

Abstract
Nitrobenzene, a potentially harmful compound found in tobacco smoke, has been largely excluded from prior analysis due to difficulties with quantification. Quantifying harmful compounds in cigarette smoke is useful to compare products, to examine the impact of design parameters on delivery, and to help estimate exposures. A sensitive high-throughput method has been developed for quantifying nitrobenzene in machine-generated mainstream cigarette smoke using isotope dilution gas chromatography-tandem mass spectrometry (ID-GC-MS/MS). This method has sufficient sensitivity to measure vapor phase nitrobenzene concentrations in the low nanogram range, with a 418 pg/cig method limit of detection. Precision estimates from two quality control cigarette products resulted in percent relative standard deviations of 11.5% and 14.9%; product variability estimates from 13 cigarette products resulted in percent relative standard deviations ranging from 2.8% to 16.9%. Nitrobenzene in the machine-generated, mainstream smoke from 15 cigarette products are reported and range from 18 to 38 ng/cig under the Health Canada Intense smoking regimen.
Keywords: Gas chromatography, Mass spectrometry, Nitrobenzene, Volatile organic compound, Tobacco, Mainstream cigarette smoke
1. Introduction
Tobacco smoke is a complex mixture of more than 7,000 components, including 93 compounds that the United States Food and Drug Administration (FDA) considers to be harmful or potentially harmful constituents (HPHCs).[1, 2] Tobacco smoke is often collected for analysis in two fractions: the particulate phase fraction, also called total particulate matter or TPM, and the vapor phase fraction, comprised primarily of volatile organic compounds (VOCs). One well-represented class of compounds in tobacco smoke are the nitro compounds, which are pyrosynthesized in situ during the smoking process by a reaction between nitrates present in the leaves of the tobacco plant and hydrocarbon radicals formed in the burning zones of the tobacco product.[3] A number of different aromatic and aliphatic nitro compounds are prevalent in cigarette smoke. However, since 1986, only three of these have been discussed with regularity in the context of toxicity, biological activity, and carcinogenicity: nitromethane, 2-nitropropane, and nitrobenzene.[4, 5] As these compounds are relatively nonpolar and low-boiling, they typically end up in the vapor phase fraction and may be counted among the volatile or semi-volatile organic components.[6] Detection of nitrobenzene in cigarettes is important because it is a possible human carcinogen, as indicated in animal, metabolic, and structure-activity relationship studies.[7–15] Previous attempts to include nitrobenzene in multi-analyte VOC methods for cigarette smoke failed, as they lacked the necessary sensitivity to quantify the low levels of this compound found in smoke;[6, 16] therefore, a separate method was developed.
To our knowledge, there are two reports dealing specifically with detection of nitrobenzene compounds in cigarettes. Hoffmann and Rathkamp published an elaborate method using impingers, distillation, liquid-liquid extraction, and column chromatography to collect, clean up, and concentrate different nitrobenzene compounds in mainstream smoke extracts before quantitation via a gas chromatograph interfaced with an electron capture detector (GC-ECD).[3, 17] This method, while appropriate for the technology available in 1970, is impractical and unnecessarily complex for present-day analysts and is certainly at odds with the modern desire for high-throughput methods. In the second method, reported by Xie et al., only the nitrobenzene content in the TPM was considered, which required that the authors smoke 20 – 80 cigarettes onto a single filter pad in order to generate sufficient nitrobenzene content for detection. A single solid-phase extraction (SPE) step was used for cleanup, followed by a 50-fold concentration step using a nitrogen blow down evaporator prior to GC-ECD analysis. To further separate the sample, the authors employed a heart-cutting step in the chromatographic analysis, in which they employed a Deans’ switch[18] to selectively capture desired portions of eluent from the first column and redirect it to a second analytical column for further separation prior to detection. In this manner, they could attain substantially greater separation between analytes that were inadequately resolved by the primary column. However, the back-to-back chromatography used in this method results in long analysis times and is impractical as a high throughput method. Also, in the Xie et al. approach only the nitrobenzene content of the TPM was analyzed; the content of the vapor phase fraction of smoke was not measured.[9]
Nitrobenzene itself is the only aromatic nitro compound currently considered a harmful or potentially harmful analyte by the FDA[2] and has presented major challenges in previous attempts at quantitation in analytical methods with mixed analyte panels; therefore it is the sole focus of this study. We developed and validated a sensitive and selective “extract and shoot” approach for the detection of nitrobenzene in mainstream smoke. This approach requires minimal solvent, no cleanup step, and a short chromatographic separation time (5.5 min) to optimize output for high-throughput analyses. Gas chromatography (GC) was chosen as the primary means of sample separation due to the semivolatile nature of nitrobenzene, and tandem mass spectrometry (MS/MS) was chosen as the detection method due to its high sensitivity and specificity. Deuterated nitrobenzene (nitrobenzene-d5) was selected as the internal standard to help account for potential handling losses, aging losses, and matrix effects.[16, 19]
2. Experimental
2.1. Chemicals and Materials
Nitrobenzene (CAS# 98–95-3, 99% extra pure grade), methylene chloride (CAS# 75–09-2, HPLC grade), and hexanes (CAS# 110–54-3, HPLC grade) were obtained from Thermo Fisher Scientific, (Waltham, MA, USA). Deuterated internal standard nitrobenzene-d5 (ISTD, CAS# 4165–60-0, isotopic purity: 99.7% atom % D, 99.9% chemical purity) was obtained from Crescent Chemical Company (Islandia, NY, USA). Methanol (MeOH, CAS# 67–56-1, CHROMASOLV HPLC grade, >99.9%) and 1 L Tedlar PLV gas sampling bags with Thermogreen LB-2 septa were purchased from Sigma Aldrich (St. Louis, MO, USA).
Thirteen different popular American cigarette products were acquired through The Lab Depot, Inc. (Dawsonville, GA, USA) representing three major domestic cigarette manufacturers: Philip Morris (six products), R.J. Reynolds (six products), and Lorillard (one product). University of Kentucky 3R4F reference cigarettes (Lexington, KY, USA) and CORESTA Monitor #6 (CM6) test pieces were used as “quality control” (QC) materials.
Prior to smoking, received cigarette products were labeled and stored in a −20 °C freezer (maintained at or below −16 °C) within 10 days of receipt in their original packaging in accordance with International Organization for Standardization (ISO, Geneva, Switzerland) guidance document ISO 3402:1999. If opened, cigarette packs returned to storage were stored in sealed bags in a −20 °C freezer within 10 days of opening. Prior to sampling, cigarette samples and Cambridge filter pads were placed in the temperature- and humidity-controlled smoking chamber and conditioned at 22 ± 1 °C and 60 ± 3% relative humidity for at least 48 hours and no more than 10 days.
2.2. Instrumentation and Method Conditions
Cigarette smoking was conducted on Cerulean SM450 20-port smoking machines (Cerulean, Richmond, VA), which were located and operated inside a temperature- and humidity-controlled smoking chamber (Parameter Generation & Control Inc., Black Mountain, NC, USA). Cigarette filter holders (44 mm) were purchased from Cerulean (Molins PLC, Milton Keynes, UK) and fitted with 44 mm Cambridge filter pads (Borgwaldt, Hamburg, Germany). A soap bubble meter obtained from Borgwaldt (Hamburg, Germany) was used to verify smoking machine puff volume. The vapor phase portion of cigarette smoke was collected using 1 L Tedlar collection bags attached to the puff engine exhaust ports using 3.5-inch lengths of PVC tubing. Cigarettes were smoked under ISO conditions (at 22 ± 1 °C and 60 ± 3% relative humidity). Two cigarettes were smoked per sample according to a modified Health Canada Intense (HCI) regimen. The intense regimen prescribes a 55 mL puff volume with a 2 s puff duration every 30 s, with 100% filter ventilation blockage. A total of three clearing puffs were collected after all cigarette coals were extinguished at the end of the smoke collection. Quality control (3R4F and CM6) cigarettes were smoked in parallel with the cigarette samples during each smoking run. Quality control samples were accepted or rejected based on a modified set of Westgard rules.[20, 21] Note that for cigarettes requiring longer smoke times, 2 L Tedlar bags can be used instead of 1 L bags without significant changes in the recovery.
Volumetric and positive-displacement repeating pipettes were obtained from Eppendorf Corporation (Hauppauge, NY, USA). Bag shaking was carried out with the help of an Eberbach 6010 fixed speed, reciprocal shaker (Eberbach Corporation, Ann Arbor, MI, USA).
An Agilent 7890B GC system interfaced to an Agilent 7000C tandem mass spectrometer (Agilent Technologies, Santa Clara, CA, USA) and a Gerstel MPS autosampler rail (GERSTEL GmbH & Co. KG, Mülheim an der Ruhr, North Rhine-Westphalia, Germany) were used for analysis. The GC inlet was fitted with an Agilent ultra-inert universal gooseneck inlet liner with glass wool and maintained at 250 °C. An 11 psi injection and a 25:1 split ratio were employed in constant flow mode. The carrier gas was research grade helium (Airgas, Inc., Radnor, PA, USA). The column was a 30 m Agilent J&W HP-5MS Ultra Inert capillary column with a 250 μm I.D. and a 0.25 μm film thickness. During chromatography, the oven was held at 110 °C for 2 minutes, then ramped to 150 °C at a rate of 20 °C/min, then to 300 °C at a rate of 100 °C/min. Mass spectrometry was carried out using electron ionization and multiple reaction monitoring (MRM), with the source heated to 230 °C and both the MS1 and MS2 quadrupoles heated to 150 °C. Ultra-high purity grade nitrogen (Airgas) was used as the collision cell gas. Two MRM transitions were selected based on abundance, with the primary (quantitation) transition selected as the transition from the molecular ion to the most abundant fragment and the secondary (confirmation) transition selected as the transition to the second most abundant fragment. A third MRM transition was monitored for the internal standard. For all transitions, the MS1 and MS2 resolution was set to “wide” and a dwell time of 80 ms was employed. The transitions and collision energies employed are summarized in Table 1. Data acquisition and analysis were carried out using Agilent MassHunter Workstation software. Analyte concentrations were calculated from the ratio of the analyte peak area to the ISTD peak area.
Table 1.
Ion transitions and collision energies
| Compound | Transition Type |
Transition Ion Masses (m/z) |
Collision Energy (V) |
|---|---|---|---|
| Nitrobenzene | Quantitation | 123 → 77 | 15 |
| Confirmation | 123 → 51 | 13 | |
| Nitrobenzene-d5 (ISTD) | Quantitation | 128 → 82 | 38 |
2.3. Screening for Nitrobenzene in Tobacco Filler
Tobacco filler from thirteen commercial cigarette products, one reference cigarette product, and one monitor test piece product was analyzed for nitrobenzene content to determine whether any nitrobenzene is present in filler prior to smoking. Tobacco filler was collected from two cigarettes, combined in a vial, spiked with a hexane-based extraction solution containing 10 ng/mL internal standard, and placed on a Barnstead Lab-line E-class orbital shaker (Dubuque, IA) at 180 rpm for 15 minutes. Extracts were purified using normal phase silica SPE tubes (Strata SI-1 silica SPE cartridges, 500 mg/3 mL, Phenomenex, Torrance, CA). SPE tubes were washed with 100% methylene chloride (DCM) and then conditioned with 100% hexanes. Samples were loaded on the conditioned SPE cartridges, washed with 5% DCM in hexanes, dried for 5 minutes under vacuum, then eluted with 20% DCM in hexanes. Eluates were analyzed by GC-MS/MS.
2.4. Screening for Nitrobenzene in Mainstream Smoke Vapor Phase and TPM
Nitrobenzene levels were assessed separately in the mainstream smoke particulate matter and vapor phases for thirteen commercial cigarette products, one reference cigarette product obtained from the University of Kentucky, and one monitor test piece product obtained from CORESTA. The particulate matter smoke fraction generated by smoking two cigarettes was collected on a Cambridge filter pad placed inside a filter holder into which the filter end of the cigarette was inserted, and the corresponding vapor fraction of the cigarette smoke was collected using a 1 L Tedlar bag. Following smoking, pads were removed from their holders, folded and placed inside vials. Hexane-based extraction solution containing 10 ng/mL internal standard was added to vials containing pads, and vials were closed and placed on a Barnstead Lab-line E-class orbital shaker at 180 rpm for 15 minutes. Extraction solution was added to bags via the push-lock valve using a repeating pipettor. The bags were then placed on an Eberbach reciprocal shaker at 180 rpm for 15 minutes. Extracts from both bags and pads were subjected to the same SPE procedure described previously for filler samples (section 2.3). The samples were analyzed by GC-MS/MS and their relative nitrobenzene contents measured.
2.5. Simplified Extraction Method
SPE was found to be unnecessary for vapor phase analysis of nitrobenzene, as the vapor phase was substantially cleaner than either the filler or TPM fraction. The sample extraction procedure was therefore simplified to an “extract and shoot” method in order to optimize throughput and minimize sample cost and waste. The vapor phase “bag-only” method involved the same smoking process described previously, except the Cambridge filter pads were discarded after smoking and only the Tedlar bags were collected for extraction and analysis. Extraction was carried out by introducing 2 mL of a methanol-based extraction solution containing 10 ng/mL internal standard into the bag via the push-lock valve. Methanol was chosen over hexanes due to easier storage and handling, as well as better solvation properties. Extraction solution was introduced within 10 minutes after termination of the smoking run to encourage rapid and complete homogenization of the internal standard and to compensate for possible sample loss in the polymeric Tedlar matrix.[16, 22] Immediately following introduction of solvent, bags were briefly shaken by hand to distribute the solvent and internal standard evenly over the interior surface of the bag. The bags were then placed on a reciprocal shaker for 15 minutes to extract and homogenize the nitrobenzene and internal standard. This extraction time was chosen based on a comparison of nitrobenzene recoveries obtained from smoked cigarettes after different time periods spent on the shaker; maximal recovery was observed after 15 minutes.
2.6. Standard Preparation
Standards with concentrations ranging from 5 – 90 ng/mL were prepared gravimetrically from neat nitrobenzene in order to comfortably encompass the full range of nitrobenzene concentrations observed in a survey of a wide variety of cigarette products. Labeled internal standard was included in calibrators and extraction solution at a concentration of 10 ng/mL; stock solutions of internal standard were prepared gravimetrically from neat nitrobenzene-d5. Standards and extraction solution were prepared in volumetric flasks using methanol for this simplified extraction method. Immediately following preparation, standards were portioned individually into amber autosampler vials and extraction solution was distributed into amber vials fitted with PTFE-faced screw caps. Aliquots of standard and extraction solutions were stored at −20 °C until use. Accuracy of calibration standards was verified from analysis of certified reference materials (CRMs) produced by third party vendors operating under ISO Guide 34 accreditation. Comparison of standards prepared from CRMs with those prepared from neat chemicals demonstrated acceptable equivalency, with calculated nitrobenzene concentrations differing by 3% on average (n = 10 calibrators prepared from each sample set).[16]
3. Results and Discussion
3.1. Extraction Efficiency
Recoveries from the Tedlar bag matrix, filter pads, and tobacco filler were assessed individually by spiking each matrix with known amounts of nitrobenzene using a gastight syringe, then extracting each of the matrices with extraction solution containing internal standard following the standard procedure. Tedlar bags were inflated with air prior to spiking and contained no tobacco smoke, and filter pads were fresh and not subjected to smoking. Recoveries were evaluated relative to “blank” (empty) vials spiked with the same amounts of nitrobenzene. Recoveries were determined in triplicate for each matrix. Average calculated percent recoveries from bags, pads, and filler were 101(± 3.3)%, 97.4(± 1.8)%, and 104(± 2.7)%, respectively, indicating adequate extraction efficiency for all matrices.
3.2. Assessment of Nitrobenzene Content in Tobacco Filler and the Vapor and Particulate Phases of Mainstream Smoke: Optimization and Simplification of Sample Preparation Procedure
None of the filler extracts contained measurable concentrations of nitrobenzene. Nitrobenzene is described as a pyrosynthesis product generated by combustion of tobacco components (and therefore nitrobenzene should not be present).[3] However, given differences in tobacco curing and processing methods, it was prudent to assess whether or not nitrobenzene is present in filler. Since none of the filler extracts contained detectable amounts of nitrobenzene, analysis of filler samples was discontinued at this point in method development. In terms of the smoke fractions, for all products analyzed, nitrobenzene was detected only in the bag; filter pad samples contained undetectable quantities (<LOD; S/N <3). Accordingly, quantification of the pad content is unnecessary for nitrobenzene within the context of this method, and particulate matter analysis was discontinued at this point in method development due to the negligible contribution of the filter pad to the total delivery. It is notable that the particulate matter and vapor phases do not represent separate routes of exposure to the smoker; rather, they are somewhat subjective groupings, useful to the tobacco analyst for categorizing the tobacco smoke fractions as obtained using common smoke collection methods.[16] The chromatographic results of a comparison of bags, pads, and filler for a representative cigarette product are shown in Figure 1.
Fig. 1.
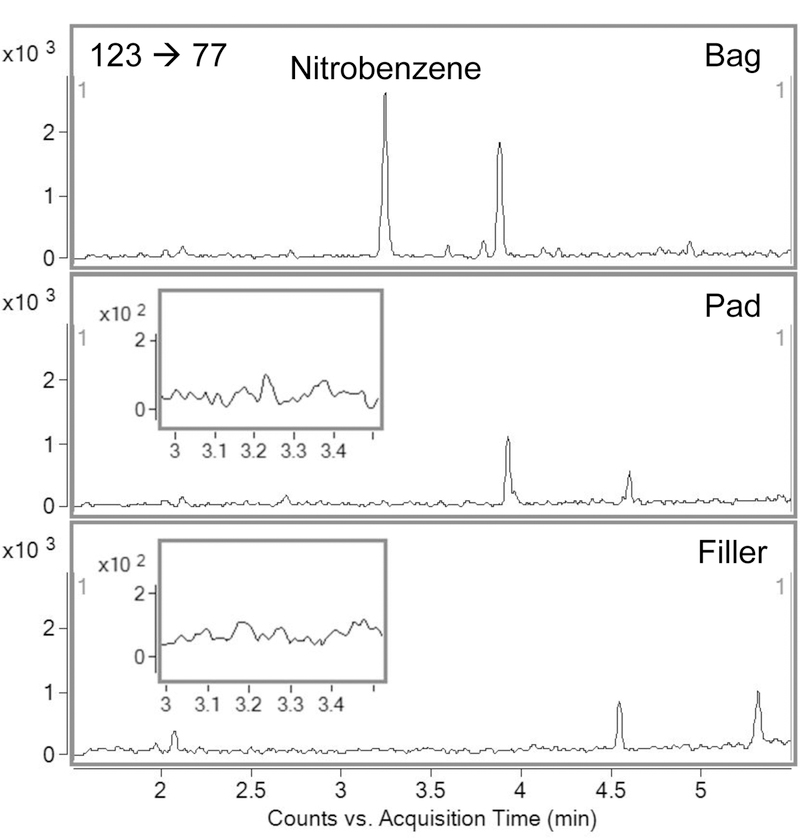
Representative chromatograms (quantitation transition, 123 77) showing relative nitrobenzene recoveries from vapor phase (bag), particulate matter (pad), and filler from a single cigarette product. Inset: magnified region for nitrobenzene quantitation transition in pad and filler illustrating lack of detectable signal.
3.3. Method Validation
Method validation was carried out to assure method quality and fitness. The parameters of dynamic range, linearity, detection limit, specificity, accuracy, precision, and ruggedness (including stability) were assessed. Matrix effects were also determined to ensure the use of solvent-based calibrators was appropriate.
3.3.1. Linear Dynamic Range
Dynamic range was assessed by serial dilutions of a standard nitrobenzene solution, for which the response (peak area ratio) was plotted as a function of concentration. The linear dynamic range was defined as the range of concentrations for which the response increased linearly with concentration. Signal increased linearly with concentration (R2 = 0.999) for nitrobenzene concentrations ranging from 1 – 100,000 ng/mL under the same sample preparation and detection conditions.
3.3.2. Limit of Detection and Linearity
The limit of detection (LOD) was estimated by evaluating the signal-to-noise (S/N) ratio for a low concentration (500 pg/mL) nitrobenzene standard injected into the instrument over the course of a five month period (n = 20). The limit of detection was extrapolated for an S/N value of 3 based on the mean S/N value of the 20 measurements.[23, 24] The limit of detection calculated in this manner was 418 pg/cig.
A calibration range was chosen such that the calibrator concentrations were greater than the limit of detection, fell within the linear dynamic range, and bracketed the range of nitrobenzene concentrations measured for a number of representative domestic products. A total of 10 calibrators were prepared in a concentration range of 5 – 90 ng/mL nitrobenzene (each containing 10 ng/mL ISTD). Analysis of the calibration plot indicated that a linear regression with 1/x weighting resulted in an optimal distribution of residuals.
3.3.3. Specificity
Specificity was demonstrated by baseline-resolved chromatograms of smoke matrix-based samples and further confirmed by the consistency of ion ratios between “blank” (solvent-based) and smoke matrix-based samples.
3.3.4. Accuracy
Accuracy was assessed by calculating percent recoveries for matrix spike samples, acquired in triplicate at high, medium, and low concentrations for a monitor test piece. The average calculated percent recoveries for the low, medium, and high spikes were 97.3%, 85.7%, and 92.1%, respectively; all values were acceptable as they fell within 20% of the known concentrations.
3.3.5. Precision
Intermediate precision was estimated by smoking and analyzing the QC materials (3R4F and CM6, n = 20 for each product, collected over the course of 20 separate smoking runs); this analysis resulted in relative standard deviations of 11.5% and 14.9%, respectively. Product variability was estimated by smoking and analyzing 13 cigarette products (n = 5 for each product) resulting in relative standard deviations ranging from 2.8 to 16.9%. The observed variability is attributable to a number of factors including the heterogeneous nature of the tobacco smoke matrix and pyrosynthetic origins of the analyte; precision was nonetheless acceptable as relative standard deviations were ≤20%.
3.3.6. Ruggedness Testing
Changes were made to five different parameters in order to test the ruggedness of the method. Testing of chromatographic settings revealed that increasing inlet temperature significantly increases nitrobenzene response and decreases internal standard response, while increasing pressure decreases both nitrobenzene and internal standard responses. Assessment of time before the addition of extraction solution to Tedlar bags revealed that as the amount of time before extraction increases, recovery significantly decreases; accordingly, smaller sample batches should be smoked to ensure quick introduction of extraction solution to all samples after termination of the smoking run. Assessment of total sample shaking (extraction) time also indicated that recoveries decrease slightly as shaking time increases. One possible explanation for this decrease is analyte adsorption into the Tedlar bag matrix, and possibly diffusion through it, as observed previously with other plastics.[6, 16, 22] An extraction time of 15 minutes was therefore selected to minimize loss of the analyte while promoting complete sample extraction and equilibration. Changes in extraction volume were evaluated by testing extraction volumes of 1, 2, 3, and 5 mL; as it was determined that the net amounts of nitrobenzene recovered using either 2, 3, or 5 mL were equivalent, a 2 mL extraction volume was selected to obtain the highest possible nitrobenzene signal. The 1 mL extraction was found to deviate significantly in apparent concentration from the others; one possible explanation is that this volume is insufficient for extraction of nitrobenzene from the sample.
3.3.7. Stability
Thermostability and photostability were assessed for three different standards (low, moderate, and high-concentration) and a smoke matrix sample under four different conditions: under bright lights at room temperature, in the dark at room temperature, in the dark at 4 °C, and in the dark at −20 °C. To avoid confounding issues resulting from simultaneous degradation of standard and internal standard, the concentrated internal standard “stock solutions” were kept separately at −70 °C under N2(g). On days of analysis, a single vial of internal standard and vials corresponding to each sample from each environment were equilibrated to room temperature. Then, each vial was spiked with internal standard solution and vortexed. Results following 100 days under the specified conditions were determined as a percentage of the original response for the sample. After 100 days at −20 °C, calibration and matrix-based samples exhibited less than a 10% change in apparent concentration, whereas under all other conditions, the changes in apparent concentrations were (on average) higher, with concomitant light exposure leading to the greatest average decreases in apparent concentration. Accordingly, samples were stored in the dark at −20 °C. Calibrators were stored for no longer than 50 days. Smoke samples were typically analyzed the day they were generated, but if storage was required, they were stored for no longer than one week.
3.3.8. Evaluation of Matrix Effects for Analyte Calibration
Measurement of matrix effects was required due to the unavailability of blank (nitrobenzene-free) smoke matrix. Matrix effects between vapor phase collected in Tedlar gas sampling bags and blank Tedlar bags were assessed by comparing the slopes of two sets of calibrators prepared in methanolic smoke vapor extract (matrix) solution and non-matrix (methanol) solution. Ten-point curves were constructed in smoke matrix and in methanol alone, equivalent to smoke samples and calibrators, respectively. Least squares slopes were calculated for 5 independent calibration curves, averaged for the matrix-based and non-matrix-based samples, and the averaged slopes were compared for both sample sets. Both matrix-based and non-matrix-based calibrators demonstrated acceptable linearity (R2 > 0.99) and matrix effects were minimal with an average difference of 0.87% between slopes. [25–27]
3.4. Analysis of Cigarette Products
Thirteen cigarette products representing three major cigarette manufacturers (R.J. Reynolds, Philip Morris, and Lorillard), one reference cigarette product obtained from the University of Kentucky, and one monitor test piece product obtained from CORESTA were analyzed for nitrobenzene content (n = 5). The results of this analysis are provided in Table 2.
Table 2.
Nitrobenzene results (ng/cig) for cigarette products analyzed
| Brand No. |
Average (ng/cig) |
Standard Deviation (ng/cig) |
%RSD | Manufacturer |
|---|---|---|---|---|
| 1 | 31 | 2.9 | 9.4 | Lorillard |
| 2 | 31 | 2.4 | 7.7 | Philip Morris |
| 3 | 33 | 4.1 | 12 | Philip Morris |
| 4 | 32 | 0.9 | 2.8 | Philip Morris |
| 5 | 28 | 1.4 | 5.0 | Philip Morris |
| 6 | 35 | 5.9 | 16.9 | Philip Morris |
| 7 | 38 | 3.2 | 8.4 | Philip Morris |
| 8 | 31 | 1.4 | 4.5 | R. J. Reynolds |
| 9 | 36 | 3.5 | 9.7 | R. J. Reynolds |
| 10 | 27 | 1.8 | 6.7 | R. J. Reynolds |
| 11 | 29 | 3.2 | 11.0 | R. J. Reynolds |
| 12 | 29 | 2.1 | 7.2 | R.J. Reynolds |
| 13 | 18 | 2.9 | 16.1 | R.J. Reynolds |
| 14 | 20 | 2.1 | 10.5 | CORESTA |
| 15 | 37 | 2.3 | 6.2 | University of Kentucky |
SP denotes soft pack; all other brands were hard packs.
Average nitrobenzene concentrations ranged from 18 to 38 ng/cig, with lower concentrations detected for the two Virginia blend cigarette products (products 13 and 14) than for the other products, which fall under the American and Turkish blend categories. American blend cigarettes are made of a blend of Burley, Virginia, and Oriental tobacco types, and Turkish blend cigarettes are made of a blend of Burley, Oriental, and Turkish tobacco, whereas Virginia blend cigarettes only contain flue-cured Virginia tobacco. Burley tobacco is often substantially higher in nitrate content than either Virginia or Oriental tobaccos, which may account for the observed difference.[28] This observation further indicates a probable positive correlation between nitrobenzene and tobacco-specific nitrosamine content and a probable negative correlation between nitrobenzene and polycyclic aromatic hydrocarbon content, which warrants further study.[3, 29, 30] Percent relative standard deviations (%RSD) ranging from 2.8 to 16.9% may be attributed to the intrinsic heterogeneity of the tobacco matrix, pyrosynthetic origins of the volatile analyte, variations in smoke sample collection, and variability in sample handling. Published nitrobenzene concentrations in the vapor phase of mainstream smoke were either unavailable by product or were declared to be below the limit of detection, precluding a direct correlation with literature values. Results from the Hoffmann and Rathkamp study indicated an average nitrobenzene yield of 25.3 ± 0.95 ng/cig from the combined vapor phase and TPM of an (unspecified) U.S. blended cigarette product, which falls within the range of values observed in this study. Results from Xie et al. indicated a yield of 2.22 ng/cig nitrobenzene in the particulate matter for 3R4F cigarettes. This concentration is above the limit of detection (but below the lowest calibrator) for our group’s method, but nitrobenzene was not observed in the particulate matter of this cigarette product when analyzed by our group. However, in the Xie manuscript, a nonselective detector was applied, no matrix effects study was mentioned despite the use of solvent-based calibrators and a structurally distinct internal standard, and co-elution with interferents cannot be ruled out. Additionally, our group smoked only two cigarettes per sample under HCI conditions, whereas the Xie group smoked 20 cigarettes per sample under HCI conditions for its analysis; it has been demonstrated in the past that the retention characteristics of Cambridge filter pads can change when increasing numbers of cigarettes are smoked through them and the TPM load is increased.[31]
4. Conclusions
The present method fills the need for a modern technique for determining nitrobenzene in mainstream cigarette smoke. The method provides for rapid high-throughput sample collection, generates minimal waste, and has demonstrated accuracy and precision fit for the purpose of quantifying levels in mainstream tobacco smoke. Throughput and instrumental limits of detection are substantially improved relative to previous methods, and analysis of the vapor phase ensures that measured nitrobenzene content is more reflective of actual exposures. Although machine-based smoke analysis cannot account for variations in individual smoking behavior, this method has been validated to provide a means for comparing tobacco products in a precise and reproducible manner.
Acknowledgements
This research was funded by the U.S. Food and Drug Administration’s Center for Tobacco Products. This project was supported in part by an appointment to the Research Participation Program for the Centers for Disease Control and Prevention, National Center for Environmental Health, Division of Laboratory Sciences (DLS), administered by the Oak Ridge Institute for Science and Education through an agreement between the U.S. Department of Energy and DLS.
Footnotes
Disclaimer
The cigarette products analyzed were based on a one-time purchase from a single product lot and do not necessarily reflect the lot-to-lot variability of nitrobenzene concentrations in these products over time. The findings and conclusions in this study are those of the authors and do not necessarily represent the official position of the U.S. Centers for Disease Control and Prevention. Use of trade names and commercial sources is for identification only and does not constitute endorsement by the U.S. Department of Health and Human Services or the U.S. Centers for Disease Control and Prevention.
Conflicts of Interest: None
References
- [1].Smoking Facts: What’s in a Cigarette? http://www.lung.org/stop-smoking/smoking-facts/whats-in-a-cigarette.html (accessed Feb 03, 2016).
- [2].“Harmful and Potentially Harmful Constituents in Tobacco Products and Tobacco Smoke; Established List,” 77 Federal Register 64 (3 April 2012), pp 20034–20037. [Google Scholar]
- [3].Hoffmann D; Rathkamp G, Quantitative determination of nitrobenzenes in cigarette smoke. Anal. Chem 1970, 42 (13), 1643–1647. [DOI] [PubMed] [Google Scholar]
- [4].Rodgman A; Perfetti T, Chemical Components of Tobacco and Tobacco Smoke CRC Press: Boca Raton, 2009. [Google Scholar]
- [5].Hoffmann D; Hoffmann I; El-Bayoumy K, The Less Harmful Cigarette: A Controversial Issue. A Tribute to Ernst L. Wynder. Chem. Res. Toxicol 2001, 14 (7), 767–790. [DOI] [PubMed] [Google Scholar]
- [6].Oldham MJ; DeSoi DJ; Rimmer LT; Wagner KA; Morton MJ, Insights from analysis for harmful and potentially harmful constituents (HPHCs) in tobacco products. Regul. Toxicol. Pharmacol 2014, 70 (1), 138–148. [DOI] [PubMed] [Google Scholar]
- [7].Holder JW, Nitrobenzene carcinogenicity in animals and human hazard evaluation. Toxicol. Ind. Health 1999, 15 (5), 445–457. [PubMed] [Google Scholar]
- [8].Holder JW, Nitrobenzene potential human cancer risk based on animal studies. Toxicol. Ind. Health 1999, 15 (5), 458–463. [PubMed] [Google Scholar]
- [9].Xie F; Shang J; Guo J; Ge Z; Zhang S, Determination of Seven Nitrobenzene Compounds in Mainstream Cigarette Smoke with Heart-Cutting Two-Dimensional Gas Chromatography. J. Chromatogr. Sci 2012, 50 (5), 387–392. [DOI] [PubMed] [Google Scholar]
- [10].Ikeda M; Kita A, Excretion of p-Nitrophenol and p-Aminophenol in the Urine of a Patient Exposed to Nitrobenzene Br. J. Ind. Med 1964, 21, 210–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Robinson D; Smith JN; Williams RT, Studies in detoxication. 40. The metabolism of nitrobenzene in the rabbit. o-, m- and p-Nitrophenols, o-, m- and p-aminophenols and 4-nitrocatechol as metabolites of nitrobenzene. Biochem. J 1951, 50 (2), 228–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Parke DV, Detoxication LXVIII. The metabolism of C14-nitrobenzene in the rabbit and guinea pig. Biochem. J 1956, 62, 339–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Salmowa J; Piotrowski J; Neuhorn U, Evaluation of Exposure to Nitrobenzene: Absorption of Nitrobenzene Vapour through Lungs and Excretion of p-Nitrophenol in Urine. Br. J. Ind. Med 1963, 20 (1), 41–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].NTP/NIH, Report on Carcinogens 12th ed.; DIANE Publishing: Darby, 2011. [Google Scholar]
- [15].IARC, Nitrobenzene. In Printing Processes and Printing Inks, Carbon Black and Some Nitro Compounds, IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, International Agency for Research on Cancer: Lyon, 1996; Vol. 65. [PMC free article] [PubMed] [Google Scholar]
- [16].Sampson MM; Chambers DM; Pazo DY; Moliere F; Blount BC; Watson CH, Simultaneous analysis of 22 volatile organic compounds in cigarette smoke using gas sampling bags for high-throughput solid-phase microextraction. Anal. Chem 2014, 86 (14), 7088–7095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hoffmann D; Rathkamp G; Nesnow S, Quantitative Determination of 9-Methylcarbazoles in Cigarette Smoke. Anal. Chem 1969, 41 (10), 1256–1259. [DOI] [PubMed] [Google Scholar]
- [18].Boeker P; Leppert J; Mysliwietz B; Lammers PS, Comprehensive Theory of the Deans’ Switch As a Variable Flow Splitter: Fluid Mechanics, Mass Balance, and System Behavior. Anal. Chem 2013, 85 (19), 9021–9030. [DOI] [PubMed] [Google Scholar]
- [19].Lanckmans K; Sarre S; Smolders I; Michotte Y, Use of a structural analogue versus a stable isotope labeled internal standard for the quantification of angiotensin IV in rat brain dialysates using nano-liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom 2007, 21 (7), 1187–1195. [DOI] [PubMed] [Google Scholar]
- [20].Caudill SP; Schleicher RL; Pirkle JL, Multi-rule quality control for the age-related eye disease study. Stat. Med 2008, 27 (20), 4094–4106. [DOI] [PubMed] [Google Scholar]
- [21].Westgard JO; Barry PL; Hunt MR; Groth T, A multi-rule Shewhart chart for quality control in clinical chemistry. Clin. Chem 1981, 27 (3), 493–501. [PubMed] [Google Scholar]
- [22].Blount BC; McElprang DO; Chambers DM; Waterhouse MG; Squibb KS; Lakind JS, Methodology for collecting, storing, and analyzing human milk for volatile organic compounds. J. Environ. Monit 2010, 12 (6), 1265–1273. [DOI] [PubMed] [Google Scholar]
- [23].2.2.46 Chromatographic Separation Techniques. In European Pharmacopoeia 5th ed.; Council of Europe: Strasbourg, France, 2005. [Google Scholar]
- [24].International Conference on Harmonization of Technical Requirements for the Registration of Pharmaceuticals for Human Use, Validation of Analytical Procedures: Text and Methodology Q2(R1) ICH: Geneva, Switzerland, 1996. [Google Scholar]
- [25].Thompson M, Standard additions: myth and reality. AMCTB No. 37 2009, 1–2. [DOI] [PubMed] [Google Scholar]
- [26].Ellison SLR; Thompson M, Standard additions: myth and reality. Analyst 2008, 133 (8), 992–997. [DOI] [PubMed] [Google Scholar]
- [27].Guidi LR; Tette PAS; Evangelista WP; Fernandes C; Glória MBA, Matrix effect on the analysis of amphenicols in fish by liquid chromatography-tandem mass spectrometry (LC-MS/MS). Journal of Physics: Conference Series 2015, 575 (1), 012036. [Google Scholar]
- [28].Fischer S; Spiegelhalder B; Preussmann R, Preformed tobacco-specific nitrosamines in tobacco—role of nitrate and influence of tobacco type. Carcinogenesis 1989, 10 (8), 1511–1517. [DOI] [PubMed] [Google Scholar]
- [29].Ding YS; Zhang L; Jain RB; Jain N; Wang RY; Ashley DL; Watson CH, Levels of Tobacco-Specific Nitrosamines and Polycyclic Aromatic Hydrocarbons in Mainstream Smoke from Different Tobacco Varieties. Cancer Epidemiology Biomarkers & Prevention 2008, 17 (12), 3366–3371. [DOI] [PubMed] [Google Scholar]
- [30].Rathkamp GH, D., Chemical Studies on Tobacco Smoke XIII. Inhibition of the Pyrosyntheses of Several Selective Smoke Constituents. Beit. Tabakforsch 1970, 5 (6), 302–306. [Google Scholar]
- [31].Cockayne L; Drake L; McAdam KG; McAughey J A review of the capability of the 44 mm Cambridge Filter Pad for trapping total particulate matter in excess of 150 mg, Proceedings of the CORESTA Smoke Science and Product Technology Meeting Sapporo, 23rd-27th September 2012; British American Tobacco: 2012. [Google Scholar]