

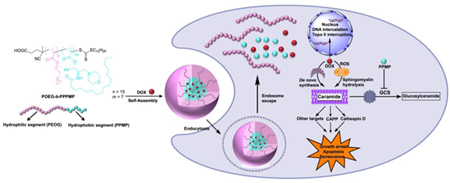
Abstract
In order to improve the efficacy of chemotherapy for cancers, we have developed a novel prodrug micellar formulation to co-deliver ceramide-generating anticancer agents and ceramide degradation inhibitor (PPMP). The prodrug nanocarrier is based on a well-defined POEG-b-PPPMP diblock copolymer. The hydrophilic block of POEG-b-PPPMP is POEG, and the hydrophobic block is composed of a number of PPMP units, which could work synergistically with loaded anticancer drugs. POEG-b-PPPMP was readily synthesized via a one-step reversible addition-fragment transfer (RAFT) polymerization from a PPMP monomer. The newly synthesized polymers were self-assembled into micelles and served as a carrier for several hydrophobic anticancer drugs including DOX, PTX and C6-ceramide.POEG-b-PPPMP prodrug polymer exhibited intrinsic antitumor activity in vitro and in vivo. In addition, POEG-b-PPPMP prodrug polymer was comparable to free PPMP in selectively enhancing the production of pro-apoptotic ceramide species as well as down-regulating the mRNA expression of GCS. DOX-loaded POEG-b-PPPMP micelles exhibited an excellent stability of 42 days at 4 °C. Moreover, DOX loaded in POEG-b-PPPMP micelles showed higher levels of cytotoxicity than DOX loaded in a pharmacologically inert polymer (POEGb-POM) and Doxil formulation in several tumor cell lines. Consistently, in a 4T1.2 murine breast cancer model, the tumor inhibition followed the order of DOX/POEG-b-PPPMP > DOX/POEG-b-POM ≥ Doxil > POEG-b-PPPMP> POEG-b-POM. Our data suggest that POEG-b-PPPMP micelles are a promising dual-functional carrier that warrants more studies in the future.
Keywords: PPMP, Ceramide, Doxorubicin, Co-delivery, Prodrug micelles
Graphical Abstract
1. Introduction
Ceramides are not only essential structural components of cell membranes regulating fluidity and subdomain stucture of lipid bilayer, but also have crucial and specific links to various aspects of cancer initiation, progression and response to chemotherapies[1, 2]. As tumor suppressor lipids, ceramides have important messenger functions mediating cell differentiation, cell cycle arrest, senescence and apoptosis[3]. Mounting evidence shows that ceramides are intimately involved in those pathways by regulating specific protein targets such as phosphatases and kinases[4]. Ceramides activate the ceramide-activated protein phosphatases (CAPPs), which comprise the serine/threonine protein phosphatases PP1 and PP2A. PP1 gives rise to the dephosphorylation of SR proteins that mediate the alternative splicing of BCL-X, while PP2A dephosphorylates and inactivates anti-apoptotic proteins such as BCL2 and AKT[5, 6]. Moreover, cathepsin D is activated by ceramides, leading to activation of the pro-apoptotic protein BID and the subsequent activation of caspase-9 and caspase-3[7]. Ceramides also activate the kinase suppressor of RAS (KSR)[8]. Proteins modulated by these pathways include telomerase, c-MYC, caspases and cyclin-dependent kinases (CDKs)[1]. All of these downstream effects can lead to changes in growth arrest, apoptosis and/or senescence. Therefore, manipulating the generation and/or accumulation of the ceramides could disarm a key mechanism for tumour viability and growth. Several ceramide mimetics and delivery systems have been developed to increase the solubility, specificity and efficacy of ceramides[9]. For example, analogues and mimics of ceramides such as C2- and C6-ceramides, which are similar to the natural metabolites of ceramides, are capable of direct activation of ceramide targets, inducing cell death in a myriad of cancer types[10]. B13, another ceramide analogue, inhibits acid CDase, inducing accumulation of ceramide and increasing apoptosis in an in vivo model of metastatic human colon cancer and in a prostate cancer xenograft model[11]. In addition, several delivery systems have been empolyed for systemic delivery of ceramides including liposomes[12], calcium phosphate nanocomposite particles (CPNPs)[13], linear dentritic nanoparticles[14], nanoemulsions[15] and others. For instance, delivery of ceramides via pegylated liposomes, which are generally more effective at crossing the cell membrane, increase accumulation of ceramides and their ability to kill cancer cells[16].
Recently it has been shown that ceramide metalolism can also be modulated by chemotherapy or radiotherapy[17]. Despite the differences in their chemical structures and the postulated major mechanisms of action, many chemotherapeutic agents are all capable of inducing ceramide generation through enhanced de novo synthesis, catabolism of sphingomyelin, or both, suggesting a role of ceramide metabolism in the overall antitumor activity of these agents [18]. Doxorubicin, etoposide, camptothecin, 4-HPR, fludarabine, cisplatin, gemcitabine, irinotican, vorinostat or radiation induces de novo ceramide genration[19, 20]. A-SMase has been implicated in mediating apoptosis induced by paclitaxel (PTX), 5-FU, daunorubicin and radiation[17]. Cytosine arabinoside (Ara-C) triggers ceramide generation through the activation of N-SMase[9]. In each case, the result is an enhanced ceramide-governed cytotoxic response.
However, the accumulation of ceramides in tumor cells could simultaneously initiate and/or up-regulate the catabolic pathways towards themselves. Glucosylceramide synthase (GCS) is one of the most pivotal metabolic enzymes to clear ceramides. GCS catalyzes ceramide glycosylation, transferring a glucose residue from UDP-glucose to ceramide for the synthesis of glucosylceramide; this process facilitates ceramide clearance and limits ceramide-induced apoptosis[21–23]. Emerging evidence shows that ceramides increase GCS enzyme activity and GCS mRNA level[24, 25]. More importantly, chemotherapeutic agents, such as DOX, which increase endogenous ceramide levels, have been demonstrated to activate GCS promoter and induce GCS gene expression[26]. Therefore, inhibition of GCS would diminish the ablility of tumor cells to detoxify ceramide and restore the sensitivity of tumor cells to anticancer drugs. PPMP (1-phenyl-2-palmitoylamino-3-morpholino-1- propanol) is a potent inhibitor of GCS and can help sustain a high level of ceramide inside tumor cells through inhibiting its conversion to bio-inactive glycosylated ceramide[27]. PPMP also inhibits l-O-acylceramide synthase, another minor ceramide metabolizing enzyme[28]. Like other inhibitors of GCS, PPMP can both induce cell death by itself and synergize with classical chemotherapeutic agents. The combination of PPMP with anticancer drugs such as PTX[29], 4-HPR[30], irinotican[29] has been reported to cause increased ceramide levels and cytotoxicity on tumor cells, such as those derived from neuroblastoma, melanoma, prostate, lung, colon, breast and pancreatic cancers.
One of the issues for the clinical translation of the combination therapy is lack of an effective strategy for selective codelivery of PPMP and the chemotherapeutic agents.
In this study, we developed a PPMP prodrug-based polymeric nanocarrier (POEG-b-PPPMP) to overcome this limitation. POEG-b-PPPMP could serve as a depot system allowing the release of active PPMP over a prolonged period of time. More importantly, POEG-b-PPPMP could self-assemble to form micelles to achieve synergistic codelivery with other anticancer drugs including DOX, PTX and C6-ceramide. We systematically evaluated the sizes, structures and drug loading efficiency of POEG-b-PPPMP-based nanocarrier. The antitumor activity of drug-free POEG-b-PPPMP and DOX-loaded POEG-b-PPPMP was also investigated in vitro and in vivo.
2. Experimental Section
2.1. Materials
PPMP·HCl was synthesized and purified following a published patent (WO 2005/049827A2). Methacryl chloride,triethylamine,4-Cyano-4-[(dodecylsulfanylthiocarbonyl)sulfanyl] pentanoic acid, oligo(ethylene glycol) methacrylate OEGMA (average Mn = 500), 2,2-Azobis (isobutyronitrile)(AIBN), Dulbecco’s Modified Eagle’s Medium (DMEM), trypsin-EDTA solution, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) were purchased from Sigma-Aldrich (MO, U.S.A).DOX·HCl was purchased from LC Laboratories (MA, U.S.A). PTX was purchased from AK Scientific Inc. (CA, U.S.A.). Fetal bovine serum (FBS), penicillin-streptomycin solution and TRIzol lysis reagent were purchased from Invitrogen (NY, U.S.A.). QuantiTect Reverse Transcription Kit was purchased from Qiagen (MD, U.S.A).
2.2. Methods
2.2.1. Synthesis of PPMP-monomer
Methacryloyl chloride (209 uL, 2 mmol) was added to the solution of PPMP·HCl salt (511 mg, 1 mmol) in 5 mL DCM, followed by triethylamine (689 uL, 5mmol) at 0 °C. The reaction mixture was stirred at 0 °C for 1 h and then kept at room temperature overnight. The completion of reaction was monitored by TLC (Petroleum ether/EtOAc = 9:1). The reaction mixture was quenched by the addition of saturated NaHCO3, and the product was extracted with DCM (3×15 mL). The organic phase was collected and washed by water and brine, dried over Na2SO4, filtered, and the solvent was then evaporated. The crude product was purified by column chromatography on silica gel eluted with petroleum ether/EtOAc (12: 1) to give the oil product PPMP monomer (450 mg, 90%).
2.2.2. Synthesis of POEG macroCTA
POEG macroCTA was synthesized and purified following a published protocol [31].
2.2.3. Synthesis of POEG-b-PPPMP
POEG macroCTA (390 mg, 0.052 mmol), PPMP-monomer (225 mg, 0.415 mmol), AIBN (4.5 mg, 0.027 mmol), and 2 mL dried 1, 4-Dioxane and THF (V/V=1/1) were added in a Schlenk tube and deoxygenated by free-pump-thawing for three times. Under N2 protection, the mixture was kept in an oil bath thermostated at 90°C for 24 h, and then quenched by cooling the tube in liquid nitrogen. The mixture was precipitated in hexane for 3 times and dried in vacuum.
2.2.4. Preparation and Characterization of Blank or Drug-loaded POEG-b-PPPMP micelles
DOX solution was first prepared by dissolving DOX·HCl in a mixture of chloroform/methanol (1:1, v/v) containing triethylamine (5 equiv) to remove HCl. Then DOX (5mg/mL in chloroform/methanol) was mixed with POEG-b-PPPMP polymers (50 mg/mL in chloroform) at different carrier/drug ratios. The solvent was removed by nitrogen flow to produce a thin film of carrier/drug mixture, which was further dried in vacuum for 2 h to remove any remaining solvent. Then the thin film was hydrated and gently vortexed in Dulbecco’s phosphate-buffered saline (DPBS) to form DOX-loaded micelles. PTX-loaded, C6-ceramide-loaded POEG-b-PPPMP micelles and drug-free micelles were prepared similarly as described above.
The average diameter and the size distribution of POEG-b-PPPMP micelles were assessed via a Zetasizer (DLS). The morphology of POEG-b-PPPMP blank micelles and drug-loaded micelles was observed by transmission electron microscopy (TEM). The drug loading efficiency of DOX and PTX was examined by Waters Alliance 2695 Separations Module combined with Waters 2475 Fluorescence Detector (excitation, 490 nm; emission, 590 nm; gain, 3; sensitivity (FUFS), 10,000) and high performance liquid chromatography (HPLC) respectively as described previously [32]. Drug loading capacity (DLC) and drug loading efficiency (DLE) were calculated according to the following equation: DLC %= [weight of drug loaded/(weight of polymer used + weight of drug used)]×100%, DLE %= (weight of loaded drug/weight of input drug)×100 %. The colloidal stability of drug-loaded micelles with various carrier/drug molar ratios at room temperature and 4°C was evaluated by following the changes in sizes of the particles or visible precipitates every hour in the first 12 h and daily after 12 h following sample preparation.
The CMC of POEG-b-PPPMP micelles was determined by using nile red as a fluorescence probe as described previously[33].
The kinetics of DOX release from DOX/ POEG-b-PPPMP was performed according to a previous report[34].
2.2.5. In Vitro Cytotoxicity Assay
Cytotoxicity assay was performed on 4T1.2 mouse breast cancer cells, MCF-7 human breast cancer cells and PC-3 human prostate cancer cells. 4T1.2 (1×103 cells/well), MCF-7 (5×103 cells/well) or PC-3 (3×103 cells/well) cells were seeded in 96-well plates followed by 24 h of incubation in DMEM with 10% FBS and 1% streptomycin/penicillin.
To evaluate the combinational effect of PPMP with other anticancer drugs, cells were treated with various concentrations of free PPMP, free DOX·HCl, and the combination of both respectively for 48 h. The combination of PPMP and PTX or C6-ceramide was evaluated similarly. To measure the cytotoxicity of blank POEG-b-PPPMP micelles, cells were challenged with various concentrations of blank POEG-b-PPPMP micelles or free PPMP at equivalent PPMP concentrations for 48 h. To explore the cytotoxicity of drug-loaded POEG-b-PPPMP micelles, a pharmacologically inert polymer, POEG-b-POM, was served as a control carrier for anticancer drugs. Six groups were tested in this experiment, including free anticancer drug, commercial formulation of anticancer drug, drug-loaded POEG-b-PPPMP micelles, drug-loaded POEG-b-POM micelles, drug-free POEG-b-PPPMP micelles and drugfree POEG-b-POM micelles. The former four drug-containing groups were compared at the same concentration of anticancer drugs. The concentrations of last two drug-free micelles were same as those of the carrier in drug-loaded groups respectively. Specifically, for testing of DOX-loaded micelles, tumor cells were challenged with various concentrations of free DOX·HCl, Doxil, POEG-b-PPPMP/DOX micelles, POEG-b-POM/DOX micelles, POEG-b-PPPMP micelles and POEG-b-POM micelles. For testing of PTX-loaded micelles, cells were incubated with PTX, Taxol, POEG-b-PPPMP/PTX micelles, POEG-b-POM/PTX micelles,POEG-b-PPPMP micelles and POEG-b-POM micelles for 48 h.
MTT assay and the calculation of cell viability were performed as described before[34]. The anti-proliferation data for single drug and combination treatment was fitted to an inhibitory, normalized dose-response model with variable slope (Y = 100/(1 + 10^((LogEC50-X) *HillSlope); (GraphPad Prism, San Diego, CA)
2.2.6. Real-time PCR
Real-time PCR studies were performed on 4T1.2 mouse breast cancer cells and MCF-7 human breast cancer cells. 4T1.2 (2×104 cells/well) or MCF-7 (6×104 cells/well) cells were seeded in 6-well plates followed by 24 h of incubation in DMEM containing 10% FBS and 1% streptomycin/penicillin. After 24 hours, medium was replaced with 2% FBS medium containing free PPMP, blank POEG-b-PPPMP, DOX●HCl, free PPMP + free DOX or POEG-b-PPPMP/DOX. Free PPMP, blank POEG-b-PPPMP and POEG-b-PPPMP/DOX were examined at the same concentration of PPMP (1.7 µM), while free DOX and POEG-b-PPPMP/DOX were compared at the same concentration of DOX (100 nM). After 48 hours, total cellular RNA was extracted using the TRIzol lysis reagent. cDNA was generated from the purified RNA using QuantiTect Reverse Transcription Kit according to the manufacturer’s instructions. The cDNAs corresponding to GCS were amplified by PCR using the specific primers (mouse GCS forward 5’-CCAGGAGGGAATGGCCTTGT-3’, mouse GCS reverse 5’-AGAGACACCAGGGAGCTTGC-3’; human GCS forward 5’- CCACCCTGTCCTCCTCCTG-3’, human GCS reverse 5’-GAAGACGGCCATTCCCTCCA-3’). Quantitative real-time PCR was performed using SYBR Green Mix on a 7900HT Fast Real-time PCR System. Relative target mRNA levels were analyzed using delta-delta-Ct calculations and normalized to GAPDH.
2.2.7. Analysis of lipids
Ceramides measurements were also carried out on 4T1.2 cells or MCF-7 cells. All cells were similarly treated as described above. After 48 h treatment, cells were washed twice with ice-cold D-PBS and lysed in 60 µL 0.2% sodium dodecyl sulfate (SDS). An aliquot (30 µL) of the cell lysates was taken for protein determination. For another aliquot (30 µL) of the cell lysates, 0.5 µg of C6-ceramide was added as the internal standard. Lipids were extracted using 400 µL of chloroform/methanol (3:1, v/v). After gentle vortex and centrifugation at 3,000 rpm at RT for 10 min, the resulting organic lower phase was collected and evaporated under a stream of nitrogen. Lipids were resuspended in 200 µL ACN/IPA/H2O (1:1:1, v/v/v) and centrifuged at 12,500 rpm for 10 min at 4 °C and 150 µL of the clear supernatant was collected in UPLC injection vials. Sphingolipids were separated on an Agilent 1200 high performance liquid chromatography system (Agilent Technologies, CA, U.S.A) and analyzed by electrospray ionization-tandem mass spectrometry on a 400 QTRAP (AB Sciex, MA,U.S.A). The peak areas for different sphingolipid subspecies (C16-ceramide, C18-ceramide, C20-ceramide, C22-ceramide and C24-ceramide) were quantified by internal standards (C6-ceramide) and then normalized to protein concentrations.
2.2.8. Plasma Pharmacokinetics and Tissue Distribution
For plasma pharmacokinetics, POEG-b-PPPMP/DOX micelles, POEG-b-POM/DOX and DOX·HCl were injected intravenously into groups of 5 tumor-free female CD1 mice at a DOX dose of 5 mg/kg, respectively. Blood samples were collected in heparinized tubes at designated time points (3min, 10 min, 30 min, 1 h, 2 h, 4 h, 8 h and 12 h) post injection. The samples were centrifuged at 12,500 rpm for 10 min at 4 °C and 20 µL of the supernatant was collected. Then 200 μL acetonitrile was added and the samples were vortexed for 1 min. The samples were centrifuged at 12,500 rpm for 10 min at 4 °C and 150 μL of the clear supernatant was collected and injected into HPLC for DOX analysis. Noncompartmental pharmacokinetic analysis was executed by WinNonlin.
For tissue distribution study, POEG-b-PPPMP/DOX micelles, POEG-b-POM/DOX and DOX·HCl were i.v. injected into female BALB/c mice bearing 4T1.2 tumors of 400–600 mm3 (n = 5) at a DOX dose of 5 mg/kg, respectively. Mice were sacrificed 24 h post injection. Major organs (including heart, liver, spleen, lung and kidney) and tumors were harvested, weighed, and stored at −80°C. One hundred mg of each organ was weighed and homogenized in 500 μL PBS on ice. Five hundred μL methanol/acetonitrile (1/1, v/v) was then added and vortexed for 3 min. The samples were centrifuged at 12,500 rpm for 10 min at 4 °C and the supernatant was transferred to clean tubes and dried under nitrogen flow. Two-hundred μL acetonitrile was added and vortexed to dissolve samples, which were then centrifuged at 12,500 rpm for 10 min at 4 °C. One hundred and fifty μL of the clear supernatant was collected and injected into HPLC for DOX analysis.
2.2.9. In Vivo Therapeutic Study
The in vivo antitumor efficacy of the DOX-loaded POEG-b-PPPMP micelles was tested in a syngeneic 4T1.2 mouse breast cancer model. 4T1.2 cells (2×105 in 20 μL DPBS) were inoculated s.c. at the right mammary fat pad of female BALB/c mice. When the tumor volume reached ~ 50 mm3, mice were randomly divided into six groups (n=5), and treated via tail vein injection with DPBS, blank POEG-b-PPPMP micelles, blank POEG-b-POM micelles, DOX-loaded POEG-b-PPPMP micelles, DOX-loaded POEG-b-POM or Doxil, respectively, at a DOX dose of 5 mg/kg. The treatments were conducted every three days for a total of 3 times. Tumor sizes were measured with the digital caliper every three days following the initiation of the treatment and calculated by the formula: (L×W2)/2, where L is the longest and W is the shortest in tumor diameters (mm). Body weights were also monitored for the indication of toxicity. On 23 days post injection, all mice were sacrificed and tumor tissues were collected for weight, photography and H&E staining.
2.2.10. Histochemical staining
The H&E staining was similarly conducted as described before[35].
2.2.10. Statistical analysis
In vitro or in vivo data are presented as mean ± standard deviation (SD) or mean± standard error of mean (SEM), respectively. Two-tailed Student’s T test or analysis of variance(ANOVA) was used to compare two groups or multiple groups, respectively. Significance was determined with Tukey simultaneous post hoc test. In all statistical analyses, P < 0.05 is considered statistically significant.
3. Results
3.1. Effect of Anticancer Drugs on GCS mRNA Expression
To evaluate the effect of DOX, PTX, or C6-ceramide on GCS expression, we treated MCF-7 cells with various concentrations of DOX, PTX and C6-ceramide, respectively. The expression of GCS mRNA was examined by RT-qPCR 48 h following the drug treatment. As shown in Fig. 1A, treatment of MCF-7 cells with either DOX or C6-ceramide led to significant increases in the mRNA expression levels of GCS in a dose-dependent manner. At a DOX concentration of 200 nM and a C6-ceramide concentration of 2 μM, the GCS mRNA expression levels in MCF-7 cells were increased by 4.2 and 6.5 fold respectively. Induction of GCS mRNA expression by DOX or C6-ceramide was similarly observed in 4T1.2 cells (Fig. 1B). However, induction of GCS expression by DOX or C6-ceramide was less dramatic in 4T1.2 cells compared to that in MCF-7 cells. Treatment with PTX also led to significant induction of GCS expression in MCF-7 cells but not in 4T1.2 cells. These results suggest that ceramides generated in tumor cells in response to chemotherapeutic stress upregulate GCS expression to detoxify ceramides and prevent cell death.
Fig. 1.
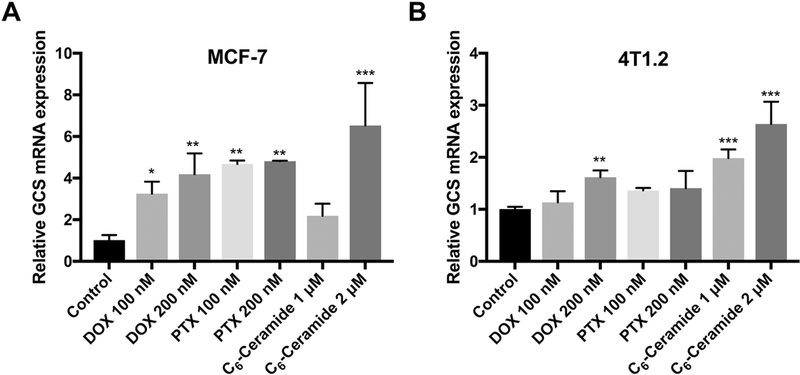
Effects of chemotherapies on GCS mRNA expression in MCF-7 (A) and 4T1.2 (B) cell after 48 h treatment. Data are presented as the means ± SD for triplicate samples. P values were generated by one-way ANOVA using the Dunnett test for multiple comparisons to one control (*P < 0.05, **P < 0.01, *** P < 0.001).
3.2. Effect of Combination of PPMP and Other Chemotherapeutic Drugs on Tumor Cell Proliferation
To assess whether GCS inhibitor PPMP could further enhance the cytotoxicity and/or restore sensitivity of tumor cells to the action of anticancer drugs, we evaluated the growth inhibitory activity of combination treament of PPMP with other anticancer drugs in MCF-7, 4T1.2 and PC-3 cell lines. As shown in Fig. 2A, PPMP or DOX alone caused a concentration-dependent inhibition of MCF-7 cell proliferation. It is also apparent that combination of the two led to a significant improvement in the level of cytotoxicity. Similar synergistic effects between PPMP and DOX were found in 4T1.2 (Fig. 2B) and PC-3 (Fig. 2C) cells. PPMP also synergized with PTX or C6-ceramide in inhibiting the tumor cell proliferation in all tumor cell lines tested (Fig. 2D~J). Combination index (CI) was then calculated to assess a potential synergy between PPMP and other anticancer drugs, by the equation CI=(d1/IC501)+(d2/IC502), with d1 or d2 being the concentration of PPMP or other anticancer agent (DOX, PTX or C6-ceramide) required to achieve 50% killing effect in co-treatment, while IC501 or IC502 being IC50 of PPMP or other anticancer agents in single treatment, respectively. All of CI values listed in Table 1 were less than 1, indicating the synergy between PPMP and DOX, PTX or C6-ceramide in all of the examined cancer cell lines.
Fig. 2.
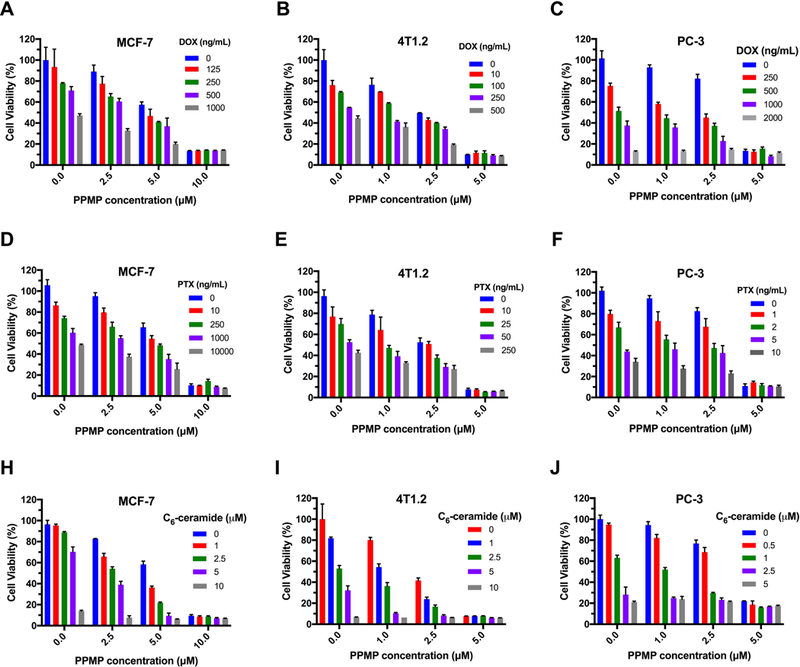
Synergistic effect between PPMP and other anticancer drugs in inhibiting the proliferation of tumor cells. (A-C) MCF-7, 4T1.2 or PC-3 cells were treated with various concentrations of free PPMP, free DOX or the combination of PPMP and DOX. (D-F) MCF-7, 4T1.2 or PC-3 cells were treated with various concentrations of free PPMP, free PTX or the combination of PPMP and PTX. (G-I) MCF-7, 4T1.2 or PC-3 cells were treated with various concentrations of free PPMP, free C6-ceramide or the combination of PPMP and C6-ceramide. After 48 h, the cytotoxicity was determined by MTT assay. The experiments was performed in triplicate and repeated three times. Data are presented as means ± SD.
Table 1.
Synergistic Antiproliferative Activity of PPMP and Other Anticancer Drugs in Cancer Cells
| Drug1 | Drug2 | Cell Lines |
d1 (μM) |
D501 (μM) |
d2 (ng/mL)a (μM)b |
D502 (ng/mL)a (μM)b |
CI |
|---|---|---|---|---|---|---|---|
| PPMP | DOX | MCF-7 | 3.8±0.6 | 5.7±0.4 | 250 | 964.3±106.6 | 0.93 |
| PPMP | DOX | 4T1.2 | 1.8±0.3 | 2.2±0.1 | 10 | 272.0±62.1 | 0.85 |
| PPMP | DOX | PC-3 | 1.5±0.3 | 3.4±0.1 | 250 | 629.4±33.6 | 0.84 |
| PPMP | PTX | MCF-7 | 4.1±0.5 | 5.7±0.4 | 250 | 7143.0±1866.0 | 0.75 |
| PPMP | PTX | 4T1.2 | 1.0±0.3 | 2.2±0.1 | 25 | 108.1±18.1 | 0.69 |
| PPMP | PTX | PC-3 | 1.5±0.4 | 3.4±0.1 | 2 | 4.3±0.2 | 0.91 |
| PPMP | Ceramide | MCF-7 | 3.6±0.1 | 5.7±0.4 | 1 | 6.2±0.2 | 0.79 |
| PPMP | Ceramide | 4T1.2 | 1.1±0.2 | 2.2±0.1 | 1 | 2.8±0.2 | 0.86 |
| PPMP | Ceramide | PC-3 | 1.1±0.4 | 3.4±0.1 | 1 | 1.6±0.1 | 0.95 |
Unit of DOX and PTX.
Unit of C6-ceramide.
Combination Index (CI) of co-treatment of PPMP with DOX, PTX and C6-ceramide in MCF-7, 4T1.2 and PC-3 cells. The cell viability was determined by MTT assay. The anti-proliferation data for single drug and combination treatment was fitted to an inhibitory, normalized doseresponse model with variable slope (Y = 100/(1 + 10^((LogEC50-X) *HillSlope); (GraphPad Prism, San Diego,CA). The CI was calculated by the formula: CI=(d1/D501)+(d2/D502), where D501 is the IC50 of PPMP in single treatment, and d1 is the concentration of PPMP required to achieve 50% inhibition effect with d2 in cotreatment. Similarly, D502 is IC50 of DOX, PTX or C6-ceramide in single treatment, and d2 is the concentration of DOX, PTX or C6-ceramide required to obtain the same 50% cell-killing effect in combination with d1. The CI values are interpreted as follows: <1.0, synergism; 1.0, additive; and >1.0, antagonism. The experiments was performed in triplicate and repeated three times.
3.3. Synthesis and Characterization of the POEG-b-PPPMP Polymers
First, PPMP·HCl was synthesized following a published patent (WO 2005/049827 A2) and its chemical identity was confirmed by 1H-NMR (Fig. S1). Then PPMP was conjugated with methacryloyl chloride through a hydrolyzable ester linkage as shown in Scheme 1. The structure of PPMP monomer was confirmed by 1H-NMR (Fig. S2). Meanwhile, the macro-chain transfer agent POEG was synthesized by RAFT polymerization of hydrophilic OEGMA monomer according to a published method [31]. POEG (MW=7500) was then used to initiate the polymerization of hydrophobic PPMP monomer, yielding the amphiphilic POEG-b-PPPMP block copolymer. The structure of POEG-b-PPPMP was confirmed by 1H-NMR (Fig. 3), and the average degree of polymerization of the PPMP monomers was calculated by comparing the intensity of Ib and Ia. The calculated molecular weight by 1H-NMR was Mn=11700. The molecular weight detected by gel permeation chromatography (GPC) was Mn=12100 with narrow polydispersity (PDI) of 1.3, which was consistent with molecular weight calculated by 1H-NMR. Therefore, each POEG-b-PPPMP molecule contained 15 units of OEGMA and 7 units of PPMP, denoted by POEG15-b-PPPMP7.
Scheme 1.
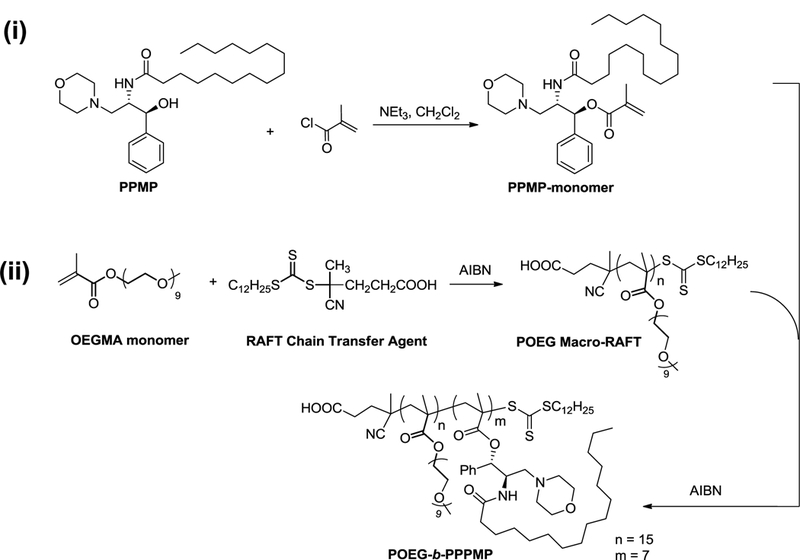
Synthesis Scheme of POEG-b-PPPMP Conjugate
Fig. 3.
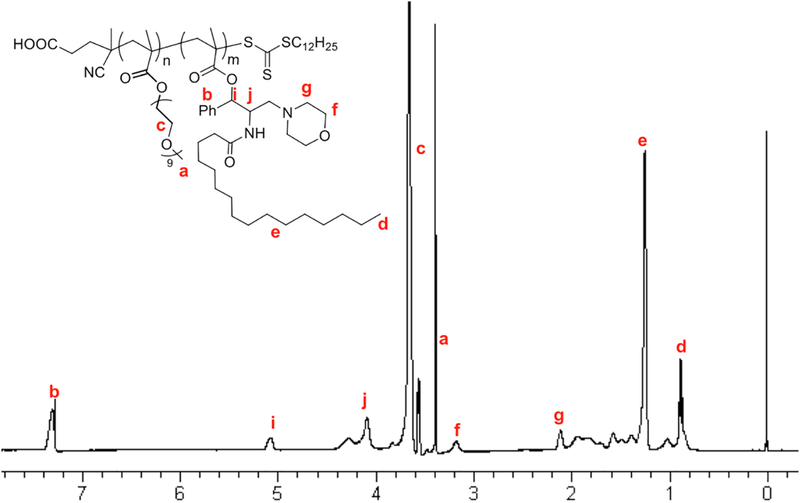
1H-NMR of POEG-b-PPPMP polymers in CDCl3.
3.4. Biophysical Characterization of Blank and Drug-loaded POEG-b-PPPMP micelles
POEG-b-PPPMP alone or POEG-b-PPPMP/drug mixture readily formed transparent micellar solution in DPBS by a simple film hydration method. Fig. 4 showed that the CMC of POEGb-PPPMP micelles was around 0.03 mg/mL. The low CMC of POEG-b-PPPMP shall provide a good stability for micelles upon dilution in blood stream after intravenous administration. The hydrodynamic sizes of blank and drug-loaded POEG-b-PPPMP micelles were examined by DLS and the data are shown in Fig. 5. POEG-b-PPPMP formed micelles with a diameter of 105 nm (Fig. 5A). Interestingly, incorporation of a drug into micelles at a carrier/drug ratio of 20:1 (mg/mg) resulted in a slight decrease in particle sizes (80~100 nm) (Fig. 5B-D). This is likely due to an enhanced interaction between the carrier and loaded drugs, leading to the formation of a more compact structure. TEM images further confirmed the spherical morphology of blank and drug-loaded POEG-b-PPPMP micelles with a uniform size distribution (Fig. 5). The size, DLC, and formulation stability of drug-loaded POEG-b-PPPMP micelles were then examined (Table 2). DOX could be loaded into POEG-b-PPPMP micelles at a carrier/drug ratio as low as 10/1 (mg/mg), at which ratio DOX was incorporated into the carrier at a DLC of 6.5 % and DOX-loaded micelles were stable with no obvious changes in size or precipitation for 6 days at 4 °C. In addition, with an increase in the carrier/drug ratio, the drug encapsulation efficiency and colloidal stability were further improved. As shown in Table 2, at a carrier/drug ratio of 50:1, DOX-loaded micelles were stable for 42 days in solution at 4 °C. In order to deliver more PPMP as well as provide good stability, a carrier/drug mass ratio of 50:1 was used for all subsequent studies. In addition to DOX, other chemotherapeutic agents such as PTX and C6-ceramide, could be effectively loaded into POEG-b-PPPMP nanocarrier. Therefore, POEG-b-PPPMP might serve as a carrier for the delivery of different types of anticancer drugs.
Fig. 4.
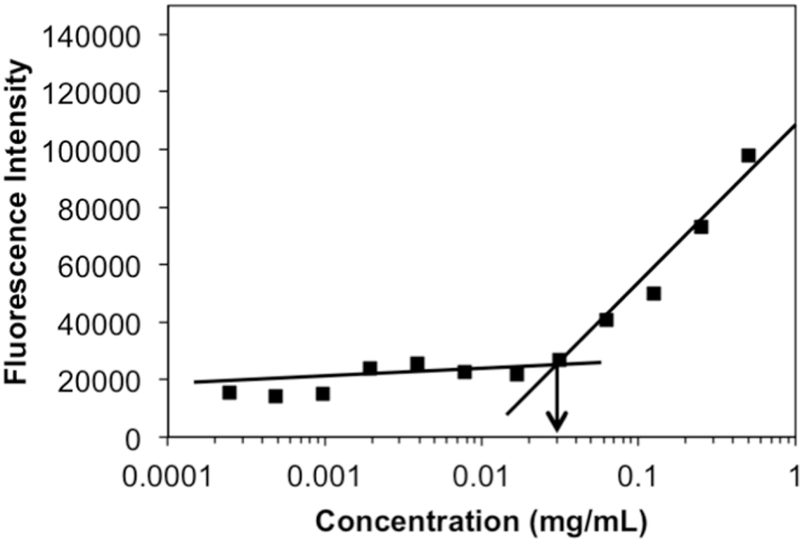
Plot of fluorescence intensity at 641 nm versus concentrations of POEG-b-PPPMP copolymers.
Fig. 5.
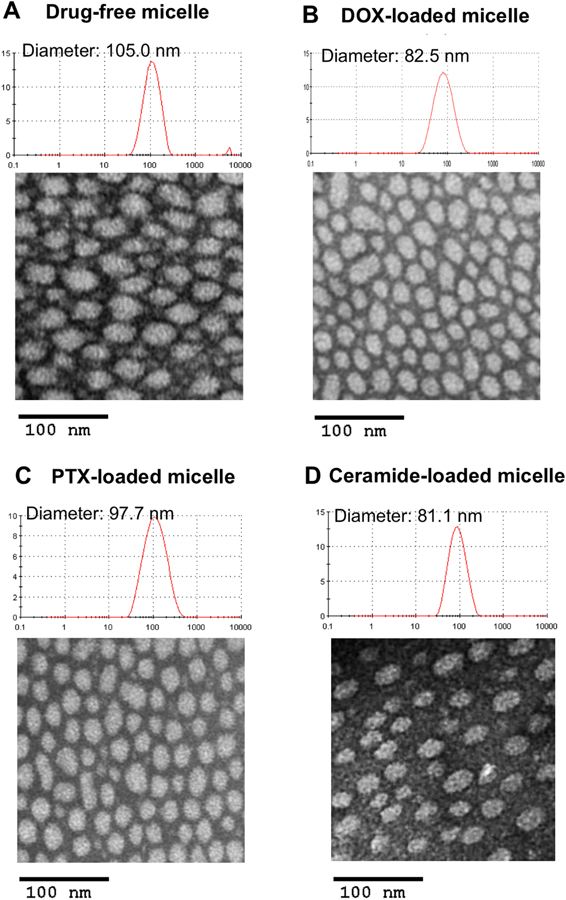
TEM images of drug-free POEG-b-PPPMP micelles (A), DOX-loaded POEG-b-PPPMP micelles (B), PTX-loaded POEG-b-PPPMP micelles (C) and ceramide-loaded POEG-b-PPPMP micelles (D) using negative staining. DOX, PTX or ceramide were loaded in to POEG-b-PPPMP micelles at carrier/drug ratio of 20/1 (mg/mg). Scale bar is 100 nm.
Table 2.
Physicochemical Characterizations of DOX-loaded and PTX-loaded POEG-b-PPPMP Micelles.
| Micelles | Mass ratio (mg: mg) a |
Size (nm) b | PDI c | DLC (%) d |
DLE (%) e |
Stability f (RT) |
Stability f (4°C) |
|---|---|---|---|---|---|---|---|
| POEG15-b-PPPMP7 | 105.0 | 0.179 | |||||
|
POEG15-b-PPPMP7 : DOX |
10:1 | 87.58 | 0.221 | 6.5 | 68.9 | 48 h | 6 d |
|
POEG15-b-PPPMP7 : DOX |
20:1 | 82.46 | 0.214 | 3.8 | 79.7 | 4 d | 25 d |
|
POEG15-b-PPPMP7 : DOX |
30:1 | 79.77 | 0.196 | 2.8 | 86.7 | 7 d | 37 d |
|
POEG15-b-PPPMP7 : DOX |
50:1 | 89.34 | 0.193 | 1.9 | 96.0 | 12 d | 42 d |
|
POEG15-b-PPPMP7 : PTX |
10: 1 | 107.1 | 0.205 | 8.2 | 90.4 | 24 h | 51 h |
|
POEG15-b-PPPMP7 : PTX |
20: 1 | 97.7 | 0.212 | 4.4 | 92.5 | 45 h | 6 d |
|
POEG15-b-PPPMP7 : PTX |
30: 1 | 95.8 | 0.217 | 3.1 | 95.2 | 53 h | 17 d |
|
POEG15-b-PPPMP7 : PTX |
50:1 | 93.2 | 0.205 | 1.9 | 95.6 | 7 d | 21 d |
DOX or PTX concentration in micelles was kept at 0.5 or 1 mg/mL respectively.
Measured by dynamic light scattering particle sizer.
PDI = polydispersity index.
DLC = drug loading capacity.
DLE = drug loading efficiency.
Data mean there was no noticeable size change and visible precipitates during the follow-up period.
3.5. In vitro Drug Release
The release profile of DOX from DOX-loaded POEG-b-PPPMP micelles was investigated by a dialysis method in DPBS and compared to that of free DOX and DOX-loaded POEG-b POM micelles. As shown in Fig. 6, DOX release from two micellar formulations was significantly slower than that from free DOX. Forty eight percent of DOX was rapidly released from free DOX in 2 h and around 83% of total drug was released within 12 h. In contrast, less than 10% DOX was released from POEG-b-PPPMP formulation in 2 h and the slow release kinetics extended for 72 h. The performance of DOX cumulative release followed the order of DOX/POEG-b-PPPMP > DOX/POEG-b-POM > free DOX.
Fig. 6.
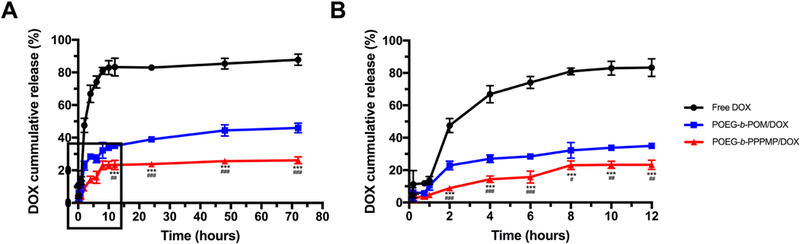
Cumulative DOX release profile from POEG-b-PPPMP micelles with free DOX and POEG-b-POM micelles as control. (A) 0 to 72 hours, (B) 0 to 12 hours. DPBS was used as the release medium. DOX concentration was fixed at 0.5 mg/mL. Values reported are the means ± SD for triplicate samples. Statistical significance was determined by two-way ANOVA using the Tukey test for multiple comparisons. *P < 0.05, **P < 0.01, *** P < 0.001 (POEG-b-PPPMP/DOX vs free DOX); #P < 0.05, ##P < 0.01, ###P < 0.001 (POEG-b-PPPMP/DOX vs POEG-b-POM/DOX).
3.6. In vitro Cytotoxicity of Prodrug Micelles and PTX/DOX/Ceramide-loaded micelles
The in vitro cytotoxicity of POEG-b-PPPMP blank micelles was tested in 4T1.2, PC-3 and MCF-7 cancer cells and compared to that of free PPMP. As shown in Fig. 7A, free PPMP inhibited the proliferation of 4T1.2 tumor cells in a dose-dependent manner. Compared to free PPMP, POEG-b-PPPMP prodrug micelles showed less cytotoxicity at equivalent amounts of PPMP in 4T1.2 cells. Similar results were shown in PC-3 and MCF-7 tumor cells (Fig. 7B & C). The cytotoxicity of POEG-b-PPPMP shall likely come from the PPMP cleaved from the prodrug polymer following the intracellular uptake. The less effectiveness of POEG-b-PPPMP was likely due to the limited PPMP cleavage and release in a relatively short period of treatment.
Fig. 7.
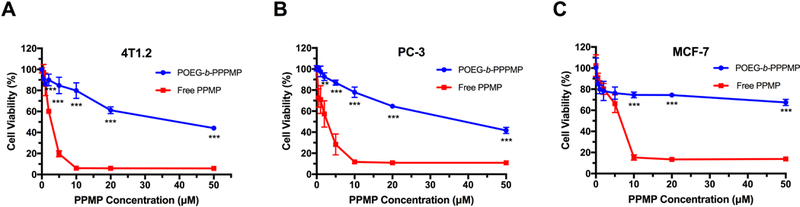
Cytotoxicity of POEG-b-PPPMP prodrug micelles in 4T1.2 mouse breast cancer cell line (A), PC-3 human prostate cancer cell line (B) and MCF-7 human breast cancer cell line (C) with free PPMP as the control.Cells were treated with different micelles for 48 h and values reported are the means ± SD for triplicate samples. P values were determined by two-tailed Student’s T test. *P < 0.05, **P < 0.01, *** P < 0.001 (POEG-b-PPPMP vs PPMP).
The cytotoxicity of DOX-loaded POEG-b-PPPMP micelles was also investigated in 4T1.2 (Fig. 8A), PC-3 (Fig. 8B) and MCF-7 (Fig. 8C) cells. DOX-loaded POEG-b-POM, DOX·HCl and Doxil were included as controls. At all doses that were used for loading DOX, POEG-b-POM showed minimal impact on the proliferation of 4T1.2 cells (Fig. 8A). POEGb-PPPMP alone showed modest effects on tumor cell proliferation at higher doses. DOX formulated in POEG-b-PPPMP was more effective than DOX·HCl and Doxil in inhibiting the proliferation of 4T1.2 cancer cells. More importantly, DOX-loaded POEG-b-PPPMP was more potent than DOX-loaded POEG-b-POM in inhibiting the proliferation of 4T1.2 cells. The improved cytotoxicity was likely due to the released PPMP from POEG-b-PPPMP prodrug copolymer, via the cleavage of the hydrolyzable ester bond by the esterase in tumor cells, resulting in synergistic action with codelivered DOX. A trend of synergistic action between POEG-b-PPPMP-based carrier and codelivered DOX was also shown in PC-3 (Fig.8B) and MCF-7 (Fig. 8C) cells. We have further shown a synergy between POEG-b-PPPMP and codelivered PTX (Fig. 8 D-F) or C6-ceramide (Fig. 8 G-I).
Fig. 8.
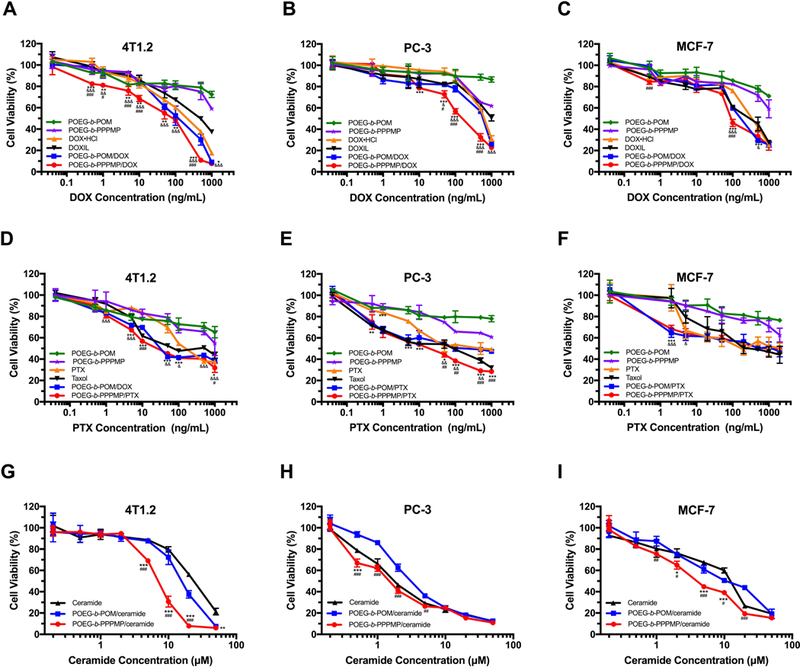
Cytotoxicity of DOX-loaded POEG-b-PPPMP micelles in 4T1.2 (A), PC-3 (B) and MCF-7 (C) cell lines, PTX-loaded POEG-b-PPPMP micelles in 4T1.2 (D), PC-3 (E) and MCF-7 (F) cell lines, and C6-ceramideloaded POEG-b-PPPMP micelles in 4T1.2 (G), PC-3 (H) and MCF-7 (I) cell lines after 48 h treatment. Data are presented as the means ± SD for triplicate samples. P values were determined by two-way ANOVA using the Tukey test for multiple comparisons. *P < 0.05, **P < 0.01, *** P < 0.001 (POEG-b-PPPMP/DOX vs DOXHCl, POEG-b-PPPMP/PTX vs PTX, POEG-b-PPPMP/ceramide vs ceramide); &P < 0.05, &P < 0.01,&&&P < 0.001 (POEG-b-PPPMP/DOX vs DOXIL, POEG-b-PPPMP/PTX vs Taxol); #P < 0.05, ##P < 0.01, ###P < 0.001 (POEG-b-PPPMP/DOX vs POEG-b-POM/DOX, POEG-b-PPPMP/PTX vs POEG-b-POM/PTX), POEGb-PPPMP/ceramide vs POEG-b-POM/ceramide).
3.7. Effect of POEG-b-PPPMP on Regulation of Ceramides Production
As a prodrug of PPMP, the ability of POEG-b-PPPMP in regulating ceramides production was investigated in 4T1.2 and MCF-7 cells. The peak areas for different sphingolipid subspecies (C16-ceramide, C18-ceramide, C20-ceramide, C22-ceramide and C24-ceramide) were quantified using a non-naturally occurring C6-ceramide as an internal standard and then normalized to protein concentrations. We focused on these species as long-chain ceramides (C16–C20) are known to have anti-proliferative and pro-apoptotic effects[36]. It has been reported that the levels of C16, C24:0 or C24:1 ceramides are significantly increased in breast cancer tissues [37]. Especially, evidence is mounting that the endogenous levels of C16:0 ceramide were increased under pro-apoptotic conditions, which played a decisive role in regulating apoptosis[38–40] As shown in Fig. 9A & G, treatment with PPMP as well as the POEG-b-PPPMP-based prodrug led to a modest increase in the amounts of C16 ceramide. As a ceramide-generating anticancer agent, DOX significantly induced C16 ceramide production in both tested cell lines, while free drug combination (DOX·HCl+PPMP) or POEG-b-PPPMP/DOX mixed micelles were most effective in promoting ceramide accumulation. Unlike its effect on C16 ceramide, our POEG-b-PPPMP/DOX formulation caused negligible changes in the amounts of C24 ceramides (Fig. 9 E, F, K & L). Interestingly C24:0 or C24:1 has been reported to have minimal effect in promoting apoptosis and could even promote the proliferation of tumor cells [41, 42]. Therefore, our POEG-b-PPPMP/DOX system appears to selectively promote the accumulation of pro-apoptotic ceramide species.
Fig. 9.
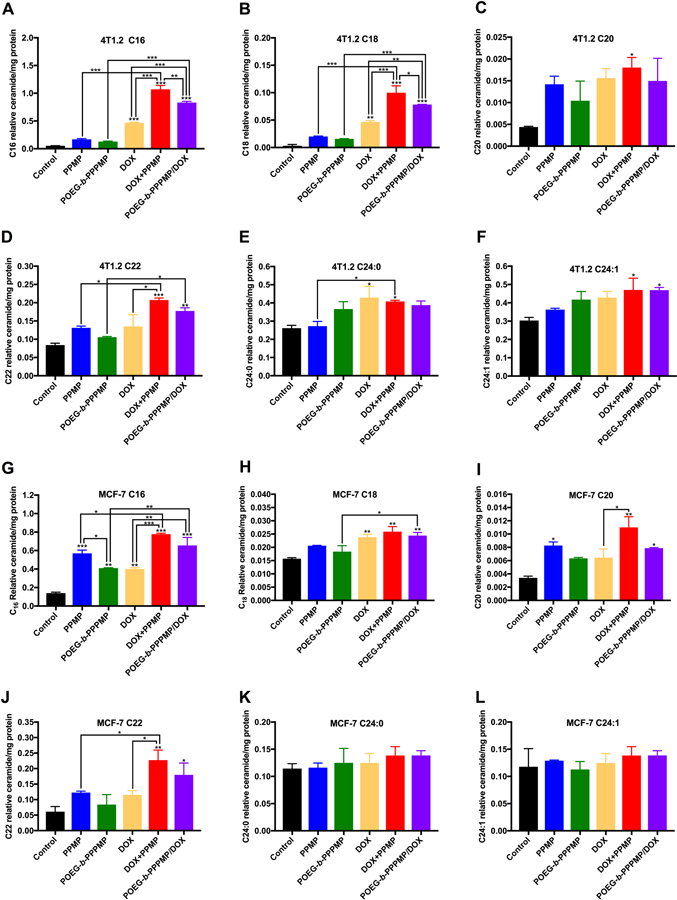
Different species of relative ceramide level in 4T1.2 or MCF-7 cells measured by LC/MS 48 h after POEG-b-PPPMP or POEG-b-PPPMP/DOX treatments compared to free drug alone or combination. Tumor cells were treated with PPMP, POEG-b-PPPMP, DOX·HCl +PPMP or POEG-b-PPPMP/DOX at the same PPMP concentration of 1.7 μM; DOX·HCl, DOX·HCl +PPMP or POEG-b-PPPMP/DOX at the same DOX concentration of 100 nM. Data are presented as the means ± SD for triplicate samples. P values were generated by one-way ANOVA using the Tukey test for multiple comparisons. *P < 0.05, **P < 0.01, *** P < 0.001.
3.8. Effect of POEG-b-PPPMP on GCS mRNA Expression:
In addition to direct inhibition of GCS enzymatic activity, PPMP has been shown to downregulate the mRNA expression of GCS in tumor cells. PPMP treatment was shown to cause decreases in GCS mRNA levels in drug-resistant cell lines with high endogenous GCS mRNA expression, including head and neck cancer (AMC-HN2, relative resistance to cisplatin) [43] and leukemia cell lines (K562/AO2, multidrug resistance)[44]. Accordingly, we examined the efficacy of POEG-b-PPPMP in downregulating GCS mRNA expression in both MCF-7 and 4T1.2 cells and compare to that of free PPMP. As shown in Fig. 10, treatment with free PPMP led to significant decreases in the basal levels of GCS mRNA in both cancer cell lines. Free PPMP was also effective in inhibiting the DOX-induced upregulation of GCS mRNA expression. POEG-b-PPPMP was comparable to free PPMP in inhibiting both basal and DOX-induced expression of GCS mRNA.
Fig. 10.
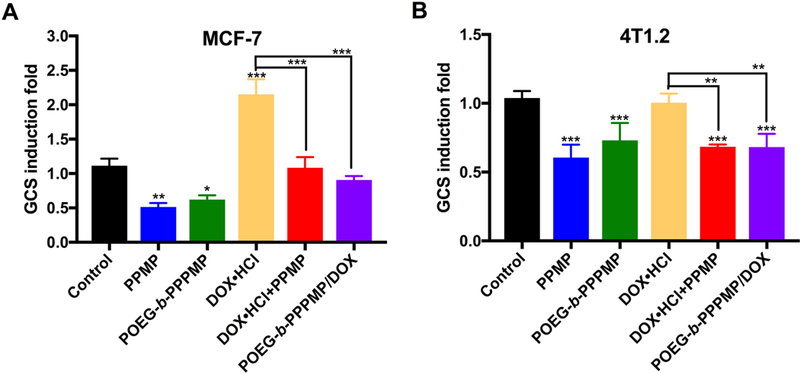
Effects of POEG-b-PPPMP and POEG-b-PPPMP/DOX on GCS mRNA expression in MCF-7 (A) or 4T1.2 (B) cells after 48 h treatment compared to free drug alone or combination. Tumor cells were treated with PPMP, POEG-b-PPPMP, DOX·HCl+PPMP or POEG-b-PPPMP/DOX at the same PPMP concentration of 1.7 μM; DOX·HCl, DOX·HCl+PPMP or POEG-b-PPPMP/DOX at the same DOX concentration of 100 nM. Data are presented as the means ± SD for triplicate samples. P values were generated by one-way ANOVA using the Tukey test for multiple comparisons. *P < 0.05, **P < 0.01, *** P < 0.001.
3.9. Plasma Pharmacokinetics and Tissue Distribution
The POEG-b-PPPMP/DOX, POEG-b-POM/DOX or DOX·HCl was injected into tumor-free mice at a DOX dose of 5mg/kg. The plasma concentrations of DOX were examined at different time points. The initial blood concentration of POEG-b-PPPMP was around 3 mg/mL, which was 100-fold higher than its CMC (0.03 mg/mL). The concentrations of DOX in the blood following i.v. injection of different DOX formulations as a function of time were illustrated in Fig. 11A. Compared to DOX·HCl, the plasma concentrations of DOX for both POEG-b-POM/DOX and POEG-b-PPPMP/DOX were significantly higher at early time points and maintained at relatively high levels until 12 h, which is attributed to the surface modification of PEG and stealth-shielding against RES system. With the same length of coated PEG, however, POEG-b-PPPMP/DOX provided higher levels of DOX in circulation than POEG-b-POM/DOX. This is likely due to a more effective interaction between POEGb-PPPMP and DOX, which is also supported by the data from in vitro release study (Fig. 3).
Fig. 11.
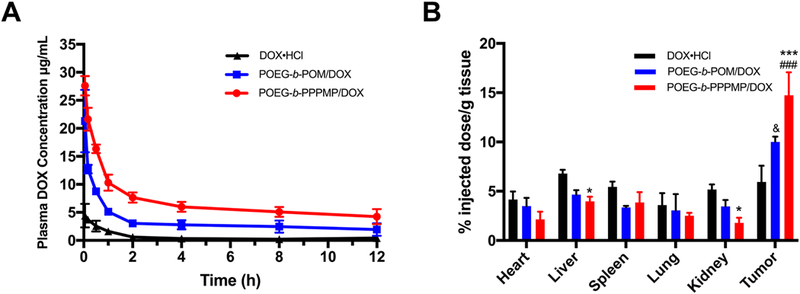
(A) Pharmacokinetics of DOX·HCl, POEG-b-POM/DOX and POEG-b-PPPMP/DOX micelles in tumor-free female CD1 mice. (B) Tissue distribution of DOX 24 h post injection in tumor-bearing female BALB/c mice treated with DOX·HCl, POEG-b-POM/DOX or POEG-b-PPPMP/DOX micelles at the same dose of 5mg DOX/kg. Values reported are the means ± SEM, n=5. P values were generated by two-way ANOVA using the Tukey test for multiple comparisons. *P < 0.05, *** P < 0.001 (POEG-b-PPPMP/DOX vs DOX·HCl; &P < 0.05 (POEG-b-POM/DOX vs DOX·HCl); ###P < 0.001 (POEG-b-PPPMP/DOX vs POEG-b-POM/DOX).
Next, the DOX tissue distribution of the three formulations was investigated in tumor-bearing mice. POEG-b-PPPMP/DOX, POEG-b-POM/DOX or DOX·HCl was injected into 4T1.2 tumor-bearing mice at a DOX dose of 5mg/kg. Twenty-four hours following the injection, major organs and tumors were harvested for the quantification of DOX. Compared to free DOX, both micellar formulations exhibited significantly greater amounts of DOX in tumor tissue, which is attributed to the EPR effect of nanoparticles. In addition, DOX formulated in POEG-b-PPPMP was more effective in tumor accumulation than in POEG-b-POM. This is consistent with the PK data and is likely ascribed to the better stability in blood stream due to the increased carrier-drug interaction between DOX and POEG-b-PPPMP. The DOX uptake in other organs was lower for POEG-b-PPPMP/DOX compared to free DOX group, although it was not statistically significant. The decreased accumulation of DOX in normal tissues, particularly in heart shall help to decrease the DOX-associated toxicity and allow higher doses of DOX to be administered to maximize the therapeutic effect.
3.10. In Vivo Therapeutic Study
The in vivo tumor growth inhibition efficiency of POEG-b-PPPMP/DOX micelles was investigated in a highly aggressive syngeneic murine breast cancer model (4T1.2, s.c. at mammary fat pad). As expected, the control carrier alone (POEG-b-POM) showed minimal antitumor activity (Fig. 12A). POEG-b-PPPMP alone exhibited a modest but significant (P<0.05) tumor inhibition effect. Doxil formulation and POEG-b-POM/DOX showed comparable and enhanced tumor inhibitory effect. Among all treatment groups, POEG-b-PPPMP/DOX was most effective in inhibiting the tumor growth. Fig. 12 B&D show the weights and images of tumor tissues that were collected at the end of the experiment, which was consistent with data of tumor growth curves (Fig. 12A). There were slight increases in body weights in all groups over the period of study, suggesting the negligible toxicity of POEG-b-PPPMP/DOX micelles in vivo (Fig. 12C).
Fig. 12.
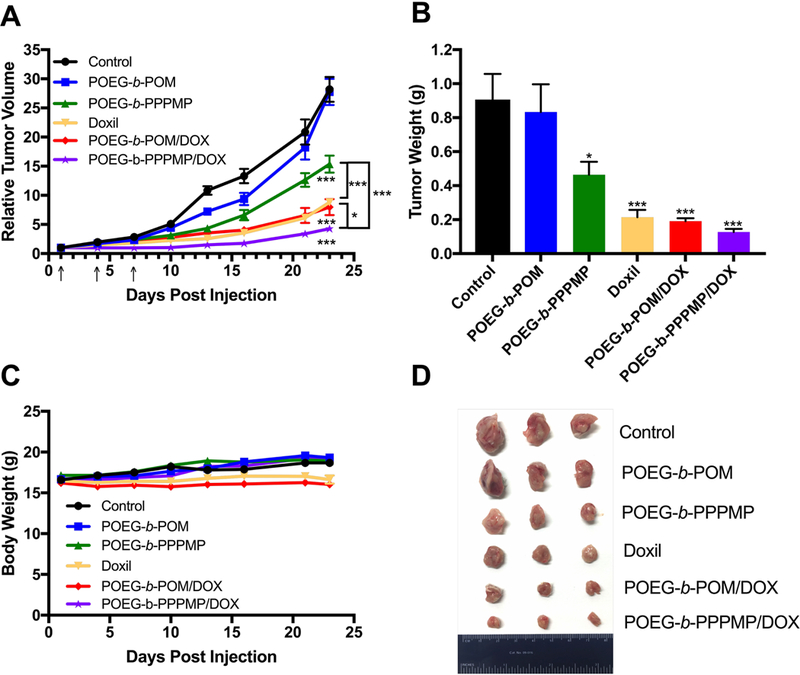
Antitumor activity of blank POEG-b-POM, blank POEG-b-PPPMP micelles, Doxil, DOX loaded POEG-b-POM and DOX loaded POEG-b-PPPMP micelles in a syngeneic murine breast cancer model (4T1.2). Three injections were given on day 1, 4 and 7. (B) Weights of tumors collected from different groups at the end of experiment. (C) Changes of body weight in mice receiving different treatments. (D) Photographs of tumors collected from different treatment groups at the end of experiment. Values reported are the means ± SEM, n = 5. P values were generated by one-way ANOVA using the Tukey test for multiple comparisons. *P < 0.05, **P <0.01, ***P < 0.001 (vs control).
3.11. Histochemical Staining
Histological analysis was conducted to further evaluate the therapeutic effect of POEG-b-PPPMP/DOX formulation. As shown in Fig. 11, tumors treated with DPBS or POEG-b-POM carrier alone exhibited abundant aggregates of neoplastic cells with conspicuous nucleoli and scant cytoplasm due to high proliferation rate. In contrast, tumors that were treated with POEG-b-PPPMP/DOX showed significantly altered morphology with shrunken nuclei and the lowest tumor cell density.
4. Discussion
As a primary approach in cancer treatment, various classes of chemotherapeutic drugs are available in clinic. Regardless of the differences in the structures of the agents, their molecular targets, and the major mechanisms of action, the eventual onset of apoptosis seems to be a common consequence in chemotherapy-induced cell death. Ceramide, the central molecule in sphingolipids family, has recently been identified as a key mediator of this process and has attracted tremendous attention in cancer therapy[45]. Mounting evidence shows that most chemotherapeutic agents are capable of inducing endogenous ceramide accumulation through either activation of sphingomyelin catabolism and/or increases in de novo synthesis[2].
Various strategies have been developed to increase the intracellular levels of ceramides in tumor cells including intracellular delivery of exogenous ceramides, enhancement of ceramide synthesis, and inhibition of ceramide catabolism[10]. As a stand-alone strategy, the first two approaches are insufficient as the increased levels of ceramides are only short-lived due to activation of the ceramide catabolism pathways[2]. Importantly, many of the ceramide metabolites have the opposite effects of ceramides, playing an important role in tumor cell survival and drug resistance[23, 46, 47].
A number of pathways are involved in the catabolism of ceramides inside cells with GCS being the dominant mechanism for ceramide catabolism[48]. Drugs that cause increases in the intracellular levels of ceramides such as DOX, PTX and C6-ceramide itself, also induce upregulation of GCS expression. However, different types of tumor cells respond differently to a ceramide-generating drug (e.g., DOX) with respect to the extent of GCS upregulation. Our data (Fig. 1) show that GCS upregulation was much more dramatic in MCF-7 cells than in 4T1.2 cells following treatment of DOX, PTX or C6-ceramide. Nonetheless, combination of a ceramide-generating drug with an inhibitor of GCS shall represent an effective strategy to improve the therapeutic outcome.
PPMP is a small molecule drug that was initially known to inhibit the enzymatic activity of GCS. PPMP was later shown to be also effective in downregulating the mRNA expression levels of GCS in tumor cells (Fig. 10). Our data show that PPMP effectively downregulated both basal and DOX-induced GCS mRNA levels in MCF-7 and 4T1.2 cells, which is consistent with what was found in other tumor cells [42,43]. Interestingly, PPMP appears to selectively upregulate the proapoptotic ceramide species while having minimal impact on those species that have no effect in promoting the apoptosis or even enhance the tumor cell proliferation (Fig. 9). The underlying mechanism is unclear and requires more studies in the future.
As a strategy to faciliate the codelivery of PPMP and another ceramide-genertaiung anticancer agent, we initially developed a dual-functional carreir that is based on PEGPPMP2. One issue with this system is the low number (2) of PPMP units in the carrier, which will limit the amounts of PPMP that can be delivered to tumor tissues. This led us to the developemnt of an improved nanocarrier, which is a POEG-b-PPPMP-based copolymer with significantly incrased number of PPMP units (7) in each molecule of polymer. A PPMP monomer was first synthesized, which allows the use of a simple and well-controlled polymerization protocol to obtain well-defined POEG-b-PPPMP polymers. This approach has an obvious advantage of simplicity compared to the strategy of post-conjugation of PPMP following the synthesis of a polymer scaffold. It also avoids the issue of the presence of some remianing reactive groups in polymers after conjugation of PPMP. After synthesis, the formulation of blank or drug-loaded POEG-b-PPPMP could be prepared through a very simple film hydration method. In addition, POEG-b-PPPMP was effective in formulating a wide range of hydrophobic anticancer drugs of different structures.
POEG-b-PPPMP well retains the pharmacological activity of free PPMP. POEG-b-PPPMP exhibited comparable activity in downregulation of basal GCS mRNA expression (Fig. 9), and similarly reversed the DOX-induced GCS upregulation as free PPMP (Fig. 10). More importantly, like free PPMP, POEG-b-PPPMP also selectively increased the accumulation of pro-apoptotic ceramides with negligible effect on anti-apoptotic ceramides species.
POEG-b-POM was synthesized as a control carrier that has the same units of hydrophilic POEG and hydrophobic oleic acid (POEG15-b-POM7) compared to POEG-b-POM. Interestingly, DOX formulated in POEG-b-POM showed a relatively slower kinetics of drug release (Fig. 6), longer stay in blood (Fig. 11A), and more effective accumulation at tumor tissues (Fig. 11B) compared to DOX formulated in the control carrier. This is likely due to a more effective interaction between POEG-b-PPPMP and DOX. In addition to hydrophobic interaction as seen with POEG-b-POM/DOX, the aromatic rings of PPMPs in POEG-b-PPPMP further interact with DOX through π-π stacking, resulting in an enhancement in the overall carrier/drug interactions.
POEG-b-PPPMP demonstrated significant antitumor activity both in vitro and in vivo. This is likely due to the release of active PPMP following delivery of POEG-b-PPPMP to tumor cells. Importantly, codelivery of DOX via POEG-b-PPPMP led to effective growth inhibition of 4T1.2 breast tumor, much more effectively than Doxil and POEG-b-PPPMP/DOX. The improved antitumor activity of POEG-b-PPPMP/DOX over POEG-b-POM/DOX might be largely attributed to the synergistic action between the released PPMP from POEG-b-PPPMP and the codelivered DOX. The improved delivery of DOX via POEG-b-PPPMP-based carrier (Fig. 11) may also play a role.
5. Conclusion
We have developed a well-characterized POEG-b-PPPMP prodrug-based micellar nanocarrier that consists of 7 units of PPMP and 15 units of POEG for efficient delivery of water insoluble anticancer drugs. POEG-b-PPPMP well retained the biological ability of PPMP. DOX loaded in POEG-b-PPPMP micelles exhibited slow release kinetics in vitro as well as a sustained PK profile in mice. Combination of DOX with POEG-b-PPPMP led to inhibition of DOX-induced upregulation of GCS, increased accumulation of pro-apoptotic ceramides, and enhanced cytotoxicity towards tumor cells. More importantly, systemic administration of DOX formulated in POEG-b-PPPMP micelles resulted in significant inhibition of 4T1.2 breast tumor, much more effectively than Doxil and POEG-b-POM/DOX.
Supplementary Material
Fig. 13.
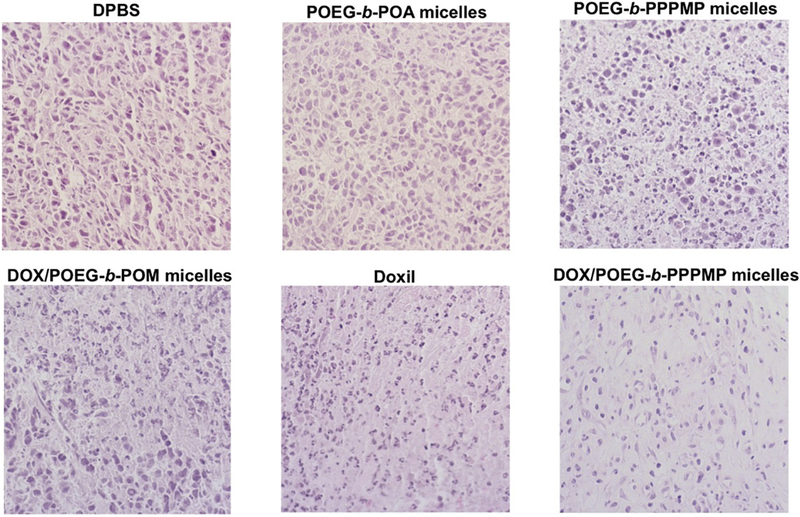
Histological analyses of tumor tissues collected from different groups at the end of in vivo therapeutic experiment using H&E staining.
Table 3.
Pharmacokinetic Parameters of DOX in Different Formulations.
| Groups | T1/2(h) | AUC0−∞ (μg/ml*h) | Cmax (μg/µL) | CL (µL/h) | Vd (µL) |
|---|---|---|---|---|---|
| DOX.HCl | 3.35 | 9.20 | 4.41 | 10.87 | 52.49 |
| POEG-b-POM/DOX | 15.96 | 84.31 | 21.30 | 1.19 | 27.31 |
| POEG-b PPPMP/DOX | 16.19 | 180.16 | 27.60 | 0.56 | 12.97 |
Highlights.
We developed a new copolymer with built-in blocks of ceramide degradation inhibitor
Our polymer formed micelles that are suitable for codelivery of other anticancer drug
Our codelivery strategy led to selective induction of proapoptotic ceramide species
We demonstrated significantly improved antitumor activity in vivo with the codelivery
Acknowledgments
Funding: This work was supported by the National Institutes of Health [R01CA216410], [R01CA223788], [R01CA219399] and [R01CA174305].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interest: none
References:
- [1].Ogretmen B, Hannun YA, Biologically active sphingolipids in cancer pathogenesis and treatment, Nature Reviews Cancer, 4 (2004) 604–616. [DOI] [PubMed] [Google Scholar]
- [2].Morad SA, Cabot MC, Ceramide-orchestrated signalling in cancer cells, Nature reviews. Cancer, 13 (2013) 51–65. [DOI] [PubMed] [Google Scholar]
- [3].Hannun YA, Functions of Ceramide in Coordinating Cellular Responses to Stress, Science, 274 (1996) 1855–1859. [DOI] [PubMed] [Google Scholar]
- [4].Galadari S, Rahman A, Pallichankandy S, Thayyullathil F, Tumor suppressive functions of ceramide: evidence and mechanisms, Apoptosis : an international journal on programmed cell death, 20 (2015) 689–711. [DOI] [PubMed] [Google Scholar]
- [5].Dobrowsky RT, Kamibayashi C, Mumby MC, Hannun YA, Ceramide activates heterotrimeric protein phosphatase 2A, The Journal of biological chemistry, 268 (1993) 15523–15530. [PubMed] [Google Scholar]
- [6].Chalfant CE, Szulc Z, Roddy P, Bielawska A, Hannun YA, The structural requirements for ceramide activation of serine-threonine protein phosphatases, Journal of lipid research, 45 (2004) 496–506. [DOI] [PubMed] [Google Scholar]
- [7].Heinrich M, Neumeyer J, Jakob M, Hallas C, Tchikov V, Winoto-Morbach S, Wickel M, Schneider-Brachert W, Trauzold A, Hethke A, Schutze S, Cathepsin D links TNFinduced acid sphingomyelinase to Bid-mediated caspase-9 and −3 activation, Cell death and differentiation, 11 (2004) 550–563. [DOI] [PubMed] [Google Scholar]
- [8].Pettus BJ, Chalfant CE, Hannun YA, Ceramide in apoptosis: an overview and current perspectives, Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids, 1585 (2002) 114–125. [DOI] [PubMed] [Google Scholar]
- [9].Barth BM, Cabot MC, Kester M, Ceramide-based therapeutics for the treatment of cancer, Anti-cancer agents in medicinal chemistry, 11 (2011) 911–919. [DOI] [PubMed] [Google Scholar]
- [10].Li F, Zhang N, Ceramide: Therapeutic Potential in Combination Therapy for Cancer Treatment, Current drug metabolism, 17 (2015) 37–51. [DOI] [PubMed] [Google Scholar]
- [11].Selzner M, Bielawska A, Morse MA, Rudiger HA, Sindram D, Hannun YA, Clavien PA, Induction of apoptotic cell death and prevention of tumor growth by ceramide analogues in metastatic human colon cancer, Cancer research, 61 (2001) 1233–1240. [PubMed] [Google Scholar]
- [12].Stover T, Kester M, Liposomal Delivery Enhances Short-Chain Ceramide-Induced Apoptosis of Breast Cancer Cells, Journal of Pharmacology and Experimental Therapeutics, 307 (2003) 468–475. [DOI] [PubMed] [Google Scholar]
- [13].Kester M, Heakal Y, Fox T, Sharma A, Robertson GP, Morgan TT, Altinoglu EI, Tabakovic A, Parette MR, Rouse SM, Ruiz-Velasco V, Adair JH, Calcium phosphate nanocomposite particles for in vitro imaging and encapsulated chemotherapeutic drug delivery to cancer cells, Nano letters, 8 (2008) 4116–4121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Stover TC, Kim YS, Lowe TL, Kester M, Thermoresponsive and biodegradable linear-dendritic nanoparticles for targeted and sustained release of a pro-apoptotic drug, Biomaterials, 29 (2008) 359–369. [DOI] [PubMed] [Google Scholar]
- [15].Ganta S, Singh A, Kulkarni P, Keeler AW, Piroyan A, Sawant RR, Patel NR, Davis B, Ferris C, O’Neal S, Zamboni W, Amiji MM, Coleman TP, EGFR Targeted Theranostic Nanoemulsion for Image-Guided Ovarian Cancer Therapy, Pharmaceutical research, 32 (2015) 2753–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Shabbits JA, Mayer LD, Intracellular delivery of ceramide lipids via liposomes enhances apoptosis in vitro, Biochimica et Biophysica Acta (BBA) - Biomembranes, 1612 (2003) 98–106. [DOI] [PubMed] [Google Scholar]
- [17].Saddoughi SA, Song P, Ogretmen B, Roles of bioactive sphingolipids in cancer biology and therapeutics, Sub-cellular biochemistry, 49 (2008) 413–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Reynolds CP, Maurer BJ, Kolesnick RN, Ceramide synthesis and metabolism as a target for cancer therapy, Cancer letters, 206 (2004) 169–180. [DOI] [PubMed] [Google Scholar]
- [19].Futerman AH, Hannun YA, The complex life of simple sphingolipids, EMBO reports, 5 (2004) 777–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Senchenkov A, Litvak DA, Cabot MC, Targeting ceramide metabolism--a strategy for overcoming drug resistance, Journal of the National Cancer Institute, 93 (2001) 347–357. [DOI] [PubMed] [Google Scholar]
- [21].Liu Y-Y, Han TY, Yu JY, Bitterman A, Le A, Giuliano AE, Cabot MC, Oligonucleotides blocking glucosylceramide synthase expression selectively reverse drug resistance in cancer cells, Journal of lipid research, 45 (2004) 933–940. [DOI] [PubMed] [Google Scholar]
- [22].Liu Y.-y., Han T.-y., Giuliano AE, CABOT MC, Ceramide glycosylation potentiates cellular multidrug resistance, The FASEB Journal, 15 (2001) 719–730. [DOI] [PubMed] [Google Scholar]
- [23].Bleicher RJ, Cabot MC, Glucosylceramide synthase and apoptosis, Biochimica et biophysica Acta (BBA)-Molecular and Cell Biology of Lipids, 1585 (2002) 172–178. [DOI] [PubMed] [Google Scholar]
- [24].Abe A, Radin NS, Shayman JA, Induction of glucosylceramide synthase by synthase inhibitors and ceramide, Biochimica et biophysica acta, 1299 (1996) 333–341. [DOI] [PubMed] [Google Scholar]
- [25].Komori H, Ichikawa S, Hirabayashi Y, Ito M, Regulation of UDP-glucose:ceramide glucosyltransferase-1 by ceramide, FEBS letters, 475 (2000) 247–250. [DOI] [PubMed] [Google Scholar]
- [26].Liu YY, Yu JY, Yin D, Patwardhan GA, Gupta V, Hirabayashi Y, Holleran WM, Giuliano AE, Jazwinski SM, Gouaze-Andersson V, Consoli DP, Cabot MC, A role for ceramide in driving cancer cell resistance to doxorubicin, FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 22 (2008) 2541–2551. [DOI] [PubMed] [Google Scholar]
- [27].Kovacs P, Pinter M, Csaba G, Effect of glucosphingolipid synthesis inhibitor (PPMP and PDMP) treatment on Tetrahymena pyriformis: data on the evolution of the signaling system, Cell biochemistry and function, 18 (2000) 269–280. [DOI] [PubMed] [Google Scholar]
- [28].Lee L, Abe A, Shayman JA, Improved inhibitors of glucosylceramide synthase, The Journal of biological chemistry, 274 (1999) 14662–14669. [DOI] [PubMed] [Google Scholar]
- [29].Litvak DA, Bilchik AJ, Cabot MC, Modulators of ceramide metabolism sensitize colorectal cancer cells to chemotherapy: a novel treatment strategy, Journal of gastrointestinal surgery, 7 (2003) 140–148. [DOI] [PubMed] [Google Scholar]
- [30].Maurer BJ, Melton L, Billups C, Cabot MC, Reynolds CP, Synergistic cytotoxicity in solid tumor cell lines between N-(4-hydroxyphenyl) retinamide and modulators of ceramide metabolism, Journal of the National Cancer Institute, 92 (2000) 1897–1909. [DOI] [PubMed] [Google Scholar]
- [31].Sun J, Chen Y, Li K, Huang Y, Fu X, Zhang X, Zhao W, Wei Y, Xu L, Zhang P, Venkataramanan R, Li S, A prodrug micellar carrier assembled from polymers with pendant farnesyl thiosalicylic acid moieties for improved delivery of paclitaxel, Acta biomaterialia, 43 (2016) 282–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Xu J, Zhang X, Chen Y, Huang Y, Wang P, Wei Y, Ma X, Li S, Improved Micellar Formulation for Enhanced Delivery for Paclitaxel, Molecular pharmaceutics, 14 (2017) 31–41. [DOI] [PubMed] [Google Scholar]
- [33].Xu J, Sun J, Wang P, Ma X, Li S, Pendant HDAC inhibitor SAHA derivatised polymer as a novel prodrug micellar carrier for anticancer drugs, Journal of drug targeting, 26 (2018) 448–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Xu J, Sun J, Wang P, Ma X, Li S, Pendant HDAC inhibitor SAHA derivatised polymer as a novel prodrug micellar carrier for anticancer drugs, Journal of drug targeting, (2017) 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zhang P, Li J, Ghazwani M, Zhao W, Huang Y, Zhang X, Venkataramanan R, Li S, Effective co-delivery of doxorubicin and dasatinib using a PEG-Fmoc nanocarrier for combination cancer chemotherapy, Biomaterials, 67 (2015) 104–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Hartmann D, Lucks J, Fuchs S, Schiffmann S, Schreiber Y, Ferreiros N, Merkens J, Marschalek R, Geisslinger G, Grosch S, Long chain ceramides and very long chain ceramides have opposite effects on human breast and colon cancer cell growth, The international journal of biochemistry & cell biology, 44 (2012) 620–628. [DOI] [PubMed] [Google Scholar]
- [37].Schiffmann S, Sandner J, Birod K, Wobst I, Angioni C, Ruckhäberle E, Kaufmann M, Ackermann H, Lötsch J, Schmidt H, Ceramide synthases and ceramide levels are increased in breast cancer tissue, Carcinogenesis, 30 (2009) 745–752. [DOI] [PubMed] [Google Scholar]
- [38].Panjarian S, Kozhaya L, Arayssi S, Yehia M, Bielawski J, Bielawska A, Usta J, Hannun YA, Obeid LM, Dbaibo GS, De novo N-palmitoylsphingosine synthesis is the major biochemical mechanism of ceramide accumulation following p53 up-regulation, Prostaglandins & other lipid mediators, 86 (2008) 41–48. [DOI] [PubMed] [Google Scholar]
- [39].Eto M, Bennouna J, Hunter OC, Hershberger PA, Kanto T, Johnson CS, Lotze MT, Amoscato AA, C16 ceramide accumulates following androgen ablation in LNCaP prostate cancer cells, The Prostate, 57 (2003) 66–79. [DOI] [PubMed] [Google Scholar]
- [40].Schiffmann S, Ziebell S, Sandner J, Birod K, Deckmann K, Hartmann D, Rode S, Schmidt H, Angioni C, Geisslinger G, Grosch S, Activation of ceramide synthase 6 by celecoxib leads to a selective induction of C16:0-ceramide, Biochemical pharmacology, 80 (2010) 1632–1640. [DOI] [PubMed] [Google Scholar]
- [41].Mesicek J, Lee H, Feldman T, Jiang X, Skobeleva A, Berdyshev EV, Haimovitz-Friedman A, Fuks Z, Kolesnick R, Ceramide synthases 2, 5, and 6 confer distinct roles in radiation-induced apoptosis in HeLa cells, Cellular signalling, 22 (2010) 1300–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Grösch S, Schiffmann S, Geisslinger G, Chain length-specific properties of ceramides, Progress in lipid research, 51 (2012) 50–62. [DOI] [PubMed] [Google Scholar]
- [43].Roh J-L, Kim EH, Park JY, Kim JW, Inhibition of Glucosylceramide Synthase Sensitizes Head and Neck Cancer to Cisplatin, Molecular cancer therapeutics, 14 (2015) 1907–1915. [DOI] [PubMed] [Google Scholar]
- [44].Liu Y, Xie K-M, Yang G-Q, Bai X-M, Shi Y-P, Mu H-J, Qiao W-Z, Zhang B, Xie P, GCS induces multidrug resistance by regulating apoptosis-related genes in K562/AO2 cell line, Cancer chemotherapy and pharmacology, 66 (2010) 433–439. [DOI] [PubMed] [Google Scholar]
- [45].Henry B, Moller C, Dimanche-Boitrel MT, Gulbins E, Becker KA, Targeting the ceramide system in cancer, Cancer letters, 332 (2013) 286–294. [DOI] [PubMed] [Google Scholar]
- [46].Abuhusain HJ, Matin A, Qiao Q, Shen H, Kain N, Day BW, Stringer BW, Daniels B, Laaksonen MA, Teo C, McDonald KL, Don AS, A metabolic shift favoring sphingosine 1-phosphate at the expense of ceramide controls glioblastoma angiogenesis, The Journal of biological chemistry, 288 (2013) 37355–37364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Gomez-Munoz A, Gangoiti P, Arana L, Ouro A, Rivera IG, Ordonez M, Trueba M, New insights on the role of ceramide 1-phosphate in inflammation, Biochimica et biophysica acta, 1831 (2013) 1060–1066. [DOI] [PubMed] [Google Scholar]
- [48].Kartal Yandim M, Apohan E, Baran Y, Therapeutic potential of targeting ceramide/glucosylceramide pathway in cancer, Cancer chemotherapy and pharmacology, 71 (2013) 13–20. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.