

SUMMARY
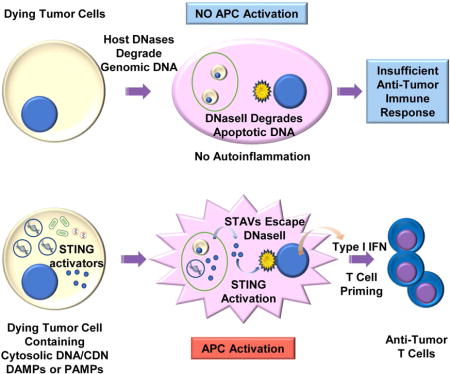
The ability of dying cells to activate antigen presenting cells (APCs) is carefully controlled to avoid unwarranted inflammatory responses. Here we show that engulfed cells containing cytosolic dsDNA species (viral or synthetic) or cyclic di-nucleotides (CDNs) are able to stimulate APCs, via extrinsic STING-signaling, to promote antigen cross-presentation. In the absence of STING agonists, dying cells were ineffectual in the stimulation of APCs in trans. Cytosolic STING activators, including CDNs, constitute cellular danger associated molecular patterns only generated by viral infection or following DNA-damage events, that rendered tumor cells highly immunogenic. Our data sheds insight into the molecular mechanisms that drive appropriate anti-tumor adaptive immune responses, while averting harmful autoinflammatory disease, and provides a therapeutic strategy for cancer treatment.
Keywords: STING, Innate Immunity, STING-dependent adjuvants (STAVs), cyclic di-nucleotides (CDNs), Antigen presenting cells (APCs), anti-tumor T cells, melanoma
INTRODUCTION
The generation of T cells that recognize specific antigens presented on tumor cells constitutes an important host defense response that has evolved to eliminate the development of cancer (Gajewski et al., 2013). The mechanisms underlining the stimulation of antigen presenting cells (APCs) and the priming of tumor specific T cells remains to be clarified but is thought to involve the generation of immune stimulatory type I interferon (IFN) and other cytokines (Diamond et al., 2011; Fuertes et al., 2011; Ma et al., 2013; Marichal et al., 2011; Zitvogel et al., 2015). Generally, non-tumorigenic cells undergoing apoptosis avoid activating APCs, an event that would otherwise cause lethal autoinflammatory disease due to chronic cytokine production (Ahn and Barber, 2014; Kawane et al., 2001; Nagata et al., 2003). By likely adopting comparable processes, tumor cells are also able to avoid the activation of APCs and thus the subsequent spontaneous generation of anti-tumor T-cells. In contrast, microbial infected cells are potently able to activate APCs following engulfment, and can robustly generate anti-pathogen T-cells (Belz et al., 2004; Smith et al., 2003). While DNA from engulfed cells is known to play a key role in stimulating APCs (Ahn et al., 2012), how phagocytes differentiate between an apoptotic/tumorigenic cell and an infected cell, all of which harbor considerable amounts of cellular DNA, remains to be fully determined.
The innate immune pathways governing the stimulation of cytokine production involve STING (stimulator of interferon genes) signaling within phagocytes such as CD8+ dendritic cells (Barber, 2015; Corrales et al., 2015; Deng et al., 2014; Woo et al., 2014). STING directly senses cyclic dinucleotides (CDNs) including c-di-GMP or c-di-AMP secreted by invading intracellular bacteria or cyclic-GMP-AMP (cGAMP) generated by the cellular synthase, Cyclic GMP-AMP synthase (cGAS) following association with cytosolic dsDNA species such as microbial DNA or even self –DNA (Ablasser et al., 2013; Barber, 2014). Generally, the cytosol of the cell is free of DNA, since it would aggravate STING-dependent cytokine production, an event that can lead to lethal autoinflammatory disease. For example, self- DNA leaked from the nucleus of cells, following cell division or following DNA damage, is prevented from activating STING signaling by the exonuclease DNase III (Trex1) (Ahn et al., 2014a). Consequently, defects in Trex1 function lead to severe autoinflammatory disease due to undigested self-DNA triggering STING activity. In addition, following the engulfment of apoptotic cells, phagocyte-dependent DNase II plays a critical role in digesting the DNA within the dead cell, to prevent it from activating STING-signaling extrinsically (Ahn et al., 2012). Loss of DNase II function is embryonic lethal in murine models due to high level cytokine production being instigated by overactive STING activity.
Thus, the eradication of apoptotic cells is designed to avoid invoking an inflammatory event. Dying cells are generally poor activators of phagocytes and immunologically indolent due to the nuclear compartmentalized genomic DNA being degraded by host DNases to prevent the intrinsic and extrinsic activation of STING. Given this, we thus postulated that apoptotic cells containing cytosolic dsDNA species or CDNs could potentially stimulate APCs, via extrinsic STING-signaling, to promote the cross presentation of antigen. Plausibly, cytosolic STING activators, including CDNs, constitute potent cellular danger associated molecular patterns (DAMPs) only generated by viral infection or following DNA damage events, that can render apoptotic and tumorigenic cells immunogenic and able to facilitate anti-tumor T cell activity. Our goals were to further gain insight into the molecular mechanisms that drive appropriate adaptive immune responses, while averting harmful autoinflammatory disease and possibly providing effective therapeutic strategies for the treatment of cancer.
RESULTS
STING-dependent adjuvants (STAVs) can stimulate the activation of macrophages in a STING-dependent manner
To further investigate the importance of STING in facilitating adaptive immune responses we generated a variety of DNA-dependent nucleic acids and examined their ability to activate STING signaling. We noted that transfected cytosolic dsDNA, modified on the 5’end to help prevent exonuclease degradation, greater than approximately 30 bp in murine cells (MEFs; murine embryonic fibroblasts) or 70 bp in human cells (human telomerase reverse transcriptase immortalized cell lines (hTERT) and primary human macrophages) were required for the efficient activation of STING (Figures S1A-S1I). The effects, following transfection appeared to be largely independent of sequence specificity and both AT or GC rich structures were readily able to trigger STING activity. As a result of these endeavors, an AT rich STING activating dsDNA ligand of 90 bp with modified 5-ends (referred to STAVs; STING-dependent adjuvants) was used for further study. Following the transfection of a variety of cells including murine B16 cells, we noted that the majority of the STAVs remained in the cytosol of the cell in as yet undefined cellular compartments (Figure 1A). Quantitation studies indicated that the cytosolic STAVs constituted approximately 1% of the total cellular DNA content (Figure S1J).
Figure 1. Activation of macrophages by exogenous cytosolic DNA (STAVs) in engulfed apoptotic cells.
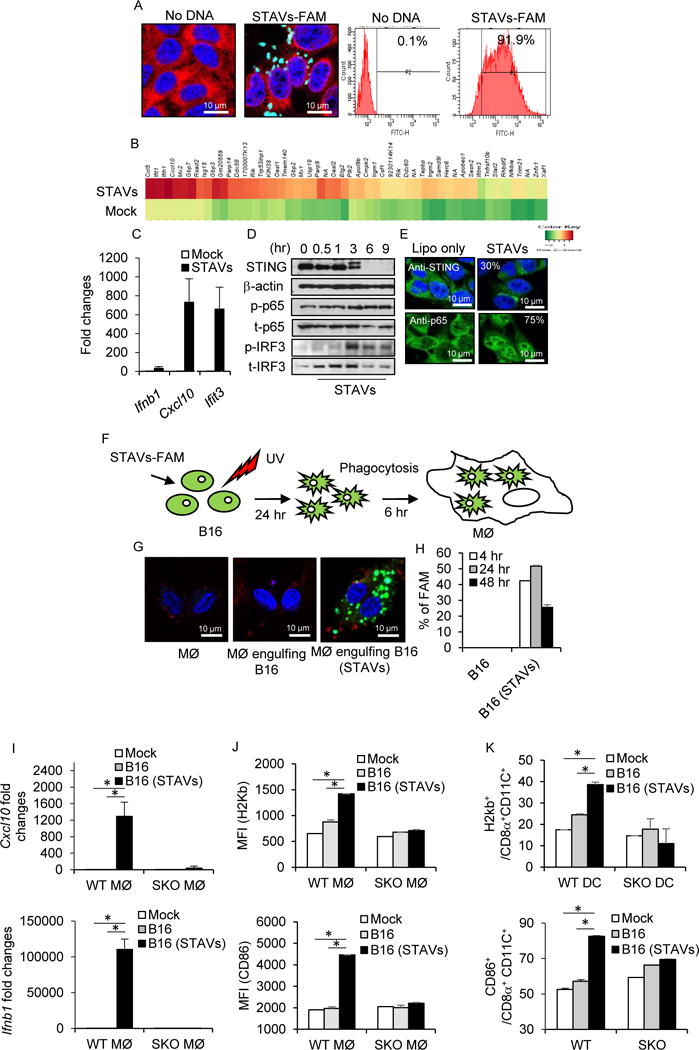
(A) Confocal analysis and flow cytometry analysis of B16 OVA cells (B16) transfected with FAM labeled STAVs (green). DAPI (blue) and anti-calreticulin (red) as counter staining; bar size, 10 μm. (B) Gene array analysis of B16 cells transfected with 3 μg/ml of STAVs for 6 hr. Highest variable inflammation-related genes are shown. (C) qRT-PCR analysis of Ifnb1, Cxcl10 and Ifit3 in B16 OVA cells same as in (B). (D) Western blot analysis of STING, p65 and IRF3 in B16 cells transfected with 3 μg/ml of STAVs and incubated for time courses as indicated. (E) Immunofluorescent Microscopy analysis using anti-STING and anti-p65 in B16 cells at 3 hr after transfection of STAVs (3 μg/ml); bar size, 10 μm. (F) Schematic representation of the phagocytosis of B16 cells by macrophages. B16 cells were transfected by 3 μg/ml of STAVs for 3 hr and irradiated by UV (120 mJ/cm). The irradiated B16 cells were feed to macrophages (MØ) at 24 hr after UV irradiation. (G, H) Confocal Microscopy analysis (G) and flow cytometry analysis (H) in macrophages following cellular engulfment of B16 cells transfected with FAM labeled STAVs. (I) qRT-PCR analysis of Cxcl10 and Ifnb1 in wild type (WT) and STING knock out (SKO) macrophages (WT MØ and SKO MØ) following engulfment of B16 cells in presence or absence of STAVs. (J) Flow cytometry for H2Kb and CD86 on macrophages following phagocytosis of B16 cells. (K) Flow cytometry for CD86 and H2Kb on CD8α+CD11C+ dendritic cells following phagocytosis of B16 cells containing STAVs. Data is representative of at least three independent experiments. Error bars indicate mean ± SD. *, p<0.05; Student’s t-test. See also Figures S1, S2, S3 and Table S1.
To evaluate the importance of STING signaling in the stimulation of APCs following cellular engulfment, we transfected B16 cells with STAVs, routinely obtaining greater than 90% transfection efficiency (Figure 1A) and confirmed that B16 cells exhibited cytosolic DNA-dependent STING signaling as determined by observing an increase in cytokine production, including Cxcl10 (Figures 1B, 1C and Table S1). This event coincided with and increased in STING and IRF3 phosphorylation (Figures 1D and S1K) and STING and NF-κB (p65) trafficking (Figure 1E). Cytokine levels were noted to be elevated in the presence of STAVs compared to unmodified dsDNA or cGAMP, perhaps due to being protected from host DNases (Figure S2). This was performed since we have previously noted that numerous types of cancer cells appear defective in STING signaling, perhaps to avoid DNA-damage mediated cytokine production that can occur via intrinsic STING signaling, which likely alerts the immune system to the vicinity of the damaged cell (Xia et al., 2016a; Xia et al., 2016b). We next fed UV treated STAVs containing cells to phagocytes (BMDM; Murine bone marrow derived macrophages from wild type (WT) or Sting knockout (SKO)) in vitro (Figure 1F). UV irradiation triggered both Annexin V and PI positive cell staining in greater than 90 % of the cells, with the cells retaining STAVs for up to 24 hr (>90 %) (Figures S3A and S3B). Approximately 50 % of the macrophages consistently engulfed the cells as determined using B16 cells transfected with fluorescently labelled STAVs (Figures 1F–1H and S3C). B16 cells containing STAVs robustly induced the production of cytokines in macrophages that was dependent on extrinsic STING signaling within the macrophages (Figures 1I and 1J). However, UV treated B16 cells alone or B16 cells containing Poly I:C failed to stimulate the macrophages as verified by measuring Cxcl10, type I IFN, macrophage maturation marker (CD86) and MHI class I (H2kD) (Figures 1I, 1J and S3D). Irradiated B16 cells harboring STAVs were also observed to activate dendritic cells (Murine bone marrow derived dendritic cells; BMDC) as verified by upregulation of the maturation markers CD86 and H2kD (Figure 1K). We confirmed that cells, containing but not containing STAVs, undergoing alternate forms of cell death, such as initiated by cisplatin or hydrogen peroxide, also induced the production of cytokines in macrophages (Figures S3E and S3F). A similar effect was observed following the phagocytosis of HEK293 cells containing STAVs (Figure 2 and Table S2). This data indicated that exogenous cytosolic DNA species present in engulfed apoptotic cells can potently stimulate the activation of macrophages in trans in a STING-dependent manner.
Figure 2. Extrinsic STING signaling dependent gene expression in macrophages.
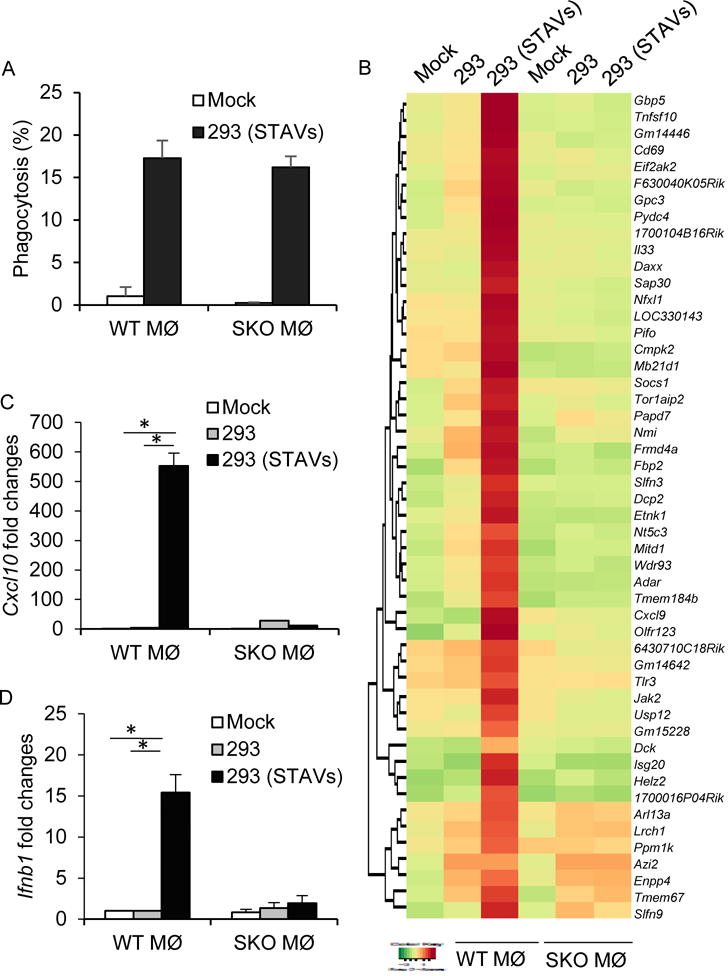
(A) Flow cytometry analysis in macrophages following cellular engulfment of UV-irradiated HEK293 cells (293) transfected with FAM labeled STAVs. (B) Gene array analysis of WT and SKO macrophages following engulfment of irradiated 293 cells with/without STAVs. Highest variable inflammation-related genes are shown. (C, D) qRT-PCR analysis of Cxcl10 (C) and Ifnb1 (D) in same as in (A). Data is representative of at least three independent experiments. Error bars indicate mean ± SD. *, p<0.05; Student’s t-test. See also Table S2.
The stimulation of engulfing macrophages can occur in trans by cytosolic DNA promoting STING signaling in macrophages
It is possible that the transfected cytosolic DNA could stimulate intrinsic STING signaling within the treated cell and facilitate the production of immunoregulatory cytokines that may provoke APC activation. Thus, we treated mouse embryonic fibroblasts (MEFs) that lacked STING or cGAS, with STAVs and confirmed that both STING and cGAS were required to produce cytokines such as type I IFN in the presence of cytosolic DNA (Figures 3A and 3B). UV treated MEFs were then incubated with macrophages to ascertain the latter’s activation (Figure 3C). Our results again indicated that only apoptotic cells containing cytosolic DNA were able to activate macrophages (Figures 3D and 3E). Indeed, MEFs lacking cGAS or STING, transfected with STAVs remained able to activate APCs, indicating that STING-dependent cytokine production within the engulfed cell was not completely essential for macrophage activation in vitro (Figures 3A, 3D, 3E, and S4A). A similar effect was observed following the phagocytosis of B16 Sting KO (B16-SKO) and B16 cGAS KO (B16-cGAS KO) generated by CRISPR technology (Figure S4B). This data suggests that exogenous cytosolic DNA but not indigenous cellular DNA, is responsible for the stimulation of the APCs including macrophages. To complement these studies, we transfected STAVs into human 293T cells that lack both cGAS and STING. Unlike normal human hTERT cells, 293T cells are consequently unable to produce type I IFN in response to STAVs (Figures 3F and S4C). We then incubated STAVs treated 293T cells with murine macrophages and observed that only 293T cells containing STAVs were able to stimulate cytokine production in engulfing macrophages (Figures 3G and 3H). This effect was dependent on STING signaling in the macrophages (Figures 3G and 3H).
Figure 3. Macrophage stimulation in trans by cytosolic DNA.
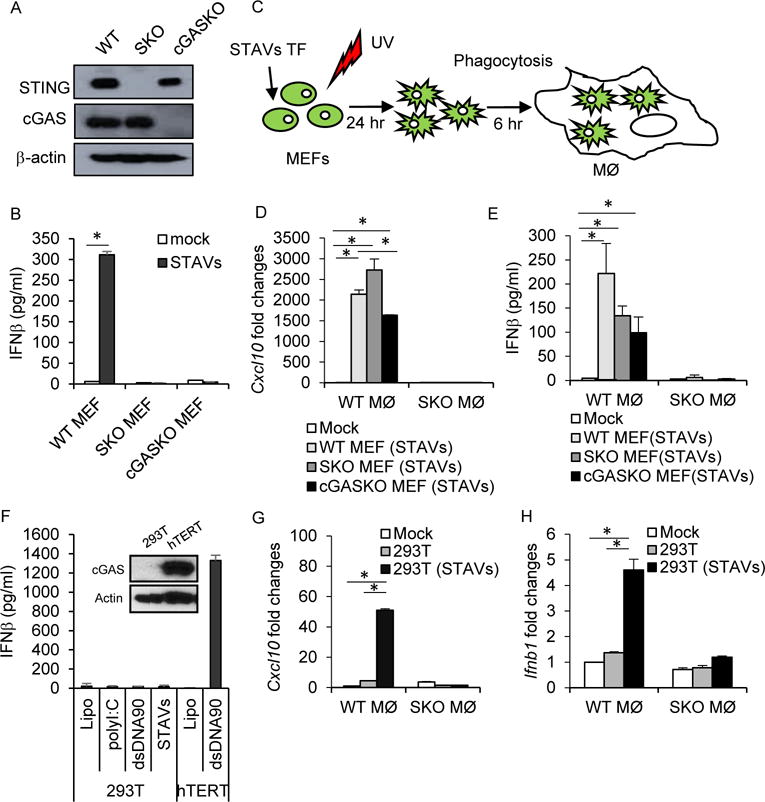
(A) Western blot analysis of STING and cGAS in mouse embryonic fibroblasts (MEFs). (B) ELISA analysis of IFNβ in WT, SKO and cGAS knock out (cGAS KO) transfected with 3 μg/ml of STAVs. (C) Schematic representation of the phagocytosis of MEFs by macrophages. (D, E) qRT-PCR analysis of Cxcl10 (D) and Ifnb1 (E) in WT and SKO macrophages following engulfment of UV irradiated WT, SKO and cGAS KO MEFs with 3 μg/ml of STAVs. (F) ELISA analysis of IFNβ in 293T and hTERT cells transfected with STAVs. (G, H) qRT-PCR analysis of Cxcl10 (G) and Ifnb1 (H) in WT and SKO macrophages following engulfment of UV irradiated 293T cells with or without STAVs. Data is representative of at least three independent experiments. Error bars indicate mean ± SD. *, p<0.05; Student’s t-test. See also Figures S4.
CDNs in engulfed cells can trans-activate STING signaling in macrophages
To extend our studies, we next further examined the importance of extrinsic STING signaling within the engulfing APC. To accomplish this, we treated B16 cells with STAVs and fed them to murine macrophages lacking cGAS or STING. This analysis surprisingly indicated that macrophages lacking cGAS or TLR9, but not STING, were readily able to be activated by B16 cells containing STAVs (Figure 4A and S4B). Thus, STING, but not cGAS, is essential for the activation of APCs following cellular engulfment. This event was also noted to not require TLR9 (Figure 4A). This data suggests that STAVs within the UV treated cell could be binding to an alternate DNA binding STING activating molecule within the APCs or conversely that CDNs were being generated within the B16 cell by cGAS, which are able to activate STING signaling extrinsically within the APC, in trans. Our analysis indeed indicated that cGAS was expressed in B16 cells and could generate CDNs in the presence of cytosolic DNA, as determined by mass spectrometry (Figure 4B). Next, we transfected 293T cells, which lack cGAS or STING, with STAVs. Unlike B16 or MEFs cells, 293T cells cannot generate CDNs (Figures 3F and S4D). We had previously confirmed that 293T cells containing cytosolic DNA (STAVs) were able to modestly stimulate wild type macrophages, but not macrophages that lacked STING (Figures 3G and 3H). We further observed that macrophages lacking cGAS were similarly unable to be activated by 293T cells containing STAVs, since the STAVs are probably unable to activate cGAS-generated CDNs production within the APC to activate STING signaling (Figure 4C). To extend this study, we therefore reconstituted 293T cells with a plasmid expressing cGAS. This experiment indicated that 293T cells expressing cGAS could readily generate CDNs, as determined by mass spectrometry and that these intrinsic CDN containing cells could activate STING signaling in macrophages, in trans (Figures 4C, 4D, S4E and S4F). These results were confirmed using a human colon cancer cell-line that similarly lacks cGAS (HT116) (Figures 4E and S4G). cGAS lacking cells containing STAVs were also observed to activate cGAS lacking phagocytes less than WT or SKO phagocytes, again suggesting that CDN’s are stimulatory in trans (Figures 3D, 3E and S4B).
Figure 4. Extrinsic activation of the cGAS/STING axis in macrophages.
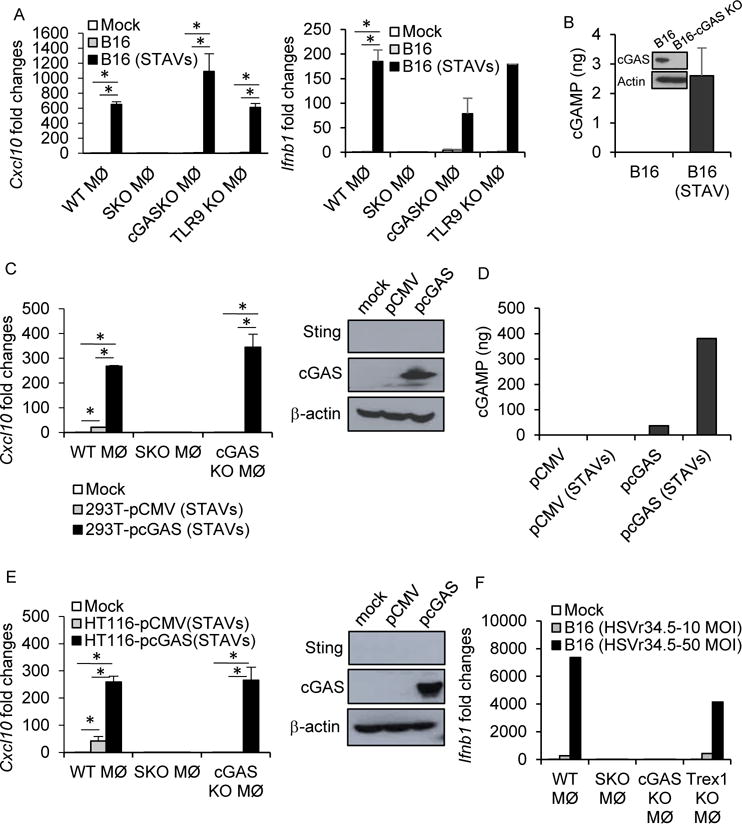
(A) qRT-PCR analysis of Cxcl10 and Ifnb1 in WT, SKO, cGAS KO and TLR9 KO macrophages following engulfment of UV-irradiated B16 cells in presence of 3 μg/ml of STAVs or absence. (B) cGAS expression by Western blot and cGAMP amount by a hybrid mass spectrometer in B16 cells. B16-cGAS knock out cells were used as negative control for WB. (C) qRT-PCR analysis of Cxcl10 in WT, SKO, and cGAS KO macrophages following engulfment of UV-irradiated 293T cells containing 3 μg/ml of STAVs. The 293T cells were reconstituted with pcGAS or pCMV as control vector. (D) Measurement of cGAMP levels by a hybrid mass spectrometer in 293T cells same as in (c). (E) qRT-PCR analysis of Cxcl10 in WT, SKO, and cGAS KO macrophages following engulfment of UV-irradiated HT116 cells containing STAVs. The HT116 cells were reconstituted with pcGAS or pCMV as control vector. (F) qRT-PCR analysis of Ifnb1 in WT, SKO, cGAS KO, and Trex1 KO macrophages following engulfment of B16 cells infected with HSVγ34.5. The HT116 cells were reconstituted with pcGAS or pCMV as control vector. Error bars indicate mean ± SD. *; p<0.05, Student’s t-test. See also Figure S4.
To complement this study, we evaluated the importance of STING signaling in phagocyte activation in relation to other forms of cell death and cytosolic DNA species. Our data indicated that the DNA virus HSV1 (γ34.5) functioned similarly to transfected STAVs following infection of B16 cells. That is, only engulfed viral-infected cells and not uninfected cells, could activate macrophages. Further, this event similarly occurred in a cGAS/STING-dependent manner (Figure 4F). We next evaluated whether STING signaling was important for the immunogenic effects not only of DNA virus infected cells but also of chemotherapeutic drugs (Cisplatin) (Galluzzi et al., 2015; Pfirschke et al., 2016). We have previously shown that Cisplatin-induced DNA damage can activate intrinsic STING signaling (Ahn et al., 2014b). B16 cells heterologously expressing cGAS were treated with Cisplatin or UV or γ-irradiation, the results of which indicated the release of host nuclear DNA into the cytosol which colocalized with cGAS (Figure S5A) (Ahn et al., 2014b; Harding et al., 2017; Mackenzie et al., 2017). STAV’s were also confirmed as co-localizing with cGAS (Figure S5B). We observed that Cisplatin treatment could generate CDN’s in a cGAS-specific manner similar to UV or γ-irradiation (Figure S5C). Engulfed Cisplatin treated B16 cells were observed to stimulate phagocyte activity as determined by measuring the upregulation of H2kb and cytokines such as Cxcl10 (Figures S5D and S5E). Finally, mice treated with Cisplatin were able to generate enhanced CD8+ T cell activity to subcutaneously growing B16 tumors that was dependent on STING signaling (Figure S5F). Our analysis would suggest that in addition to UV and γ-irradiation, DNA virus infected cells and alternate forms of DNA damage that generates cytosolic DNA species can stimulate immune activity through extrinsic STING-dependent signaling in phagocytes. Collectively, our data indicate that STAVs transfected into cells can extrinsically activate the cGAS/STING axis in phagocytes. Second, STAVs and other cytosolic DNA species generated from DNA damage events can generate CDNs within the treated cell, which can also act in trans to stimulate extrinsic STING signaling in APCs. Further, reconstitution of cGAS, for example within a tumor cell, can generate CDNs that are able to similarly act in trans to stimulate the activation of phagocytes via STING. Finally, host macrophages can distinguish between a viral infected and uninfected dying cell predominantly through cGAS/STING detection of viral cytoplasmic DNA, analogous to STAVs.
Apoptotic cells containing STAVs escape degradation by DNase II and stimulate extrinsic cGAS/STING signaling
Our data thus indicate that cytosolic DNA (STAVs) or CDNs can activate APCs, directly or indirectly, and facilitate antigen cross presentation (Figures S6A-6D). It remained unclear why cytosolic DNA (STAVs) and not indigenous cellular DNA is able to stimulate APCs in trans. However, nuclear DNA, and plausibly mitochondrial DNA, undergoes degradation during the apoptotic process. The responsible nucleases within the nuclei that cleaves genomic DNA between nucleosomes involves CAD (caspase activated DNase) (McIlroy et al., 2000; Nagata et al., 2003). Thus, fragments of nuclear DNA sufficient to activate STING signaling may not be generated, nor escape into the cytosol. Following engulfment by macrophages, the remainder of the DNA is likely degraded by additional DNases (DNase II) within the lysosomal compartment of APCs (Ahn et al., 2012; Kawane et al., 2001). It is thus plausible that cytosolic DNA species (STAVs/viral DNA) escape cellular degradation within the apoptotic cell and following engulfment in APCs is more readily available to escape the lysosomal compartment and stimulate extrinsic cGAS/STING signaling in the APC. To explore this further, we retrieved macrophages from mice lacking DNase I (Figures 5A and 5B), II (Figures 5A and 5C), or III (Trex1) (Figures 5A and 5D) and fed them B16 cells containing or lacking STAVs. Our data again indicated that apoptotic cells poorly activate normal macrophages, perhaps explaining why tumor cells are generally non-immunogenic. However, APCs lacking DNase II, but not DNase I or DNase III, exhibited an increase in cytokine production following the engulfment untreated apoptotic cells (Figures 5 and S6E). This event was greatly augmented in STAVs containing cells (Figure 5). This data confirms that DNases such as CAD efficiently degrade self-DNA within apoptotic cells to help prevent the activation of macrophages (Nagata and Tanaka, 2017). However, DNase II in APCs predominantly ensures that any engulfed apoptotic DNA that escapes degradation is broken down in lysosomal compartments into non-STING activating nucleotides. Significantly, cells containing cytosolic DNA (STAVs) or cytoplasmic viral DNA likely escape degradation in the apoptotic cell and are able to stimulate extrinsic cGAS/STING signaling in the APCs, prior to degradation of by DNase II.
Figure 5. Apoptotic cells containing STAVs escape degradation by DNase II.
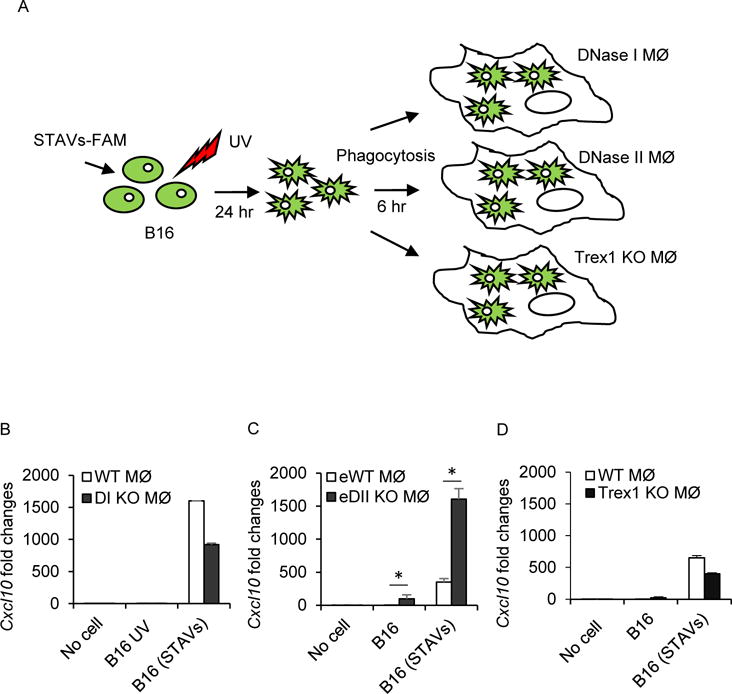
(A) Schematic representation of the phagocytosis of B16 cells by DNase I, DNase II, or Trex1 knockout macrophages. B16 cells were transfected by STAVs for 3 hr and irradiated by UV (120 mJ/cm). The irradiated B16 cells were feed to three different genotypes of macrophages (MØ) at 24 hr after UV irradiation. (B-D) qRT-PCR analysis of Cxcl10 in DNase I KO (B), DNase II KO (C) and Trex1 KO (D) macrophages at 6 hr following engulfment of B16 cells containing STAVs. B16; UV irradiated, B16 (STAVs); Transfected with STAVs and UV irradiated, DI KO, DNase I KO; eWT, WT embryo; eDII KO, DNase II KO embryo. Error bars indicate mean ± SD. *, p<0.05; Student’s t-test.
STAVs induce anti-tumor immunity involved the generation of cytotoxic T cell activity
Our data indicates that cells have devised efficient ways to eliminate the lethal possibility of self-DNA activating innate immune sensor pathways such as those governed by STING (Ahn et al., 2012; Ahn et al., 2014a). Likely, tumor cells also utilize this process to remain immunologically indolent. To determine whether STAVs could render tumor cells immunogenic in vivo, we intratumorally inoculated B16-OVA melanoma cells containing or lacking STAVs into immunocompetent C57BL/6J mice (Figure 6A). We observed that B16-OVA cells treated with STAVs exhibited less growth compared to mice inoculated with untreated cells (Figure 6B). In addition, the STAVs containing cells did not exert any anti-tumor activity in the absence of STING in the recipient mice (Figure 6C). The ability of the STAVs to inhibit tumor growth involved the generation of anti-tumor CTL to the tumor, as determined by measuring anti-SIINFEKL specific CD8+ T cells (Figure 6D). To start to evaluate the importance of dendritic cells in this process, we utilized C57/BL6 syngeneic mice lacking STING in CD11c+ cells (CD11C-SKO). B16 SIY cells (expressing the peptide SIYRYYGL) were inoculated into the flanks of syngeneic C57/BL6 CD11C-SKO mice. After 9 days, STAVs were injected intratumorally. CD8+ T cells were isolated from splenocytes and analyzed by ELISPOT to evaluate anti-tumor T cell responses. This analysis indicated that mice lacking STING specifically in CD11c+ dendritic cells generated less anti-tumor T cell activity compared to wild type mice thus implying a key role for STING signaling in T cell priming and these subsets of APCs (Figures 6E and 6F). Further, our data would indicate that tumor cells containing STAVs could be potent stimulators of anti-tumor immunity.
Figure 6. Anti-tumor activity of STAVs in B16 OVA melanomas bearing mice.
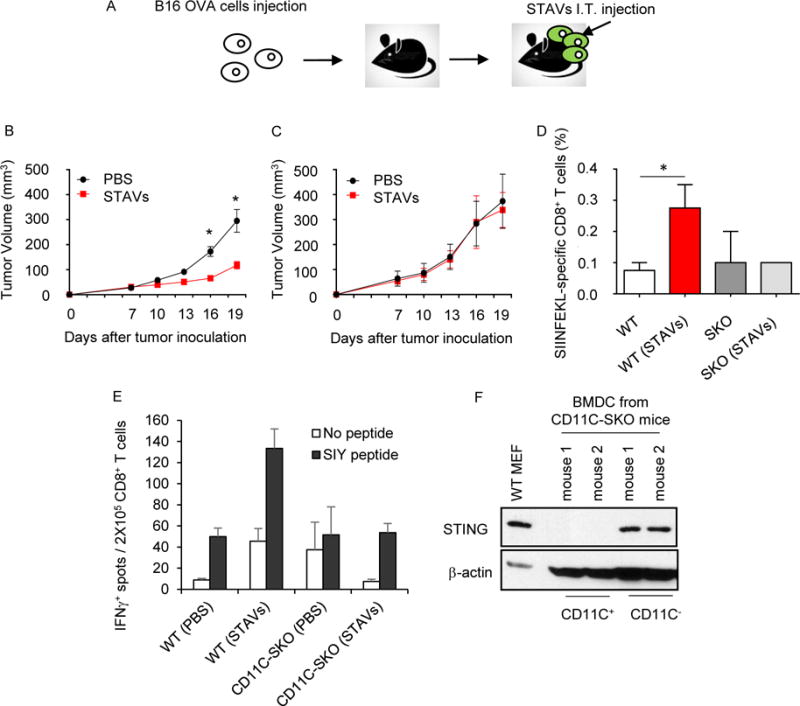
(A) Schematic representation of intratumoral injection of STAVs in B16 OVA melanoma bearing mice. The mice were subcutaneously injected with B16-OVA cells on the flank. 10 μg of STAVs was injected intratumorally (i.t.) every three days. (B, C) Tumor volumes from WT (n=7/group) (B) and SKO mice (n=7/group) (C) were measured on the indicated days. (D) Frequency of OVA specific CD8+ T cells in the spleen from WT (n=4/group) and SKO (n=4/group) mice injected with STAV or PBS as control. (E) IFNγ ELISPOT assay in CD8+ T cells from WT or CD11C-cre;Stingloxp (CD11C-SKO) mice. The mice were subcutaneously injected with B16-SIY cells on the flank. 10 μg of STAVs was injected intratumorally (i.t.) every three days. CD8+ T cell priming was evaluated by IFNγ ELISPOT. (F) STING expression in CD11C+ bone marrow derived dendritic cells (BMDC) from the CD11C-SKO mice. CD11C+ cells were selected by CD11C microbeads (CD11C+), lysed and analyzed for STING expression by western blot. The unlabeled cell fraction was used as a control (CD11C−).
Error bars indicate mean ± SD. *, p<0.05; Student’s t-test. See also Figure S6.
STAVs containing cells provide an effective immunotherapeutic cell-based therapy against melanoma metastasis
To additionally evaluate whether STAVs containing cells were able to generate immune responses in murine models, B16 cells were loaded with STAVs, irradiated and used to immunize C57BL/6J mice (Figure 7A). This study indicated that STAVs containing cells were competent to stimulate CD8+ T cell priming and the generation of type II IFN, a further indicator of CTL activity (Figures 7B and S7A-S7C). STAVs dependent type II IFN production was dependent of STING and partially dependent of cGAS, but not TLR9. This data would indicate that tumor cells containing STAVs could be potent stimulators of anti-tumor immunity, through STING signaling.
Figure 7. Protection of lung metastasis by B16 OVA requires STING.
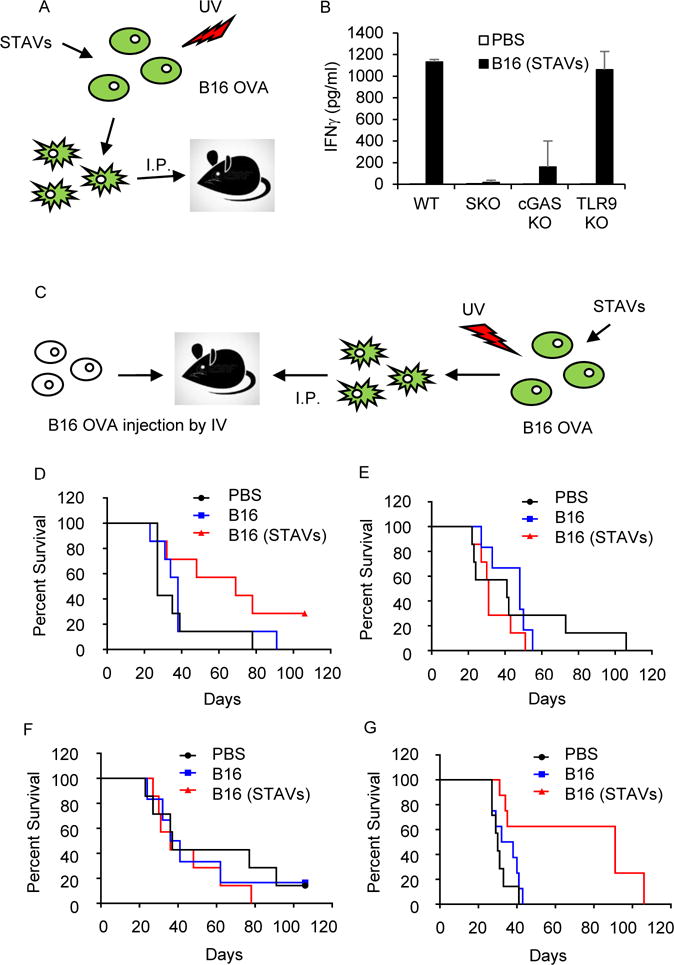
(A) Schematic representation of the dead cell immunization. B16 OVA cells were transfected by STAVs for 3 hr and irradiated by UV (120 mJ/cm). After 24 hr, WT, SKO, TLR9 KO, and cGAS KO mice were intraperitoneally (i.p.) injected with irradiated B16 cells with/without STAV, twice every week. (B) IFNγ measurement in splenocytes from WT, SKO, TLR9 KO, and cGAS KO mice at 7 days after the second immunization. Error bars indicate mean ± SD. (C) Schematic representation of post-vaccination for B16 OVA mediated lung metastasis. WT, TLR9KO, SKO, and cGAS KO mice were intravenously (i.v.) injected with B16 OVA cells (5×104 cells/mouse). On day 1, 3, 7, and 14, the mice were I.P injected by UV irradiated B16 OVA cells (1×106 cells/mouse) with STAVs. (D-G) Survival rates from WT (p=0.0429, n=7/group) (D), SKO (p=0.2616, n=7/group) (E), cGAS KO (p=0.4075, n=7/group) (F), and TLR9KO (p=0.0012 n=7/group) (G) mice were monitored. PBS: Control group treated with PBS, B16: post-vaccinated group with UV-irradiated B16 cells, B16 (STAVs): post-vaccinated group with UV-irradiated B16 cells with STAVs. p values are based on Log-rank test, with p<0.05 considered statistically significant. See also Figure S7.
To examine this further, we inoculated C57BL/6J mice with B16 OVA melanoma cells, intravenously, which induces metastatic disease. We subsequently vaccinated the tumor bearing mice with B16 OVA cells loaded with STAVs (Figure 7C). We found that the B16 OVA tumors killed the majority of mice within 40 days in WT mice (Figure 7D). However, mice treated with B16 OVA STAVs had a median life of 70 days, with 40% of the mice alive after 100 days (Figure 7D). Mice lacking STING (Figure 7E) or cGAS (Figure 7F), but not TLR9 (Figure 7G), however, succumbed to lethal disease similar to wild type mice, indicating the importance of STING signaling in combatting cancer metastasis (Figures 7E–7G). Finally, we tested whether the blockade of the immunosuppressive receptor programmed cell death-1 (PD-1) could enhance the antitumor activity of STAV containing tumor cells. We thus vaccinated tumor bearing mice with B16 OVA cells loaded with STAVs, in the presence or absence of anti-PD-1 antibody. We observed that survival rates significantly increased in mice treated with both anti-PD-1 and STAV containing cells (Figure S7D). This data indicates that anti-PD-1 could improve the therapeutic efficacy of STAVs. To confirm our anti-tumor strategy, we used another syngeneic tumor model, namely Balb/c mice bearing STAV treated or untreated TS/A (breast adenocarcinoma)-Luc cells. Similarly, we observed that mice carrying metastatic TS/A survived longer when treated with TS/A-luc cells loaded with STAVs (Figures S7E and S7F). Significantly less luciferase expressing metastatic TS/A cells were detected in the STAV treated mice, using in vivo imaging systems (IVIS) (Figure S7G). The immunized mice with TS/A (STAVs) were re-challenged with TS/A cells in the flank of the mice, 153 days after the first exposure of the metastatic tumor. Tumor growth in the immunized mice with TS/A (STAVs) was clearly shown to be significantly reduced or prevented entirely (Figure S7H). Thus, cells containing STAVs may provide an effective immunotherapeutic cell-based therapy for the treatment of cancer.
DISCUSSION
STING-signaling has become a key mechanism for stimulating innate and adaptive immune responses in response to sensing DNA species in the cytosol. Generally, STING functions as a sensor to detect microbial invasion, although leaked self-DNA generated following DNA damage events or cell division can also trigger STING activity and cytokine production (Barber, 2015). These responses would presumably alert the immune system to the damaged area, with the cells being eliminated by phagocytosis. However, overactive STING activity is now known to cause lethal autoinflammatory disease (Ahn and Barber, 2014; Barber, 2015; Liu et al., 2014; Nagata and Tanaka, 2017). Thus, STING signaling is rigidly controlled to avoid autoimmune malaise. Indeed, defects in cellular DNases that are accountable for degrading cytosolic self-DNA species are responsible for instigating severe autoinflammatory disease caused via STING signaling (Ahn et al., 2012; Ahn et al., 2014a; Rodero and Crow, 2016).
Our data here, demonstrates that cytosolic dsDNA species present within a dying cell can activate extrinsic STING signaling in phagocytes likely following association with cGAS which would generate CDNs. It is likely that the cytosolic DNA species avoid being degraded by nuclear DNases, responsible for degrading genomic DNA. Such cytosolic species appear significantly more capable of activating STING in phagocytes, as a result of avoiding being degraded within the dying cell and/or by phagocytes following engulfment. DNase resistant cytosolic species were thus more competent at activating STING in trans. In addition, CDNs generated from an infected or damaged cell are able to directly activate STING-dependent signaling in APCs in trans, to trigger immune responses, the cross-presentation of antigen and the generation of T cells. CDNs are only generated following DNA-damaging events, or after infection and are thus effective DAMPs. CDNs generated within a dying cell are also predominantly resistant to the activity of DNases within the engulfed phagocyte, unlike susceptible cellular DNA, which makes them highly efficient at stimulating extrinsic STING signaling, although they are likely susceptible to other forms of negative regulation. It is likely that tumorigenic cells closely mimic normal cells undergoing cell death and avoid triggering STING signaling. Thus, tumorigenic cells are predominantly non-immunogenic.
We suggest that intrinsic STING signaling is likely important within a stressed cell (DNA damage, or microbial infection) to alert APCs to the damaged region, while the extrinsic STING signaling component within phagocytes is critical for the production of cytokines such as type I IFN that facilitate cross presentation events. Since, self DNA from the apoptotic/tumor cell is efficiently degraded to prevent autoinflammatory events, tumor cells are, as a consequence, immunologically indolent (Nagata and Tanaka, 2017). Our data indicates that CD8αCD11C+ APC’s play a key role in facilitating STING-dependent T-cell priming (Belz et al., 2004). However, Macrophages (CD11b+) cells may also play a role and are presently under investigation. Our data also indicates that numerous tumor cells exhibit defective STING signaling through loss of cGAS and/or STING (Xia et al., 2016a; Xia et al., 2016b). This would presumably enable the DNA damaged cells to avoid alerting the immune system for elimination. However, our analyses also indicate that by suppressing CDN production, a tumor cell or infected cell would also additionally evade the activation of the APC itself, in trans. Our data further indicates that reconstitution of CDNs and STING signaling may not only alert the immune system to the tumor cell, but also stimulate the adaptive immune responses by activating phagocytes in trans. It has been suggested that certain forms of cell death are more immunostimulatory than other types (Deng et al., 2014; Dou et al., 2017; Galluzzi et al., 2015; Harding et al., 2017; Kono and Rock, 2008; Pfirschke et al., 2016; Russell and Peng, 2017; Yatim et al., 2017). However, a variety of studies have shown that the immunomodulatory effects are modest and generally most dying cells are relatively non-immunogenic, to avoid autoinflammation, as described (Nagata and Tanaka, 2017; Woo et al., 2014). Our data here indicates that STAVs, viral or DNA-damage generated self-dsDNA species within a dying cell, or CDNs can greatly enhance the immunogenicity of tumor cells. The use of STAVs to treat cancer, either directly into tumors, or carried inside tumor cells, or as vaccine adjuvants may provide powerful anti-tumor therapies. Moreover, the generation of CDNs within a tumor cell may also provide a valid approach to stimulate anti-tumor T cell responses. It is possible to consider that loss of cGAS (CDNs) and/or STING in tumors may help explain resistance to radiation treatment, which exerts its anti-tumor effects in part through the stimulation STING-dependent immune responses (Harding et al., 2017; Mackenzie et al., 2017). Thus loss of STING may help explain resistance not only to radiation treatment but also to the immunological benefits of chemotherapy. Suppression of STING signaling may also help explain mechanisms of DNA-virus-mediated oncolysis. Activators of STING–signaling (STAVs) may be useful for the immuno-treatment of cancer following intratumoral inoculation, similar to strategies using CDNs (Woo et al., 2014). Alternatively, or in combination, tumor cells loaded with STAVs may also be a potent mechanism for stimulating anti-tumor T cell responses. Reconstitution of cGAS-STING signaling in cancer cells may increase the efficacy of a diversity of immuno-oncology-related tumor treatments in addition to the use of checkpoint inhibitors (Rivera Vargas et al., 2017). Indeed, it is plausible that complementary treatments consisting of a variety of methods to augment STING signaling may have significant benefits for the treatment of cancer. Finally, our data provides a plausible explanation for how phagocytes are able to distinguish between an uninfected versus a microbial infected cell.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Glen Barber (gbarber@med.miami.edu).
EXPETIMENTAL MODEL AND SUBJECT DETAILS
Mice
STING knockout mice (SKO) were generated in our laboratory (Ishikawa and Barber, Nature 2008) and have backcrossed with a C57BL/6J. Wild type C57BL/6J mice (WT) were purchased from Jackson Laboratory. cGAS Knockout mice (cGAS KO) were kindly provided by Dr. Herbert W. Virgin IV (Washington University, School of Medicine). TLR9 Knockout mice (TLR9KO) were purchased from Jackson Laboratory. Balb/C mice were purchased from Jackson Laboratory. To generate the conditioning Sting knockout mice, we developed animals with the STING gene floxed. In brief, the exons 1-5 were flanked with loxp sites in C57/BL6 derived embryonic stem (ES) cells in order to render STING susceptible to Cre-mediated recombination. The floxed STING mice were crossed to mice expressing Cre under a cell specific promoter (CD11C-Cre) to generate STINGloxp/CD11C-cre (CD11C-SKO) mice. Mice care and study were conducted under approval from the Institutional Animal Care and Use Committee of the University of Miami. Mouse genotypes from tail biopsies were determined by real-time PCR with specific probes designed for each gene by commercial vendor (Transnetyx).
In Vivo Tumor Models
For anti-tumor effects, mice were subcutaneously injected with 5×105 cells of B16-OVA or B16-SIY on the right flank. One week later, when tumors were 50mm3 in volume, 10 μg of STAVs was injected intratumorally (i.t.) every three days 3 times. The tumor volume was measured using calipers and calculated with the formula V=(length × width2)/2. For post-vaccination in tumor bearing mice, mice were intravenously (i.v.) injected with B16 OVA cells (1 × 105 cells/mouse), or TS/A-luc cells (1 × 105 cells/mouse). At 1, 3, 7, and 14 days, mice were injected intraperitoneally (i.p.) with UV irradiated B16 OVA cells (> 1 × 106 cells/mouse) or TS/A-luc cells (> 4×106 cells/mouse). Survival rates were monitored for 110 days. B16-OVA cells or TS/A-luc cells were transfected with STAVs at 3 μg/ml for 3 hr and irradiated by a UVC500 UV crosslinker at 120 mJ/cm2 followed by 24 hr incubation. Antibodies were administered simultaneously with irradiated cells at 100 mg/mouse using isotype control IgG (BE0091 BioXcell) or anti PD-1 (J43 BE0033-2 BioXcell).
Cell Lines
B16-OVA cells (B16) were kindly provided by Dr. Eli Gilboa (University of Miami) and B16 SIY cells were generously provided by the Weichselbaum lab (University of Chicago). Both cells were cultured in complete Dolbecco’s Modified Eagle Medium (DMEM, Invitrogen) including 10% heat-inactivated fetal calf serum (FCS, Invitrogen). HEK293 (293), 293T cells were purchased from ATCC and cultured in their appropriate media. hTERT-BJ1 cells were originally purchased from CLONTECH and cultured in its appropriate medium following manufacturer’s instruction. TS/A and TS/A-luc cells were maintained in RPMI 1640 supplemented with 10% FBS, 5% penicillin-streptomycin, and 10 mg/ml puromycin for TS/A-luc.
Primary Cell Cultures
Mouse embryonic fibroblasts (MEFs) were obtained from e13.5 embryos by a standard procedure. Bone Marrow Derived Macrophages (BMDM; MØ) and Bone Marrow Derived Dendritic cells (BMDC; DCs) were isolated from hind-limb femurs of 8-10 weeks old WT, SKO, cGASKO, TLR9KO, DNase I KO, DNase II KO, or DNase III (Trex1) KO mice. The hematopoietic cells from the bone marrow were cultured in complete DMEM (Invitrogen) including 10 ng/ml of Mouse Recombinant Colony-Stimulating Factor (M-CSF, R&D Systems) or Recombinant Muse GM-CSF (GM-CSF, R&D Systems) for 10 to 14 days.
METHOD DETAILS
STING-Dependent Adjuvants (STAVs)
An ssDNA sense strand modified with Phosphorothioates (S-oligos) at the end (polyA90mer) was annealed to ssDNA antisense strand with same modification as anti-sense strand (polyT90mer); STAVs. Each oligos were synthesized and modified from TriLink Biotechnologies (Table S3).
In Vitro Phagocytosis
MØ was cultured as described above and seeded on 12 well plates (2×105 cells/well). B16-OVA cells were transfected with STAVs at 3 μg/ml for 3 hr and irradiated by a UVC500 UV crosslinker at 120 mJ/cm2 followed by 24 hr incubation. The treated B16-OVA cells were added to macrophages for 6 hr. Following vigorous washing to remove unengulfed B16-OVA cells, the macrophages were harvested for RNA extraction for qPCR and Gene array analysis. To check phagocytosis efficiency, macrophages were collected at 4 hr after phagocytosis and stained using APC-anti-mouse CD11b antibody (eBiosciences). Percentage of phagocytosed cells was assessed by flow cytometry using a LSRII instrument (Becton Dickinson, USA).
Gene Array Analysis
Total RNA was isolated from cells or tissues with RNeasy Mini kit (Qiagen). RNA quality was analyzed by Bioanalyzer RNA 6000 Nano (Agilent Technologies). Gene array analysis was examined by Affymetrix Mouse Gene array (2.0 ST Array) at the Center for Genome Technology, John P. Hussman Institute for Human Genomics, University of Miami. Gene expression profiles and statistical analysis was performed by Biostatistics & Bioinformatics Shared Resource at Sylvester Comprehensive Cancer Center, University of Miami.
Quantitative Real-Time PCR (qPCR)
Total RNA was extracted from cells using Trizol method and reverse transcribed by M-MLV reverse transcriptase (Promega). Real-time PCR was performed with the TaqMan gene Expression Assay.
Cell Death Induction and Analysis
B16-OVA cells were transfected with 3 μg/mL STAVs for 3 hr and then were treated by UV light (120 mJ/cm2). After 24 hr at 37°C, 5% CO2, cells were collected and fed to bone marrow-derived macrophages for 6 hr. B16-OVA cells viability was assessed by flow cytometric analysis with a FACSCantoII (Becton Dickinson, USA) after staining with 1 μg/mL Propidium Iodide (eBiosciences) and APC-labeled Annexin V (eBiosciences).
Transfection Efficiency
STAVs transfection efficiency into B16-OVA or 293T cells was evaluated by Flow cytometry analysis. The cells were transfected with 3 μg/mL of FAM-labelled STAVs. After 24 hr, cells were collected and analyzed by flow cytometry using a LSRII instrument (Becton Dickinson, USA).
Antigen Presentation Assay
Bone marrow-derived macrophages were transfected with 3 μg/mL of STAVs for 24 hr and then pulsed with or without SIINFEKL (3 μM). After 2 hr, cells were stained with APC-labeled anti-H-2Kb-SIINFEKL (clone 25-D1.16) (Biolegend) and FITC-labeled anti-mouse CD11b (eBiosciences). Presentation of SIINFEKL on H-2Kb was evaluated by flow cytometry using a LSRII instrument (Becton Dickinson, USA).
Immunofluorescence Staining
Cells were fixed with 4 % paraformaldehyde in DMEM for 15 min at 37°C and were permeabilized with 0.2 % Triton X-100. Fixed and permeabilized cells were blocked with 10 % BSA in PBS, incubated with primary antibodies in 2 % BSA in PBS and then incubated with fluorophore-conjugated secondary antibodies (with DAPI counterstaining). After staining, cells were mounted in anti-fade mounting solution (Invitrogen) and examined under Leica SP5 spectral confocal inverted microscope.
Liquid Chromatography Mass Spectrometry (LC-MS) Analysis
The cells were transfected with 3 μg/mL of STAVs for 3 hr or 1 μg/mL of pCMV or pcGAS plasmids for 24 hr. Following appropriate treatment, 1×107 cells were pelleted and snap-frozen in liquid nitrogen and stored at -80 °C before further processing. To extract cGAMP, frozen cells were thawed on ice and lysed in cold 80% (vol/vol) methanol with 2% (vol/vol) acetic acid (HAc). Cyclic-di-GMP was supplemented as internal standard. Cell lysates were cleared by centrifugation at 4 °C, 10,000 × g for 10 min. Pellets were further extracted in 20% (vol/vol) methanol with 2% HAc twice and all extracts were pooled. cGAMP was then enriched by solid-phase extraction (SPE) using HyperSep Aminopropyl SPE Columns (Thermo Scientific) as previously described in Gao et al, 2015. Briefly, columns were activated by 100 % methanol and washed twice with 2% HAc; after drawing through the extracts, columns were washed twice with 2% HAc and once with 80% methanol, and finally eluted with 2% (vol/vol) ammonium hydroxide in 80% methanol. The eluents were spin-vacuumed to dryness, reconstituted in liquid chromatography (LC)/MS-grade water and stored at −20 °C before subject to LC/MS analysis. LC/MS analysis was performed by R. Marshall Wilson Mass Spectrometry Facility at the University of Cincinnati. Chromatography was performed using a Thermo Scientific Surveyor MS Pump Plus pump and Micro AS autosampler. The separation was isocratic on a Water’s XBridge Amide column (3.5 um, 2.1×100 mm) at 200 μl/min using 18:82 water:acetonitrile 6.3 mM ammonium hydroxide and 6.3 mM ammonium bicarbonate. The samples were introduced into a Thermo Scientific LTQ-FT, a hybrid mass spectrometer consisting of a linear ion trap and a Fourier transform ion cyclotron resonance mass spectrometer. The standard electrospray source was used operated in negative ion mode. cGAMP was quantitated using the m/z 522 product ion from the collision-induced dissociation of the deprotonated parent ion at m/z 673. An external calibration curve derived from eight standards was used in the quantitation and acquired before and after the samples were analyzed. The c-di-GMP component was quantitated from the m/z 344 product ion originating from the deprotonated m/z 689 parent.
In Vivo Imaging of Mice
Tumor-bearing Balb/C mice were transferred to the UM in vivo imaging system (IVIS) facility for imaging at 35 days after TS/A-luc administration. During imaging, mice were injected with luciferin (Caliper Life Science; 150 mg/kg diluted in PBS), anesthetized using isoflurane, and imaged 5 min after luciferin injection with the time post-injection matching between groups.
OVA Specific CD8+ T cell Analysis and Ifnγ ELISA
1-2 × 106 splenocytes were stained with H-2Kb/SIINFEKL Pro5® Pentamer (ProImmune, UK) for 10 minutes at 22°C. Cells were then washed twice and incubated with FITC-labeled anti-mouse CD8 (eBiosciences) and PECy5-labeled anti-mouse CD19 (ProImmune, UK) antibodies for 20 minutes on ice, shielded from light. Following two further washes, cells were resuspended in fixative and analyzed by flow cytometry using a LSRII instrument (Becton Dickinson, USA). For IFNγ ELISA, 1 × 106 splenocytes were plated and stimulated with SIINFEKEL peptide (10 μg/ml). At 2 days after stimulation, the supernatant was harvested and IFNγ production was estimated by BD mouse Ifnγ kit.
IFNγ γELISPOT assay
For tumor-specific CD8+ T cells functional assay, one day after the last STAVs injection, CD8+ T cells were isolated from spleens by using magnetic beads (Miltenyl Biotec). 2×105 CD8+ T cells were stimulated with 10 μg/ml of SIY peptide for 3 days. ELISPOT assays were performed to detect the cytokine spots for IFNγ according to product protocol (R&D).
DimerX Analysis
H-2Kb SIY peptide was purchased from MBL medical. SIY peptide loaded H-2K[b]:Ig fusion protein was prepared by incubation of the BD DimerX I H-2K[b]:Ig dimer with 320 molar excess SIY peptide in PBS at 37 ℃ overnight. For immunofluorescent staining, isolated splenocytes were incubated with peptide loaded H-2K[b]:Ig protein on ice for 1 hr after treatment with anti-mouse CD16/32 FcBlocker (BD Pharmingen). After washing, cells were incubated with PE-conjugated A85-1 mAb (BD Pharmingen), and co-stained with α-CD3e (clone 145-2C11, BD Pharmingen) and α-CD8α (clone 53-6.7, BD Pharmingen) antibodies. Samples were acquired using FACS Canto II flow cytometry (BD Biosciences), and analyzed by FlowJo software V10 (TreeStar).
QUANTIFICATION AND STATISTICAL ANALYSES
Comparisons of differences between 2 groups were analyzed by Student’s t test. The Log-rank test was used to evaluate the significant difference of survival curve. The data were considered to be significantly different when P < 0.05 (*).
DATA AND SOFTWARE AVAILABILITY
Microarray gene expression data have been deposited to GEO (accession no: GSE110931). It can be accessed at https://na01.safelinks.protection.outlook.com/?url=https%3A%2F%2Fwww.ncbi.nlm.nih.gov%2Fgeo%2 Fquery%2Facc.cgi%3Facc%3DGSE110931&data=02%7C01%7Cagriswold%40med.miami.edu%7C1ce4b4d 6a68c49f9892808d57936c37f%7C2a144b72f23942d48c0e6f0f17c48e33%7C0%7C0%7C6365481984737 92397&sdata=4SKmgwYmFQOP2RA1v%2BcmsbZwCVNTWITl54AbhzEOo%2BQ%3D&reserved=0.
Supplementary Material
SIGNIFICANCE.
Tumor cells are notoriously non-immunogenic and acquire properties that enable them to evade the immunosurveillance system. Here we demonstrate that defects in cytosolic DNA activated innate immune signaling pathways, controlled by STING, enable precancerous cells to escape DNA-damage mediated cytokine production. We further demonstrate that tumor cells additionally avoid aggravating antigen presenting cells (APCs) by efficiently simulating regular dying cells which following phagocytosis are prevented from initiating autoinflammatory responses. However, dying tumor cells containing exogenous cytosolic DNA, viral DNA or cyclic di-nucleotides potently activate APCs in trans through extrinsic STING-signaling to generate cytotoxic T lymphocytes (CTLs) activity. Our data provides an explanation as to how tumor cells avoid triggering immune responses and provides a therapeutic strategy to stimulate anti-tumor immunity.
HIGHLIGHTS.
Defective STING signaling helps tumor cells to escape the immunosurveillance system.
Phagocytosed dying cells are immunologically indolent to avoid autoinflammation.
Tumor cells mimic these properties to avoid anti-tumor T cell activity.
STING agonists in tumor cells potently increase tumor immunogenicity in trans.
Acknowledgments
We thank Ms. Delia Gutman and Ms. Auristela Rivera for mouse maintaining and genotyping; Dr. Hyelim Park for assisting flow cytometry analysis; Dr. Eli Gilboa for providing B16 OVA cells; Dr. Ralph R. Weichselbaum at University of Chicago for providing B16 SIY cells; Biostatistics & Bioinformatics Shared Resource at Sylvester Comprehensive Cancer Center for gene array analysis; Dr. Larry Sallans at University of Cincinnati for Mass Spectrometry analysis. This study was supported by the NIH grants (5R01AI079336-10).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
J.A. carried out the majority of experiments and data analysis; T.X. participated in designing STAVs and Mass spectrometry analysis; A.R.C performed flow cytometry analysis and helped with animal experiments; D.B performed Balb/c metastasis experiments; and G.N.B wrote the manuscript and supervised the study.
DECLARATION OF INTERESTS
The authors declare no competing interests.
The authors have a patent related to this work. Application number: 15/120,694/Undergoing Examination.
Ahn et al. show that dying tumor cells that contain STING-dependent adjuvants activate STING signaling in engulfing macrophages to trigger immune responses, cross-presentation of antigens, and generation of cytotoxic T cells, providing a potential cancer therapeutic strategy.
References
- Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Rohl I, Hopfner KP, Ludwig J, Hornung V. cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature. 2013;498:380–384. doi: 10.1038/nature12306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn J, Barber GN. Self-DNA, STING-dependent signaling and the origins of autoinflammatory disease. Curr Opin Immunol. 2014;31:121–126. doi: 10.1016/j.coi.2014.10.009. [DOI] [PubMed] [Google Scholar]
- Ahn J, Gutman D, Saijo S, Barber GN. STING manifests self DNA-dependent inflammatory disease. Proc Natl Acad Sci U S A. 2012;109:19386–19391. doi: 10.1073/pnas.1215006109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn J, Ruiz P, Barber GN. Intrinsic self-DNA triggers inflammatory disease dependent on STING. J Immunol. 2014a;193:4634–4642. doi: 10.4049/jimmunol.1401337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn J, Xia T, Konno H, Konno K, Ruiz P, Barber GN. Inflammation-driven carcinogenesis is mediated through STING. Nat Commun. 2014b;5:5166. doi: 10.1038/ncomms6166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber GN. STING-dependent cytosolic DNA sensing pathways. Trends Immunol. 2014;35:88–93. doi: 10.1016/j.it.2013.10.010. [DOI] [PubMed] [Google Scholar]
- Barber GN. STING: infection, inflammation and cancer. Nat Rev Immunol. 2015;15:760–770. doi: 10.1038/nri3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belz GT, Smith CM, Eichner D, Shortman K, Karupiah G, Carbone FR, Heath WR. Cutting edge: conventional CD8 alpha+ dendritic cells are generally involved in priming CTL immunity to viruses. J Immunol. 2004;172:1996–2000. doi: 10.4049/jimmunol.172.4.1996. [DOI] [PubMed] [Google Scholar]
- Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, Woo SR, Lemmens E, Banda T, Leong JJ, et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep. 2015;11:1018–1030. doi: 10.1016/j.celrep.2015.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, Li XD, Mauceri H, Beckett M, Darga T, et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity. 2014;41:843–852. doi: 10.1016/j.immuni.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, Lee H, Arthur CD, White JM, Kalinke U, et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med. 2011;208:1989–2003. doi: 10.1084/jem.20101158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, Simithy J, Lan Y, Lin Y, Zhou Z, et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature. 2017;550:402–406. doi: 10.1038/nature24050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, Gajewski TF. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med. 2011;208:2005–2016. doi: 10.1084/jem.20101159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14:1014–1022. doi: 10.1038/ni.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunological Effects of Conventional Chemotherapy and Targeted Anticancer Agents. Cancer Cell. 2015;28:690–714. doi: 10.1016/j.ccell.2015.10.012. [DOI] [PubMed] [Google Scholar]
- Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature. 2017;548:466–470. doi: 10.1038/nature23470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawane K, Fukuyama H, Kondoh G, Takeda J, Ohsawa Y, Uchiyama Y, Nagata S. Requirement of DNase II for definitive erythropoiesis in the mouse fetal liver. Science. 2001;292:1546–1549. doi: 10.1126/science.292.5521.1546. [DOI] [PubMed] [Google Scholar]
- Kono H, Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol. 2008;8:279–289. doi: 10.1038/nri2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Montealegre Sanchez GA, Tenbrock K, Wittkowski H, Jones OY, Kuehn HS, et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med. 2014;371:507–518. doi: 10.1056/NEJMoa1312625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Adjemian S, Mattarollo SR, Yamazaki T, Aymeric L, Yang H, Portela Catani JP, Hannani D, Duret H, Steegh K, et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity. 2013;38:729–741. doi: 10.1016/j.immuni.2013.03.003. [DOI] [PubMed] [Google Scholar]
- Mackenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, Olova N, Sutcliffe H, Rainger JK, Leitch A, et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature. 2017;548:461–465. doi: 10.1038/nature23449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marichal T, Ohata K, Bedoret D, Mesnil C, Sabatel C, Kobiyama K, Lekeux P, Coban C, Akira S, Ishii KJ, et al. DNA released from dying host cells mediates aluminum adjuvant activity. Nat Med. 2011;17:996–1002. doi: 10.1038/nm.2403. [DOI] [PubMed] [Google Scholar]
- McIlroy D, Tanaka M, Sakahira H, Fukuyama H, Suzuki M, Yamamura K, Ohsawa Y, Uchiyama Y, Nagata S. An auxiliary mode of apoptotic DNA fragmentation provided by phagocytes. Genes Dev. 2000;14:549–558. [PMC free article] [PubMed] [Google Scholar]
- Nagata S, Nagase H, Kawane K, Mukae N, Fukuyama H. Degradation of chromosomal DNA during apoptosis. Cell Death Differ. 2003;10:108–116. doi: 10.1038/sj.cdd.4401161. [DOI] [PubMed] [Google Scholar]
- Nagata S, Tanaka M. Programmed cell death and the immune system. Nat Rev Immunol. 2017;17:333–340. doi: 10.1038/nri.2016.153. [DOI] [PubMed] [Google Scholar]
- Pfirschke C, Engblom C, Rickelt S, Cortez-Retamozo V, Garris C, Pucci F, Yamazaki T, Poirier-Colame V, Newton A, Redouane Y, et al. Immunogenic Chemotherapy Sensitizes Tumors to Checkpoint Blockade Therapy. Immunity. 2016;44:343–354. doi: 10.1016/j.immuni.2015.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera Vargas T, Benoit-Lizon I, Apetoh L. Rationale for stimulator of interferon genes-targeted cancer immunotherapy. Eur J Cancer. 2017;75:86–97. doi: 10.1016/j.ejca.2016.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodero MP, Crow YJ. Type I interferon-mediated monogenic autoinflammation: The type I interferonopathies, a conceptual overview. J Exp Med. 2016;213:2527–2538. doi: 10.1084/jem.20161596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell SJ, Peng KW. Oncolytic Virotherapy: A Contest between Apples and Oranges. Mol Ther. 2017;25:1107–1116. doi: 10.1016/j.ymthe.2017.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CM, Belz GT, Wilson NS, Villadangos JA, Shortman K, Carbone FR, Heath WR. Cutting edge: conventional CD8 alpha+ dendritic cells are preferentially involved in CTL priming after footpad infection with herpes simplex virus-1. J Immunol. 2003;170:4437–4440. doi: 10.4049/jimmunol.170.9.4437. [DOI] [PubMed] [Google Scholar]
- Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, Duggan R, Wang Y, Barber GN, Fitzgerald KA, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. 2014;41:830–842. doi: 10.1016/j.immuni.2014.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia T, Konno H, Ahn J, Barber GN. Deregulation of STING Signaling in Colorectal Carcinoma Constrains DNA Damage Responses and Correlates With Tumorigenesis. Cell Rep. 2016a;14:282–297. doi: 10.1016/j.celrep.2015.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia T, Konno H, Barber GN. Recurrent Loss of STING Signaling in Melanoma Correlates with Susceptibility to Viral Oncolysis. Cancer Res. 2016b doi: 10.1158/0008-5472.CAN-16-1404. [DOI] [PubMed] [Google Scholar]
- Yatim N, Cullen S, Albert ML. Dying cells actively regulate adaptive immune responses. Nat Rev Immunol. 2017;17:262–275. doi: 10.1038/nri.2017.9. [DOI] [PubMed] [Google Scholar]
- Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G. Type I interferons in anticancer immunity. Nat Rev Immunol. 2015;15:405–414. doi: 10.1038/nri3845. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.