

Abstract
This work describes a nanoparticle system for systemic delivery of therapeutics that incorporates a means of tracking the fate of the nanocarrier and its residual drug payload in vivo by photoluminescence. Porous silicon nanoparticles (PSiNPs) containing the proapoptotic antimicrobial peptide payload, D[KLAKLAK]2, are monitored by measurement of the intrinsic photoluminescence (PL) intensity and the PL lifetime of the nanoparticles. The PL lifetime of the PSiNPs is on the order of microseconds, substantially longer than the nanosecond lifetimes typically exhibited by conventional fluorescent tags or by autofluorescence from cells and tissues; thus emission from the nanoparticles is readily discerned in the time-resolved PL spectrum. It is found that the luminescence lifetime of the PSiNP host decreases as the nanoparticle dissolves in phosphate buffered saline solution (37 °C), and this correlates with the extent of release of the peptide payload. The time-resolved PL measurement allows tracking of the in vivo fate of PSiNPs injected (via tail vein) into mice. Clearance of the nanoparticles through the liver, kidneys, and lungs of the animals is observed. The luminescence lifetime of the PSiNPs decreases with increasing residence time in the mice, providing a measure of half-life for degradation of the drug nano-carriers.
Keywords: Theranostics, bioimaging, biodegradation, time-gated luminescence imaging, Photoluminescence lifetime
Table of contents
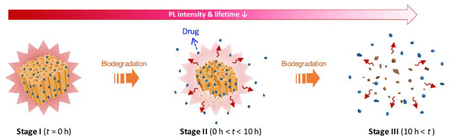
The intrinsic photoluminescence lifetime of porous silicon nanoparticles provides a means to track the status and fate of this drug delivery system. Their long-lived photoluminescence discriminates the nanoparticles from natural tissue autofluorescence, and the biodistribution and in vivo elimination can be assessed by time-gated photoluminescence imaging.
Graphical Abstract
In the context of drug delivery, one of the primary tasks of a nanoparticle is to release its drug payload and then disappear–either by excretion or by dissolution into harmless components. In order to probe the fate of nanoparticle drug delivery vehicles (liposomes, silica, polymers, etc.), it is currently a common practice to attach a fluorescent reporter dye to the superstructure or to the payload, or to both. Fluorescent molecular dyes are often used to visualize biodistribution, systemic elimination, and pharmacokinetics of a nanocarrier because they provide high fidelity spatial and temporal information.[1–7] While they have proven very useful, conjugated dyes have two significant limitations: first, they can become detached from the nanoparticle carrier and so there is often a question as to whether one is monitoring an intact nanoparticle construct or just the free dye molecule. Second, a fluorescent probe has little utility in determining the status of the nano-carrier; in particular, whether or not the nanoparticle has released its payload, and to what extent the nano-carrier has degraded or dissolved. These are important questions that are difficult to answer either in vivo or in vitro with most nanotherapeutic systems.[8–14]
Porous silicon nanoparticles (PSiNPs) offer a solution to both of the above challenges. PSiNPs consist of relatively large (200 nm) and highly porous (50–80% porosity) nanoparticles that contain a high spatial density of emissive silicon nanocrystallite domains (< 3 nm) within their skeleton. Because photoluminescence (PL) is intrinsic to the silicon skeleton,[15] the PL image directly and conclusively reports on the location of the nanoparticle carrier. The indirect band gap of silicon gives it a very long-lived excited state, and in silicon nanocrystals the PL lifetime can be in the range of hundreds of nanoseconds to many tens of microseconds.[16–17] This feature allows the use of time-gating to eliminate short-lived tissue autofluorescence when imaging the materials in vivo, and it has been shown to provide as much as 100-fold enhancement in signal to noise.[18] Porous silicon is attractive because, in addition to the PL imaging potential, the pores can be loaded with small molecule, protein, or nucleic acid therapeutics for treatment of various diseases.[19–23] The material has been demonstrated as a minimally toxic, bioresorbable,[24–26] and versatile platform for specific tissue targeting, drug delivery, and imaging in various in vitro and in vivo disease models.[27–30]
In addition to the ability of the intrinsic PL signal to directly report the location of the nano-carrier, the PL emission energy and the excited state lifetime both depend on the size of the nanostructure, so these spectral features can provide information on the extent of degradation of the material. It is already well established that as a silicon nanostructure dissolves in an aqueous medium, it displays a spectral blueshift and a decrease in lifetime of the excited state.[31–33] Also, prior work has reported correlations between these spectral properties and payload release from PSiNPs in vitro, which has been described as a ‘self-reporting’ characteristic.[34–35] In this work we show how analysis of a combination of the intensity and the lifetime of PL allows both tracking and assessment of the state of degradation of PSiNPs, and we provide the first demonstration of the application of this concept in an animal model.
The PSiNPs were prepared by electrochemical etching of highly doped p-type single crystalline silicon wafers in ethanolic hydrofluoric acid electrolyte, followed by lift-off, ultrasonic fracture, and activation of PL as reported previously.[36–37] Use of a “perforation etching” anodization waveform[36] yielded PSiNPs with a well-defined mesoporous structure (pore size ~10 nm) and narrow nanoparticle size distribution with a mean diameter of ~180 nm (Figure 1a). Photoluminescence was activated by growth of a native oxide on the porous silicon skeleton, generating a Si-SiO2 core-shell type of structure that passivates non-radiative surface defects and increases quantum yield from the quantum-confined crystalline silicon core.[38–39] The resulting PSiNPs displayed a relatively broad PL emission spectrum in the red/near-infrared region (500–950 nm, Figure 1b).
Figure 1. Evolution of photoluminescence characteristics of PSiNPs during dissolution.

(a) Transmission electron microscope (TEM) image of PSiNPs. Inset: photograph of PSiNP solution under UV irradiation (λex: 365 nm). (b) Time series of steady-state PL emission spectra (λex: 365 nm) of PSiNPs incubated in PBS (pH 7.4, 37 °C), and (c) intensity (red solid squares) and wavelength (blue open circles) of the emission maximum as a function of incubation time. (d) Family of time-resolved PL emission spectra from PSiNPs in PBS, measured at 10 μs increments post-excitation. Spectra were obtained immediately after dispersion of PSiNPs in PBS, corresponding to the 0 h sample in (b). (e) Time-resolved PL emission decays taken from the family of emission spectra in (d). Each trace represents PL intensity as a function of time after the pulsed excitation, measured at the indicated emission wavelength. Inset: normalized PL decay trace presented on a log(intensity) scale, showing the increase in PL lifetime with longer emission wavelength characteristic of Si nanoparticles. A complete set of PL spectra and intensity decay plots for nanoparticles dispersed in PBS, measured periodically over a period of 18 h are given in Supporting Information, Figures S1 and S2. (f) The PL excited state lifetime, τ, plotted as a function of emission wavelength and measured after the indicated incubation times in PBS. The value of τ was obtained from single-exponential fits of the PL intensity-time traces (Supporting Information, Figure S2). (g) Total intensity of emitted PL from PSiNPs, integrated from the PL spectra over the wavelength range 500 –950 nm and plotted as a function of time immersed in PBS at 37 °C. The raw data are given in Supporting Information, Figure S1. Inset: normalized integrated PL intensity presented on a log(intensity) scale, as a function of time. (h) Corresponding integrated PL emission lifetime (τ) extracted from the integrated PL decay traces in (g). Because the decays were obtained from spectra integrated over the wavelength range 500 –950 nm, the τ values represent intensity-weighted average lifetimes for the entire ensemble of emitters in the PSiNP sample. The average PL lifetime decreases with increasing incubation time (n=3, error bars represent standard deviation).
Consistent with prior observations,[37] incubation of the PSiNPs in aqueous phosphate-buffered saline (PBS) solution (pH 7.4, 37 °C, Figure 1b) led to slow dissolution of the nanoparticles, resulting in a steady decrease in intensity of the intrinsic PL from the nanoparticles. A monotonic blue shift of the peak wavelength of PL was also observed as the PSiNPs dissolved (Figure 1c). As noted previously,[16, 40] dissolution of a Si nanoparticle results in a blue shift in its emission spectrum due to a reduction in size of the quantum-confined nanocrystal.[41] The aqueous dissolution process can be described as a simultaneous hydrolysis of the SiO2 shell and oxidation of the crystalline Si skeleton (Scheme 1). Under the present experimental conditions, quantifiable PL signals could be obtained for up to 8 h of incubation; PL was almost undetectable after 18 h of incubation.
Scheme 1.

The process by which luminescent porous silicon nanoparticles dissolve in aqueous media involves a combination of hydrolysis and oxidation. In the hydrolysis step the SiO2 shell surrounding the silicon skeleton dissolves (top). Removal of this oxide exposes elemental silicon from the skeleton core, which then is oxidized by water to replace the SiO2 shell (bottom). The result of this continuous cycle is the gradual shrinking in size of the quantum-confined Si domains, generating a blue shift in the photoluminescence spectrum. Eventually the porous Si nanoparticles dissolve completely, resulting in total loss of photoluminescence.
We next measured the time-resolved emission spectra of the PSiNPs while they were undergoing dissolution in aqueous buffer. A spectrometer fitted with an intensified CCD detector was used to capture PL spectra in 10 μs increments after a pulse of incident UV excitation (Figure 1d, 1e, and Figure S1). Consistent with previous results, the emission decays of the PSiNPs were slower at the longer wavelengths.[42] The time-resolved emission spectrum can yield the excited state lifetime for emission, τ, which is inversely proportional to the sum of the radiative and non-radiative decay constants (see Supplemental Discussion S1, Supporting Information).[43] In the present case, we found that on the microsecond timescale the emission decays could be well fit with a single-exponential function. The wavelength-dependent emission lifetime (τ) values obtained from single-exponential fits of wavelength slices extracted from the family of time-resolved spectra (Figure 1e and Figure S2) showed a pronounced increase in lifetime with increasing emission wavelength, ranging from 17 μs at λem = 550 nm, to 118 μs at λem = 850 nm.[44–46] Sets of wavelength-dependent lifetime values were captured periodically from the PSiNPs as they underwent slow dissolution in phosphate-buffered saline. Notably, the PL lifetime at a given emission wavelength remained nearly constant during dissolution (Figure 1f). Because the emission wavelength is characteristic of size of the quantum-confined domains in the silicon skeleton of the nanoparticle,[47] this result indicates that, for a given nanocrystallite size, the dissolution process did not introduce additional non-radiative electron-hole recombination sites–in other words, the emission quantum yield was more dependent on the size of the silicon domains in the nanoparticle than on the length of time it had been exposed to the dissolution conditions.
As the lifetime measured at a given PL emission wavelength did not change appreciably during dissolution, the lifetime data provided a characteristic signature, similar to emission wavelength, that reported on the size of the nanocrystallites. Thus we next attempted to determine if the lifetime data could be used to determine the extent of degradation of the porous nanostructure host. One advantage of measurement in the time domain rather than the spectral domain is that autofluorescence and other endogenous fluorophores in tissue samples can emit at the same wavelength as a Si nanostructure, but the very short-lived excited states (typically < 5 ns) of most natural fluorophores are readily distinguished from the microsecond lifetime of the excited state of a silicon nanoparticle. A disadvantage of the time domain measurement is that the signals tend to be weaker and limited by instrumental sensitivity, which can be a particular challenge for in vivo measurements. In order to increase the signal strength from the time-resolved measurements in the present work, we explored the possibility of measuring lifetime from the entire emission spectrum, rather than from individual wavelength slices. Decay curves obtained by integrating the PL signal in the wavelength range 500–950 nm are presented in Figure 1g. Because of the wavelength dependence of the emission lifetime, these decays represent intensity-weighted average lifetimes for the entire ensemble of emitters in the PSiNP sample. The values of τ extracted from this data (fit to a single exponential) are given in Figure 1h. They show a consistent, steady decrease in PL emission lifetime as the nanoparticles dissolve. We tested a number of different PSiNP samples, prepared using various PL activation conditions. The samples displayed a relatively wide range of PL lifetimes initially (Figure S3 and Table S1), which can be expected based on the different conditions used in their preparation. For all the samples, the measured lifetime values decreased with increasing incubation time in the PBS solution. The change in lifetime was found to fit a simple exponential decay function (see Supplemental Discussion S2, Supporting Information), which yielded an empirical expression relating the age of the nanoparticles (x) with the measured emission lifetime values:
| (1) |
where τ is the PL emission lifetime measured at a given point in time, τ0 is the PL lifetime measured initially, prior to incubation in the aqueous medium, x is the time (in hours) the nanoparticles have been exposed to the incubation medium, and td is a constant representing the time constant (in hours) for degradation in the aqueous medium. The functional form of this expression is consistent with the reported kinetics of dissolution of solid particles for materials such as silica.[48–50] For the PSiNPs exposed to aqueous PBS at 37°C, the average time constant td for all the nanoparticle preparations was found to be 10.6 ± 0.3 hours. Thus once an initial decay constant (τ0) was acquired (prior to dissolution), the measured value of τ and eq. 1 could be used to predict the length of time that the particle had been exposed to the dissolution conditions. These results indicate that the PL lifetime is a physically intensive property, similar to the emission wavelength, that can be used to quantify the extent of degradation of PSiNPs. This contrasts with the measured steady-state intensity of PL, which depends on both the emission quantum yield and the concentration of emissive species being observed, and so is not effective at determining nanoparticle status. In other words, a decrease in PL intensity from an ensemble of PSiNPs could indicate either that a fixed number of nanoparticles have degraded, or that some of the nanoparticles being measured have diffused out of the field of observation. The PL emission lifetime measurement does not depend on the number density of nanoparticles, and so it is a more direct probe of the status of the PSiNPs.
Next, we evaluated the potential to use the PL lifetime data to monitor drug delivery with PSiNPs. A proapoptotic antimicrobial peptide, D[KLAKLAK]2, was labeled with fluorescein (6-FAM) to allow independent tracking of this model drug.[51–53] The polycationic peptide was then loaded into the negatively charged PSiNPs by electrostatic interactions. The loading capacity was 6 wt%, and no obvious aggregation or size change was observed for the peptide-loaded PSiNPs (Figure S4). In vitro release experiments performed in PBS at 37 °C displayed a time progression in the steady-state PL spectra (Figure S5) that was quite similar to the behavior of the empty PSiNPs (Figure 1b-c). The release kinetics of the peptide payload, determined by measuring fluorescence intensity from the 6-FAM label in the supernatant (after separation from the PSiNPs by centrifugation), matched the aqueous degradation profile of the PSiNPs: the loss in PL intensity from the PSiNPs correlated linearly with the appearance of fluorescence from the FAM label (Figure S5c). This correlation between drug release and decrease in steady-state PL intensity has been reported previously for the release of siRNA, protein, and small molecule payloads from porous Si particles.[30, 34–35, 54] However, the time-resolved PL spectrum has not previously been used to monitor payload release from a porous Si delivery vehicle.
The time-resolved PL spectral data provided a means to separate the short-lived emission of the FAM label on the drug payload from the longer-lived emission of the PSiNP carrier. While the emission maximum of the 6-FAM-labeled peptide (λem = 520 nm) was distinct from the nanoparticle emission maximum (λem ~ 700–800 nm), the relatively broad bands of the two emitters showed significant spectral overlap (Figure S5a). For the purpose of discerning if the nanoparticles could be distinguished from the molecular fluorophore in the time domain, we integrated the emission spectra over the wavelength range (λem = 500–950 nm), and measured this integrated intensity as a function of time post-excitation by the pulsed light source. The resulting time-resolved data showed a prompt decay component (τ1), corresponding to a combination of the short-lived (nanoseconds) fluorescence from the 6-FAM label and the shorter-lived portion of the emission from the PSiNP ensemble, and a longer decay component (τ2) corresponding solely to the PSiNPs (Figure S6). The time resolution of the PL decays measured in these experiments was 10 μs, so the short-lived component could be effectively removed simply by ignoring the first time point in the decay curves (Figure 2b). The longer-lived portion of each decay curve was then fit to a single exponential function, yielding the value τ2. The τ2 values from the peptide-loaded nanoparticles (Figure 2c) showed the same behavior as the τ values for the empty nanoparticles when subjected to the in vitro dissolution conditions (Figure 1h). The quantity of drug released from the peptide-loaded nanoparticles showed a correlation with this measured τ2 value (Figure 2c, inset), although the correlation was not as linear as the correlation of drug release with steady-state PL signal (Figure S5c). When fit to the degradation model of eq. 1, the degradation of the drug-loaded PSiNPs in aqueous PBS buffer at 37 °C displayed a similar time constant td for degradation as seen with the empty PSiNPs (10.5 vs 10.6 hours, respectively; s.d. = 0.3). From the time-resolved and the steady-state PL data we conclude that the dissolution kinetics of the drug-loaded PSiNPs did not substantially differ from the empty PSiNPs, at least under the in vitro conditions of this portion of the study.
Figure 2. Self-reporting payload delivery based on photoluminescence lifetime from PSiNPs.

(a) Schematic illustration of the self-reporting release of drug from the PSiNP carrier. (b) Integrated PL (λem = 500–950 nm) of the peptide-loaded PSiNPs as a function time; the family of curves represent sequential measurements during degradation in PBS at 37°C. Each PL spectral decay was measured after a pulse of incident excitation (λex: 365 nm), and each trace represents a decay measured at the incubation times indicated. The fluorescence spectrum of the 6-FAM-labeled peptide payload in these nanoparticles overlapped with the PL spectrum of the PSiNPs (Figure S5a), and thus the integrated PL intensity showed a prompt decay component corresponding primarily to the short-lived fluorescence of 6-FAM. The PL decay τ2 which corresponds to the long-lived PL decay of PSiNPs is plotted. The PL decay traces measured after 6 h of incubation were of low intensity and did not provide reliable τ values. Inset: normalized PL intensitiy presented on a log(intensity) scale (c) Representative PL lifetime values for the τ2 component, obtained from the PL decays in (b), and the cumulative amount of peptide released from the PSiNPs, both measured as a function of incubation time. The supernatant was separated by centrifugation and the released peptide was quantified by measurement of the fluorescence intensity from the 6-FAM-label on the peptide (λex: 480 nm/λem: 520 nm). Inset: correlation of cumulative peptide released with PSiNP PL lifetime, measured during the course of PSiNP dissolution.
We next evaluated the feasibility of harnessing the PL emission decay from the nanocarrier as a self-reporting probe in ex vivo animal tissues (Figure 3). The experiment was configured to determine if the relative age of the PSiNPs could be discerned from the emission decay data. Two types of peptide-loaded PSiNPs were prepared and directly injected at separate locations in liver tissue harvested from a mouse: the first type was an as-prepared PSiNP sample similar to the samples discussed above (“PSiNPs-1”, 30 μg/mL, 5 μL), and the second was “pre-aged” PSiNP sample which was taken from the same batch of nanoparticles as PSiNPs-1, but which had then been incubated in PBS at 37 °C for 4 h prior to injection (“PSiNPs-2”, 120 μg/mL, 10 μL). The PSiNPs-2 sample was aged to the point that the nanoparticles had released ~76% of their original peptide payload prior to injection into the liver tissue (Figure S7). The PL properties of both PSiNP samples were then measured at their respective sites of injection, and they displayed the characteristic long-lived luminescence of PSiNPs (Figure 3c). As expected, the emission lifetime of the as-prepared PSiNPs-1 sample was longer than the lifetime of the pre-aged PSiNPs-2 sample. We chose to perform these experiments in a mouse liver–the most challenging medium for imaging due to its high level of natural tissue autofluorescence relative to most other organs. Consequently, steady-state PL images of the liver prior to PSiNP injection showed strong tissue autofluorescence from the entire organ (Figure 3a), and this background made it impossible to discern the PL signal from injected PSiNPs (Figure 3d). Time-gated images (TGI) were acquired from the same organ, in the time window from 10 to 100 μs post-excitation. As reported previously,[55] the prompt (< 5 ns) autofluorescence from the endogenous fluorophores in the native tissues was eliminated in the TGI, and the non-injected liver showed no substantial signal (Figure 3b). However, the organ injected with PSiNPs showed distinctive signals at both the injection points PSiNPs-1 and PSiNPs-2 (Figure 3e).
Figure 3. Continuous wave images (CWI) and time-gated images (TGI) of PSiNPs before and after localized injection into mouse liver tissue and analyzed ex vivo (𝜆ex = 365 nm, 𝜆em > 460 nm, using a long-pass filter).

(a, b) Prior to introduction of the PSiNPs, substantial tissue autofluorescence from endogenous fluorophores in the liver is observed in the CWI (a), while this short-lived autofluorescence is eliminated in the TGI (b). Two types of PSiNPs were then locally injected in different regions of the same liver (indicated with the dotted line in d–f). Sample designated as PSiNP-1: as-prepared, injected dose: 30 μg/mL, 5 μL; Sample PSiNP-2: nanoparticle preparation similar to PSiNP-1 but aged in PBS at 37°C for 4 h prior to injection, injected dose: 120 μg/mL, 10 μL. (c) Time-resolved PL emission intensity decays for the two injected PSiNP samples and for the native liver tissue. Data were acquired from the region of interest (ROI) as indicated in (e). The excitation source is turned off at time = 0, and so the traces prior to time = 0 represent emission intensity under continuous excitation. Both PSiNP samples display long-lived luminescence, while the liver displays a very rapid decrease characteristic of tissue autofluorescence. (d) CWI and (e, f) TG images of the PL emission, extracted from the different acquisition time periods as indicated in the color panels of (c).
Analysis of the time-resolved ex vivo liver images allowed for the discrimination of nanoparticle age. To accomplish this, we acquired two time-gated images using different time gates, one between 10–100 μs (Figure 3e) and the other between 100–500 μs (Figure 3f). The time gates that were used are depicted schematically in Figure 3c, labeled TGI-A and TGI-B corresponding to an early and a late time gate, respectively. The image using the early gate therefore captured more of the PSiNPs with short emission lifetimes, corresponding to the aged, more extensively degraded PSiNPs-2, whereas the late gate captured more of the PSiNPs with the longer emission lifetimes, corresponding to the younger, less degraded PSiNPs-1. In the experiments of Figure 3 the injected dose of PSiNPs-2 was set to be 8-fold larger than that of PSiNPs-1, such that the TGI PL signal intensity in the early time-gated image of the site of PSiNPs-2 injection was substantially larger than at the PSiNP-1 injection site (Figure 3e). While the aged particles appeared brighter than the young particles in the early time-gated image, they appeared substantially dimmer than the young particles in the late time-gated image (Figure 3f). This is consistent with the differing emission lifetimes of the young versus the aged nanoparticles. The emission lifetimes were verified at the two injection sites by acquisition of decay traces and exponential fits of the data (Figure S7). Thus the ex vivo measurements establish that the time-gated imaging method can be used to discriminate the extent of degradation of PSiNPs, which is related to the amount of drug payload that remains to be delivered in a given nanoparticle ensemble. This is a unique and remarkable feature of PSiNPs that derives from the quantum confined nature of the porous silicon skeleton. While here we have used the terms “young” and “aged” to describe the two PSiNP types compared in the experiment, “extent of degradation” may be the more appropriate term, as the time-resolved data do not provide the true chronological age of the nanoparticles. As reflected in the value of td from eq. 1, the extent of degradation, or “age” of the PSiNPs is expected to be highly dependent on the environment to which the nanoparticles have been subjected.
Finally, we demonstrated the utility of the time-gated measurement to track the fate of PSiNPs injected into systemic circulation in live mice. Mice were injected with the peptide-loaded PSiNPs via tail vein, the PSiNPs were allowed to circulate, and then the mice were sacrificed at specific time points and the major organs harvested for imaging. Organ images were obtained as soon as feasible after sacrifice to minimize degradation of the PSiNPs post-mortem. Although numerous studies of PSiNPs have used various chemical coatings to avoid MPS (mononuclear phagocyte system) uptake or to target specific organs or diseased tissues,[28, 37, 56–58] in this study we purposely did not functionalize the nanoparticles with targeting or stealth coatings, in order to induce and then track MPS uptake. Most injected PSiNPs of the size range used in this study accumulate in the liver because liver sinusoidal endothelial cells and Kupffer cells quickly sequester the administered nanoparticles.[59–62] In the present case, images of the liver acquired by conventional continuous wave imaging (CWI) could not discern the presence of PSiNPs due to substantial tissue autofluorescence (Figure 4a). However, as was seen with the ex vivo experiments of Figure 3, the PSiNPs were readily identified in the liver as well as in the lungs and kidney by TGI. Notable liver uptake was observed within 1 h of injection, and the strongest time-gated PL signals were observed at 2 h post-injection, followed by a decreasing signal from organs harvested at later time points (Figure 4c). The emission lifetime of the nanoparticles in the liver, kidney, and lung tissues was quantified at each time point (Figure 4b and Figure S8); a steady drop in τ2 was observed from all these organs, indicative of degradation of the PSiNPs. The observed decrease in τ2 with increasing residence time in vivo was fit to eq. 1, yielding a time constant td for degradation of 9.4 ± 0.3 hours. This number is somewhat smaller than what was obtained with the in vitro experiments (10.5 ± 0.3), suggesting that the nanoparticles degrade faster in the in vivo environment.
Figure 4. Tracking the fate of PSiNPs in vivo based on their PL characteristics.

(a) Representative CWI and TGI of mouse organs harvested after in vivo injection and circulation of PSiNPs for the indicated times (𝜆ex = 365 nm, 𝜆em = 460 nm long-pass filter). Note that tissue autofluorescence is removed in the TGI, enabling clear identification of the PSiNPs. Dotted white lines are drawn to outline the organs in the TGI. The “0 h” images indicate a control set, obtained from a mouse that was not injected with PSiNPs. (b) Normalized PL decay curves obtained from livers harvested from treated mice. The times indicated represent hours post-injection. Trace labeled “PSiNP” represents the PSiNP dispersion in PBS prior to injection. Trace labeled “Liver” represents the liver of a healthy mouse that was not injected with PSiNPs. The long-lived component of the PL emission lifetime, τ2, represents the PL emission lifetime that can be definitively assigned to PSiNPs. (c) Plots of total PL intensity from the liver, obtained from the TGI images in (a), and emission decay lifetime (τ2) as a function of in vivo circulation time. Each data point represents mean ± standard deviation (n = 3). Note that the PL lifetime at 0 h (green open circle) was obtained from the decay curve of the PSiNPs in PBS solution (orange diamonds in (b)), fit to a single exponential function.
The combination of PL intensity and lifetime data extracted from the time-gated images afford a higher fidelity picture of the fate of a nanoparticle in vivo (Figure S9). The data can answer questions relating to the mechanism of clearance of the nanoparticles. In particular, the observation that the emission lifetime decreases with increasing time in circulation means that the nanoparticles are continuously degrading in the organs, rather than becoming sequestered and then being eliminated intact. This is not surprising and it is consistent with the in vitro results, although it should be pointed out that immobilizing the nanoparticles within cellular compartments or coating them with opsonizing proteins could easily be expected to decrease the rate of dissolution of the PSiNPs. This contrasts with what was observed in the present work, where the in vivo rate of degradation was slightly faster than what was observed with a simple buffer solution. PL signals from the PSiNPs were also observed in the kidneys, and the signals displayed emission lifetimes similar to those measured in the other organs. This suggests that the PSiNPs had not been filtered but were residing in the renal corpuscle and tubule system of the kidney. If the PSiNPs had been filtered into the urinary space they would be expected to be of a size < 5nm, substantially smaller than the injected PSiNPs (180 nm, Figure S4).[63–67]
Self-reporting systems employing measurement of fluorescence intensity have shown promise as a noninvasive means of assessing drug delivery status.[1–4] However, fluorescent labels conjugated to drug nanocarriers can become detached from the carrier host during circulation, complicating the interpretation. In addition, small differences in surface chemistry used to attach the fluorescent label can influence the in vivo fate in unpredictable ways. As shown in this work, the unique ability of the PL lifetime to report on the status of PSiNPs provides a new, high fidelity means to assess the status and fate of a delivery system. This non-toxic nanocarrier harnesses the intrinsic PL of the silicon nanoparticles within its skeletal structure, and the long-lived PL lifetime enabled clear discrimination of the silicon nanoparticles from tissue autofluorescence. We showed that the combination of intensity and lifetime analyses of the time-gated images afford signals that are directly attributable to the nanoparticles, enabling more accurate assessment of biodistribution and in vivo elimination.
Supplementary Material
Acknowledgments
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Education (Grant No. 2017R1D1A1B03035525), the Korea Health Technology R&D Project though the Korea Health Industry Development Institute (KHIDI) funded by the Ministry of Health & Welfare (Grant No. HI14C1090), the Bio & Medical Technology Development Program of the National Research Foundation funded by the Ministry of Science and ICT (2017M3A9C6032056), and grants (2016–7016, 2017–7016, 2018–7016, 2018–7045) from the Asan Institute for Life Sciences, Asan Medical Center, Seoul, Korea. This research was also supported in part by the National Science Foundation grant No. CBET-1603177 and by the National Institutes of Health, through contracts R01 AI132413–01, R01 CA152327, and R01 CA214550. D. Kim thanks the Basic Science Research Program through the Korea NRF funded by the Ministry of Education (NRF-2018R1A6A1A03025124). Y. Jin and D. Kim contributed equally to this work.
Footnotes
Author Disclosure of Competing Interests
MJS is a scientific founder of Spinnaker Biosciences, Inc., a member of the Board of Directors, and has an equity interest in the company. Although one or more of the grants that supported this research has been identified for conflict of interest management based on the overall scope of the project and its potential benefit to Spinnaker Biosciences, Inc., the research findings included in this particular publication may not necessarily relate to the interests of Spinnaker Biosciences, Inc. The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies. The other authors declare no competing interests.
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Dr. Yusung Jin, Biomedical Engineering Research Center, Asan Institute for Life Sciences, Asan Medical Center, Seoul 05505, Rep. of Korea Department of Convergence Medicine, Asan Medical Center and University of Ulsan College of Medicine, Seoul 05505, Rep. of Korea.
Prof. Dokyoung Kim, Department of Anatomy and Neurobiology, College of Medicine, and Biomedical Science Institute, Kyung Hee University, Seoul 02447, Rep. of Korea
Hajung Roh, Biomedical Engineering Research Center, Asan Institute for Life Sciences, Asan Medical Center, Seoul 05505, Rep. of Korea; Department of Chemical Engineering, Pohang University of Science and Technology, Pohang 37673, Rep. of Korea.
Sojeong Kim, Biomedical Engineering Research Center, Asan Institute for Life Sciences, Asan Medical Center, Seoul 05505, Rep. of Korea; Department of Chemistry, Pohang University of Science and Technology, Pohang 37673, Rep. of Korea.
Dr. Sazid Hussain, Cancer Research Center, Sanford Burnham Prebys Medical Discovery Institute, La Jolla, CA 92037, USA
Jinyoung Kang, Department of Nanoengineering, University of California, San Diego, La Jolla, CA 92037, USA.
Prof. Chan-Gi Pack, Department of Convergence Medicine, Asan Medical Center and University of Ulsan College of Medicine, Seoul 05505, Rep. of Korea
Prof. Jun Ki Kim, Biomedical Engineering Research Center, Asan Institute for Life Sciences, Asan Medical Center, Seoul 05505, Rep. of Korea Department of Convergence Medicine, Asan Medical Center and University of Ulsan College of Medicine, Seoul 05505, Rep. of Korea.
Prof. Seung-Jae Myung, Department of Gastroenterology, University of Ulsan College of Medicine, Asan Medical Center, Seoul 05505, Rep. of Korea
Prof. Erkki Ruoslahti, Cancer Research Center, Sanford Burnham Prebys Medical Discovery Institute, La Jolla, CA 92037, USA and Center for Nanomedicine, and Department of Cell, Molecular and Developmental Biology, University of California, Santa Barbara, CA 93106, USA
Prof. Michael J. Sailor, Department of Nanoengineering, University of California, San Diego, La Jolla, CA 92037, USA msailor@ucsd.edu.
Prof. Song Cheol Kim, Biomedical Engineering Research Center, Asan Institute for Life Sciences, Asan Medical Center, Seoul 05505, Rep. of Korea drksc@amc.seoul.kr; Department of Surgery, University of Ulsan College of Medicine, Asan Medical Center, Seoul 05505, Rep. of Korea.
Prof. Jinmyoung Joo, Biomedical Engineering Research Center, Asan Institute for Life Sciences, Asan Medical Center, Seoul 05505, Rep. of Korea joo@amc.seoul.kr; Department of Convergence Medicine, Asan Medical Center and University of Ulsan College of Medicine, Seoul 05505, Rep. of Korea.
References
- [1].Wolfbeis OS, Chem. Soc. Rev 2015, 44, 4743–4768. [DOI] [PubMed] [Google Scholar]
- [2].Wang KM, He XX, Yang XH, Shi H, Acc. Chem. Res 2013, 46, 1367–1376. [DOI] [PubMed] [Google Scholar]
- [3].Reisch A, Klymchenko AS, Small 2016, 12, 1968–1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Chen WS, Huang QY, Ou WZ, Hao YQ, Wang LQ, Zeng K, Guo HY, Li J, Liu YN, Small 2014, 10, 1261–1265. [Google Scholar]
- [5].Luk BT, Zhang LF, ACS Appl. Mater. Inter 2014, 6, 21859–21873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lee JH, Kim ES, Cho MH, Son M, Yeon SI, Shin JS, Cheon J, Angew. Chem. Int. Edit 2010, 49, 5698–5702. [DOI] [PubMed] [Google Scholar]
- [7].Cui LY, Lin QY, Jin CS, Jiang WL, Huang H, Ding LL, Muhanna N, Irish JC, Wang F, Chen J, Zheng G, ACS Nano 2015, 9, 4484–4495. [DOI] [PubMed] [Google Scholar]
- [8].Zheng J, Yang RH, Shi ML, Wu CC, Fang XH, Li YH, Li JH, Tan WH, Chem. Soc. Rev 2015, 44, 3036–3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lai JP, Shah BP, Garfunkel E, Lee KB, ACS Nano 2013, 7, 2741–2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Li SY, Liu LH, Rong L, Qiu WX, Jia HZ, Li B, Li F, Zhang XZ, Adv. Funct. Mater 2015, 25, 7317–7326. [Google Scholar]
- [11].Chen HT, Kim SW, Li L, Wang SY, Park K, Cheng JX, Proc. Natl. Acad. Sci. USA 2008, 105, 6596–6601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Huang J, Li YC, Orza A, Lu Q, Guo P, Wang LY, Yang L, Mao H, Adv. Funct. Mater 2016, 26, 3818–3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ulbrich K, Hola K, Subr V, Bakandritsos A, Tucek J, Zboril R, Chem. Rev 2016, 116, 5338–5431. [DOI] [PubMed] [Google Scholar]
- [14].Mazuel F, Espinosa A, Luciani N, Reffay M, Le Borgne R, Motte L, Desboeufs K, Michel A, Pellegrino T, Lalatonne Y, Wilhelm C, ACS Nano 2016, 10, 7627–7638. [DOI] [PubMed] [Google Scholar]
- [15].Canham LT, Appl. Phys. Lett 1990, 57, 1046–1048. [Google Scholar]
- [16].Sa’ar A, Nanophotonics J 2009, 3, 032501. [Google Scholar]
- [17].Kocevski V, Eriksson O, Rusz J, Phys. Rev. B 2013, 87. [Google Scholar]
- [18].Gu L, Hall DJ, Qin ZT, Anglin E, Joo J, Mooney DJ, Howell SB, Sailor MJ, Nat. Commun 2013, 4, 2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Fan DM, De Rosa E, Murphy MB, Peng Y, Smid CA, Chiappini C, Liu XW, Simmons P, Weiner BK, Ferrari M, Tasciotti E, Adv. Funct. Mater 2012, 22, 282–293. [Google Scholar]
- [20].Low SP, Voelcker NH, Canham LT, Williams KA, Biomaterials 2009, 30, 2873–2880. [DOI] [PubMed] [Google Scholar]
- [21].Bimbo LM, Sarparanta M, Santos HA, Airaksinen AJ, Makila E, Laaksonen T, Peltonen L, Lehto VP, Hirvonen J, Salonen J, ACS Nano 2010, 4, 3023–3032. [DOI] [PubMed] [Google Scholar]
- [22].Bayliss SC, Heald R, Fletcher DI, Buckberry LD, Adv. Mater 1999, 11, 318–321. [Google Scholar]
- [23].Kang J, Joo J, Kwon EJ, Skalak M, Hussain S, She Z-G, Ruoslahti E, Bhatia SN, Sailor MJ, Adv. Mater 2016, 28, 7962–7969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Granitzer P, Rumpf K, Roca AG, Morales MP, Poelt P, Albu M, J. Magn. Magn. Mater 2010, 322, 1343–1346. [Google Scholar]
- [25].Erogbogbo F, Yong KT, Roy I, Xu GX, Prasad PN, Swihart MT, ACS Nano 2008, 2, 873–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Shen J, Xu R, Mai J, Kim H-C, Guo X, Qin G, Yang Y, Wolfram J, Mu C, Xia X, Gu J, Liu X, Mao Z-W, Ferrari M, Shen H, ACS Nano 2013, 7, 9867–9880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Salonen J, Kaukonen AM, Hirvonen J, Lehto VP, J. Pharm. Sci-US 2008, 97, 632–653. [DOI] [PubMed] [Google Scholar]
- [28].Tasciotti E, Liu XW, Bhavane R, Plant K, Leonard AD, Price BK, Cheng MMC, Decuzzi P, Tour JM, Robertson F, Ferrari M, Nat. Nanotechnol 2008, 3, 151–157. [DOI] [PubMed] [Google Scholar]
- [29].Mann AP, Scodeller P, Hussain S, Joo J, Kwon E, Braun GB, Molder T, She Z-G, Kotamraju VR, Ranscht B, Krajewski S, Teesalu T, Bhatia S, Sailor MJ, Ruoslahti E, Nat. Commun 2016, 7, 11980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Hussain S, Joo J, Kang J, Kim B, Braun GB, She Z-G, Kim D, Mann AP, Mölder T, Teesalu T, Carnazza S, Guglielmino S, Sailor MJ, Ruoslahti E, Nat. Biomed. Eng 2018, 2, 95–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Dasog M, Yang ZY, Regli S, Atkins TM, Faramus A, Singh MP, Muthuswamy E, Kauzlarich SM, Tilley RD, Veinot JGC, ACS Nano 2013, 7, 2676–2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ghosh B, Takeguchi M, Nakamura J, Nemoto Y, Hamaoka T, Chandra S, Shirahata N, Sci. Rep.-UK 2016, 6, 36951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Li Q, Jin RC, Nanotechnol Rev 2017, 6, 601–612. [Google Scholar]
- [34].Wang J, Kumeria T, Bezem MT, Wang J, Sailor MJ, ACS Appl. Mater. Inter 2017, 10, 3200–3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Maher S, Alsawat M, Kumeria T, Fathalla D, Fetih G, Santos A, Habib F, Losic D, Adv. Funct. Mater 2015, 25, 5107–5116. [Google Scholar]
- [36].Qin Z, Joo J, Gu L, Sailor MJ, Part. Part. Syst. Charact 2014, 31, 252–256. [Google Scholar]
- [37].Park JH, Gu L, von Maltzahn G, Ruoslahti E, Bhatia SN, Sailor MJ, Nat. Mater 2009, 8, 331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Ray M, Sarkar S, Bandyopadhyay NR, Hossain SM, Pramanick AK, J. Appl. Phys 2009, 105. [Google Scholar]
- [39].Gelloz B, Koshida N, J. Appl. Phys 2005, 98. [Google Scholar]
- [40].Joo J, Cruz JF, Vijayakumar S, Grondek J, Sailor MJ, Adv. Funct. Mater 2014, 24, 5688–5694. [Google Scholar]
- [41].Sychugov I, Valenta J, Linnros J, Nanotechnology 2017, 28. [DOI] [PubMed] [Google Scholar]
- [42].Joo J, Defforge T, Loni A, Kim D, Li ZY, Sailor MJ, Gautier G, Canham LT, Appl. Phys. Lett 2016, 108. [Google Scholar]
- [43].Turro NJ, Ramamurthy V, Scaiano JC, Photochem. Photobiol 2012, 88, 1033–1033. [Google Scholar]
- [44].Li Q, He Y, Chang J, Wang L, Chen HZ, Tan YW, Wang HY, Shao ZZ, J. Am. Chem. Soc 2013, 135, 14924–14927. [DOI] [PubMed] [Google Scholar]
- [45].Timmerman D, Gregorkiewicz T, Nanoscale Res. Lett 2012, 7, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Matsumoto K, Nishio R, Nomura T, Kamiya K, Inada M, Suzuki S, Jpn. J. Appl. Phys 2015, 54. [Google Scholar]
- [47].Yu YX, Fan G, Fermi A, Mazzaro R, Morandi V, Ceroni P, Smilgies DM, Korgel BA, J. Phys. Chem. C 2017, 121, 23240–23248. [Google Scholar]
- [48].Jendoubi F, Mgaidi A, El Maaoui M, Can. J. Chem. Eng 1998, 76, 233–238. [Google Scholar]
- [49].Knauss KG, Wolery TJ, Geochim. Cosmochim. Ac 1988, 52, 43–53. [Google Scholar]
- [50].StÖBer W, in Equilibrium Concepts in Natural Water Systems, Vol. 67 (Ed.: Stumm), American Chemical Society, 1967, pp. 161–182. [Google Scholar]
- [51].Kwon EJ, Skalak M, Bertucci A, Braun G, Ricci F, Ruoslahti E, Sailor MJ, Bhatia SN, Adv. Mater 2017, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Johnson GA, Muthukrishnan N, Pellois JP, Bioconjugate Chem 2013, 24, 114–123. [DOI] [PubMed] [Google Scholar]
- [53].Javadpour MM, Juban MM, Lo WCJ, Bishop SM, Alberty JB, Cowell SM, Becker CL, McLaughlin ML, J. Med. Chem 1996, 39, 3107–3113. [DOI] [PubMed] [Google Scholar]
- [54].Joo J, Kwon EJ, Kang JY, Skalak M, Anglin EJ, Mann AP, Ruoslahti E, Bhatia SN, Sailor MJ, Nanoscale Horiz 2016, 1, 407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Joo J, Liu X, Kotamraju VR, Ruoslahti E, Nam Y, Sailor MJ, ACS Nano 2015, 9, 6233–6241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Serda RE, Ferrati S, Godin B, Tasciotti E, Liu XW, Ferrari M, Nanoscale 2009, 1, 250–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Decuzzi P, Godin B, Tanaka T, Lee SY, Chiappini C, Liu X, Ferrari M, Control J Release 2010, 141, 320–327. [DOI] [PubMed] [Google Scholar]
- [58].Ferrati S, Mack A, Chiappini C, Liu XW, Bean AJ, Ferrari M, Serda RE, Nanoscale 2010, 2, 1512–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Huang J, Bu LH, Xie J, Chen K, Cheng Z, Li XG, Chen XY, ACS Nano 2010, 4, 7151–7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Zhang YN, Poon W, Tavares AJ, McGilvray ID, Chan WCW, Control J Release 2016, 240, 332–348. [DOI] [PubMed] [Google Scholar]
- [61].Tsoi KM, MacParland SA, Ma XZ, Spetzler VN, Echeverri J, Ouyang B, Fadel SM, Sykes EA, Goldaracena N, Kaths JM, Conneely JB, Alman BA, Selzner M, Ostrowski MA, Adeyi OA, Zilman A, McGilvray ID, Chan WCW, Nat. Mater 2016, 15, 1212–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].MacParland SA, Tsoi KM, Ouyang B, Ma XZ, Manuel J, Fawaz A, Ostrowski MA, Alman BA, Zilman A, Chan WCW, McGilvray ID, ACS Nano 2017, 11, 2428–2443. [DOI] [PubMed] [Google Scholar]
- [63].Scown TM, van Aerle R, Johnston BD, Cumberland S, Lead JR, Owen R, Tyler CR, Toxicol. Sci 2009, 109, 372–380. [DOI] [PubMed] [Google Scholar]
- [64].Zuckerman JE, Choi CHJ, Han H, Davis ME, Proc. Natl. Acad. Sci. U S A 2012, 109, 3137–3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Gao S, Hein S, Dagnaes-Hansen F, Weyer K, Yang C, Nielsen R, Christensen EI, Fenton RA, Kjems J, Theranostics 2014, 4, 1039–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Williams RM, Shah J, Ng BD, Minton DR, Gudas LJ, Park CY, Heller DA, Nano Lett 2015, 15, 2358–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Choi CH, Zuckerman JE, Webster P, Davis ME, Proc. Natl. Acad. Sci. U S A 2011, 108, 6656–6661. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.