

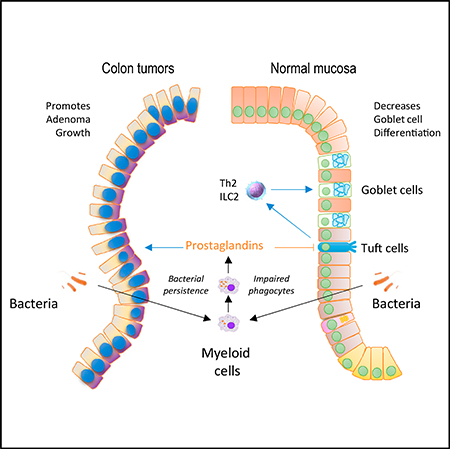
SUMMARY
Physiologic microbe-host interactions in the intestine require the maintenance of the microbiota in a luminal compartment through a complex interplay between epithelial and immune cells. However, the roles of mucosal myeloid cells in this process remain incompletely understood. In this study, we identified that decreased myeloid cell phagocytic activity promotes colon tumorigenesis. We show that this is due to bacterial accumulation in the lamina propria and present evidence that the underlying mechanism is bacterial induction of prostaglandin production by myeloid cells. Moreover, we show that similar events in the normal colonic mucosa lead to reductions in Tuft cells, goblet cells, and the mucus barrier of the colonic epithelium. These alterations are again linked to the induction of prostaglandin production in response to bacterial penetration of the mucosa. Altogether, our work highlights immune cell-epithelial cell interactions triggered by the microbiota that control intestinal immunity, epithelial differentiation, and carcinogenesis.
In Brief
Miyata et al. find that defective bacterial elimination by intestinal myeloid cells promotes prostaglandin production and drives excess colonic neoplasia in a genetic mouse model. Moreover, in the normal mucosa, similar prostaglandin overproduction suppresses differentiation of mucus-producing goblet cells through direct effects on Tuft cells, a regulator of goblet cells.
Graphical Abstract
INTRODUCTION
The intestine is the largest mucosal surface in the human body and is a niche for the most abundant and diverse microbiota (Walter and Ley, 2011). The vast numbers of microorganisms harbored in the intestine play a number of crucial roles in human physiology, including the regulation of host metabolism and immune function (Honda and Littman, 2016; Koppel and Balskus, 2016; Thaiss et al., 2016; Walter and Ley, 2011). While the microbiota are not pathogenic, per se, their penetration past the mucosal barrier has the potential to cause harm; therefore, a delicate balance is in place between the host’s intestinal mucosa and the microbiota that allows for the persistent presence of these organisms in a compartmentalized manner.
The maintenance of this compartment is mediated by a variety of factors, including the cellular barrier created by the intestinal epithelium and the protective properties of its secreted products (Hooper, 2015), including mucins, which are produced by specialized epithelial cells known as goblet cells (Johansson et al., 2013). These proteins are essential for the formation of the mucus barrier, which plays a central role in creating physical separation between most luminal bacteria and the apical surface of the epithelium. In addition to the intestinal epithelium, cells of the mucosal immune system play a number of roles in the maintenance of a physiologic compartment for the microbiota (Honda and Littman, 2016; Thaiss et al., 2016). These include innate immune defense mechanisms mediated by professional phagocytic cells, such as macrophages and dendritic cells, as well as adaptive immune processes, including the production of secretory immunoglobulin A (IgA).
In the context of intestinal neoplasia, disruption of these protective mechanisms results in increased penetration of bacteria into the lamina propria. An impaired epithelial barrier due to poor cell-cell contacts among neoplastic cells is part of this process (Grivennikov et al., 2012). Bacterial penetration is not only a consequence of the neoplastic process but it is known to also promote tumor development through the effects of local inflammation and the resulting cytokines and other products that are released in the tumor microenvironment, which have been linked in several instances to macrophages (Grivennikov et al., 2010).
In order to produce a functional mucosal barrier, these protective mechanisms are coordinated through specific immune cellepithelial cell interactions. Recent studies have identified intricate cellular responses in the intestinal mucosa that drive goblet cell differentiation. An epithelial cell type, known as the Tuft cell, is thought to be at the apex of the response (Gerbe et al., 2016; Howitt et al., 2016; von Moltke et al., 2016). These cells comprise less than 1% of the epithelial cell mass and display features of quiescent stem cells (Chandrakesan et al., 2015; Gagliardi et al., 2012; Gerbe et al., 2011; Nakanishi et al., 2013). At the same time, they are thought to be endowed with the ability to detect luminal factors, including helminth- and protozoa-derived products (Howitt et al., 2016). Tuft cells are the predominant source for interleukin (IL)-25 in the intestinal mucosa, and the production of this cytokine recruits Th2 and ILC2 cells to the lamina propria. The products of these cells, particularly IL-4 and IL-13, are required for anti-helminth immunity (Roediger and Weninger, 2015) and promote goblet cell differentiation and mucus production, which are critical components of type 2 immune responses (Gerbe et al., 2016; Howitt et al., 2016; von Moltke et al., 2016). Whether other components of the immune system—particularly, innate immune cells of the myeloid lineage—can affect intestinal epithelial differentiation in response to changes in the intestinal microbiota is not as well understood.
In this study, we examined intestinal phenotypes in animals lacking Commd1 in the myeloid lineage. This gene, which encodes a prototypical member of the COMMD protein family, previously linked to the regulation of copper homeostasis and nuclear factor kB (NF-kB) signaling (Maine et al., 2007; van de Sluis et al., 2002), was found here to also be required for optimal myeloid cell phagocytic activity and bacterial clearance by macrophages. Studies in this mouse and other mouse strains with defective phagocyte activity uncovered that bacterial elimination by mucosal myeloid cells can affect epithelial tumor development and the differentiation programs of the intestinal epithelium that control goblet cells in the intestinal mucosa. Altogether, this work highlights important aspects of immune cell-epithelial cell interactions that regulate intestinal homeostasis.
RESULTS
Commd1 Depletion Affects Bacterial Handling by Myeloid Cells
COMMD1, the prototypical member of the COMMD protein family, was identified as a component of a conserved complex referred to as the COMMD/CCDC22/CCDC93, or CCC, complex (Bartuzi et al., 2016; Li et al., 2015; Phillips-Krawczak et al., 2015). This complex is recruited to endosomes to regulate the recycling of cargo proteins to other subcellular destinations. In other recent studies, COMMD proteins were identified by proteomics to be associated with the phagosome, an endosome-like structure of myeloid-derived phagocytes (Dill et al., 2015). Immunofluorescence staining of bone marrow-derived macrophages (BMDMs) confirmed that key components of this complex— namely, Ccdc22, Ccdc93, and Vps35l (formerly C16orf62)—are recruited with high frequency to phagolysosomes (Figure 1A). This prompted us to examine the role of the CCC complex in macrophages. To that end, we investigated Commd1-deficient bone marrow-derived macrophages (BMDMs) derived from mice with myeloid-specific deletion of this gene (Commd1fl/fl, Lys2Cre, referred to hereinafter as Commd1ΔMye). In agreement with prior work indicating that COMMD1 deficiency destabilizes the CCC complex (Phillips-Krawczak et al., 2015), loss of Commd1 led to dramatically reduced expression of several CCC complex components, whereas Commd9 deficiency in BMDMs (Commd9fl/fl, Lys2Cre) resulted in a more modest effect (Figure 1B). Thus, Commd1 deficiency can be viewed as a model of severe CCC complex depletion.
Figure 1. The CCC Complex Regulates Phagocytic Activity.

(A) Immunofluorescence staining of BMDMs for the indicated CCC complex components (red) and nuclei (DAPI, blue). The cells had been pulsed with fluorescent E. coli particles (Alexa 488, green). Magnified subcellular regions are shown (insets). The percentage of E. coli particles that colocalize with the indicated CCC complex component is also indicated (right lower corner of each panel). Scale bars, 5 μm (1 μm in insets). Data are representative of 2 independent experiments.
(B) Immunoblots for CCC complex subunits, including various COMMD family members, in cell lysates from BMDMs, as indicated (representative of over 3 prior iterations).
(C and D) Quantification of BMDMs positive for pHRodo-labeled E. coli particles. Commd1ΔMye (C) or Commd9ΔMye (D) BMDMs were compared to “floxed” control macrophages. Data displayed indicate mean and SEM; these results are representative of 3 independent experiments. Technical duplicates were used; cells were derived from one or two mice of each genotype per group for each experiment. *p < 0.05 (unpaired Student’s t test comparing Commd1fl/fl to Commd1ΔMye). ns, not significant.
(E) Imaging of BMDMs pulsed with GFP-expressing E. coli (green) that had been labeled with a copper-sensitive dye (CF4, red). The percentage of E. coli that stain for CF4 is also indicated (left upper corner). Scale bars, 10 μm. Data are representative of 2 independent experiments. **p < 0.01 (χ2 test).
See also Figures S1A and S1B.
Next, we assessed whether Commd1-deficient BMDMs had altered phagocytic activity. When exposed to pHRodo-labeled E. coli particles, Commd1-deficient BMDMs had about twice as many particle-positive cells (Figure 1C), and, in contrast, Commd9-deficient BMDMs displayed no differences (Figure 1D). A similar increase was observed when using live GFP-positive E. coli (Figure S1A), and when pulsed with Alexa-594-labeled E. coli particles, moreCommd1-deficient macrophages remained positive over time than wild-type control cells (Figure S1B), which is indicative of a reduced capacity to clear intracellular bacteria.
It has been reported that the copper transporter ATP7A is mobilized to phagolysosomes, where it contributes to bacterial killing through the transport of copper to the vesicular lumen (White et al., 2009). Interestingly, COMMD1 and the CCC complex are known to regulate the intracellular movement of ATP7A in epithelial cells (Phillips-Krawczak et al., 2015; Vonk et al., 2012). Therefore, we examined the possibility that copper flux into the phagolysosome could be impaired in Commd1-deficient macrophages. To this end, we loaded GFP-expressing bacteria with a copper-specific sensor, CF4, and offered these bacteria to BMDMs. As expected, the CF4 fluorescence signal was detected in the phagolysosomes of wild-type BMDMs, but the signal was significantly reduced in Commd1-deficient macrophages, which is indicative of a defect in copper transport (Figure 1E). Altogether, these findings indicated that Commd1 deficiency results in reduced phagocytic clearance of penetrating bacteria, which is explained, at least in part, by its effects on copper delivery to the phagolysosome.
Commd1 Deficiency Results in Reduced Bacterial Clearance In Vivo
Commd1ΔMye mice are more susceptible to sepsis following intestinal perforation, but these animals have not been noted to have any phenotype at baseline (Li et al., 2014). In view of the decreased clearance of bacteria noted in Commd1-deficient macrophages, we examined whether there was evidence of pathologic bacterial tissue penetration.
Bacterial penetration is known to occur in the lamina propria of colonic adenomas as a result of poor barrier function of the neoplastic epithelium (Grivennikov et al., 2012). Mice carrying the Min mutation in the Apc gene (so-called ApcMin mice) develop spontaneous intestinal adenomas (Su et al., 1992) and were used to study this phenomenon. With fluorescence in situ hybridization (FISH) to detect bacterial tissue penetration, we observed an abundant accumulation of bacteria in the lamina propria of colonic adenomas, and the signal was in nearly complete colocalization with the macrophage marker F4/80 (Figure 2A), indicating that penetrating bacteria are efficiently engulfed by lamina propria myeloid cells. Next, ApcMin, Commd1ΔMye mice were generated; and, by FISH analysis, these animals were found to display a significant increase in bacteria in the lamina propria compared to littermate controls (Figures 2B and 2C), a finding confirmed by qPCR designed to amplify common eubacterial sequences (Figure S2A). Altogether, these findings indicate that COMMD1 is required for macrophages to function effectively in the elimination of penetrating bacteria in the lamina propria.
Figure 2. Commd1 Deficiency Impairs Bacterial Clearance in the Colonic Lamina Propria.

(A) Representative images (from over 5 independent iterations) of a polypstained by FISH with a eubacterial probe (green) and concurrently stained for F4/80 (red) and nuclei (blue). Scale bar, 100 μm.
(B) Representative images of FISH staining of polyps (from over 5 independentiterations) demonstrating bacterial penetration into the colonic lamina propria in Commd1fl/fl or Commd1ΔMye mice carrying the ApcMin allele. Scale bars, 100 μm.
(C) Quantification of the imaging shown in (B), expressed as fold over the Commd1fl/fl group (three animals per group). Data displayed indicate mean and SEM; **p < 0.01 (unpaired Student’s t test).
See also Figure S2A.
Defective Bacterial Clearance by Myeloid Cells Promotes Colonic Adenoma Formation
Bacterial penetration in the lamina propria of colonic adenomas is thought to promote neoplastic development through various effects on the tumor microenvironment (Grivennikov et al., 2012). Therefore, we examined the consequence of myeloid deficiency of Commd1, and its resulting decreased myeloid clearance of bacteria, for the development of intestinal neoplasia. ApcMin, Commd1ΔMye mice had a significant increase in the number of colonic tumors without an appreciable effect on tumor burden in the small intestine (Figures 3A and 3B). Interestingly, this increase was driven specifically by an increased rate of distal colonic polyps in ApcMin, Commd1ΔMye mice (Figure S2B). This was associated with worse anemia in ApcMin, Commd1ΔMye mice, a marker of more significant tumor burden (Figure S2C, left).
Figure 3. Impaired Phagocytic Activity of Myeloid Cells Promotes Colon Adenoma Formation.

(A) Tumor count in the colon or small bowel of Commd1fl/fl (n = 20) or Commd1ΔMye (n = 13) animals carrying the ApcMin allele. Data displayed indicate mean and SEM; these are the aggregate of two independent experiments. *p < 0.05; ns, not significant (unpaired Student’s t test). Additional data are displayed in Figure S2B.
(B) Representative images of colon tumor burdendepicted in (A). Scale bars, 10 μm.
(C) Tumor count in the colon and small intestine of Cybb+ (n = 7) and Cybb− (n = 7) animals carrying the ApcMin allele. Data displayed indicate mean and SEM (aggregate of 2 experiments). *p < 0.05; ns, not significant (unpaired Student’s t test).
(D) Representative images of colonic tumor burden depicted in (C). Scale bars, 10 μm.
(E) Bacterial penetration into the colonic laminapropria of polyps in Cybb+ and Cybb− mice carrying the ApcMin allele, quantified by FISH staining (expressed as fold over the Cybb+ group; three animals per group). Data displayed indicate mean and SEM. *p < 0.05 (unpaired Student’s t test); ns, not significant.
(F) Schematic depicting the antibiotic treatment course for animals depicted in (G) and (H).
(G) Tumor count in the colon of Commd1fl/fl (n = 10) or Commd1ΔMye (n = 10) animals carrying the ApcMin allele that had been treated with oral antibiotics. Data displayed indicate mean and SEM. These are the aggregate of two independent experiments; ns, not significant (unpaired Student’s t test).
(H) Representative images of colon tumor burdendepicted in (G). Scale bars, 10 mm.
See also Figures S2B and S2C.
Next, we evaluated a different model of defective myeloid cell function: mice carrying a defect in Cybb, an X-linked gene encoding a critical subunit of NADPH oxidase, which resembles aspects of chronic granulomatous disease in humans (Chiriaco et al., 2016). When crossed with ApcMin mice, Cybb− male progeny had excess colonic adenomas compared to Cybb+ controls, without changes in small bowel adenoma burden (Figures 3C and 3D), resembling both aspects of the phenotype of ApcMin, Commd1ΔMye mice. Moreover, these mice also displayed increased bacterial penetration into the lamina propria of colonic adenomas (Figure 3E). To confirm that these phenotypes were, indeed, driven by intestinal bacteria, ApcMin, Commd1ΔMye mice were treated with oral antibiotics (Figure 3F). This treatment attenuated the formation of intestinal adenomas, so that there were no longer any differences in total colonic adenoma burden (Figures 3G and 3H) or hematocrit (Figure S2C, right) between ApcMin, Commd1ΔMye mice and control animals. Therefore, we concluded that bacteria are required for the increased tumor multiplicity observed in animals with impaired phagocytic activity. In aggregate, these data indicate that defective bacterial elimination by myeloid-derived phagocytes is associated with increased colon tumor burden in a genetic model of intestinal cancer.
Prostaglandin Induction Links Bacterial Penetration and Tumor Development
Next, we set out to understand the mechanisms connecting bacterial penetration into the lamina propria, their elimination by myeloid cells, and tumor development. First, histologic assessment of inflammatory activity in these adenomas did not reveal an increase in histologic features of inflammation (Figures S3A and S3B). Immune cell infiltration of these polyps by immunofluorescence staining, using myeloid (F4/80, Ly6G) and T cell markers (CD3), showed no significant difference in colonic adenomas from ApcMin, Commd1ΔMye and control mice (Figure S3C).
Next, we profiled the expression of a number of pro-inflammatory genes. While most genes were expressed at comparable levels, a set of genes were increased in expression in adenomas from ApcMin, Commd1ΔMye mice; namely, Il12b, Il6, Cxcl2, Il23a, and Ptgs2 (Figure 4A). Notably, Ptgs2, which encodes the enzyme cyclooxygenase 2 (COX-2) involved in prostaglandin generation, has been extensively implicated in early adenoma development (Moreira and Castells, 2011; Oshima et al., 1996; Williams et al., 1999). Interestingly, expression of Ptgs1, which encodes the other cyclooxygenase isoform COX-1, was not increased (Figure 4A). In agreement with the RNA expression data, immunofluorescence staining demonstrated an increased frequency of COX-2-positive cells in colonic adenomas from ApcMin, Commd1ΔMye mice (Figure 4B). Using a combination of FISH (to delineate bacterial localization in the tissue) and immunofluorescence staining, we found that most COX-2+ cells are F4/80+ myeloid cells (68%) and that, conversely, almost all F4/ 80-positive cells in the lamina propria were COX-2 positive (92%) (Figure 4C). This observation is in line with prior studies that demonstrated that macrophages are the main source of COX-2 expression in adenomas from ApcMin mice, while COX-1 is mostly derived from non-myelomonocytic cells (Hull et al., 2006). Furthermore, over half (56%) of COX-2+ cells also stained positive for the eubacterial FISH probe, consistent with prior reports that bacterial persistence in myeloid cells triggers increased expression of COX-2 (Miller et al., 2015). To test the possibility that bacteria are responsible for COX-2 induction, we examined the consequence of antibiotic decontamination (Figure 3F) on Ptgs2 expression in colonic adenomas. Under these conditions, Ptgs2 expression was identical in ApcMin, Commd1ΔMye and control mice (Figure 4D), supporting the notion that bacterial penetration plays a key role in inducing COX-2 expression.
Figure 4. COX-2 Induction Links Bacterial Penetration and Tumor Development.

(A) mRNA expression of the indicated genes by real-time qPCR analysis in adenomas from Commd1fl/fl (n = 10) or Commd1ΔMye (n = 10) animals carrying the ApcMin allele. Data displayed indicate mean and SEM. These are the aggregate of 3 experiments. *p < 0.05; **p < 0.01; ns, not significant (unpaired Student’s t test). Additional data are displayed in Figures S3A and S3B.
(B) Quantification of COX-2 signal intensity in immunofluorescence staining of colonic adenomas, expressed as fold over the Commd1fl/fl group. The data displayed indicate the mean and SEM (three animals per group). *p < 0.05 (unpaired Student’s t test).
(C) Representative images (from over 5 independent iterations) of colon polyps stained by FISH with a eubacterial probe (green) and concurrently immunofluorescence stained for F4/80 (magenta), COX-2 (red), and nuclei (blue). Scale bars, 100 μm for low-magnification images (top) and 50 μm for high-magnification images (bottom).
(D) Real-time qPCR for Ptgs2 in colonic adenomas of Commd1fl/fl (n = 3) or Commd1ΔMye (n = 3) mice carrying the ApcMin allele; the animals had received oral antibiotics, as depicted in Figure 3F. The data displayed indicate the mean and SEM. ns, not significant (unpaired Student’s t test).
(E) Schematic depicting aspirin treatment course for animals depicted in (F).
(F) Quantification of tumors in the colon of Commd1fl/fl (n = 10) or Commd1ΔMye (n = 10) animals carrying the ApcMin allele; mice had been treated with aspirin. The data displayed indicate the mean and SEM. ns, not significant (unpaired Student’s t test). Additional data are displayed in Figures S3C–S3E.
See also Figure S3.
To assess the importance of COX-2 in tumor induction in ApcMin, Commd1ΔMye mice, we treated the animals with aspirin, a non-selective COX-1 and COX-2 inhibitor (Figure 4E). Aspirintreated ApcMin, Commd1ΔMye mice had comparable numbers of colonic adenomas (Figure 4F) and degrees of anemia that were similar (Figure S3D) to those of control mice. Interestingly, bacterial penetration into the tumor lamina propria was still greater in ApcMin, Commd1ΔMye mice treated with aspirin (Figure S3E), indicating that bacterial accumulation is a primary defect of Commd1 deficiency that occurs independently of COX-2 activity.
Defective Bacterial Clearance by Myeloid Cells Is Associated with Altered Epithelial Differentiation
Next, we examined whether these macrophage defects would lead to increased bacterial penetration in the normal colon. FISH revealed the presence of occasional bacteria in the colonic lamina propria, which were more common in Commd1ΔMye mice than in control animals (Figure 5A), which was not explained by changes in overall intestinal permeability (Figure S4A). An analysis of the architecture of the epithelium revealed a significant reduction in goblet cells in the colon (Figures 5B and 5C). A topographical analysis indicated that the deficiency in goblet cells was most pronounced in the distal colon of Commd1ΔMye (Figures S4B and S4C) and not observed in the small intestine. Temporally, this phenotype was evident by 16 weeks of age, when bacterial penetration in the lamina propria was also significantly higher in Commd1ΔMye mice (Figures S4D and S4E), but was not present soon after weaning (6 weeks of age), when bacterial and goblet cell counts were comparable between the groups. Interestingly, Commd9ΔMye had a minimal elevation in bacteria in the lamina propria and displayed no goblet cell phenotype (Figures 5D–5F). In contrast, Cybb animals displayed significant bacterial penetration in the colonic lamina propria and, akin to Commd1ΔMye, had a significant decrease in goblet cell density in the colon (Figures 5G–5I). In contrast, enteroendocrine cells, another differentiated epithelial lineage, were unaffected in Commd1ΔMye mice (Figures S5A and S5B).
Figure 5. Myeloid Defects Are Associated with Goblet Cell Alterations.

(A) Quantification of bacterial penetration into the colonic lamina propria of Commd1fl/fl or Commd1ΔMye, expressed as fold over the Commd1fl/fl group (based on FISH staining). Additional data are presented in Figures S4A and S4D. Data displayed are mean and SEM.
(B) Representative images of Alcian blue staining of the distal colon of Commd1fl/fl or Commd1ΔMye mice (scale bars, 100 μm). Additional data are presented in Figures S4B and S4C.
(C) Quantification of goblet cell density in the distal colon of Commd1fl/fl or Commd1ΔMye (5 mice per group). Data displayed indicate mean and SEM. Additional data are presented in Figure S4E.
(D) Same as in (A), but in Commd9fl/fl or Commd9ΔMye mice (6 animals per group).
(E) Same as in (B), but in Commd9fl/fl or Commd9ΔMye mice.
(F) Same as in (C), but in Commd9fl/fl or Commd9ΔMye mice (6 animals per group).
(G) Same as in (A), but in Cybb+ (n = 4) or Cybb− (n = 5) mice.
(H) Same as in (B), but in Cybb+ or Cybb- mice.
(I) Same as in (C), but in Cybb+ or Cybb− mice (5 animals per group).
(J) Representative images of immunofluorescence staining for Muc2 (red), UAE-1 lectin (green), and nuclei (Hoechst, blue) in colonic tissue (from over 3 separateiterations). Scale bars, 100 μm (50 μm in insets).
(K) Representative images (from over 5 separate iterations) of fluorescence in situ hybridization (FISH) for eubacteria in colonic tissue (treated with Carnoy’s fixative to preserve the mucus layer). Scale bars, 50 μm.
(L) Quantification of the distance between the top of the epithelium and luminal bacteria is depicted in Commd1fl/fl or Commd1ΔMye mice (5 animals per group).Data displayed are mean and SEM. *p < 0.05; **p < 0.01; ns, not significant (unpaired Student’s t test). Figure S4.
Immunostaining for the mucin protein Muc2 also confirmed the reduction in goblet cells seen in Commd1ΔMye mice (Figure 5J). Moreover, Commd1ΔMye had a thinner mucus layer and decreased distance between the epithelium and luminal bacteria (Figures 5K and 5L). Interestingly, there was not only a reduction in numbers but also altered morphology of goblet cells, which, in contrast to control animals, stained poorly for glycoproteins using the plant lectin UAE-1 (Figure 5J). This morphologic feature was consistent with reduced goblet cell maturation (Nowarski et al., 2015). Altogether, these findings indicated that defective myeloid cell function has a significant impact on goblet cell differentiation and function in the colon.
Goblet Cell Depletion Is Linked to Colonic Microbiota and Reduced Tuft Cell Density
It has been shown that type 2 cytokines derived from ILC2 and Th2 cells, have direct trophic effects on goblet cells (Gerbe et al., 2016; Howitt et al., 2016; von Moltke et al., 2016). Moreover, the recruitment of these cells to the mucosa is controlled by Tuft cells through their production of IL-25 (Gerbe et al., 2016; Howitt et al., 2016; von Moltke et al., 2016). We started by assessing gene expression in the colon for the Tuft cell marker genes Dclk1 and Il25, as well as the type 2 cytokines Il4 and Il13. In every case, gene expression was reduced in colonic samples from the Commd1ΔMye mice compared to those from control mice (Figure 6A). Furthermore, immunofluorescence staining indicated a significant reduction in Tuft cell density in Commd1ΔMye mice as well as Cybb− mice (Figures 6C and 6D). To test the possibility that bacterial penetration in the lamina propria of the colon might be linked to the observed phenotypes, we treated mice with oral antibiotics known to reduce bacterial counts in the colon (Figure 6E). This led to the normalization of gene expression of Tuft cell (Dclk1, Il25) and ILC2/Th2 cell (Il4, Il13) markers (Figure S6A) and the normalization of Tuft cell and goblet cell density in Commd1ΔMye mice (Figures 6F–6I). Importantly, these results were reproduced using a different antibiotic combination devoid of metronidazole (Figures S6B and S6C), an antibacterial agent that also has anti-protozoal properties, which can affect goblet cell density (Howitt et al., 2016). Thus, altered phagocytic function can disturb the Tuft cell-goblet cell axis.
Figure 6. Reduced Tuft Cell Density Is Associated with Goblet Cell Depletion and Is Linked to Colonic Microbiota.

(A) Real-time qPCR analysis for the indicated genes in colonic tissue of Commd1fl/fl (n = 3) or Commd1ΔMye mice (n = 4). Data are the aggregate of 2 experiments and indicate the mean and SEM. *p < 0.05; **p < 0.01 (unpaired Student’s t test).
(B) Quantification of DCLK-1-positive Tuft cells in colonic tissue of Commd1fl/fl or Commd1ΔMye mice (3 animals per group). Data displayed indicate mean and SEM. ***p < 0.001 (unpaired Student’s t test). Additional data are presented in Figures S5A and S5B.
(C) Quantification of DCLK-1-positive Tuft cells in colonic tissue of Cybb+ and Cybb− mice (5 animals per group). Data displayed indicate mean and SEM. ***p < 0.001 (unpaired Student’s t test).
(D) Representative immunofluorescence staining of colonic tissues (from over 5 repeat iterations) for DCLK-1 (green) and nuclei (Hoechst, blue) in Commd1fl/fl or Commd1DMye (left images) and in Cybb+ or Cybb− mice (right images). Scale bars, 50 μm.
(E) Schematic depicting antibiotic treatment course for animals depicted in (F), (G), (H), and (I).
(F) Representative images (from over 5 separate iterations) of immunofluorescence staining for DCLK-1 (green) and nuclei (Hoechst, blue) in colonic tissues of Commd1fl/fl or Commd1ΔMye mice that had been treated with oral antibiotics. Scale bars, 50 μm.
(G) Quantification of images shown in (F) for DCLK-1-positive cells in colonic tissue of Commd1fl/fl or Commd1ΔMye mice treated with antibiotics (3 animals per group). Data displayed indicate mean and SEM. ns, not significant (unpaired Student’s t test). Additional data are presented in Figures S6A–S6C.
(H) Representative images of Alcian blue staining (from over 5 separate iterations) of colonic tissues from Commd1fl/fl or Commd1ΔMye mice treated with oral antibiotics. Scale bars, 100 mm. Additional data are presented in Figure S6C.
(I) Quantification of images shown in (H) for Alcian-blue-positive goblet cells in colonic tissue of Commd1fl/fl or Commd1ΔMye mice treated with antibiotics (3 animals per group). Quantitative data indicate mean and SEM. ns, not significant (unpaired Student’s t test). Additional data are presented in Figure S6B. Figures S5 and S6.
Prostaglandins Have a Direct Effect on Tuft Cell Differentiation
Our next goal was to identify a mediator released by macrophages that could link defective bacterial handling and alterations in Tuft cell density. Given the role of COX-2 induction in the development of excess colonic adenomas in these mouse models (Figure 4), we postulated that the same signal might be involved in the epithelial differentiation phenotypes. First, we determined that Ptgs2 expression was increased in the colonic mucosa of Commd1ΔMye mice, akin to the observation in polyps from ApcMin, Commd1ΔMye mice (Figure 7A). In line with our observations in the adenoma models, Ptgs1 expression was not increased, further suggesting a preferential role for COX-2. To assess the importance of prostaglandin production in the phenotypes of the colonic epithelium, animals were treated with aspirin (Figure 7B), which led to the normalization of Tuft cell and goblet cell densities in Commd1ΔMye mice (Figures 7C–7F). To confirm whether these effects were specifically due to COX-2, a similar experiment was performed using a COX-2 selective inhibitor, celecoxib, which also led to the normalization of goblet cell density in Commd1ΔMye mice (Figures S7A and S7B).
Figure 7. Prostaglandins Affect Tuft Cell Density and Epithelial Differentiation.

(A) Real-time qPCR analysis for the indicated genes in colonic tissue of Commd1fl/fl (n = 3) or Commd1ΔMye mice (n = 4). Data are the aggregate of 2 experiments and indicate the mean and SEM. *p < 0.05; **p < 0.01 (unpaired Student’s t test).
(B) Schematic depicting the aspirin treatment course for data shown in (B), (C), (D), and (E).
(C) Representative images (from over 5 independent iterations) of immunofluorescence staining for DCLK-1 (green) and nuclei (Hoechst, blue) in colonic tissues of Commd1fl/fl or Commd1ΔMye mice treated with aspirin. Scale bars, 50 μm.
(D) Quantification of images shown in (C) for Commd1fl/fl or Commd1ΔMye mice (5 and 4 animals, respectively) that had been treated with aspirin. Quantitative data indicate mean and SEM. ns, not significant (unpaired Student’s t test).
(E) Representative images of Alcian blue staining of colonic tissues from Commd1fl/fl or Commd1ΔMye mice treated with aspirin. Scale bars, 100 μm.
(F) Quantification of goblet cell density from images shown in (E) for Commd1fl/fl or Commd1DMye mice (5 and 4 animals, respectively) that had been treated with aspirin. Quantitative data indicate mean and SEM. ns, not significant (unpaired Student’s t test).
(G) Quantification of Tuft cell density in colonic2D organoids supplemented with PGE-2 (top) or aspirin (bottom) in the growth media. Data displayed indicate mean and SEM (representative of 2 experiments with four technical replicates). *p < 0.05; ns, not significant (unpaired Student’s t test).
(H) Model highlighting how bacterial penetration in the mucosa can affect adenoma formation and epithelial differentiation. Stimulatory signals are indicated by blue arrows; inhibitory signals are noted by red arrows.
The in vivo effects of COX inhibition could result from the effects of prostaglandins on a number of potential targets, including immune cells such as ILC2 cells, or on epithelial cells themselves. To try to dissect its potential downstream target, we examined the effects on colonic organoids of prostaglandin supplementation or COX inhibition. Organoids were seeded on a 2D format designed for optimal high-throughput imaging and supplemented with either prostaglandin E2 (PGE-2) or aspirin. Tuft cell density by immunofluorescence staining was significantly reduced by PGE-2 supplementation and augmented by COX inhibition by aspirin (Figure 7G). This indicates that Tuft cell differentiation is affected by prostaglandin production, suggesting that COX-2 induction in lamina propria myeloid cells can directly modulate this differentiation program.
DISCUSSION
COMMD1 has known regulatory functions on NF-kB and hypoxia-inducible factor (HIF) activation (Mackenzie et al., 2016; Maine et al., 2007; van de Sluis et al., 2010), and more recently, it has been found to regulate the sorting of receptors and transporters that traverse through the endosomal compartment (PhillipsKrawczak et al., 2015). The defect in microbial handling observed in Commd1-deficient macrophages establishes that COMMD1 and the CCC complex are important regulators of phagocytic activity. Although this finding was initially unexpected, it is in agreement with the localization of COMMD proteins and other components of the CCC complex on phagolysosomes, as reported previously (Dill et al., 2015) and directly confirmed here. Moreover, our experiments further demonstrate that COMMD1 is required for copper transport into the phagolysosome lumen, an event required for optimal bactericidal activity (White et al., 2009). The impairment in copper transport is in agreement with the role of the CCC complex in ATP7A trafficking in the endosomal compartment (Phillips-Krawczak et al., 2015) and is likely one of several mechanisms that may regulate phagolysosome function.
The studies presented here also indicate that, in general terms, the phagocytic proficiency of myeloid cells in the intestinal lamina propria regulates intestinal tumor development (Figure 7H, left). The persistence of intracellular bacteria has been previously shown to potentiate COX-2 induction in macrophages (Miller et al., 2015). In the experiments presented here, we show that defects in bacterial elimination are associated with increased COX-2 induction in vivo, and our data highlight the importance of this event in adenoma development, in agreement with extensive prior literature linking COX-2 to neoplastic development in the intestinal tract (Moreira and Castells, 2011; Williams et al., 1999). It has been recognized for some time that macrophages are a principal source for increased COX-2 expression in intestinal adenomas (Hull et al., 1999). Moreover, the same group previously showed that overexpression of COX-2 in the myeloid compartment (using the same Lys2Cre driver utilized here) can lead to increased intestinal tumorigenesis (Hull et al., 2017). However, in other studies, conditional Ptgs2 gene deletion only resulted in reduced intestinal polyp burden when the epithelial compartment was targeted in female mice, with no effects seen in males or after myeloid cell deletion of the gene (Cherukuri et al., 2014). These discrepancies between the various models remain unaccounted for in the field.
In addition to its role in tumorigenesis, we show that the balance of cell lineages in the intestinal epithelium is also regulated in response to penetration of the lamina propria by luminal bacteria. Impaired bacterial elimination by resident myeloid cells in the lamina propria, as seen in mice with myeloid-specific deletion of Commd1 or deficient in NADPH oxidase activity (Cybb null animals), results in decreased Tuft cell density. Overall, our data indicate that these effects are mediated by COX-2 induction and prostaglandin production (Figure 7H, right). These findings are not seen in Commd9DMye mice, which display less severe disruptions of the CCC complex and no appreciable deficits in macrophage activity, at least against gram-negative bacteria. A series of additional effects are observed in Commd1DMye animals, such as the reduction in Th2 cytokine expression in the colon, that conform well to recent studies that place Tuft cells at the apex of a regulatory cascade that acts on Th2/ILC2 cells in the lamina propria to, ultimately, regulate goblet cell differentiation (Gerbe et al., 2016; Howitt et al., 2016; von Moltke et al., 2016). While prior studies had suggested that myeloid cells play a role in regulating goblet cell density and anti-helminthic responses (Bowcutt et al., 2014), the precise mechanisms behind this or any indication of an effect on the Tuft cell compartment, had not been elucidated prior to our studies. It is interesting to note that the density of macrophages is highest in the large intestine, with a progressive increase in these cells toward the distal mouse colon (Mowat and Agace, 2014), which may be linked to the regional nature of the phenotypes observed here.
The experiments included here also indicate that PGE-2 can directly affect Tuft cells in in vitro systems. Interestingly, Tuft cells are the main epithelial cell type expressing COX-1 (Gerbe et al., 2011), an enzyme isoform also involved in prostaglandin synthesis, suggesting the possibility that prostaglandins can play an autocrine role in regulating Tuft cell density in the mucosa. Indeed, the effects of COX inhibition by aspirin on isolated colonic crypts in culture shown here strongly suggest that prostaglandins can also be derived from epithelial sources. Finally, recent studies have demonstrated that PGE-2 production promotes mucosal barrier function; therefore, the induction of COX-2 as a result of bacterial penetration into the lamina propria could be understood as a protective response (Duffin et al., 2016).
Altogether, our studies indicate that myeloid cells in the intestinal lamina propria play a critical role in microbial-immune interactions that affect epithelial differentiation and intestinal homeostasis. In addition to the Tuft cell-ILC2/Th2-goblet cell axis that has been recently described, we uncovered a regulatory loop triggered by penetrating bacteria that involves lamina propria macrophages, PGE-2 production, and Tuft cells. We find that alterations in these pathways can promote colon cancer development, thus establishing additional links between innate immune pathways in the myeloid cell compartment and tumor development. The role that natural variation in innate immune function plays in adenoma development in the human population is an open question that merits future investigation.
EXPERIMENTAL PROCEDURES
Animals
Mice were housed in barrier facilities and fed an irradiated standard diet. All animalprocedureswereapprovedbytheInstitutionalAnimalCareandUseCommittee (IACUC) and were under the oversight of the UT Southwestern Animal Resource Center (ARC). The conditional Commd1 allele (Commd1fl/fl) was previously described (Vonk et al., 2011). To delete this gene in the myeloid compartment (Commd1ΔMye), Commd1fl/fl were bred with mice expressing Cre under the control of the Lys2 gene (Lys2Cre knockin mice), which were obtained from The Jackson Laboratory (stock #004781, Bar Harbor, ME, USA). Commd9fl/fl mice have been previously described (Li et al., 2015); myeloid-specific deletion of Commd9 was performed as in the case of Commd1. In all experiments, animals with only one knockin allele were utilized (i.e., a heterozygote for the knockin and the wild-type Lyz2 alleles). Mice with NAPDH oxidase deficiency as a result of Cybb gene inactivation were also obtained from The Jackson Laboratory (stock #002365, Bar Harbor, ME, USA). All animals were backcrossed for at least 7 generations into the C57BL/6 background. All experiments were performed in adult mice (over the age of 6 weeks) and in both genders, with the exception of Cybb− mice, which were all males, given that the mutation is X-linked.
BMDMs
BMDMs were generated through the isolation of bone marrow from the femurs and tibias of mice. Marrow cells were cultured in RPMI containing 10% fetal bovine serum (FBS), antibiotics (100 μg/mL penicillin G and 100 μg/mL streptomycin), and 20 ng/mL granulocyte-macrophage colony-stimulating factor (GM-CSF) (Peprotech, Rocky Hill, NJ, USA). Differentiation of myeloid cells was allowed to proceed for 10 days. Cells were given fresh media on days 3 and 6 following bone marrow isolation.
Western Blot
Cell lysate preparation, SDS-PAGE, western blot transfer, and immunoblotting were performed as previously described (Burstein et al., 2004). The antibodies used for western blotting in this study are described in Table S1.
Phagocytosis Assays
BMDMs were plated onto chambered slides for imaging experiments and 6-well plates for flow cytometry experiments. After 48 hr, cells were given fresh RPMI containing 10% FBS in the absence or presence of fluorescently labeled bacterial targets for 1 hr at 37°C. Bacterial targets included E. coli (K-12 strain) bioparticles labeled with pH-Rodo or Alexa Fluor 488 conjugate (Thermo Fisher Scientific, Waltham, MA, USA) or live E. coli (BL21 strain) transformed with an isopropyl β-D-1-thiogalactopyranoside (IPTG)-inducible plasmid to express GFP. After exposure to these targets, cells were stained and imaged for confocal microscopy, as described later. In other instances, cells were subjected to fluorescence-activated cell sorting (FACS). In that case, BMDMs were washed twice in FACS media buffer (PBS/0.01% sodium azide/2.5% FBS) and harvested into 0.5 mL FACS media buffer. Flow-cytometric analysis was performed using BD FACSCanto (BD Biosciences, Mountain View, CA, USA) and analyzed with FlowJo software.
Copper Mobilization Assay
Wild-type and Commd1 knockout BMDMs were plated on coverslips at 300,000 cells per well in 12-well plates. After 16–24 hr, macrophages were treated with or without 250 mM CuCl2 overnight. E. coli cells (BL21 strain, Thermo Fisher Scientific, Waltham, MA, USA) were transformed with pET-HisGFP and incubated in Luria-Bertani (LB) broth on a shaker at 37°C overnight, and isopropyl β-D-1-thiogalac- topyranoside (IPTG) was added to bacteria cultures for 3–4 hr to induce expression of GFP. Bacteria were pelleted and resuspended in 500 μL sterile cold PBS. Resuspended solutions were treated with or without 2 μL CF4 probe (kindly provided by Chris Chang, University of California, Berkeley) and then placed on a shaker at 37°C for 30 min (Lim et al., 2015). Bacteria were pelleted and washed twice with cold PBS. CF4-labeled or unlabeled E. coli cells were added to macrophages at an MOI of 10:1. Cell culture plates were spun at 500 × g for 5 min at room temperature and placed in an incubator for 10 min to allow for phagocytosis of bacteria. Hoechst stain was added to the coverslips for 2 min. Coverslips were washed twice briefly in PBS prior to being mounted onto microscope slides and immediately imaged using a Zeiss confocal microscope (LSM 700).
Adenoma Model
Intestinal adenoma formation was studied in ApcMin mice, which were obtained from The Jackson Laboratory (Stock #002020, Bar Harbor, ME, USA). These animals were successively bred with Commd1ΔMye to derive ApcMin, Commd1DMye and littermate ApcMin, Commd1fl/fl control mice. Given that the Cybb gene is located in the X chromosome, male ApcMin mice were bred with female Cybb−/− or wild-type mice to derive male mice with NAPDH oxidase deficiency (ApcMin, Cybb−) or control animals (wild-type for Cybb). Tumor phenotypes were determined using a stereoscopic dissection microscope (Stemi 2000-c, Carl Zeiss, Oberkochen, Germany). Briefly, the intestine was dissected and opened along the mesenteric side and inspected under the microscope to count the number of adenomas and determine their dimensions. Complete blood counts were performed by the clinical laboratory of the Animal Resource Center at UT Southwestern.
Tissue Processing, Staining, and Imaging
Tissues were fixed in 4% paraformaldehyde (PFA) overnight at 4°C, washed 3 times with PBS, and placed in 70% ethanol. Tissue processing (paraffin embedding and sectioning) and routine histologic staining (H&E, Alcian blue) were performed by the UT Southwestern Molecular Pathology Core. For immunofluorescence staining of tissues, paraffin-embedded slides were deparaffinized, and antigen retrieval was performed by incubation in citrate buffer (Sigma-Aldrich, St. Louis, MO, USA) and short heating in a microwave oven. The samples were then incubated in blocking buffer, consisting of PBS supplemented with either normal goat or rat serum (depending on the species of primary antibody) for 30 min. Samples were incubated in a humidifier chamber overnight at 4°C with primary antibodies diluted in blocking buffer. The UAE-1 lectin (1:150 dilution; Sigma-Aldrich, St. Louis, MO, USA) was applied in a manner similar to the application of a primary antibody at this stage in the staining process. Following 3 washes in PBS, tissue samples were incubated with secondary antibodies, which were incubated and then washed three times in PBS, followed by the addition of Hoechst 33342 nuclear stain and coverslip mounting with SlowFade Gold Antifade reagent (Life Technologies, Grand Island, NY, USA). Immunofluorescence staining of cells was performed as previously reported (Phillips-Krawczak et al., 2015). Details regarding the primary antibodies used in this study are presented in Table S1.
Aspirin, Antibiotic, and Celecoxib Treatments
The administration of aspirin to mice was performed by providing a diet in which 0.05% aspirin was blended into AIN-76A rodent diet (Envigo, Loughborough, UK). Administration of celecoxib to mice was performed through providing an AIN-76A rodent diet (Envigo, Loughborough, UK) supplemented with 0.1% celecoxib (Alembic Pharmaceuticals, Vadodara, India). Mice were given the antibiotics in the drinking water for 3 or 15 weeks, as indicated. The following combinations were used: (1) ampicillin (1 g/L), neomycin (1 g/L), metronidazole (1 g/L), vancomycin (500 mg/L), and (2) a combination in which metronidazole was replaced by clindamycin (1 g/L). Ampicillin, metronidazole, clindamycin, and vancomycin were obtained from Sigma-Aldrich (St. Louis, MO, USA); neomycin was obtained from Thermo Fisher Scientific (Waltham, MA, USA). Treatment duration is indicated in each experiment.
FITC-Dextran Permeability Assay
Intestinal permeability was assessed by the administration of fluorescein isothiocyanate (FITC)-dextran (average molecular weight, 4000; Sigma-Aldrich, St. Louis, MO). After a 4-hr fast, mice were orally gavaged with FITC-dextran (20 mg/100 g body weight). Whole blood was obtained by cheek bleeding, and fluorescence was measured in the serum by a fluorometer (BioTek, Winooski, VT, USA).
FISH
To examine the mucus barrier, tissues were fixed in Carnoy’s solution (Ricca Chemical, Arlington, TX, USA). Analysis of bacteria in the lamina propria of adenomas or normal colonic mucosa was performed in PFA-fixed tissue sections. Paraffin-embedded slides were de-paraffinized and hybridized to universal eubacterial probe (5’-GCTGCCTCCCGTAGGAGT-3’) or a control probe (5’-CGACGGAGGGCATCCTCA-3’) labeled with Alexa 488. All probes were obtained from IDT Technologies (Coralville, IA, USA). Hybridization was performed overnight at 56C, followed by washing and counterstaining with the nuclear dye Hoechst 33342. For concurrent FISH and immunofluorescence staining, slides were stained for eubacteria FISH probe, as described earlier. The antigen retrieval step was excluded. COX-2 and F4/80 staining was performed as follows. After a 30-min blocking step with BSA, COX-2 and F4/80 antibodies were diluted, applied to the samples, and incubated in a humidifier chamber overnight at 4°C. Alexa Fluor 647 secondary anti-rat (Thermo Fisher Scientific) or Alexa Fluor 555 anti-rabbit antibodies (Thermo Fisher Scientific) were added and incubated at room temperature for 30 min. After 3 washes in PBS and the addition of Hoechst 33342 nuclear stain, coverslips were mounted on slides.
Imaging Analysis and Quantification
Bright-field and fluorescent images were acquired with an Optronics Microfire CCD (charge-coupled device) camera on a Leica DM2000 Upright Compound Microscope. Confocal imaging was performed with an LSM 700 (403 oil immersion objective lens; Carl Zeiss) or A1R (60×/1.4 oil immersion objective lens; Nikon). Images were analyzed using either ZEN software or FUJI ImageJ (NIH, Bethesda, MD, USA; https://imagej.nih.gov/ij/). Tuft cells (Dckl1+ epithelial cells), enteroendocrine cells (CHGA+ epithelial cells), and goblet cell (morphologically identified in Alcian-blue-stained sections) were manually counted and normalized to the number of crypt units. Quantification of bacterial penetration into the normal mucosa was performed in a similar manner. Quantification of bacterial penetration into polyps was performed by measuring the area of fluorescent bacteria per image and expressing it as a ratio to nuclear fluorescent area and expressed as fold change from wild-type.
Histologic Scoring
The assessment of inflammation was performed in a blinded fashion by a gastrointestinal (GI) pathologist. H&E-stained sections were rated for two features of chronic inflammation (homogeneous diffuse increase in inflammatory cells in the lamina propria or basally located lymphoid aggregates) and one feature of acute inflammation (presence of crypt abscesses). Each feature received 1 point (for a possible maximum of 3 points), with normal mucosa without these findings receiving only 1 point.
RNA Extraction and real-time qPCR
Tissue samples were stabilized with RNAlater (QIAGEN), and RNA was extracted using the RNeasy Mini Kit (QIAGEN) following the manufacturer’s instructions. For cDNA synthesis, 5 μg of the total RNA was reverse transcribed using SuperScript III Reverse Transcriptase (Invitrogen). Real-time qRT-PCR was performed using SYBR Green-based detection (Invitrogen) and a Mastercycler (Eppendorf, Germany). Technical triplicates were used, and data were normalized to the housekeeping gene Gapdh; the relative abundance of transcripts was calculated by the comparative DDCt method. Primer sequences are provided in Table S2.
Colonic Organoid Cultures
2D colonic organoid cultures were prepared as recently described (Thorne et al., 2018). Large intestines were removed from freshly sacrificed 4- to 16-week-old mice. For imaging experiments, cells were plated in Falcon 96-Well, Black/Clear Tissue Culture-Treated Plates (flat bottom with lid; catalog #353219). All plates were imaged using a GE INCell 6000 automated microscope, with a 10× objective lens, and DAPI, FITC, and tetramethylrhod-amine isothiocyanate (TRITC) filter sets. For each experimental well, 16 nonoverlapping images were taken per well, covering the entire well.
Statistical Analysis
Data for continuous variables involving two groups were analyzed by unpaired Student’s t test, while non-parametric data were evaluated with the χ2 test using built-in functions in Microsoft Excel. p values less than 0.05 were considered significant. Statistical analysis of RNA-seq and microbiome analysis data are described earlier.
Supplementary Material
Highlights.
Commd1 is required for optimal myeloid cell phagocytic activity
Poor bacterial clearance by intestinal myeloid cells promotes colonic neoplasia
Myeloid cells also regulate epithelial differentiation in response to bacteria
COX-2 induction in intestinal macrophages links bacteria to these effects
ACKNOWLEDGMENTS
We are grateful to Bart van de Sluis, who developed the Commd1 floxed mice and graciously shared them with us. We are grateful to Chris Chang for providing the CF4 copper probe and to Michael Petris for giving general advice about its use in macrophages. We want to also thank Shuiqing Hu for help with F4/80 staining, as well as John Shelton and the staff of the Molecular Pathology Core at UT Southwestern. This work was supported by NIH through the following grants: R01DK073639 (to E.B.), R01DK107733 (to E.B. and D.D.B.), T32DK007745 (to L.L.M.), K08DK107886 (to E.T.), K01DK106346 (to A.S.) and P30CA142543 (to the UT Southwestern Live Cell Imaging Facility). Additional support was provided by the Cancer Prevention Research Institute of Texas (CPRIT) through grant RP130409 (to E.B.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and two tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.07.066.
REFERENCES
- Bartuzi P, Billadeau DD, Favier R, Rong S, Dekker D, Fedoseienko A, Fieten H, Wijers M, Levels JH, Huijkman N, et al. (2016). CCC- and WASH-mediated endosomal sorting of LDLR is required for normal clearance of circulating LDL. Nat. Commun 7, 10961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowcutt R, Bramhall M, Logunova L, Wilson J, Booth C, Carding SR, Grencis R, and Cruickshank S (2014). A role for the pattern recognition receptor Nod2 in promoting recruitment of CD103+ dendritic cells to the colon in response to Trichuris muris infection. Mucosal Immunol. 7, 1094–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burstein E, Ganesh L, Dick RD, van De Sluis B, Wilkinson JC, Klomp LW, Wijmenga C, Brewer GJ, Nabel GJ, and Duckett CS (2004). A novel role for XIAP in copper homeostasis through regulation of MURR1. EMBO J. 23, 244–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrakesan P, May R, Qu D, Weygant N, Taylor VE, Li JD, Ali N, Sureban SM, Qante M, Wang TC, et al. (2015). Dclk1+ small intestinal epithelial tuft cells display the hallmarks of quiescence and self-renewal. Oncotarget 6, 30876–30886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherukuri DP, Ishikawa TO, Chun P, Catapang A, Elashoff D, Grogan TR, Bugni J, and Herschman HR (2014). Targeted Cox2 gene deletion in intestinal epithelial cells decreases tumorigenesis in female, but not male, ApcMin/+ mice. Mol. Oncol 8, 169–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiriaco M, Salfa I, Di Matteo G, Rossi P, and Finocchi A (2016). Chronic granulomatous disease: Clinical, molecular, and therapeutic aspects. Pediatr. Allergy Immunol 27, 242–253. [DOI] [PubMed] [Google Scholar]
- Dill BD, Gierlinski M, Härtlova A, Arandilla AG, Guo M, Clarke RG, and Trost M (2015). Quantitative proteome analysis of temporally resolved phagosomes following uptake via key phagocytic receptors. Mol. Cell. Proteomics 14, 1334–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffin R, O’Connor RA, Crittenden S, Forster T, Yu C, Zheng X, Smyth D, Robb CT, Rossi F, Skouras C, et al. (2016). Prostaglandin E2 constrains systemic inflammation through an innate lymphoid cell-IL-22 axis. Science 351, 1333–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagliardi G, Moroz K, and Bellows CF (2012). Immunolocalization of DCAMKL-1, a putative intestinal stem cell marker, in normal colonic tissue. Pathol. Res. Pract 208, 475–479. [DOI] [PubMed] [Google Scholar]
- Gerbe F, van Es JH, Makrini L, Brulin B, Mellitzer G, Robine S, Romagnolo B, Shroyer NF, Bourgaux JF, Pignodel C, et al. (2011). Distinct ATOH1 and Neurog3 requirements define tuft cells as a new secretory cell type in the intestinal epithelium. J. Cell Biol 192, 767–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerbe F, Sidot E, Smyth DJ, Ohmoto M, Matsumoto I, Dardalhon V, Cesses P, Garnier L, Pouzolles M, Brulin B, et al. (2016). Intestinal epithelial tuft cells initiate type 2 mucosal immunity to helminth parasites. Nature 529, 226–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikov SI, Greten FR, and Karin M (2010). Immunity, inflammation, and cancer. Cell 140, 883–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikov SI, Wang K, Mucida D, Stewart CA, Schnabl B, Jauch D, Taniguchi K, Yu GY, Osterreicher CH, Hung KE, et al. (2012). Adenomalinked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 491, 254–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda K, and Littman DR (2016). The microbiota in adaptive immune homeostasis and disease. Nature 535, 75–84. [DOI] [PubMed] [Google Scholar]
- Hooper LV (2015). Epithelial cell contributions to intestinal immunity. Adv. Immunol 126, 129–172. [DOI] [PubMed] [Google Scholar]
- Howitt MR, Lavoie S, Michaud M, Blum AM, Tran SV, Weinstock JV, Gallini CA, Redding K, Margolskee RF, Osborne LC, et al. (2016). Tuft cells, taste-chemosensory cells, orchestrate parasite type 2 immunity in the gut. Science 351, 1329–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull MA, Booth JK, Tisbury A, Scott N, Bonifer C, Markham AF, and Coletta PL (1999). Cyclooxygenase 2 is up-regulated and localized to macrophages in the intestine of Min mice. Br. J. Cancer 79, 1399–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull MA, Faluyi OO, Ko CW, Holwell S, Scott DJ, Cuthbert RJ, Poulsom R, Goodlad R, Bonifer C, Markham AF, and Coletta PL (2006). Regulation of stromal cell cyclooxygenase-2 in the ApcMin/+ mouse model of intestinal tumorigenesis. Carcinogenesis 27, 382–391. [DOI] [PubMed] [Google Scholar]
- Hull MA, Cuthbert RJ, Ko CWS, Scott DJ, Cartwright EJ, Hawcroft G, Perry SL, Ingram N, Carr IM, Markham AF, et al. (2017). Paracrine cyclooxygenase-2 activity by macrophages drives colorectal adenoma progression in the Apc Min/+ mouse model of intestinal tumorigenesis. Sci. Rep 7, 6074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson ME, Sjövall H, and Hansson GC (2013). The gastrointestinal mucus system in health and disease. Nat. Rev. Gastroenterol. Hepatol 10, 352–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppel N, and Balskus EP (2016). Exploring and understanding the biochemical diversity of the human microbiota. Cell Chem. Biol 23, 18–30. [DOI] [PubMed] [Google Scholar]
- Li H, Chan L, Bartuzi P, Melton SD, Weber A, Ben-Shlomo S, Varol C, Raetz M, Mao X, Starokadomskyy P, et al. (2014). Copper metabolism domain-containing 1 represses genes that promote inflammation and protects mice from colitis and colitis-associated cancer. Gastroenterology 147, 184–195.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Koo Y, Mao X, Sifuentes-Dominguez L, Morris LL, Jia D, Miyata N, Faulkner RA, van Deursen JM, Vooijs M, et al. (2015). Endosomal sorting of Notch receptors through COMMD9-dependent pathways modulates Notch signaling. J. Cell Biol 211, 605–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim YW, Sanz LA, Xu X, Hartono SR, and Che´ din F (2015). Genomewide DNA hypomethylation and RNA:DNA hybrid accumulation in Aicardi-Goutie` res syndrome. eLife 4, e08007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie KJ, Carroll P, Lettice L, Tarnauskaite_ Z, Reddy K, Dix F, Revuelta A, Abbondati E, Rigby RE, Rabe B, et al. (2016). Ribonuclease H2 mutations induce a cGAS/STING-dependent innate immune response. EMBO J. 35, 831–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maine GN, Mao X, Komarck CM, and Burstein E (2007). COMMD1 promotes the ubiquitination of NF-kappaB subunits through a cullin-containing ubiquitin ligase. EMBO J. 26, 436–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MS, Rialdi A, Ho JS, Tilove M, Martinez-Gil L, Moshkina NP, Peralta Z, Noel J, Melegari C, Maestre AM, et al. (2015). Senataxin suppresses the antiviral transcriptional response and controls viral biogenesis. Nat. Immunol 16, 485–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira L, and Castells A (2011). Cyclooxygenase as a target for colorectal cancer chemoprevention. Curr. Drug Targets 12, 1888–1894. [DOI] [PubMed] [Google Scholar]
- Mowat AM, and Agace WW (2014). Regional specialization within the intestinal immune system. Nat. Rev. Immunol 14, 667–685. [DOI] [PubMed] [Google Scholar]
- Nakanishi Y, Seno H, Fukuoka A, Ueo T, Yamaga Y, Maruno T, Nakanishi N, Kanda K, Komekado H, Kawada M, et al. (2013). Dclk1 distinguishes between tumor and normal stem cells in the intestine. Nat. Genet 45, 98–103. [DOI] [PubMed] [Google Scholar]
- Nowarski R, Jackson R, Gagliani N, de Zoete MR, Palm NW, Bailis W, Low JS, Harman CC, Graham M, Elinav E, and Flavell RA (2015). Epithelial IL-18 equilibrium controls barrier function in colitis. Cell 163, 1444–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima M, Dinchuk JE, Kargman SL, Oshima H, Hancock B, Kwong E, Trzaskos JM, Evans JF, and Taketo MM (1996). Suppression of intestinal polyposis in Apc delta716 knockout mice by inhibition of cyclooxygenase 2 (COX-2). Cell 87, 803–809. [DOI] [PubMed] [Google Scholar]
- Phillips-Krawczak CA, Singla A, Starokadomskyy P, Deng Z, Osborne DG, Li H, Dick CJ, Gomez TS, Koenecke M, Zhang JS, et al. (2015). COMMD1 is linked to the WASH complex and regulates endosomal trafficking of the copper transporter ATP7A. Mol. Biol. Cell 26, 91–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roediger B, and Weninger W (2015). Group 2 innate lymphoid cells in the regulation of immune responses. Adv. Immunol 125, 111–154. [DOI] [PubMed] [Google Scholar]
- Su LK, Kinzler KW, Vogelstein B, Preisinger AC, Moser AR, Luongo C, Gould KA, and Dove WF (1992). Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science 256, 668–670. [DOI] [PubMed] [Google Scholar]
- Thaiss CA, Zmora N, Levy M, and Elinav E (2016). The microbiome and innate immunity. Nature 535, 65–74. [DOI] [PubMed] [Google Scholar]
- Thorne CA, Chen IW, Sanman LE, Cobb MH, Wu LF, and Altschuler SJ (2018). Enteroid monolayers reveal an autonomous WNT and BMP circuit controlling intestinal epithelial growth and organization. Dev. Cell 44, 624–633.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Sluis B, Rothuizen J, Pearson PL, van Oost BA, and Wijmenga C (2002). Identification of a new copper metabolism gene by positional cloning in a purebred dog population. Hum. Mol. Genet 11, 165–173. [DOI] [PubMed] [Google Scholar]
- van de Sluis B, Mao X, Zhai Y, Groot AJ, Vermeulen JF, van der Wall E, van Diest PJ, Hofker MH, Wijmenga C, Klomp LW, et al. (2010). COMMD1 disrupts HIF-1a/b dimerization and inhibits human tumor cell invasion. J. Clin. Invest 120, 2119–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Moltke J, Ji M, Liang HE, and Locksley RM (2016). Tuft-cell-derived IL-25 regulates an intestinal ILC2-epithelial response circuit. Nature 529, 221–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonk WI, Bartuzi P, de Bie P, Kloosterhuis N, Wichers CG, Berger R, Haywood S, Klomp LW, Wijmenga C, and van de Sluis B (2011). Liverspecific Commd1 knockout mice are susceptible to hepatic copper accumulation. PLoS ONE 6, e29183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonk WI, de Bie P, Wichers CG, van den Berghe PV, van der Plaats R, Berger R, Wijmenga C, Klomp LW, and van de Sluis B (2012). The copper-transporting capacity of ATP7A mutants associated with Menkes disease is ameliorated by COMMD1 as a result of improved protein expression. Cell. Mol. Life Sci 69, 149–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter J, and Ley R (2011). The human gut microbiome: ecology and recent evolutionary changes. Annu. Rev. Microbiol 65, 411–429. [DOI] [PubMed] [Google Scholar]
- White C, Lee J, Kambe T, Fritsche K, and Petris MJ (2009). A role for the ATP7A copper-transporting ATPase in macrophage bactericidal activity. J. Biol. Chem 284, 33949–33956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams CS, Mann M, and DuBois RN (1999). The role of cyclooxygenases in inflammation, cancer, and development. Oncogene 18, 7908–7916. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.