A) Substrate entry area. The carbon atoms in the superimposed structures of complexes A and B are tan and blue, respectively. The corresponding orientation of VNI in the structures of A.fumigatus CYP51B (PDB ID 4uyl, gray lines) and Trypanosoma brucei CYP51 (PDB ID 3gw9, purple lines) are shown for comparison. The A. fumigatus CYP51 substrate entry-defining secondary structural elements are presented as gray ribbon and labeled. The semitransparent ribbon of the (longer) F/G loop area in the protozoan CYP51 (3gw9) is purple, and the F′′ helix and the protozoan-specific G′ helix are labeled. (B) Overall view in semitransparent surface representation. (C) New ligand/enzyme interactions formed as a result of the VNI modification. Atoms added to the VNI structure are presented as spheres. The amino acid residues of A. fumigatus CYP51B involved in the additional interactions are shown as stick models and labeled, and selected distances are displayed.
DISCUSSION AND CONCLUSIONS
It has been known for years that antifungal azoles, both clinical drugs and agricultural fungicides, kill fungal cells by inhibiting their sterol biosynthesis at the stage of 14α−demethylation of sterol precursors.20 This is a three-step reaction catalyzed by a single cytochrome P450 monooxygenase, named sterol 14α− demethylase21 (CYP51). This target enzyme, being a highly hydrophobic membrane-bound protein, has never actually been directly utilized in the process of drug discovery or development.7
In part because of this, the process of antifungal drug discovery has been slow, costly, and low-efficient.6 The six clinical systemic azoles, which represent the major portion of the current arsenal of clinical antifungal drugs, are derivatives of two inhibitory scaffolds: ketoconazole (itraconazole and posaconazole) and fluconazole (voriconazole and isavuconazole) (Figure S1A). The third, a tetrazole-based scaffold from Viamet (Figure S1B), is currently in trials. All of these drugs were discovered rather empirically by monitoring effects of hundreds of compounds on fungal cell growth. Our study shows that direct analysis of CYP51 enzyme inhibition can be utilized to shift the traditional drug discovery paradigm, making it potentially much more effective.
All of the 15 compounds synthesized and analyzed in this work, including the parent compound VNI,9 inhibit the initial rate of fungal CYP51 reactions when added at an equimolar ratio to the enzyme, thus producing IC50 values of ~0.25 μM, corresponding to one-half of the enzyme concentration in the reaction mixture (data not shown). However, as seen in Table 1, with time some of the inhibitors are replaced by the substrate in the fungal CYP51 active site more easily than the others, although most of the (5-phenyl) ring substituents (presumably positioned in close proximity to the CYP51 substrate entry) increase potencies of the inhibitors to hold the enzyme in the catalytically inactive state.
Azoles are generally known as competitive reversible heme coordinating inhibitors of cytochromes P450.22 However, as it was first observed in 1987 for CYP51 from S. cerevisiae23 and later rediscovered by our research group for protozoan CYP51,16 some azole-based inhibitors can act in a functionally irreversible manner due to their high affinity. Added at a concentration equimolar to the enzyme, they entirely inhibit CYP51 catalysis and cannot be replaced by the substrate for an extended period of time, even though no covalent bonds with the protein are formed.7 VNI is an example of such an inhibitor for trypanosomal CYP51, while VT-1161, VT-1598, and posaconazole can serve as examples of such inhibitors for fungal CYP51.
The very strong affinity of some of the azoles appears to be somehow connected with the strict functional phylogenetic conservation of CYP51 and the three-step stereoselective oxidation reaction sequence, which together require high structural rigidity of the enzyme active site.24 Indeed, none of more than 30 crystal structures of microbial CYP51s bound to various inhibitory chemotypes that we have determined have displayed any large-scale rearrangements in the active site backbone upon inhibitor binding. Fortunately, for antifungal drug design, the active site of human CYP51 appears to be much more flexible,25–27 rendering the human enzyme naturally resistant to inhibition, at least with all ever tested antimicrobial azoles, both commercial and experimental,26 a favorable feature that enables pathogen-selectivity of CYP51-targeting antimicrobial agents.
In the protozoan CYP51 structures, VNI12 and its derivatives VFV and VNT18 all form hydrogen bonds with the amino acid residues in the central, catalytic area of the CYP51 active site. The VT tetrazoles form H-bonds with the fungal-specific histidine (His-374 in A. fumigatus and His-377 in C. albicans) that lines the CYP51 substrate access channel.10,11 CYP51 complexes with posaconazole do not involve any H-bond formation but display multiple enzyme—inhibitor contacts around the substrate entry.10
Binding of VNI to A. fumigatus CYP51 does not produce any H-bonds, and its three ring arm is not long enough to reach the substrate entry site. Nevertheless, in addition to the strong (2.0 Å) coordination bond between its imidazole (N3) nitrogen and the enzyme heme iron, VNI forms van der Waals contacts with 19 amino acid residues, including the sandwich π—π stacking interactions with Phe-234.9 This bonding provides a structural background for the strong inhibitory effect of VNI on the initial fungal CYP51 reaction and the reversibility of inhibition in the presence of the substrate. The results support our hypothesis that elongation of the VNI arm is likely to increase its potency as fungal CYP51 inhibitor.
The crystal structure of A. fumigatus CYP51 complexed with compound 3 (ligand PDB ID LFV) shows that the stronger enzyme inhibition is indeed due to the new interactions around the substrate entry (Figure 3B) and also suggests that larger substituents here might be even more efficient. In both conformations, LFV forms contacts with an additional seven amino acid residues of the enzyme (Figure 3C). In molecule A (CYP51-common portion of the substrate entry), Thr-65, Tyr-68, Gly-69 (helix A′), Leu-91, Leu-92 (β1 hairpin), and phe-234 (helix F′) interact with the trifluoroethoxy moiety, and Val-121 (helix B′) interacts with the fluorine atom inserted into the ortho position relative to the oxadiazole ring. In molecule B (fungal CYP51-specific portion of the substrate entry), Asn-233, Phe-234, Met-235, Leu-236, Pro-237, and Trp-238 (helix F′′) interact with the trifluoroethoxy moiety, while Val-121 and Phe-124 (helix B′) interact with the additional orthofluorine. Interestingly, in both complexes, Phe-234 “follows” the inhibitor orientation, preserving the π—π stacking interactions with its 5-phenyl aromatic ring (see also table of content graphic, right panel). It remains to be clarified if one of the two LFV conformations warrants a higher inhibitory potency, but it appears that such a probability for complex B may be higher. In this complex, a fluorine atom of the LFV trifluoroethoxy moiety displays a propensity toform H-bond like interactions with the main chain oxygen of Met-235 (2.3 Å) and with the indole nitrogen of Trp-238 (3.2 Å), while the ethoxy oxygen is positioned only 3.5 Å from the main chain oxygen of Phe-234 (Figure S3). All of these three residues are in helix F′′, the CYP51 area that is most likely to serve as part of the “lid” for the substrate entry. Assuming that binding of the substrate requires opening of the FG-lid, these contacts with the ligand are likely to restrict the required movement. No such gatekeeping interactions are seen in complex A.
Further efforts directed toward strengthening fungal CYP51—VNI scaffold interactions are in progress, with our ultimate goal being to design very high affinity compounds that will bind to the fungal CYP51 most efficiently, keeping the enzyme in its catalytically inactive state and preventing a conformational switch required for the interaction with the electron donor partner NADPH-cytochrome P450 reductase.
Overall, the results of this study prove that structure-based design is useful for the target fungal CYP51, meaning that novel potent inhibitory scaffolds can be rationally built. Minor modifications may be sufficient to optimize pharmacokinetics and thus to improve the IFIs treatment efficiency and retard drug resistance. The availability of various alternative CYP51 inhibitory scaffolds should be extremely useful in overcoming situations in which patients succumb tofungal sepsis simply because they must be temporarily immunosuppressed and there are no efficient drugs available.
EXPERIMENTAL SECTION
Chemical Synthesis.
All reagents and solvents were commercial grade and were purchased from Fisher Scientific, Sigma-Aldrich, Santa Cruz, or Alpha Aesar. Microwave reactions were performed with a Biotage Initiator Microwave System. 1H NMR spectra were acquired on a Bruker AVANCE-400 MHz spectrometer in CDCl3 and DMSO-d6 at 25 °C. Chemical shift values are given in δ (ppm) relative to TMS as internal standard. Coupling constants are given in Hz. The following abbreviations are used to set multiplicities: s = singlet, d = doublet, dd = double of doublets, t = triplet, q = quartet, m = multiplet, br = broad. Mass spectrometry was done with a LTQ Orbitrap XL Hybrid Ion Trap-Orbitrap mass spectrometer. The molecular peaks (m/z) was observed as [M + H]+or [M — H]− ions. Compounds were purified by flash chromatography using silica gel high-purity grade, pore size 60 Å/230–400 mesh/particle size 40–63 μm (Sigma-Aldrich). The final compounds were filtered through HF Bond Elut-SCX cartridges (500 mg, 6 mL, Agilent Technologies), using a 2.0 M ammonia solution in methanol (Sigma-Aldrich). LC — MS (UV and ESI—MS) and reverse-phase HPLC were used for analyzing the purity of the compounds (>95%). Analytical LC—MS was performed on an Agilent 1200 series with UV detection at 214 and 254 nm, along with ELSD detection. General LC—MS parameters were as follow: Phenomenex-C18 Kinetex column, 50 mm × 2.1 mm, 2 min gradient, 5% (0.1% TFA/CH3CN)/95% (0.1% TFA/H2O) to 100% (0.1% TFA/CH3CN) (all v/v). The reversed-phase HPLC system (Waters) was equipped with a dual-wavelength UV 2489 detector set at 250 and 291 nm (VNI-specific absorption maximum, ε291 = 36 mM−1 cm−1) and a Symmetry C18 (3.5 μm) 4.6 mm × 75 mm column.13 The mobile phase was 55% 0.015 M ammonium acetate solution (pH 7.2) and 45% CH3CN (v/v) with an isocratic flow rate of 1.0 mL/min. The enantiomeric excess (ee) of the final compounds was evaluated by analytical chiral chromatography on a Chiralpak IB-3 column, 250 mm × 4.6 mm i.d. (Daicel Corp., Japan) using a Waters HPLC system with the detector set at 250 and 291 nm. As eluent, a solution of hexane, ethanol, diethylamine, and ethylenediamine (ratio 70:30:0.1:0.1, v/v) was used, and the flow rate was 0.7 mL/min. High-resolution mass spectra were obtained on an Agilent 6540 UHD Q-TOF with ESI source. MS parameters were as follows: fragmentor, 150; capillary voltage, 3500 V; nebulizer pressure, 60 psig; drying gas flow, 13 L/m in; drying gas temperature, 275 °C. Samples were introduced via an Agilent 1290 UHPLC comprised of a G4220A binary pump, G4226A ALS, G1316C TCC, and G4212A DAD with ULD flow cell. UV absorption was observed at 215 and 254 nm with a 4 nm bandwidth. Column: Waters Acquity BEH C18, 1.0 × 50 mm, 1.7 um. Gradient conditions: 5% to 95% CH3CN in H2O (0.1% formic acid) over 1.25 min, hold at 95% CH3CN for 0.25 min, 0.3 mL/min, 40 °C.
Preparation of (R)-1-(2,4-Dichlorophenyl)-2-(1H−imidazol-1-yl)ethan-1-amine.
Synthesis of 2-Bromo-1-(2,4-dichlorophenyl)-ethan-1-one (Step 1).
CuBr2 (11.8 g, 53 mmol) was ground with a mortar and pestle to ensure a large surface area for the reaction and placed in a round-bottom flask. EtOAc (10 mL) was added, and the solution was heated until reflux. Subsequently, 2′,4′-dichloroaceto-phenone (5 g, 26.5 mmol) was dissolved in CHO3 (10 mL), stirred at 40 °C, and added dropwise to the CuBr2 solution. The resulting mixture was stirred for 2 h under reflux. The reaction was allowed to cool at room temperature, filtrated, diluted with EtOAc, and washed with aqueous NH4Cl. The organic layer was then washed with brine, dried over anhydrous Na2SO4, filtered, and the solvent was removed in vacuo. The crude product was purified by flash column chromatography (hexane/EtOAc, 8:2, v/v) affording the title product as a yellow oil in quantitative yield. 1H NMR (CDCI3): δ ppm 7.56 (d, J = 8.4 Hz, 1H), 7.48 (d, J = 1.9 Hz, 1H), 7.36 (dd, J = 8.4, 1.9 Hz, 1H), 4.50 (s, 2H). ESI-MS: mass calcd for C8H5BrCl2O [M + H]+, 267.89; found, m/z 267.90.
Synthesis of 1-(2,4-Dichlorophenyl)-2-(1H-imidazol-1-yl)ethan-1-one (Step 2).
To a solution of imidazole (9 g, 133 mmol) in DMF (10 mL) was added (dropwise) 2-bromo-1-(2,4-dichlorophenyl)-ethan-1-one (7 g, 27 mmol). The reaction was stirred for 2 h at 0 °C. The reaction mixture was diluted with H2O, extracted into EtOAc, washed with LiCl (5%, w/v) and brine, and dried over anhydrous Na2SO4, filtered, and the solvent was removed in vacuo. The crude product was purified by flash column chromatography (CH2Cl2/CH3OH, 9:1, v/v) affording the title product as a white solid, isolated in 60% yield. 1H NMR (CDCl3): δ ppm 7.56 (d, J = 8.9 Hz, 1H), 7.49 (d, J = 1.9 Hz, 2H), 7.36 (dd, J = 8.4, 1.9 Hz, 1H), 7.11 (s, 1H), 6.93 (s, 1H), 5.32 (s, 2H). ESI-MS: mass calcd for C11H8Cl2N2O [M + H]+, 255.10; found, m/z 255.3.
Synthesis of (S)-1-(2,4-Dichlorophenyl)-2-(1H-imidazol-1-yl)-ethan-1-ol (Step 3).
1-(2,4-Dichlorophenyl)-2-(1H−imidazol-1-yl)-ethan-1-ol was prepared as previously described using 1-(2,4-dichlorophenyl)-2-(1H−imidazol-1-yl)ethan-1-one.28 The title compound was isolated as a white solid in quantitative yield. 1H NMR (CDCl3): δ ppm 7.59 (d, J = 8.4 Hz, 1H), 7.39 (d, J = 2.0 Hz, 1H), 7.37 (s, 1H), 7.30 (dd, J = 8.4, 2.0 Hz, 1H), 6.89 (s, 1H), 6.82 (s, 1H), 5.21 (dd, J = 8.3, 2.0 H, 1H), 4.19 (dd, J = 14.0, 2.2 Hz, 1H), 3.84 (dd, J = 14.0, 8.3 Hz, 1H). ESI-MS: mass calcd for C11H10Cl2N2O [M + H]+, 257.02; found, m/z 256.9.
Synthesis of (R)-1-(2-Azido-2-(2,4-dichlorophenyl)ethyl)-1H-imi-dazole (Step 4).
To a cold solution of (S)-1-(2,4-dichlorophenyl)-2-(1H−imidazol-1-yl)ethan-1-ol (230 mg, 0.9 mmol) and diphenylphos-phoryl azide (DPPA) (194 μL, 0.9 mmol) in anhydrous DMF (1.8 mL) was added dropwise 1,5-diazabiciclo[5.4.0]undec-5-ene (DBU) (188 μL, 1.26 mmol). The solution was stirred at 0 °C for 2 h and then at room temperature overnight. The reaction mixture then was diluted with water, extracted into EtOAc, washed with LiCl (5%, w/v) and brine, and dried over anhydrous Na2SO4, filtered, and the solvent removed in vacuo. The crude product was purified by flash column chromatography (CH2Cl2/CH3OH, 9:1, v/v) affording the title product as a white solid, isolated in 70% yield. 1H NMR (CDCl3): δ ppm 7.43 (d, J = 2.0 Hz, 1H), 7.36–7.29 (m, 3H), 7.05 (s, 1H), 6.90 (s, 1H), 5.24 (dd, J = 7.6, 3.2 Hz, 1H), 4.21 (dd, J = 14.4, 3.2 Hz, 1H), 4.01 (dd, J = 14.4, 7.6 Hz, 1H). ESI-MS: mass calcd for C11H9Cl2N5 [M + H]+, 282.02; found, m/z 282.0.
Synthesis of (R)-1-(2,4-Dichlorophenyl)-2-(1H-imidazol-1-yl)-ethan-1-amine (Step 5).
To a suspension of LiAlH4 (87.5 mg, 2.30 mmol) in dry THF (3 mL) was added (R)-1-(2-azido-2-(2,4-dichlorophenyl)ethyl)-1H−imidazole (542 mg, 1.92 mmol) (dissolved in 2 mL of dry THF) dropwise at 0 °C. The suspension was warmed to room temperature and stirred overnight. The reaction mixture was quenched by Rochelle’s salt and filtered through Celite. The crude product was extracted into EtOAc, washed with brine, dried on anhydrous Na2SO4, filtered, and the solvent removed in vacuo. The crude product was purified by flash chromatography (CH2Cl2/CH3OH, 9:1, v/v) to give a white solid in quantitative yield. 1H NMR (CDCl3): δ ppm 7.44−7.40 (m, 3H), 7.27 (d, J = 2.0 Hz, 1H), 7.06 (s, 1H), 6.90 (s, 1H), 4.72 (dd, J = 8.0, 3.7 Hz, 1H), 4.19 (dd, J = 14.0, 3.7 Hz, 1H), 3.94 (dd, J = 14.0, 8.0 Hz, 1H). ESI-MS: mass calcd for C11H11Cl2N3 [M + H]+, 256.03; found, m/z 256.0.
Preparation of the Side Chains.
Intermediates 1b, 2a, 5a, 6b, 7a, 8a, and 14a were commercially available.
General Synthesis of Intermediates (3a and 4a).
To a mixture of selected acid (1 equiv) and K2CO3 (5 equiv) in DMF (0.5 M) was added 2,2,2-trifluoroethyl trifluoromethanesulfonate (1.5 equiv), and the solution was stirred at 80 °C for 2 h. The reaction mixture was cooled to room temperature, poured into water (100 mL), and the aqueous layer was extracted by EtOAc, washed with brine, dried with anhydrous Na2SO4, and filtered, and the solvent removed in vacuo. The crude product was purified by flash chromatography (CH2Cl2/CH3OH, 9:1) to give a white solid.
2-Fluoro-4-(2,2,2-trifluoroethoxy)benzoic Acid (3a).
Yield 78%. 1H NMR (DMSO-d6): δ 11.0 (br, 1H), 7.77 (t, J = 8.8 Hz, 1H), 6.73 (dd, J = 8.7, 2.3 Hz, 1H), 6.65 (dd, J = 13.1, 2.2 Hz, 1H), 4.90 (q, J = 8.8 Hz, 2H). ESI-MS: mass calcd for C9H6F4O3 [M–H]−, 237.03; found, m/z 236.9.
3-Fluoro-5-(2,2,2-trifluoroethoxy)benzoic Acid (4a).
Yield 50%. 1H NMR (DMSO-d6): δ 11.0 (br, 1H), 7.20 (s, 1H), 7.07 (d, J = 8.8 Hz, 1H), 6.90 (d, J = 10.6 Hz, 1H), 4.92 (q, J = 9.0 Hz, 2H). ESI-MS: mass calcd for C9H6F4O3 [M–H]−, 237.03; found, m/z 237.0.
General Synthesis of Intermediates (9a-13a).
The selected boronic acid (1 equiv), 2-bromopyridine (1 equiv), [1,1′-bis-(diphenylphosphino)ferrocene]dichloropalladium(II) (Pd(dppf)Cl2) (0.6 mol %), dioxane, and H2O (ratio 4:1, v/v) were stirred under argon. The reaction was performed in the microwave reactor for 30 min at 120 °C. The solvent was evaporated, and the crude residue was extracted with EtOAc, washed with brine, and dried over anhydrous Na2SO4, filtered, and the solvent removed in vacuo. The crude product was purified by flash chromatography (hexanes/EtOAc, 4:1, v/v) to give a white solid in 5 0 −9 0 % yield.
Methyl 3-(Pyridin-2-yl)benzoate (9a).
Yield 60%. 1H NMR (CDCl3): δ ppm 8.74−8.72 (m, 1H), 8.65−8.64 (m, 1H), 8.27−8.24 (m, 1H), 8.11−8.09 (m, 1H), 7.80 (m, 2H), 7.57 (t, J = 7.8 Hz, 1H), 7.30−7.27 (m, 1H), 3.96 (s, 3H). ESI−MS: mass calcd for C13H11NO2 [M + H]+, 214.08; found, m/z 214.10.
Methyl 4-(Pyridin-2-yl)benzoate (10a).
Yield 50%. 1H NMR (CDCl3): δ ppm 8.74–8.72 (m, 1H), 8.15 (d, J = 8.6 Hz, 2H), 8.08 (d, J = 8.6 Hz, 2H), 7.80–7.79 (m, 2H), 7.31 −7.28 (m, 1H), 3.95 (s, 3H). ESI-MS: mass calcd for C13H11NO2 [M + H]+, 214.08; found, m/z 214.05.
Methyl 3-Fluoro-5-(pyridin-2-yl)benzoate (11a).
Yield 70%. 1H NM R (CDCl3): δ ppm 8.73 −8.72 (m, 1H), 8.45–8.44 (m, 1H), 8.03–7.99 (m, 1H), 7.82–7.75 (m, 3H), 7.33–7.30 (m, 1H), 3.97 (s, 3H). ESI-MS: mass calcd for C13H10FNO2 [M + H]+, 232.07; found, m/z 232.1.
Methyl 2-Fluoro-4-(pyridin-2-yl)benzoate (12a).
Yield 50%. 1H NMR (CDCl3): δ ppm 8.73–8.71 (m, 1H), 8.05–7.95 (m, 1H), 7.86 (s, 1H), 7.84–7.76 (m, 3H), 7.34–7.31 (m, 1H), 3.96 (s, 3H). ESI-MS: mass calcd for C13H10FNO2 [M + H]+, 232.07; found, m/z 232.1.
Methyl 2-Fluoro-5-(pyridin-2-yl)benzoate (13a).
Yield 60%. 1H NMR (CDCl3): δ ppm 8.69–8.67 (m, 1H), 8.54 (dd, J = 9.4, 2.5 Hz, 1H), 8.23–8.19 (m, 1H), 7.79–7.72 (m, 2H), 7.27–7.26 (m, 1H), 7.24 (d, J = 1.5 Hz, 1H), 3.96 (s, 3H). ESI-MS: mass calcd for C13H10FNO2 [M + H]+, 232.07; found, m/z 232.09.
Methyl 3-Fluoro-5-(5-fluoropyrimidin-4-yl)benzoate (15a).
Yield 92%. 1H NMR (CDCl3): δ ppm 9.13 (d, J = 3.0 Hz), 8.72 (s, 1H), 8.65 (s, 1H), 8.12–8.09 (m, 1H), 7.91–7.88 (m, 1H), 3.4 (s, 3H). ESI-MS: mass calcd for C12H8F2N2O2 [M + H]+, 251.06; found, m/z 251.10.
General Synthesis of Intermediates b and c.
To a solution of substituted benzoic acid (1 equiv) in EtOH were added hydrazine monohydrate (2 equiv) and a few drops of concentrated H2SO4. The reaction mixture was heated under reflux for 6 h, and then, after cooling, the white solid was filtered to give the desired product (intermediate b) in quantitative yield, which was used for the next step. (The same procedure was performed with substituted methyl benzoate without adding concentrated H2SO4.)
Subsequently, methyl 4-(chlorocarbonyl)benzoate, dissolved in anhydrous THF, was added dropwise to a dried round-bottom flask containing the appropriate intermediate b, under an argon flow. The mixture was stirred at room temperature for 6 h. The solvent then was removed under reduced pressure. The corresponding N′−benzoyl-(hydrazinocarbonyl)esters, differently substituted, were used without further purification in the next step, when POCl3 was added dropwise. The mixture was heated at reflux overnight (85 °C), then cooled to room temperature and poured into ice and solid NaHCO3. The resulting solution was extracted with EtOAc, washed with H2O, and dried with Na2SO4. Differently substituted methyl 4-(5-phenyl-1,3,4-oxadiazol-2-yl)benzoates were purified by flash chromatography (hexane/EtOAc, 3:1, v/v). The hydrolysis of the benzoate ester to benzoic acid was performed by adding, to a suspension of the benzoate in THF, LiOH H2O and CH3OH (THF/CH3OH/H2O, 3:1:1, v/v/v). The reaction mixture was stirred at room temperature for 3 h, and then the solvent was removed under reduced pressure, and the residue was extracted with an aqueous HCl solution and EtOAc. The solvent was evaporated, and a white pure solid was obtained in quantitative yield (intermediate c).
General Procedure for Amide Coupling.
(Step 6). (R)-1-(2,4-Dichlorophenyl)-2-(1H−imidazol-1-yl)ethan-1-amine (1.2 equiv) was dissolved in CH2Cl2 (4 mL), and the corresponding acid (intermediate c, 1.0 equiv) was added to the round-bottom flask, which was cooled to 0 °C before N−(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC/HCl) (1.0 equiv) and 4-dimethylaminopyridine (DMAP) were added. The reaction mixture was warmed to room temperature and stirred overnight. The mixture was diluted with CH2Cl2, washed with NaHCO3, and brine dried on Na2SO4. The solvent was evaporated, and the final compound was purified by flash chromatography (CH2Cl2/CH3OH, 9:1, v/v). The yields were calculated as pure compounds, obtained as a white solid.
(R)-N-(1-(2,4-Dichlorophenyl)-2-(1H-imidazol-1-yl)ethyl)-4-(5-(3,4-difluorophenyl)-1,3,4-oxadiazol-2-yl)benzamide (Compound 1).
Yield 50%. 1H NMR (CDCl3): δ ppm 9.97 (s, 1H), 9.55 (d, J = 8.8 Hz, 1H), 8.04 (d, J = 8.4 Hz, 2H), 7.98—7.89 (m, 2H), 7.79 (dd, J = 8.3, 5.2 Hz, 3H), 7.47—7.45 (m, 2H), 7.40—7.35 (m, 3H), 6.08—6.04 (m, 1H), 5.27 (dd, J = 13.8, 12.0 Hz, 1H), 4.41 (dd, J = 14.0, 3.2 Hz, 1H). LC—MS: found [M + H]+ m/z 539.8; ESI—MS: mass calcd for C26H17Cl2F2N5O2 [M]+, 539.0727; found, Mobs 539.0733. HPLC, tR 12.6 min, purity 98%. Enantiomeric excess >97%.
(R)-N-(1-(2,4-Dichlorophenyl)-2-(1H-imidazol-1-yl)ethyl)-4-(5-(3-fluoro-4-(trifluoromethyl)phenyl)-1,3,4-oxadiazol-2-yl)benzamide (Compound 2).
Yield 50%. 1H NMR (CDCl3): δ ppm 9.92 (s, 1H), 9.49 (d, J = 8.5 Hz, 1H), 8.08 (d, J = 8.4 Hz, 2H), 8.04 (d, J = 8.2 Hz, 1H), 7.97 (d, J = 10.2 Hz, 1H), 7.82—7.77 (m, 4H), 7.47—7.45 (m, 2H), 7.40 (s, 1H), 7.35 (dd, J = 8.4, 2.0 Hz, 1H), 6.09—6.04 (m, 1H), 5.26 (dd, J = 14.0, 12.0 Hz, 1H), 4.42 (dd, J = 14.0, 3.3 Hz, 1H). LC — MS found, [M + H]+ m /z 590.18; ESI—MS: mass calcd for C27H17Cl2F4N5O2 [M]+, 598.0695; found, Mobs 598.0696. HPLC, tR 11.4 min, purity 99%. Enantiomeric excess >97%.
(R)-N-(1-(2,4-Dichlorophenyl)-2-(1H-imidazol-1-yl)ethyl)-4-(5-(2-fluoro-4-(2,2,2-trifluoroethoxy)phenyl)-1,3,4-oxadiazol-2-yl)-benzamide (Compound 3).
Yield 40%. 1H NMR (CDCl3): δ ppm 9.79 (s, 1H), 9.47 (d, J = 8.4 Hz, 1H), 8.13 (t, J = 8.4 Hz, 1H), 8.02 (d, J = 8.3 Hz, 2H), 7.80 (dd, J = 8.3, 3.4 Hz, 3H), 7.45 (d, J = 2.0 Hz, 1H), 7.44 (s, 1H), 7.38 (s, 1H), 7.34 (dd, J = 8.4, 2.0 Hz, 1H), 6.94—6.86 (m, 2H), 6.07—6.02 (m, 1H), 5.23 (dd, J = 13.6, 12.4 Hz, 1H), 4.49—4.39 (m, 3H). LC—MS: found [M + H]+ m /z 618.10; ESI MS: mass calcd for C28H19Cl2F4N5O3 [M]+, 619.0801; found, Mobs 619.0805. HPLC, tR 17.6 min, purity 99%. Enantiomeric excess >97%.
(R)-N-(1-(2,4-Dichlorophenyl)-2-(1H-imidazol-1-yl)ethyl)-4-(5-(3-fluoro-5-(2,2,2-trifluoroethoxy)phenyl)-1,3,4-oxadiazol-2-yl)-benzamide (Compound 4).
Yield 30%. 1H NMR (CDCl3): δ ppm 10.0 (s, 1H), 9.55 (d, J = 9.0 Hz, 1H), 8.04 (d, J = 8.3 Hz, 2H), 7.78 (dd, J = 11.4, 8.4 Hz, 3H), 7.55—7.53 (m, 2H), 7.46—7.45 (m, 2H), 7.42 (s, 1H), 7.36 (dd, J = 8.4, 2.0 Hz, 1H), 6.93—6.90 (m, 1H), 6.08—6.04 (m, 1H), 5.30 (dd, J = 14.0, 11.8 Hz, 1H), 4.49—4.45 (m, 2H), 4.42—4.38 (m, 1H). LC—MS: found [M + H]+ m /z 620.09; ESI MS: mass calcd for C28H19Cl2F4N5O3 [M]+, 619.0801; found, Mobs 619.0802. HPLC, tR 13.4 min, purity 98%. Enantiomeric excess >97%.
(R)-4-(5-(3,4-Dichlorophenyl)-1,3,4-oxadiazol-2-yl)-N-(1-(2,4-di-chlorophenyl)-2-(1H-imidazol-1-yl)ethyl)benzamide (Compound 5).
Yield 20%. 1H NMR (CDCl3): δ ppm 8.21 (br, 1H), 8.16 (d, J = 2.0 Hz, 1H), 8.04 (d, J = 8.4 Hz, 2H), 7.96 (d, J = 8.4 Hz, 2H), 7.93 (dd, J = 8.4, 2.0 Hz, 1H), 7.61 (d, J = 8.4 Hz, 1H), 7.45 (d, J = 2.0 Hz, 1H), 7.42 (d, J = 8.4 Hz, 1H), 7.23 (dd, J = 8.4, 2.0 Hz, 1H), 7.09 (s, 1H), 7.05 (s, 1H), 5.92—5.87 (m, 1H), 4.76 (dd, J = 14.0, 8.9 Hz, 1H), 4.26 (dd, J = 14.0, 5.1 Hz, 1H). LCx2014;MS: found [M + H]+ m /z 573.91; ESI—MS: mass calcd for C26H17Cl4N5O2 [M]+, 571.1036; found, Mobs 571.1046. HPLC, tR 13.4 min, purity 98%. Enantiomeric excess >97%.
(R)-4-(5-(3-Bromophenyl)-1,3,4-oxadiazol-2-yl)-N-(1-(2,4-di-chlorophenyl)-2-(1H-imidazol-1-yl)ethyl)benzamide (Compound 6).
Yield 50%. 1H NMR (CDCl3): δ ppm 8.30 (s, 1H), 8.24 (d, J = 7.4 Hz, 2H), 8.11 (d, J = 7.5 Hz, 1H), 7.91 (d, J = 7.5 Hz, 2H), 7.72 (d, J = 7.8 Hz, 1H), 7.51 (s, 1H), 7.45 (m, 1H), 7.35 (s, 1H), 7.12 (d, J = 7.8 Hz, 1H), 7.06 (s, 1H), 6.95—6.93 (m, 1H), 6.86 (s, 1H), 5.79 (m, 1H), 4.56 (d, J = 6.48 Hz, 2H). LC—MS: found [M + H]+ m /z 581.9; ESI MS: mass calcd for C26H18BrCl2N5O2 [M]+, 581.0021; found, Mobs 581.0026. HPLC, tR 17.0 min, purity 98%. Enantiomeric excess >97%.
(R)-N-(1-(2,4-Dichlorophenyl)-2-(1H-imidazol-1-yl)ethyl)-4-(5-(3-morpholinophenyl)-1,3,4-oxadiazol-2-yl)benzamide (Compound 7).
Yield 60%. 1H NMR (CDCl3): δ ppm 8.06 (d, J = 7.6 Hz, 2H), 7.91 (d, J = 7.6 Hz, 1H), 7.87 (d, J = 7.6 Hz, 2H), 7.60 (s, 1H) 7.53 (d, J = 7.0 Hz, 1H), 7.45 (s, 1H), 7.41—7.29 (m, 3H), 7.22 (d, J = 8.2 Hz, 1H), 7.08 (d, J = 7.5 Hz, 1H), 7.0 (s, 1H), 6.92 (s, 1H), 5.88—5.79 (m, 1H), 4.55 (dd, J = 13.7, 6.8 Hz, 1H), 4.45 (dd, J = 13.0, 4.4 Hz, 1H), 3.87 (s, 4H), 3.22 (s, 4H). LC—MS: found [M + H]+ m /z 589.6. HPLC, tR 7.3 min, purity 98%. Enantiomeric excess >97%.
(R)-N-(1-(2,4-Dichlorophenyl)-2-(1H-imidazol-1-yl)ethyl)-4-(5-(4-morpholinophenyl)-1,3,4-oxadiazol-2-yl)benzamide (Compound 8).
Yield 40%. 1H NMR (CDCl3): δ ppm 8.27 (br, 1H), 8.04 (d, J = 8.2 Hz, 2H), 7.98—7.95 (m, 4H), 7.48 (s, 1H), 7.45 (d, J = 2.0 Hz, 2H), 7.24 (dd, J = 8.4, 2.0 Hz, 1H), 7.08 (br, 2H), 6.97 (s, 1H), 6.95 (s, 1H), 5.95—5.90 (m, 1H), 4.79 (dd, J = 13.2, 8.1 Hz, 1H), 4.45 (dd, J = 14.0, 4.4 Hz, 1H), 3.87 (m, 4H), 3.22 (m, 4H). LC—MS: found [M + H]+ m /z 589.23. HPLC, tR 6.8 min, purity 98%. Enantiomeric excess >97%.
(R)-N-(1-(2,4-Dichlorophenyl)-2-(1H-imidazol-1-yl)ethyl)-4-(5-(3-(pyridin-2-yl)phenyl)-1,3,4-oxadiazol-2-yl)benzamide (Compound 9).
Yield 20%. 1H NMR (CDCl3): δ ppm 8.75—8.74 (m, 2H), 8.48 (s, 1H), 8.38 (s, 1H), 8.21—8.18 (m, 2H), 8.04 (d, J = 8.4 Hz, 2H), 7.99 (d, J = 8.4 Hz, 2H), 7.84—7.82 (m, 2H), 7.65 (t, J = 7.8 Hz, 1H), 7.50 (d, J = 8.6 Hz, 1H), 7.44 (d, J = 2.0 Hz, 1H), 7.33—7.30 (m, 1H), 7.28 (d, J = 2.2 Hz, 1H), 7.20 (s, 1H), 5.96—5.90 (m, 1H), 4.89 (dd, J = 14.0, 9.5 Hz, 1H), 4.46 (dd, J = 14.0, 4.9 Hz, 1H). (One signal was overlapped with CDCl3.) LC—MS: found [M + H]+ m /z 581.2; ESI—MS: mass calcd for C31H22Cl2N6O2 [M]+, 580.1181; found, Mobs 580.1192. HPLC, tR 11.0 min, purity 99%. Enantiomeric excess >97%.
(R)-N-(1-(2,4-Dichlorophenyl)-2-(1H-imidazol-1-yl)ethyl)-4-(5-(4-(pyridin-2-yl)phenyl)-1,3,4-oxadiazol-2-yl)benzamide (Compound 10).
Yield 30%. 1H NMR (CDCl3): δ ppm 8.75 (d, J = 4.5 Hz, 1H), 8.67—8.62 (m, 2H), 8.23—8.17 (m, 4H), 8.05 (s, 3H), 7.82—7.81 (m, 2H), 7.58 (d, J = 8.4 Hz, 1H), 7.46 (d, J = 2.0 Hz, 1H), 7.33—7.28 (m, 2H), 7.19 (s, 2H), 6.00—5.95 (m, 1H), 4.95 (dd, J = 14.0, 9.6 Hz, 1H), 4.45 (dd, J = 14.0, 4.4 Hz, 1H). (One signal is overlapped with CDCl3.) LC—MS: found [M + H]+ m /z 581.09; ESI MS: mass calcd for C31H22Cl2N6O2 [M]+, 580.1181; found, Mobs 580.1195. HPLC, tR 10.9 min, purity 99%. Enantiomeric excess >97%.
(R)-N-(1-(2,4-Dichlorophenyl)-2-(1H-imidazol-1-yl)ethyl)-4-(5-(3-fluoro-5-(pyridin-2-yl)phenyl)-1,3,4-oxadiazol-2-yl)benzam ide (Compound 11).
Yield 20%. 1H NMR (CDCl3): δ ppm 10 (s, 1H), 9.58 (d, J = 8.7 Hz, 1H), 8.75 (d, J = 4.6 Hz, 1H), 8.59 (s, 1H), 8.04 (d, J = 8.4 Hz, 2H), 7.88—7.86 (m, 2H), 7.82—7.75 (m, 4H), 7.69 (s, 1H), 7.46 (s, 1H), 7.42 (d, J = 2.0 Hz, 1H), 7.38—7.32 (m, 3H), 6.09—6.03 (m, 1H), 5.30 (dd, J = 13.4, 12.0 Hz, 1H), 4.41 (dd, J = 13.6, 3.0 Hz, 1H). LC—MS: found [M + H]+ m /z 599.09; ESI—MS: mass calcd for C31H21Cl2FN6O2 [M + H]+, 598.1087; found, Mobs 5998.1092. HPLC, tR 8.5 min, purity 98%. Enantiomeric excess >97%.
(R)-N-(1-(2/4-Dichlorophenyl)-2-(1H-imidazol-1-yl)ethyl)-4-(5-(4-(3-fluoropyridin-2-yl)phenyl)-1,3,4-oxadiazol-2-yl)benzam ide (Compound 12).
Yield 40%. 1H NMR (CDCI3): δ ppm 10.0 (s, 1H), 9.55 (d, J = 8.7 Hz, 1H), 8.76 (d, J = 4.6 Hz, 1H), 8.28 (t, J = 7.8 Hz, 1H), 8.05–8.01 (m, 3H), 7.98 (dd, J = 8.2, 1.4 Hz, 1H), 7.86–7.77 (m, 5H), 7.52 (s, 1H), 7.44 (d, J = 2.0 Hz, 2H), 7.38–7.33 (m, 2H), 6.09–6.03 (m, 1H), 5.24 (dd, J = 13.8, 12.5 Hz, 1H), 4.43 (dd, J = 14.0, 3.2 Hz, 1H). LC-MS: found [M + H]+ m /z 599.14; ESI-MS: mass calcd for C28H19CI2F4N5O3 [M]+, 619.0801; found, Mobs 619.0802. HPLC, tR 6.2 min, purity 99%. Enantiomeric excess >97%.
(R)-N-(l-(2,4-Dichlorophenyl)-2-(TH-imidazol-l-yl)ethyl)-4-(5-(2-fluoro-5-(pyridin-2-yl)phenyl)-1,3,4-oxadiazol-2-yl)benzam ide (Compound 13).
Yield 20%. 1H NMR (CDCl3): δ ppm 10.0 (s, 1H), 9.77 (d, J = 9.6 Hz, 1H), 8.96 (d, J = 5.6 Hz, 1H), 8.90 (dd, J = 6.0, 2.2 Hz, 1H), 8.45–8.41 (m, 1H), 8.35 −8.30 (m, 1H), 8.18 (d, J = 8.4 Hz, 1H), 8.07 (d, J = 8.3 Hz, 2H), 8.01 (s, 1H), 7.86 (dd, J = 8.4, 4.0 Hz, 3H), 7.53–7.50 (m, 2H), 7.46 (s, 1H), 7.42 (d, J = 2.0 Hz, 1H), 7.33 (dd, J = 8.5, 2.0 Hz, 1H), 6.09–6.03 (m, 1H), 5.34 (dd, J = 13.8, 12.5 Hz, 1H), 4.43 (dd, J = 13.2, 2.5 Hz, 1H). LCMS: found [M + H]+ m /z 599.18; ESI-MS: mass calcd for C31H21Cl2FN6O2 [M]+, 598.1087; found, Mobs 598.1090. HPLC, tR 10.3 min, purity 99%. Enantiomeric excess >97%.
(R)-4-(5-(3-Chloro-4-(methylsulfonyl)phenyl)-1,3,4-oxadiazol-2-yl)-N-(1-(2,4-dichlorophenyl)-2-(1H-imidazol-1-yl)ethyl)benzamide (Compound 14).
Yield 50%. 1H NMR (CDCI3): δ ppm 10.0 (s, 1H), 9.57 (d, J = 8.6 Hz, 1H), 8.35 (s, 1H), 8.34–8.33 (m, 1H), 8.22 (dd, J = 8.2, 1.5 Hz, 1H), 8.07 (d, J = 8.3 Hz, 2H), 7.78 (dd, J = 8.2, 5.04 Hz, 3H), 7.50 (s, 1H), 7.47 (d, J = 2.0 Hz, 1H), 7.44 (s, 1H), 6.106.05 (m, 1H), 5.30 (dd, J = 13.8, 12.4 Hz, 1H), 4.42 (dd, J = 13.6, 2.6 Hz, 1H), 3.35 (s, 3H). LC-MS: found [M + H]+ m /z 617.7; ESI-MS: mass calcd for C27H20Cl3N5O4S [M]+, 6175.0302; found, Mobs 615.0315. HPLC, tR 5.8 min, purity 99%. Enantiomeric excess >97%.
(R)-N-(1-(2,4-Dichlorophenyl)-2-(1H-imidazol-1-yl)ethyl)-4-(5-(3-fluoro-5-(5-fluoropyrim idin-4-yl)phenyl)-1,3,4-oxadiazol-2-yl)-benzamide (Compound 15).
Yield 30%. 1H NMR (CDCI3): δ ppm 10.04 (s, 1H), 9.54 (d, J = 8.7 Hz, 1H), 9.16 (d, J = 3.0 Hz, 1H), 8.76 (d, J = 3.4 Hz, 1H), 8.73 (s, 1H), 8.11–8.08 (m, 1H), 8.06 (d, J = 8.5 Hz, 2H), 8.02–7.99 (m, 1H), 7.80 (dd, J = 8.4, 3.0 Hz, 3H), 7.55 (s, 1H), 7.45 (d, J = 2.0 Hz, 2H), 7.35 (dd, J = 8.4, 2.0 Hz, 1H), 6.096.03 (m, 1H), 5.30 (dd, J = 14.0, 12.0 Hz, 1H), 4.42 (dd, J = 14.0, 3.0 Hz). LC-MS: found [M + H]+ m /z 617.8; ESI-MS: mass calcd for C30H19C12F2N7O2 [M]+, 617.0945; found, Mobs 617.0949. HPLC, tR 13.0 min, purity 99%. Enantiomeric excess >97%.
CYP51 Activity Assay.
A. fumigatus and C. albicans CYP51 were expressed in Escherichia coli and purified as described earlier.9,10 The enzymatic reaction mixture contained 0.5 μM A. fumigatus9 or C. albicans10 CYP51 and 1.0 μM rat NADPH-cytochrome P450 reductase, 100 μM L-α−1,2-dilauroyl-sn-glycerophosphocholine, 0.4 mg/mL isocitrate dehydrogenase, and 25 mM sodium isocitrate in 50 mM potassium phosphate buffer (pH 7.2) containing 10% glycerol (v/v) and 0.1% Triton X-100 (v/v). After addition of the radiolabeled ([3-3H]) sterol substrate (eburicol for A.fum igatus and lanosterol for C. albicans CYP51), final concentration 25 μM, and an inhibitor, final concentration 0.75 μM, the mixture was preincubated for 60 s at 37 °C in a shaking water bath, and the reaction was initiated by the addition of 100 μM NADPH and stopped by extraction of the sterols with EtOAc. The extracted sterols were dried, dissolved in CH3OH, and analyzed using a reversed-phase HPLC system (W aters) equipped with a β−RAM detector (INUS Systems, Inc.) using a NovaPak octadecylsilane (C18) column (particle size 4 μm, 3.9 mm × 150 mm) and a linear gradient of H2O, CH3CN, and CH3OH.9,10 The inhibitory potencies of VNI and derivatives were compared as percentage of inhibition of sterol 14α−demethylation in 60 min reactions;16,18 the molar ratio inhibitor/enzyme/substrate in the reaction mixture was 1.5:1:50. Two clinical systemic antifungal drugs, fluconazole and posaconazole, were used as controls. The experiments were performed in triplicate, and the results are presented as means ± SD.
CYP51 Ligand Binding Assay.
Spectral titrations of C. albicans and A. fumigatus CYP51 with selected VNI derivatives were carried out in 5 cm optical path length cuvettes at 0.5 μM P450 concentration as previously described.15 The ligands were added from 0.2 mM stock solutions in DMSO, and the titration range was 0.1–0.8 μM. The apparent dissociation constants of the enzyme-inhibitor complex were calculated as red shifts in the P450 Soret band maximum (from 417 to 424 nm29) using GraphPad Prism 6 software by fitting the data for the ligand-induced absorbance changes in the difference spectra Δ(Amax–Amin) versus ligand concentration to the quadratic eq 1 (tight-binding ligands):14
| (1) |
where [L] and [E] are the total concentrations of ligand and enzyme used for the titration, respectively.
Microsomal Stability Assay.
For intrinsic clearance in hepatic microsomes (human, rat, mouse), human (mixed gender), rat (male Sprague Dawley), or mouse (male CD-1) pooled liver microsomes (0.5 mg/mL, BD Biosciences) and 1 μM test compound were incubated in 100 mM potassium phosphate buffer (pH 7.4) with 3 mM MgCl2 at 37 °C with constant shaking. After a 5 min preincubation, each reaction was initiated by the addition of 1 mM NADPH. At selected time intervals (0, 3, 7, 15, 25, and 45 min), 50 μL aliquots were taken and subsequently placed into a 96-well plate containing 150 μL of cold CH3CN with internal standard (50 ng/mL carbamazepine). Plates were then centrifuged at 3000g (4 °C) for 10 min, and the supernatants were transferred to a separate 96-well plate and diluted 1:1 (v/v) with H2Ofor LC/MS/MS analysis. The experiments were performed in triplicate. The in vitro half-life (T1/2, min, eq 2), estimated intrinsic clearance (CLINT, mL/min/kg, eq 3), and subsequent predicted hepatic clearance (CLHEP, mL/min/kg, eq 4) were determined employing the following equations:
| (2) |
where k represents the slope from linear regression analysis of the natural log percent remaining of test compound as a function of incubation time:
| (3) |
where “a” is the scaling factor.
| (4) |
where Qh (hepatic blood flow) is 21 mL/min/kg for human, 70 mL/min/kg for rat, and 90 mL/m in/kg for mouse.
Antifungal Drug Susceptibility Test.
The minimal inhibitory concentration (MIC) of each drug against the various A. fumigatus strains was investigated using the Clinical and Laboratory Standards Institute broth microdilution method, document M 38-A2.30 The tests were performed in triplicate. Various strains of A. fumigatus, 1022, 32820, and MYA626 (ATCC MP-12 kit), were grown according to the protocol.30 Briefly, the strains were initially plated onto potato dextrose agarose plates for 5–6 days at 35 °C to generate conidias. The agar plate surfaces were washed with 5 mL of sterile 1× phosphate-buffered saline with 0.05% Tween-80 (v/v) to obtain the independent conidias, and the number of conidias were counted with a hemocytometer. The conidias were further diluted in RPMI media to obtain 1 × 104 conidias/mL and seeded into each well of 96-well plates. The testing media was RPMI 1640 (Gibco-BRL, Uxbridge, UK) with L-glutamine (without sodium bicarbonate, pH 7.0) adjusted with 0.165 M morpholinepropanesulfonic acid (MOPS).31 Serial base 2× dilutions of various drugs (from 50 to 0.006 mg/L) were prepared in RPMI-1640 and added to the wells of microtiter plates in 100 μL aliquots. Media alone (200 μL) was used as positive control of mycelia growth, and 200 μL of media alone without conidias was used as negative control. The plates were further incubated for 48 h at 35 °C and observed under light microscopy for the growth of mycelia. MICs were the lowest drug concentrations that produced complete growth inhibition (100%) at 48 h of incubation.
Protein Crystallization, Data Collection, Structure Refinement, and Analysis.
For crystallographic experiments, A. fumigatus CYP51B was purified in three steps, including anion exchange chromatography on DEAE-Sepharose, affinity chromatography on Ni2+−NTA agarose, and cation exchange chromatography on CM-Sepharose.9 A 10 μM solution of compound 3 was added to all of the buffers at the CM-Sepharose stage of the purification procedure. The cocrystals were obtained by the hanging drop vapor diffusion technique and were grown at 16 °C. Equal volumes of complex solution preincubated with 10 mM cyclohexylpentanoyl-N−hydrox-yethylglucamide (Anagrade) were mixed with well solution (0.2 M lithium acetate (pH 7.4) with 19% PEG 3350 (w/v)). Crystals appeared after 3 days and were cryoprotected in 40% glycerol (v/v) and flash-cooled in liquid nitrogen.
The data were collected at 100 K using synchrotron radiation on the insertion device of the Life Sciences Collaborative Access Team at the Advanced Photon Source, Argonne National Laboratory (Argonne, IL), beamline 21-ID-F at a wavelength of 0.9787 Å and using a Rayonix MX300 CCD detector. The diffraction images were processed with HKL-2000 in the hexagonal space group P31 to a maximum resolution of 2.38 Å, and the structure was solved in Phaser MR (CCP4 Program Suite version 7.0.050) using atomic coordinates of the voriconazole-bound A. fumigatus CYP51 (PDB ID 4UYM). Iterative models of the protein—inhibitor complexes were built with Coot32 and refined with Refmac5 in the CCP4 Suite.33
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by funding from the National Institutes of Health (GM067871, G.I.L.). Vanderbilt University is a member institution of the Life Sciences Collaborative Access Team at Sector 21 of the Advanced Photon Source (Argonne, IL). Use of the Advanced Photon Source at Argonne National Laboratory was supported by the United States Department of Energy, Office of Science, Office of Basic Energy Sciences, under contract DE-AC02–06CH11357. We also would like to thank Prof. Stephen W. Fesik’s group for providing access to their Agilent LC—MS and Biotage Initiator Microwave System.
ABBREVIATIONS USED
- CYP
cytochrome P450
- CYP51
sterol 14α−demethylase
- IC50
inhibitor concentration that decreases enzyme turnover rate by 50%
- MIC
minimal inhibitory concentration
- IFIs
invasive fungal infections
- DMF
N,N−dimethylformamide
- DPPA
diphenyl phosphorazidate
- DBU
1,5-diazabiciclo[5.4.0]-undec-7-ene
- THF
tetrahydrofuran
- HPLC
high-performance liquid chromatography
- Pd(dppf) Cl2
[1,1’-bis-(diphenylphosphino)ferrocene]dichloropalladium(II)
- LC — MS
liquid chromatography-mass spectrometry
- ESI
electrospray ionization
- MS
mass spectrometry
- DMSO
dimethyl sulfoxide
- TFA
trifluoroacetic acid
- ELSD
evaporative light scattering detector
- rt
room temperature
- equiv
equivalent
- h
hours
- Hz
hertz
- J
coupling constant (in NMR spectrometry)
- tR
retention time (in chromatography)
- TMS
tetramethylsi-lane
- NMR
nuclear magnetic resonance
- MHz
megahertz
- DMAP
4-(N,N−ddimethylamino)pyridine
- MW
microwave reactor
- EDC/HCl
N−(3-dimethylaminopropyl)-N’−ethylcar-bodiimide hydrochloride
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jm ed-chem.8b00641.
Table S1 (data collection and refinement statistics) and six supplementary figures: Figure S1, structural formulas of systemic antifungal agents; Figure S2, apparent ligand binding affinity versus inhibition of CYP51 activity; Figure S3, gatekeeping interactions of LFV in complexes A and B; Figure S4, 1H NMR spectra; Figure S5, HRMS spectra; and Figure S6, HPLC chromatograms (DOCX) Molecular formula strings (CSV)
Accession Codes
The coordinates and structure factors of A. fumigatus CYP51 in complex with compound 3 have been deposited to PDB under accession code 6CR2. The atomic coordinates and experimental data will be released upon acceptance of this Article for publication.
Notes
The authors declare no competing financial interest.
REFERENCES
- (1). Bongomin F; Gago S; Oladele RO.; Denning DW Global and multi-national prevalence of fungal diseases-estimate precision. J. Fungi (Basel, Switzerland) 2017, 3 (4), 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2). Pappas PG; Kauffman CA; Andes DR; Clancy CJ; Marr KA; Ostrosky-Zeichner L; Reboli AC; Schuster MG; Vazquez JA; Walsh TJ; Zaoutis TE; Sobel JD Clinical practice guideline for the management of candidiasis: 2016 Update by the infectious diseases society of America. Clin. Infect. Dis 2016, 62, 1–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3). Brown GD; Denning DW; Gow NAR; Levitz SM; Netea MG; White TC Hidden killers: human fungal infections. Sci. Transl. Med 2012, 4 (165), 165rv13. [DOI] [PubMed] [Google Scholar]
- (4). Denning DW.; Hope WW Therapy for fungal diseases: opportunities and priorities. Trends Microbiol. 2010, 18 (5), 195–204. [DOI] [PubMed] [Google Scholar]
- (5). Kullberg BJ; Arendrup MC Invasive candidiasis. N. Engl. J. Med 2015, 373 (15), 1445–1456. [DOI] [PubMed] [Google Scholar]
- (6). Denning DW; Bromley MJ How to bolster the antifungal pipeline. Science 2015, 347 (6229), 1414–1416. [DOI] [PubMed] [Google Scholar]
- (7). Lepesheva GI; Friggeri L; Waterman MR CYP51 as drug targets for fungi and protozoan parasites: past, present and future. Parasitology 2018, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8). Maertens JA; Raad II; Marr KA; Patterson TF; Kontoyiannis DP; Cornely OA; Bow EJ; Rahav G; Neofytos D; Aoun M; Baddley JW; Giladi M; Heinz WJ; Herbrecht R; Hope W.; Karthaus M; Lee D-G; Lortholary O; Morrison VA; Oren I; Selleslag D; Shoham S; Thompson GR; Lee M; Maher RM; Schmitt-Hoffmann A-H; Zeiher B; Ullmann AJ Isavuconazole versus voriconazole for primary treatment of invasive mould disease caused by Aspergillus and other filamentous fungi (SECU RE): a phase 3, randomised-controlled, non-inferiority trial. Lancet 2016, 387 (10020), 760–769. [DOI] [PubMed] [Google Scholar]
- (9). Hargrove TY; Wawrzak Z; Lamb DC; Guengerich FP; Lepesheva GI Structure-Functional Characterization of Cytochrome P450 Sterol 14alpha-Demethylase (CYP51B) from Aspergillus f umigatus and molecular basis for the development of antifungal drugs. J.. Biol. Chem 2015, 290 (39), 23916–23934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10). Hargrove TY; Friggeri L; Wawrzak Z; Qi A; Hoekstra WJ; Schotzinger RJ; York JD; Guengerich FP; Lepesheva GI Structural analyses of Candida albicans sterol 14alpha-demethylase complexed with azole drugs address the molecular basis of azole-mediated inhibition of fungal sterol biosynthesis. J. Biol. Chem 2017, 292 (16), 6728–6743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11). Hargrove TY; Garvey EP; Hoekstra WJ; Yates CM; Wawrzak Z; Rachakonda G; Villalta F; Lepesheva GI Crystal structure of the new investigational drug candidate VT-1598 in complex with Aspergillus fum igatus sterol 14alpha-demethylase provides insights into its broad-spectrum antifungal activity. Antimicrob. Agents Chemother. 2017, 61 (7), e00570–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12). Lepesheva GI; Park HW; Hargrove TY; Vanhollebeke B; Wawrzak Z; Harp JM; Sundaramoorthy M; Nes WD; Pays E; Chaudhuri M; Villalta F; Waterman MR Crystal structures of Trypanosoma brucei sterol 14α-demethylase and implications for selective treatment of human infections. J. Biol. Chem 2010, 285 (3), 1773–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13). Villalta F; Dobish MC; Nde PN; Kleshchenko YY; Hargrove TY; Johnson CA; Waterman MR; Johnston JN; Lepesheva GI VNI cures acute and chronic experimental Chagas disease. J. Infect. Dis 2013, 208 (3), 504–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14). Friggeri L; Hargrove TY; Rachakonda G; Williams AD; Wawrzak Z; Di Santo R; De Vita D; Waterman MR; Tortorella S; Villalta F; Lepesheva GI Structural basis for rational design of inhibitors targeting Trypanosoma cruzi sterol 14α−demethylase: Two regions of the enzyme molecule potentiate its inhibition. J. Med. Chem 2014, 57 (15), 6704–6717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15). Hargrove TY; Wawrzak Z; Alexander PW; Chaplin JH; Keenan M; Charman SA; Perez CJ; Waterman MR; Chatelain E; Lepesheva GI Complexes of Trypanosoma cruzi sterol 14α-demethylase (C YP51) with two pyridine-based drug candidates for Chagas disease: Structural basis for pathogen selectivity. J. Biol. Chem 2013, 288 (44), 31602–31615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16). Lepesheva GI; Ott RD; Hargrove TY; Kleshchenko YY; Schuster I; Nes WD; Hill GC; Villalta F; Waterman MR Sterol 14α−demethylase as a potential target for antitrypanosomal therapy: Enzyme inhibition and parasite cell growth. Chem. Biol 2007, 14 (11), 1283–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17). Andriani G; Amata E; Beatty J; Clements Z; Coffey BJ; Courtemanche G; Devine W.; Erath J; Juda CE; Wawrzak Z; Wood JT; Lepesheva GI; Rodriguez A; Pollastri MP Antitrypanosomal lead discovery: Identification of a ligand-efficient inhibitor of Trypanosoma cruzi CYP51 and parasite growth. J. Med. Chem 2013, 56 (6), 2556–2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18). Lepesheva GI; Hargrove TY; Rachakonda G; Wawrzak Z; Pomel S; Cojean S; Nde PN; Nes WD; Locuson CW; Calcutt MW.; Waterman MR; Daniels JS; Loiseau PM; Villalta F VFV as a new effective CYP51 structure-derived drug candidate for Chagas disease and visceral leishmaniasis. J. Infect. Dis 2015, 212 (9), 1439–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19). Heeres J; Meerpoel L; Lewi P Conazoles. Molecules 2010,15 (6), 4129–4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20). Van den Bossche H; Willemsens G; Cools W; Cornelissen F; Lauwers WF; van Cutsem JM In vitro and in vivo effects of the antimycotic drug ketoconazole on sterol synthesis. Antimicrob. Agents Chemother. 1980, 17 (6), 922–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21). Aoyama Y; Yoshida Y; Sato R Yeast cytochrome P-450 catalyzing lanosterol 14α−demethylation. II. Lanosterol metabolism by purified P-450(14)DM and by intact microsomes. J. Biol. Chem 1984, 259, 1661–1666. [PubMed] [Google Scholar]
- (22). Correia MA; Ortiz de Montellano PR Inhibition of Cytochrom e P 450 Enzymes In Cytochrom e P 450: Structure, Mechanism, and Biochemistry, 3rd ed.; Ortiz de Montellano PR, Ed.; Plenum Publishing Corp: New York, 2005; pp 246–322. [Google Scholar]
- (23). Yoshida Y; Aoyama Y Interaction of azole antifungal agents with cytochrome P-45014DM purified from Saccharomyces cerevisiae microsomes. Biochem. Pharmacol 1987, 36 (2), 229–235. [DOI] [PubMed] [Google Scholar]
- (24). Lepesheva GI; Waterman MR Structural basis for conservation in the CYP51 family. Biochim. Biophys. Acta, Proteins Proteomics 2011, 1814 (1), 88–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25). Lepesheva GI; Waterman MR Sterol 14α−demethylase (C YP51) as a therapeutic target for human trypanosomiasis and leishmaniasis. Curr. Top. Med. Chem 2011, 11 (16), 2060–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26). Hargrove TY; Friggeri L; Wawrzak Z; Sivakumaran S; Yazlovitskaya EM; Hiebert SW.; Guengerich FP; Waterman MR; Lepesheva GI Human sterol 14α−demethylase as a target for anticancer chemotherapy: towards structure-aided drug design. J. Lipid Res. 2016, 5 7 (8), 1552–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27). Yu X; Nandekar P; Mustafa G; Cojocaru V; Lepesheva GI; Wade RC Ligand tunnels in T. brucei and human CYP51: Insights for parasite-specific drug design. Biochim. Biophys. Acta, Gen. Subj 2016, 1860 (1, Part A), 67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28). Friggeri L; Scipione L; Costi R; Kaiser M; Moraca F; Zamperini C; Botta B; Di Santo R; De Vita D; Brun R; Tortorella S New promising compounds with in vitro nanomolar activity against Trypanosoma cruzi. ACS Med. Chem. Lett 2013, 4 (6), 538–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29). Schenkman JB; Remmer H; Estabrook RW Spectral studies of drug interaction with hepatic microsomal cytochrome. Mol. Pharmacol 1967, 3, 113–123. [PubMed] [Google Scholar]
- (30). Wayne P CLSI Reference M ethod fo r Broth Dilution Antifungal Ausceptibility Testing of Filamentous Fungi, 3rd ed.; CLSI Standard M 38. Clinical and Laboratory Standards Institute, 2017. [Google Scholar]
- (31). Espinel-Ingroff A; Turnidge J; Alastruey-Izquierdo A; Dannaoui E; Garcia-Effron G; Guinea J; Kidd S; Pelaez T; Sanguinetti M; Meletiadis J; Botterel F; Bustamante B; Chen YC; Chakrabarti A; Chowdhary A; Chryssanthou E; Cordoba S; Gonzalez GM; Guarro J; Johnson EM; Kus JV; Lass-Florl C; Linares-Sicilia MJ; Martin-Mazuelos E; Negri CE; Pfaller MA; Tortorano AM Posaconazole MIC distributions for Aspergillus fum igatus Species complex by four methods: Impact of CYP51A mutations on estimation of epidemiological cutoff values. Antimicrob. Agents Chemother. 2018, 62 (4), e01916–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32). Emsley P; Lohkamp B; Scott WG; Cowtan K Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66 (4), 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33). Potterton E; Briggs P; Turkenburg M; Dodson E A graphical user interface to the CCP4 program suite. Acta Crystallogr., Sect. D: Biol. Crystallogr 2003, 59 (Pt 7), 1131–1137. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.