

ABSTRACT
The exchange of mobile genomic islands (MGIs) between microorganisms is often mediated by phages, which may provide benefits to the phage’s host. The present study started with the identification of Enterobacter cloacae, Klebsiella pneumoniae and Escherichia coli isolates with exceptional cephalosporin and carbapenem resistance phenotypes from patients in a neonatal ward. To identify possible molecular connections between these isolates and their β-lactam resistance phenotypes, the respective bacterial genome sequences were compared. This unveiled the existence of a family of ancient MGIs that were probably exchanged before the species E. cloacae, K. pneumoniae and E. coli emerged from their common ancestry. A representative MGI from E. cloacae was named MIR17-GI, because it harbors the novel β-lactamase gene variant blaMIR17. Importantly, our observations show that the MIR17-GI-like MGIs harbor genes associated with high-level resistance to cephalosporins. Among them, MIR17-GI stands out because MIR17 also displays carbapenemase activity. As shown by mass spectrometry, the MIR17 carbapenemase is among the most abundantly expressed proteins of the respective E. cloacae isolate. Further, we show that MIR17-GI-like islands are associated with integrated P4-like prophages. This implicates phages in the spread of cephalosporin and carbapenem resistance amongst Enterobacteriaceae. The discovery of an ancient family of MGIs, mediating the spread of cephalosporinase and carbapenemase genes, is of high clinical relevance, because high-level cephalosporin and carbapenem resistance have serious implications for the treatment of patients with enterobacteriaceal infections.
KEYWORDS: Enterobacter cloacae, proteome, carbapenemase, cephalosporinase, mobile genomic island
Introduction
The shape of bacterial genomes, as we know them today, is the outcome of many successive evolutionary events that occurred ever since the respective bacterial species branched off from their common ancestors [1]. This is clearly evidenced through comparisons of the genome sequences of individual bacterial species, where a distinction can be made between the core genome and the accessory genome. Here, the accessory genome reflects those elements that were either lost through genomic erosion, or gained through horizontal gene transfer [2–4]. Of note, the accessory genome is only the echo of those events that happened relatively recently on the evolutionary timeline, namely after the particular species emerged. In this context, it is frequently overlooked that also the core genome has been shaped from elements that were recruited through genetic exchanges, but before speciation occurred. Thus, also the core genome is the offspring of evolution and includes ancient mobile genomic elements (MGIs), which turned out beneficial for the species within its ecological niche to the extent that they are no longer readily lost [5–7]. As a consequence of their co-evolution with the species, such ancient MGIs may have lost particular traits of present-day MGIs, such as a difference in the GC content. Yet, other traits can still be discerned, such as the typical sites of integration and an overrepresentation of certain classes of genes [8]. In the present study we focus attention on an ancient family of MGIs, first discovered within the “core genome” of Enterobacter cloacae.
E. cloacae is a rod-shaped, non-spore forming, facultative anaerobic, Gram-negative bacterium belonging to the family of Enterobacteriaceae. The occurrence of E. cloacae is widespread, ranging from soil and sewage to the human gastrointestinal tract, where it is a frequent component of the gut microbiota. Importantly, E. cloacae can cause opportunistic infections and has recently emerged as a nosocomial pathogen, especially in intensive care units (9). In the clinical setting, E. cloacae has been identified as the causative agent of skin and soft tissue infections, respiratory and urinary tract infections, intra-abdominal infections, bacteremia, endocarditis, septic arthritis, and osteomyelitis in immunocompromised patients [9,10]. The antimicrobial therapy of patients with E. cloacae infections often faces complications due to the intrinsic drug resistance of this bacterium, and its propensity to acquire multiple resistance genes. Thus, resistance has been reported against ampicillin, amoxicillin, first-generation cephalosporins and cefoxitin owing to constitutive expression of the AmpC β-lactamase. E. cloacae also exhibits a high frequency of enzymatic resistance to broad-spectrum cephalosporins [11], which is typically caused by overproduction of AmpC β-lactamases. Such extended spectrum cephalosporinases (e.g. ACT, CMY and MIR) confer resistance to third-generation cephalosporins, and they are not inhibited by common β-lactamase inhibitors [12]. Until now, fourth- and fifth-generation cephalosporins maintain reasonable activity against such strains. While ACT and MIR in Enterobacter species are encoded by intrinsic chromosomal genes, CMY genes may be plasmid-borne [13,14]. In general, extended spectrum cephalosporinases of Enterobacteriaceae have been associated with mobile genomic elements.
In contrast to cephalosporinases, acquired carbapenamase genes are still fairly uncommon in E. cloacae on a global scale [12,15]. However, many extended spectrum cephalosporinases have a weak affinity for carbapenems and can hydrolyze carbapenems with low efficiency. As a consequence, clinical resistance of E. cloacae to these last-line β-lactam antibiotics is mostly brought about by outer membrane permeability defects combined with a derepression of constitutive AmpC cephalosporinases [11,12,16].
The present study started with the identification of E. cloacae, K. pneumoniae and E. coli isolates with exceptional β-lactam resistance phenotypes, including one E. cloacae isolate with decreased susceptibility for carbapenems, in patients on a neonatal ward of the University Medical Center Groningen (UMCG). To assess possible molecular connections between these isolates and their antibiotic resistance phenotypes, the respective bacterial genomes were compared. This led to the identification of four highly conserved MGIs of ~ 140 kb in the investigated isolates. This observation is of general importance, since it focuses attention on a family of genomic islands carrying a diversity of cephalosporinase-encoding genes that was already widely transduced before the species E. cloacae, K. pneumoniae and E. coli emerged from their common ancestry.
Results
Description of the study isolates and clinical background
In the present study, we investigated four epidemiologically linked Gram-negative bacterial isolates with decreased susceptibility to carbapenems as reflected by minimal inhibitory concentration (MIC) values for meropenem and/or imipenem ≥ 0.5 mg/L. Table 1 summarizes the results of the antibiotic susceptibility testing in these four isolates.
Table 1.
Antibiotic susceptibility of the bacterial study isolates.
| Isolate 1 Patient 1 E. cloacae ST-232 |
Isolate 2 Patient 1 E. cloacae ST-97 |
Isolate 3 Patient 1 K. pneumoniae ST-20 |
Isolate 4 Patient 2 E. coli ST-131 |
|
|---|---|---|---|---|
| Antibiotic | MIC (mg/L) | MIC (mg/L) | MIC (mg/L) | MIC (mg/L) |
| Amoxicillin+ clavulanic acid | > 32 | > 32 | > 32 | > 32 |
| Cefuroxime | > 64 | 32 | > 64 | > 64 |
| Cefotaxime | > 64 | < 1.0 | 16 | > 64 |
| Ceftazidime | > 64 | < 1.0 | 1 | > 64 |
| Cefoxitin | > 64 | > 64 | > 64 | 32 |
| Cefepime | 2 | < 1.0 | 2 | 16 |
| Meropenem | 8 | < 0.25# | 0.032 | 4 |
| Imipenem | 8 | < 0.25# | 0.5 | 0.75 |
| ESBL* | negative | negative | negative | positive |
Antibiotic susceptibility of the four bacterial isolates described in this study. All four isolates carried a MIR17-GI-related MGI. In three of the four isolates, a cephalosporinase gene (ampC) was integrated in this island.
#Carbapenem-resistant colony variants grew into the carbapenem inhibition zone.
*ESBL production was tested by E-tests and indicated by a > 8-fold reduction in MIC in the presence of clavulanic acid compared to cefepime, ceftazidim or cefotaxim alone.
The first study isolate (isolate 1) was a carbapenem-resistant E. cloacae that was obtained from a rectal swab of a neonate (patient 1), who had been repatriated from Curaçao to the UMCG. Prior to admission in the UMCG, the patient had been treated with meropenem. Isolate 1 showed an atypical growth phenotype resulting in small fatty colonies on Blood Agar (BA) and Mueller Hinton Agar (MHA) plates. Automated resistance analysis with the VITEK 2 system revealed increased MIC values to the carbapenems meropenem (8 mg/L) and imipenem (8 mg/L), which were subsequently confirmed by Etests. Imipenem Etests on MHA with or without 250 mg/L cloxacillin revealed that imipenem resistance was significantly reduced in the presence of cloxacillin, which is an inhibitor of β-lactamases of the AmpC-type [17]. Specifically, the MIC was reduced to 0.125 mg/L (Figure 1). This showed that the reduced carbapenem sensitivity of isolate 1 is largely due to the production of a carbapenem-degrading enzyme. Of note, confluent plating of isolate 1 on MHA results in a patchy growth phenotype that is suppressed in the presence of cloxacillin (Figure 1). Multi-locus sequence type (MLST) analysis assigns isolate 1 to the sequence type (ST-)232 (dnaA88, fusA25, gyrB49, leuS72, pyrG49, rplB12), where dnaA88 represents a novel dnaA allele. Phenotypic ESBL tests with isolate 1 were negative. Whole-genome sequencing revealed a novel β-lactamase gene variant, which is homologous to the MIR lineage. In fact, this gene is 99.3% identical to the blaMIR-1 gene from Klebsiella pneumoniae (ENA accession M37839.2) and it was thus designated blaMIR-17 (NCBI accession CEA29752.1). Specifically, MIR17 is distinct from MIR1 due to five amino acid substitutions.
Figure 1.
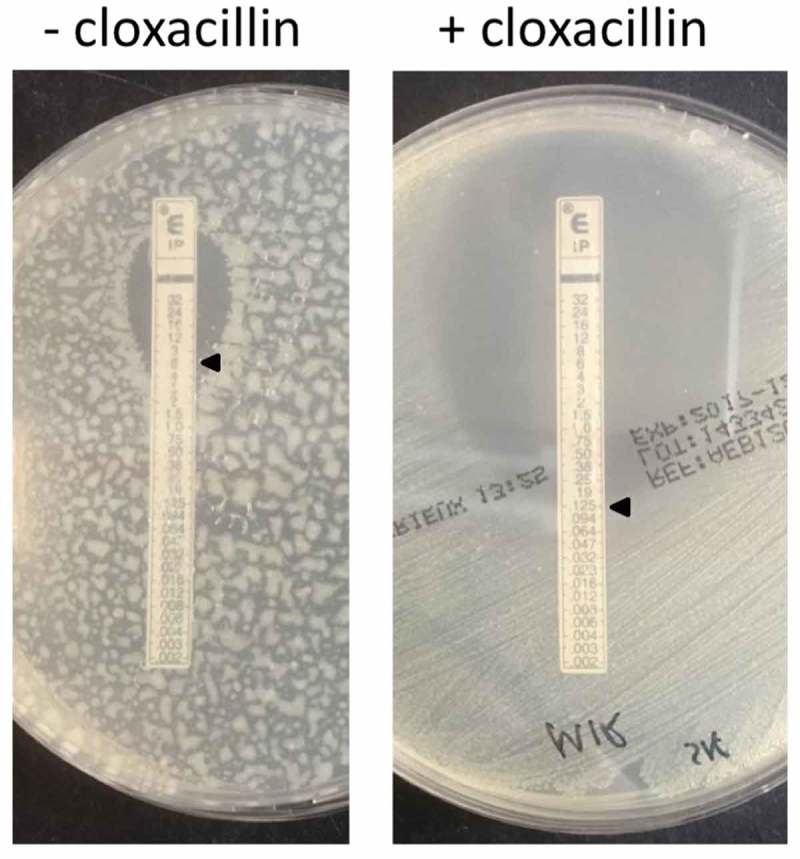
Imipenem Etest for E. cloacae study isolate 1 on Muller Hinton agar with or without cloxacillin.
Upon withdrawal of meropenem treatment for 2 weeks, a carbapenem-sensitive E. cloacae isolate (i.e. isolate 2) was obtained from an intravascular catheter tip from patient 1. This isolate 2 was susceptible to third-generation cephalosporins and carbapenems, and the sensitivity to imipenem was not influenced by the presence of cloxacillin (not shown). Genome sequencing revealed the presence of an ampC gene encoding the ACT-5 cephalosporinase. Further, MLST analysis showed that isolate 2 belongs to ST-97.
After 3 months of hospitalization, patient 1 acquired a K. pneumoniae (i.e. isolate 3) with a remarkable antibiotic resistance pattern. Specifically, isolate 3 was shown to be resistant to cefuroxime and cefotaxime, but susceptible to ceftazidime. Phenotypic ESBL-testing was negative. For the molecular detection of resistance genes, first a microarray analysis was performed and, subsequently, isolate 3 was subjected to whole genome sequencing followed by screening of the sequence against the Resfinder database. This showed the presence of a β-lactamase gene variant with 99% identity to blaSHV-140 of K. pneumoniae. This gene was therefore designated blaSHV-187 (NCBI accession LN515533.1). Specifically, SHV187 is distinct from SHV140 due to two N-terminal amino acid substitutions. MLST showed that isolate 3 belongs to ST-20.
Patient 2 was hospitalized in the same ward of the UMCG as patient 1. During this hospital stay, patient 2 acquired an E. coli (isolate 4) resistant to cefoxitin and third-generation cephalosporins. Further, isolate 4 displayed a meropenem MIC value of 4 mg/L. ESBL-tests were positive and, upon whole genome sequencing, Resfinder detected two acquired β-lactamase gene variants designated blaTEM-1b and blaCTX-M-147. Additionally, an EC-6 AmpC β-lactamase-encoding gene was detected [18]. Isolate 4 was shown to belong to ST-131.
The genomic neighborhood of blaMIR-17 is conserved in Enterobacteriaceae
Since the cephalosporinase genes identified in the four study isolates were distinct, the respective antibiotic resistance phenotypes could not be directly related to recent horizontal gene transfer events between these isolates. However, comparison of the whole genome sequencing data uncovered ~ 140-kb regions of high similarity in the four isolates, which harbor ampC genes in the case of isolates 1, 2 and 4 (Figure 2(a)). This prompted us to investigate the spread of this conserved region among Enterobacteriaceae. As shown in Figure 3 and Supplemental Figure S1, the ampC–containing region, as present in E. coli, is highly conserved in the genera Citrobacter, Escherichia, Enterobacter, Salmonella, Shigella and Klebsiella, where the sequences cluster according to the respective species. Of note, ampC genes are absent from the respective conserved regions in Salmonella and Klebsiella species, as is the case in our K. pneumoniae study isolate 3 (Figure 2(a)). Further, we observed that parts of this conserved region are present in other Enterobacteriaceae, such as Cronobacter, Dickya, Proteus, Providencia, Serratia and Yersinia species, where it lacks ampC and can be positioned at different chromosomal loci (not shown). The conserved region is completely absent from non-enterobacteriaceal Gram-negative bacteria, such as Acinetobacter, Haemophilus, Pasteurella, Prevotella and Pseudomonas species. Combined with previous studies on the phylogeny of Enterobacteriaceae [19–23], these observations are indicative of an ancient MGI that spread among the enterobacteriaceal ancestry, before genera like Enterobacter, Escherichia, and Klebsiella evolved.
Figure 2.
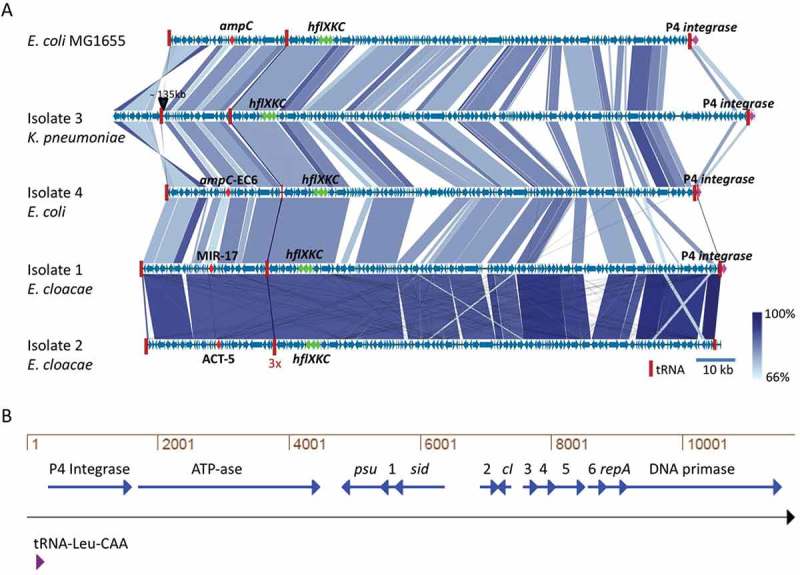
Similarity among MIR17-GI-like MGIs and schematic representation of the P4-like prophage at the 3ʹ end of MIR17-GI. (a) MIR17-GI-like MGIs identified in the four study isolates and E. coli K12 MG1655. The alignment of MIR17-GI-like MGIs was performed with Easyfig (http://mjsull.github.io/Easyfig/). The positions of relevant genes are indicated. (b) Genetic map of the early operon of the P4-like prophage associated with the 3ʹ end of MIR17-GI, following the tRNA-Leu-CAA gene. The relative positions of the genes for the phage integrase, an ATP-ase of unknown function (ATP-ase), the phage polarity suppression protein (psu), the phage capsid and scaffold protein (sid), the Ash family secondary immunity repressor (cI); the DNA replication protein (repA), and the DNA primase are indicated. Numbers correspond to the following gene annotations: 1, late gene regulator; 2, phage DNA binding protein; 3, phage immunity repressor protein; 4, immunity derepression protein; 5, phage protein of unknown function; 6, hypothetical protein for heme transfer during cytochrome c biogenesis.
Figure 3.
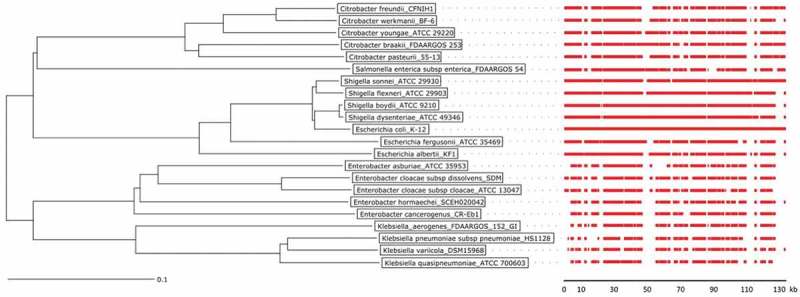
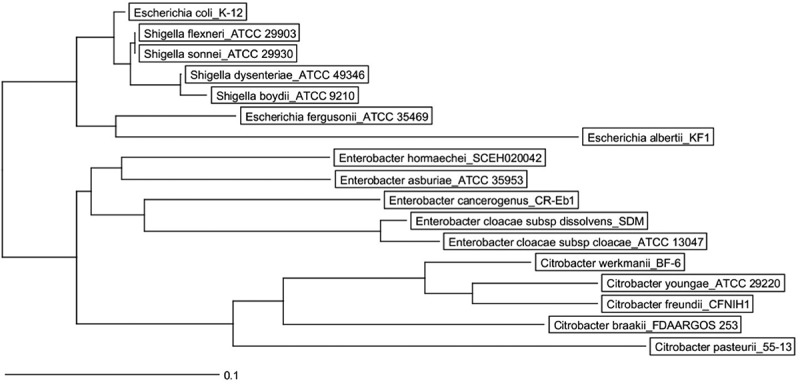
Phylogenetic tree of Citrobacter, Enterobacter, Escherichia, Klebsiella, Salmonella, and Shigella species and conservation of MIR17-GI-like MGIs.
The phylogenetic tree of was created with the Ridom SeqSphere+ software v4.1.9 using a neighbor joining algorithm. It is based on a SNP analysis of 39430 targets of the MGI from the reference genome of the E. coli K12 strain MG1655. The scale bar under the tree represents the phylogenetic distance (in %). The red bars indicate the conservation of the MIR17-GI using the respective MGI of E. coli K12 strain MG1655 as the reference. For the MGI comparisons, an 80% DNA similarity cut-off was used in DNA plotter. The scale bar under the MGI alignment indicates the sequence position of the reference MGI. Bacterial isolates and the respective genome sequence accession codes are presented in Supplemental Table S2.
The blaMIR-17 β-lactamase gene of E. cloacae isolate 1 is located on an ancient mobile genomic island
Analysis of the conserved genomic neighborhood of the blaMIR-17 gene in E. cloacae isolate 1 revealed the presence of several genes that are commonly found on MGIs, as described in detail in the following paragraph. The idea that blaMIR-17 could be part of a MGI was further corroborated by inspection of the genome sequence of E. cloacae isolate 1, which revealed that indeed a 140-kb MGI including blaMIR-17 had integrated in the Phe-GAA tRNA gene. Of note, this tRNA gene contains a phage P4-associated attachment site (GAGTCCGGCCTTCGGCACCA) [24] in the 3ʹ-5ʹ direction (Supplemental Figure S2). Since the identified MGI carries the blaMIR-17 gene, we named it MIR17-GI. At the 3ʹ end of the MIR17-GI, downstream of a Leu-CAA tRNA gene, an integrated P4 prophage was detected (Figure 2(b)). A BLAST-x analysis of the replicative helicase gene of this P4 prophage indicated that it is most closely related to a P4 bacteriophage of Salmonella enterica (GI:380464247, 87% identity, 100% coverage).
The 5ʹ side of MIR17-GI is schematically represented in Supplemental Figure S2. The blaMIR-17 gene located on the reverse strand is flanked by an ampR regulator gene on the forward strand. The blaMIR-17-ampR genes are positioned within a 32-kb region of MIR17-GI that is flanked by the Phe-GAA tRNA gene with the P4 attachment site at the 5ʹ end, and a triplet of Gly-GCC tRNA genes at the 3ʹ end. Of note, this region contains several additional resistance genes, potentially providing resistance to cations and heavy metals (cutA1, cutA2) [25], or quaternary ammonium compounds (sugE) [26]. Further, it includes the gene for a mechano-sensitive potassium efflux pump (kefA) that could be involved in osmo-protection [27], and genes for two outer membrane lipocalins (blc, yjel) implicated in the transport of small hydrophobic molecules [28]. The first gene of the island encodes a regulator of the TetR transcription regulator family, and this gene is located downstream of the cutA1 and cutA2 genes. Regulators of the TetR family repress gene transcription, and transcription is derepressed in response to stress [29].
MIR17-GI also carries several genes that are known to have housekeeping functions. In particular, on the forward strand, the groES-groEL genes encode chaperones implicated in protein folding and cell cycle regulation [30]. On the reverse strand of MIR17-GI, there are the frdABCD genes potentially involved in anaerobic respiration. Such frd genes were previously implicated in transduction by phages [31]. Lastly, MIR17-GI carries genes involved in maintenance of DNA and mobile genetic elements. The ecnA and ecnB genes located downstream of blaMIR-17 encode the Entericidin A and B toxin-antitoxin pair previously reported to prevent loss of plasmids [32]. Also located downstream of blaMIR-17, the fxsA gene encodes a polytopic membrane protein that prevents bacteriophages from exclusion [33–35]. Upstream of blaMIR-17, the DNA repair gene mutL and the high frequency of lysogeny operon hflQXKC are located (Supplemental Figure S2).
Notably, the above-listed genes represent the common context of chromosomal ampC genes in Citrobacter, Escherichia, Enterobacter, and Shigella species, which implies that MIR17-GI is an ancient mobile genomic element acquired before speciation. The latter view is supported by the fact that the GC content of the MIR17-GI and its left and right flanking sequences is quite similar among Enterobacteriacaea, as exemplified in Figure 4 for the E. cloacae study isolate 1 and the E. coli K12 reference strain MG1655. This is consistent with the notion that horizontally transferred DNA will adapt to the host genome over time, a process known as genome amelioration [36].
Figure 4.
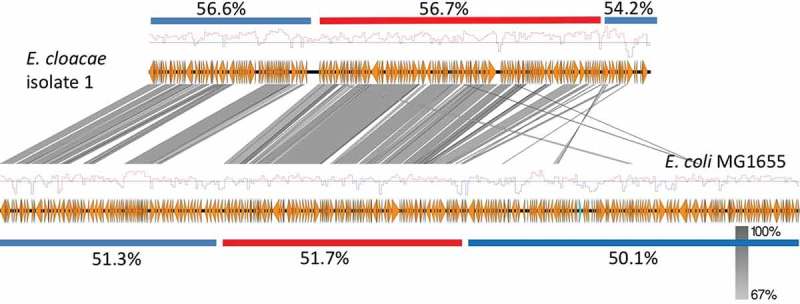
GC content of the MIR17-GI from E. cloacae and the related MGI from E. coli and their flanking regions.
Red bars indicate the position of the respective MGI and blue bars the different flanking regions. The overall GC% per region is indicated and the red-blue diagrams mark variations in the respective GC profiles.
Landmark features of MIR17-gi-like MGIs in Enterobacteriaceae
Several “landmark” features of the MIR17-GI homologous MGIs can be distinguished. In the first place, the respective tRNA genes are highly conserved in all these islands, starting with the Phe-GAA tRNA gene at the 5ʹ end, followed by the triplet Gly-GCC tRNA genes, and ending with the Leu-CAA tRNA gene at the 3ʹ end. The Phe-GAA tRNA and the Leu-CAA tRNA genes define the borders of the MGIs, which vary in length from 136 to 148 kb in E. coli, E. cloacae and K. pneumoniae (Figure 2(a)). Further, these MGIs share the afore-mentioned fxsA, groES/EL, encB, frdABCD and hflQXKC genes, as well as a trehalose operon, a putative sugar transport gene cluster, a primosomal replication protein N gene cluster, and an L-ascorbate utilization gene cluster. Lastly, a P4-associated integrase gene is located next to the LEU-CAA tRNA gene (Figure 2).
Diversity of ampC genes in MIR17-GI-like MGIs
The ampC genes of MIR17-GI-like MGIs are integrated at the same location in all Citrobacter, Escherichia, Enterobacter, and Shigella genomes, next to sugE-blc genes. Nevertheless, the sequences of these ampC genes are quite diverse. As shown in the phylogenetic tree in Figure 5, the ampC genes of Citrobacter and Enterobacter species are relatively closely related to each other, forming a distinct cluster from the ampC genes encountered in Escherichia and Shigella species. Within E. coli and Shigella, the clustering of ampC genes corresponds well with the different phylogenetic groups and species, although the ampC genes of E. coli isolates belonging to phylogenetic group A are divided over four clusters (Supplemental Figure S3). These findings are indicative of separate acquisitions of ampC by the MIR17-GI-like MGIs over time.
Figure 5.
Neighbor joining tree of the ampC genes in Citrobacter, Escherichia, Enterobacter and Shigella species.
The phylogenetic tree of ampC genes in Citrobacter, Escherichia, Enterobacter and Shigella species was created with the Ridom SeqSphere+ software v4.1.9 using a neighbor joining algorithm. The scale bar represents the % difference among 77 nucleotides of ampC, using the ampC gene of the E. coli K12 strain MG1655 as a reference. Bacterial isolates and the respective genome sequence accession codes are presented in Supplemental Table S2.
Expression profile of MGI-encoded proteins from isolates 1 and 2
The frequent occurrence of MIR17-GI-like MGIs in Enterobacteriaceae raised the question to what extent proteins encoded by such MGIs are expressed. To approximate protein expression from MIR17-GI-like islands, cells of the E. cloacae study isolates 1 and 2 were investigated using liquid chromatography and tandem mass spectrometry (LC-MS/MS). A total number of 1300 different E. cloacae proteins was identified for both strains, including 857 proteins of isolate 1 and 1116 proteins of isolate 2 (Supplementary Table S1). The MS analysis identified the largest numbers of different proteins in the stationary phase of growth (Figure 6; Supplemental Figure S4). Further, label-free quantification of the proteins identified by MS (i.e. spectral counting) showed similar patterns of representation of the proteins detected at the highest levels among the two investigated isolates (Figure 7(a)). In contrast, there was substantially more variation in the identified proteins from both isolates that were detectable at relatively low levels. Importantly, for isolate 1, eleven of the 45 MIR17-GI-encoded proteins were identified, including the MIR17 carbapenemase that was expressed at the highest levels in the stationary growth phase (Figure 7(b,c)). In fact, judged by spectral counting, MIR17 is one of the 40 most abundant proteins that were identified in isolate 1. Further, the MIR17-GI-encoded GroEL protein was the second most abundant protein identified by MS. Nine proteins encoded by the MIR17-GI-like MGI of isolate 2 were identified. Of note, the ACT-5 β-lactamase of isolate 2 remained undetected, which is consistent with the susceptibility of this isolate to third-generation cephalosporins and carbapenems.
Figure 6.
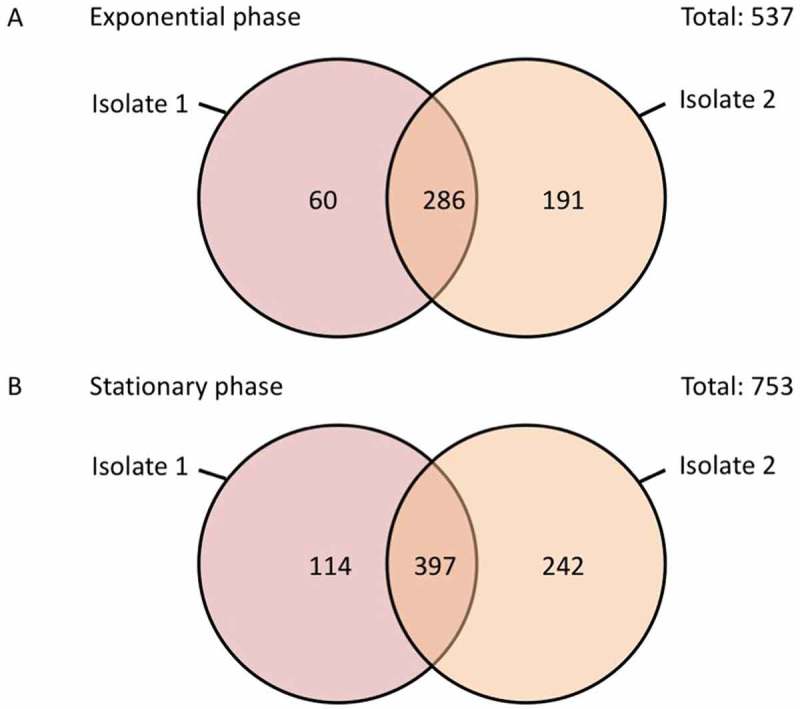
Numbers of consistently or uniquely identified proteins in the E. cloacae study isolates 1 and 2 during the exponential and stationary growth phases. The diagrams were created using the Venn diagram web tool of the VIB and the University of Gent in Belgium (http://bioinformatics.psb.ugent.be/webtools/Venn/).
Figure 7.
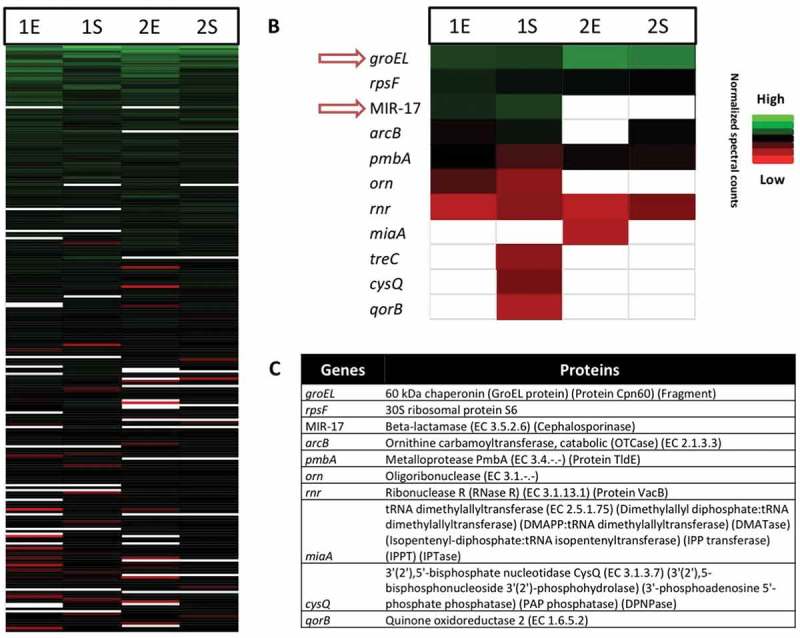
Proteomic profiles of the investigated E. cloacae study isolates 1 and 2 in the exponential and stationary growth phases. (a) Bar diagram depicting the relative amounts of 500/1300 identified E. cloacae proteins in the exponential (E) and stationary (S) growth phases based on normalized spectral counts. Relative amounts of identified proteins encoded by the MIR17-GI (b), and their respective functions (c).
Discussion
The present study highlights the identification of an ancient MGI-family integrating ampC-like cephalosporinase genes in a wide range of Enterobacteriaceae. This MGI was first identified in a carbapenem-resistant clinical isolate of E. cloacae, where it was shown to carry the blaMIR-17 gene for an AmpC-like cephalosporinase. Accordingly, the respective MGI was named MIR17-GI. Further inspection of MIR17-GI revealed a high abundance of different resistance genes of which the MIR17 enzyme was found to be highly expressed. Importantly, MIR17 displays carbapenemase activity that can be inhibited with cloxacillin, suggesting that the carbapenem resistance of isolate 1 has to be attributed to this enzyme.
Interestingly, the MIR17-GI of study isolate 1 was found to be associated with a P4 prophage. This suggests that the transmission of MIR17-GI-like MGIs between Enterobacteriaceae may have been phage-mediated. Consistent with this idea, all MIR17-GI-like MGIs were found to be integrated into Phe-GAA tRNA genes that include a phage P4-associated attachment site at the 5ʹ end of the MGI. Moreover, they are all flanked by a conserved P4 integrase gene, with the Leu-CAA tRNA gene as attachment site at the 3ʹ end. These features are typical for chromosomal P4-like gene clusters [24].
Resistance genes are frequently exchanged between microorganisms via MGIs and this seems to be the case also for the MIR17-GI-like islands of which most, but not all, were found to carry an ampC-like cephalosporinase gene. Such resistance genes that are carried and integrated into the bacterial chromosome by phages are sometimes referred to as “morons”. In general, morons are composed of one or more genes that have no particular function in the phage’s lysogenic cycle, but that may provide benefits to the phage’s host. This coevolution of prophages and bacteria would be important for species to adapt to various environmental niches [37]. In the case of MIR17, the benefit for the E. cloacae host would be the acquisition of resistance to cephalosporins or even carbapenems. In this respect, it is important to note that such antibiotics are produced in nature by fungi and Streptomycetes that may compete for the same ecological niches as E. cloacae. Morons are frequently flanked by tRNAs and this was also found to be the case for blaMIR-17 . The idea that blaMIR-17 is a moron is further supported by the observation that it is localized in the vicinity of the bacterial high frequency lysogeny operon hflQXKC [38].
Lastly, we observed a diversity of genes encoding cephalosporinases among the identified MIR17-GI-like mobile genomic islands. The integration of broad spectrum cephalosporinase genes, as found in the E. cloacae study isolates is of major clinical importance. Particularly, upon high-level expression, both the ACT- and the MIR-families of β-lactamases are capable of hydrolyzing third-generation cephalosporins [11]. Moreover, our study shows that the carbapenem resistance of E. cloacae study isolate 1 can be attributed to the new allele blaMIR-17 and the high-level production of the encoded MIR17 enzyme. The carbapenem resistance of study isolate 1 may be further enhanced by a change in outer membrane protein expression. In particular, the ompF gene of isolate 1, homologous to ompK35, contains two point mutations compared to reference ompF gene of E. cloacae MGH 54 (aeebl-supercont1.1.C3), one of which results in a stop codon at position 200. Therefore, an additional factor contributing to the carbapenem resistance of isolate 1 could be the absence of an outer membrane porin, especially OmpF. The latter would be consistent with previous studies on the role of porins in the resistance of Enterobacteriaceae to antibiotics [29,39,40].
In conclusion, we identified a novel family of MGIs harboring genes associated with the resistance to cephalosporins, phage immunity, and accessory metabolic functions. These MIR17-GI-like islands are present in various Enterobacteriaceae, including K. pneumoniae, E. coli, and E. cloacae. Importantly, the MIR17-GI-like islands are associated with integrated P4-like prophages, which implicate phages in the spread of high-level cephalosporin and carbapenem resistance amongst Enterobacteriaceae. In view of the serious consequences of high-level cephalosporin and carbapenem resistance for treatment of patients with enterobacteriaceal infections, the discovery of an ancient mobile genomic island that has facilitated the spread of “cephalosporinase and carbapenemase morons” is of high clinical relevance.
Materials and methods
Bacterial isolates
The bacterial isolates used in this study and their antibiotic resistances are summarized in Table 1. Species determination was performed by matrix-assisted laser desorption/ionization-time-of-flight (MALDI-TOF) MS, using a Bruker Microflex (Bruker Corporation, Billerica, USA).
Antibiotic susceptibility testing
Antibiotic susceptibility was routinely determined with the VITEK 2 system with an ASTn344 card (bioMérieux, Marcy l’Etoil, France). The VITEK 2 minimum inhibitory concentration results were interpreted according to the advanced expert system following EUCAST guidelines (www.eucast.org). The susceptibility to meropenem or imipenem was subsequently verified on MHA using the AB Biodisk Etest according to manufacturer’s guidelines (AB Biodisk, Mannheim, Germany). The presence of genes for carbapenemase, ESBL and AmpC was verified with the Check-MDR CT103 XL microarray assay (Check-Points, Wageningen, the Netherlands).
DNA sequence analyses
Nanopore sequencing
Bacteria were grown overnight at 37°C on Blood Agar (BA) plates. Then single colonies were picked for overnight culture at 37°C on BA plates again. For DNA extraction the DNeasy UltraClean Microbial Kit (Qiagen) was used with minor modifications. A 10 µl-loopful of bacteria was directly transferred into a tube with microbeads and microbeads solution. The incubation period was prolonged to 20 min, instead of 5 min. The quality and quantity of isolated DNA was determined using a Qubit® 2.0 fluorometer (ThermoFisher Scientific), an Agilent Tapestation 2200 (Agilent) and a NanoDrop (ThermoFisher Scientific). Libraries were prepared without shearing to maximise sequencing read length. The library was prepared using the 2D ligation sequencing kit (SQK-LSK208). The protocol for 2D ligation sequencing kit was followed as described by the manufacturer. The final library was loaded onto an FLO-MIN106 R9.4 flow cell. The run was performed on a MinION device using the NC_48Hr_Sequencing_Run_FLO-MIN107_SQKLSK208 protocol with 976 available pores (464, 314, 161 and 37 pores per group). The run proceeded for the full 48 hours. Base calling was performed after the run, using Albacore v1.2.2 (Nanopore) with the r94_250bps_2d.cfg workflow. Lastly, the quality of the data was analysed with Poretools v0.6.0 [41] and the fast5 files were transformed into a fastq file.
Illumina sequencing
Total DNA extraction for whole-genome sequencing was performed directly from colonies of the respective isolates using the Ultraclean Microbial DNA Isolation Kit (MO BIO Laboratories, Carlsbad, CA, US) according to the manufacturer’s protocol. DNA concentrations were determined using a Qubit® 2.0 fluorometer and the dsDNA HS and/or BR assay kit (Life technologies, Carlsbad, CA, US). Subsequently, DNA libraries were prepared using the Nextera XT v3 kit (Illumina, San Diego, CA, US) according to the manufacturer’s instructions. Sequence analysis was performed with an Illumina Miseq System generating paired-end reads of 300 bp as described previously [42]. De novo assembly of paired-end reads was performed using CLC Genomics Workbench v8.5.3 (QIAGEN, Hilden, Germany) after quality trimming (Qs ≥ 20) establishing a word size of 30. This resulted in 159 and 66 contigs (≥ 500 bp) with an average coverage of 72-fold. The acquired antimicrobial resistance genes and multi-locus sequence types (MLST) were identified by uploading the assembled genome sequences onto the Resfinder server v2.1. [43] and the MLST 1.7 server [44], respectively. The raw WGS datasets generated in the current study are available in the European Nucleotide Archive (ENA) repository under Bioproject PRJEB22119 (http://www.ebi.ac.uk/ena/).
Hybrid assembly
The hybrid assembly of the Illumina short reads and the MinION long reads of isolate 339389L was performed using SPAdes version: 3.10.1 (http://bioinf.spbau.ru/en). The resulting assembly was submitted to NCBI under accession number CP026536. Sequence similarity analyses between MIR17 and MGIs from Escherichia coli were performed using the rapid annotation using subsystem technology (RAST) server 4.0 [45].
Phylogenetic analyses
Phylogenetic trees based on single nucleotide polymorphisms (SNPs) were built using the SNP comparison tool in Ridom SeqSphere v4.1.9 (Münster, Germany) with default settings [46]. Properties of the enterobacteriaceal genomes used for phylogenetic analyses are summarized in Supplemental Table S2. The relatedness of MIR17-GI-like MGIs was analyzed using a local ad hoc cgMLST scheme based on 124 genes of the respective MGI from E. coli K12 strain MG1655 (Supplemental Table S3). For visualization of the MGI comparisons, BLAST+ 2.6.0 (NCBI, Bethesda, USA) and DNA plotter v 1.11 (Welcome Sanger Institute, Cambrigde, UK) were used with default settings.
Identification of orthologous proteins
To identify orthologous proteins, reciprocal best hits (RBHs) were calculated. Galaxy, a python script [47], was used to perform reciprocal protein BLAST searches (NCBI BLAST+ v. 2.3.0 [48]. Default parameters (minimum percentage identity: 70%; minimum High Scoring Pair (HSP) coverage: 50%) were used and all redundancies were removed. RBHs were calculated by blasting: (i) the two whole genome sequences; (ii) the MGI from isolate 1 and the whole genome of isolate 2; (iii) the MGI from isolate 2 and the whole genome of isolate 1; and (iv) the two MGIs.
Proteome analyses
E. cloacae isolates 1 and 2 were cultured in triplicate in brain heart infusion broth (BHI; Oxoid, Basingstoke, UK) at 37°C with vigorous shaking at 250 rpm. For sample preparation, cells were collected in the mid-exponential and stationary growth phases by centrifugation and disrupted by bead-beating with glass beads (~ 0.1 mm diameter) in a Precellys 24 homogenisator (Bertin Technologies, France) as described previously [49,50]. Glass beads and cell debris were removed by centrifugation (21,000 × g, 10 min, 4°C). The cell extract protein fraction was prepared and analyzed by LC-MS/MS using an Orbitrap Velos Pro mass spectrometer (ThermoFisher, Waltham, MA USA) as described previously [49]. Briefly, proteins were concentrated with Strataclean beads, subsequently reduced and alkylated, digested with trypsin and then purified through StageTip purification. Desalted peptides were loaded on an EASY-nLC™ II nano-flow LC system (ThermoFisher) with 10 µl buffer A (0.1% (v/v) acetic acid) and a constant flow rate of 0.5 µL/min. Afterwards, the peptides were separated by reversed phase chromatography with a 155 min non-linear gradient from 1 to 50% buffer B (0.1% (v/v) acetic acid in acetonitrile) with a constant flow rate of 0.3 µl/min and injected online into the mass spectrometer. The 20 most abundant precursor ions were selected for collision-induced dissociation (CID) fragmentation after a survey scan in the Orbitrap with a resolution of 60,000 and activated lockmass correction. MS/MS scans were recorded in the dual pressure linear ion trap after fragmentation was performed for 10 msec with a normalized collision energy of 35.
Data analysis was performed according to Bonn et al. [49,51]. In brief, database searching was done with Sorcerer-SEQUEST 4 (Sage-N Research, Milpitas, USA). After data extraction from raw files, the *.dta files were searched with Sequest against a target-decoy database with a set of common laboratory contaminants. A non-redundant database for peptide/protein searches was created from the genome sequences of isolate 1 (339389L) and isolate 2 (141024K), the genome sequence of the E. cloacae type strain ATCC 13047 as downloaded from Uniprot (http://www.uniprot.org; 23rd of October 2015), plus five additional genome sequences from unrelated clinical E. cloacae isolates. The used database includes protein sequences that differ in at least 1 amino acid, and it contains 30486 proteins in total. Only strict tryptic peptides with up to two missed cleavages were used for the database search. Fixed modifications were not considered. Oxidation of methionine and carbamidomethylation of cysteine were considered as variable modifications. Mass tolerance for precursor ions was set to 10 ppm, and for fragment ions to 0.5 Da. Validation of the MS/MS-based peptide and protein identification was performed with Scaffold v.4.4.1.1 (Proteome Software, Portland, USA). Peptide identifications were only accepted if they exceeded the following specific database search engine thresholds: the SEQUEST identifications required at least deltaCn scores of > 0.1 and XCorr scores of > 2.2, 3.3 and 3.7 for doubly, triply and all higher charged peptides, respectively. Protein identifications (Supplemental Table S1) were accepted if at least 2 identified peptides were detected with above mentioned filter criteria in 2 out of 3 biological replicates. This resulted in a false-positive discovery rate (FDR) below 0.2% on protein level as was verified by a search against a concatenated target-pseudoreversed decoy database. All MS data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD007113 [52].
Funding Statement
This work was supported by the Graduate School for Medical Sciences of the University of Groningen (to S.N. and T.S.), the People Programme (Marie Skłodowska-Curie Actions) of the European Union’s Horizon 2020 Programme under REA grant agreement [no. 642836 to S.G., D.B and J.M.v.D.)], the Deutsche Forschungsgemeinschaft (SFB/TRR 34 framework to D.B., F.B. and A.O.), the German Federal Ministry of Education and Research (Septomics to F.B.), and the Fundamental Research Funds for the Central Universities [2016FZA7008 to KZ]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Acknowledgments
We thank Giorgio Gabarrini and Eleni Tsompanidou for helpful discussions, and Monika Chlebowicz and Natacha Couto for support with data submission.
Disclosure statement
No potential conflict of interest was reported by the authors.
Ethics statement
The bacterial isolates used for the present analyses were collected in the course of routine diagnostics and infection prevention control. Oral consent for the use of such clinical samples for research purposes is routinely obtained upon patient admission to the UMCG, in accordance with the guidelines of the Medical Ethics Committee of the University Medical Center Groningen. All experiments were performed in accordance with the guidelines of the Declaration of Helsinki and the institutional regulations, and all samples were anonymized.
Supplementary Material
Supplemental data for this article can be accessed here.
References
- [1].Darmon E, Leach DRF.. Bacterial genome instability. Microbiol Mol Biol Rev. 2014;78:1–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Arber W. Genetic variation: molecular mechanisms and impact on microbial evolution. FEMS Microbiol Rev. 2000;24:1–7. [DOI] [PubMed] [Google Scholar]
- [3].Bobay LM, Ochman H. The evolution of bacterial genome architecture. Front Genet. 2017;8 DOI: 10.3389/fgene.2017.00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hacker J, Hochhut B, Middendorf B, et al. Pathogenomics of mobile genetic elements of toxigenic bacteria. Int J Med Microbiol. 2004;293:453–461. [DOI] [PubMed] [Google Scholar]
- [5].Hall JPJ, Brockhurst MA, Harrison E. Sampling the mobile gene pool: innovation via horizontal gene transfer in bacteria. Philos Trans R Soc Lond B Biol Sci. 2017;372:20160424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Penades JR, Chen J, Quiles-Puchalt N, et al. Bacteriophage-mediated spread of bacterial virulence genes. Curr Opin Microbiol. 2015;23:171–178. [DOI] [PubMed] [Google Scholar]
- [7].Bellanger X, Payot S, Leblond-Bourget N, et al. Conjugative and mobilizable genomic islands in bacteria: evolution and diversity. FEMS Microbiol Rev. 2014;38:720–760. [DOI] [PubMed] [Google Scholar]
- [8].Canchaya C, Proux C, Fournous G, et al. Prophage genomics. Microbiol Mol Biol Rev. 2003;67:238–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Mezzatesta ML, Gona F, Stefani S. Enterobacter cloacae complex: clinical impact and emerging antibiotic resistance. Future Microbiol. 2012;7:887–902. [DOI] [PubMed] [Google Scholar]
- [10].Fata F, Chittivelu S, Tessler S, et al. Gas gangrene of the arm due to Enterobacter cloacae in a neutropenic patient. South Med J. 1996;89:1095–1096. [DOI] [PubMed] [Google Scholar]
- [11].Papanicolaou GA, Medeiros AA, Jacoby GA. Novel plasmid-mediated beta-lactamase (MIR-1) conferring resistance to oxyimino- and alpha-methoxy beta-lactams in clinical isolates of Klebsiella pneumoniae. Antimicrob Agents Chemother. 1990;34:2200–2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lee EH, Nicolas MH, Kitzis MD, et al. Association of two resistance mechanisms in a clinical isolate of Enterobacter cloacae with high-level resistance to imipenem. Antimicrob Agents Chemother. 1991;35:1093–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Perez-Perez FJ, Hanson ND. Detection of plasmid-mediated AmpC beta-lactamase genes in clinical isolates by using multiplex PCR. J Clin Microbiol. 2002;40:2153–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Philippon A, Arlet G, Jacoby GA. Plasmid-determined AmpC-type beta-lactamases. Antimicrob Agents Chemother. 2002;46:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Chavda KD, Chen L, Fouts DE, et al. Comprehensive genome analysis of carbapenemase-producing Enterobacter spp.: new insights into phylogeny, population structure, and resistance mechanisms. MBio. 2016;7:e02093–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Conceicao T, Faria N, Pimentel M, et al. New chromosomal AmpC beta-lactamase in Enterobacter cloacae. Antimicrob Agents Chemother. 2004;48:1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Jacoby GA. AmpC beta-Lactamases. Clin Microbiol Rev. 2009;22:161–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Mammeri H, Poirel L, Fortineau N, et al. Naturally occurring extended-spectrum cephalosporinases in Escherichia coli. Antimicrob Agents Chemother. 2006;50:2573–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lawrence JG, Ochman H. Molecular archaeology of the Escherichia coli genome. Proc Natl Acad Sci U S A. 1998;95:9413–9417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Pham HN, Ohkusu K, Mishima N, et al. Phylogeny and species identification of the family Enterobacteriaceae based on dnaJ sequences. Diagn Microbiol Infect Dis. 2007;58:153–161. [DOI] [PubMed] [Google Scholar]
- [21].Clermont O, Gordon D, Denamur E. Guide to the various phylogenetic classification schemes for Escherichia coli and the correspondence among schemes. Microbiology. 2015;161:980–988. [DOI] [PubMed] [Google Scholar]
- [22].Paradis S, Boissinot M, Paquette N, et al. Phylogeny of the Enterobacteriaceae based on genes encoding elongation factor Tu and F-ATPase beta-subunit. Int J Syst Evol Microbiol. 2005;55:2013–2025. [DOI] [PubMed] [Google Scholar]
- [23].Hata H, Natori T, Mizuno T, et al. Phylogenetics of family Enterobacteriaceae and proposal to reclassify Escherichia hermannii and Salmonella subterranea as Atlantibacter hermannii and Atlantibacter subterranea gen nov., comb. nov. Microbiol Immunol. 2016;60:303–311. [DOI] [PubMed] [Google Scholar]
- [24].Pierson LS 3rd, Kahn ML. Integration of satellite bacteriophage P4 in Escherichia coli. DNA sequences of the phage and host regions involved in site-specific recombination. J Mol Biol. 1987;196:487–496. [DOI] [PubMed] [Google Scholar]
- [25].Tanaka Y, Tsumoto K, Nakanishi T, et al. Structural implications for heavy metal-induced reversible assembly and aggregation of a protein: the case of Pyrococcus horikoshii CutA. FEBS Lett. 2004;556:167–174. [DOI] [PubMed] [Google Scholar]
- [26].Chung YJ, Saier MH Jr. Overexpression of the Escherichia coli sugE gene confers resistance to a narrow range of quaternary ammonium compounds. J Bacteriol. 2002;184:2543–2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Cui C, Adler J. Effect of mutation of potassium-efflux system, KefA, on mechanosensitive channels in the cytoplasmic membrane of Escherichia coli. J Membr Biol. 1996;150:143–152. [DOI] [PubMed] [Google Scholar]
- [28].Bishop RE. The bacterial lipocalins. Biochim Biophys Acta. 2000;1482:73–83. [DOI] [PubMed] [Google Scholar]
- [29].Liu YF, Yan JJ, Lei HY, et al. Loss of outer membrane protein C in Escherichia coli contributes to both antibiotic resistance and escaping antibody-dependent bactericidal activity. Infect Immun. 2012;80:1815–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Gupta P, Aggarwal N, Batra P, et al. Co-expression of chaperonin GroEL/GroES enhances in vivo folding of yeast mitochondrial aconitase and alters the growth characteristics of Escherichia coli. Int J Biochem Cell Biol. 2006;38:1975–1985. [DOI] [PubMed] [Google Scholar]
- [31].Cole ST, Guest JR. Genetic and physical characterization of lambda transducing phages (lambda frdA) containing the fumarate reductase gene of Escherichia coli K12. Mol Gen Genet. 1980;178:409–418. [DOI] [PubMed] [Google Scholar]
- [32].Bishop RE, Leskiw BK, Hodges RS, et al. The entericidin locus of Escherichia coli and its implications for programmed bacterial cell death. J Mol Biol. 1998;280:583–596. [DOI] [PubMed] [Google Scholar]
- [33].Cheng X, Wang W, Molineux IJ. F exclusion of bacteriophage T7 occurs at the cell membrane. Virology. 2004;326:340–352. [DOI] [PubMed] [Google Scholar]
- [34].Coudron PE, Hanson ND, Climo MW. Occurrence of extended-spectrum and AmpC beta-lactamases in bloodstream isolates of Klebsiella pneumoniae: isolates harbor plasmid-mediated FOX-5 and ACT-1 AmpC beta-lactamases. J Clin Microbiol. 2003;41:772–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Susin MF, Baldini RL, Gueiros-Filho F, et al. GroES/GroEL and DnaK/DnaJ have distinct roles in stress responses and during cell cycle progression in Caulobacter crescentus. J Bacteriol. 2006;188:8044–8053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lawrence JG, Ochman H. Amelioration of bacterial genomes: rates of change and exchange. J Mol Evol. 1997;44:383–397. [DOI] [PubMed] [Google Scholar]
- [37].Cumby N, Davidson AR, Maxwell KL. The moron comes of age. Bacteriophage. 2012;2:225–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kihara A, Akiyama Y, Ito K. Host regulation of lysogenic decision in bacteriophage lambda: transmembrane modulation of FtsH (HflB), the cII degrading protease, by HflKC (HflA). Proc Natl Acad Sci USA. 1997;94:5544–5549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Bajaj H, Scorciapino MA, Moynie L, et al. Molecular basis of filtering carbapenems by porins from beta-lactam-resistant clinical strains of Escherichia coli. J Biol Chem. 2016;291:2837–2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Majewski P, Wieczorek P, Ojdana D, et al. Altered outer membrane transcriptome balance with ampc overexpression in carbapenem-resistant Enterobacter cloacae. Front Microbiol. 2016;7:2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Loman NJ, Quinlan AR. Poretools: a toolkit for analyzing nanopore sequence data. Bioinformatics. 2014;30:3399–3401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Zhou K, Ferdous M, de Boer RF, et al. The mosaic genome structure and phylogeny of Shiga toxin-producing Escherichia coli O104: h4is driven by short-term adaptation. Clin Microbiol Infect. 2015;21:468.e7–468.18. [DOI] [PubMed] [Google Scholar]
- [43].Zankari E, Hasman H, Cosentino S, et al. Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother. 2012;67:2640–2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Larsen MV, Cosentino S, Rasmussen S, et al. Multilocus sequence typing of total-genome-sequenced bacteria. J Clin Microbiol. 2012;50:1355–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Aziz RK, Bartels D, Best AA, et al. The RAST server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Leopold SR, Goering RV, Witten A, et al. Bacterial whole-genome sequencing revisited: portable, scalable, and standardized analysis for typing and detection of virulence and antibiotic resistance genes. J Clin Microbiol. 2014;52:2365–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Cock PJ, Chilton JM, Gruning B, et al. NCBI BLAST+ integrated into galaxy. Gigascience. 2015;4:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Camacho C, Coulouris G, Avagyan V, et al. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Bonn F, Bartel J, Buttner K, et al. Picking vanished proteins from the void: how to collect and ship/share extremely dilute proteins in a reproducible and highly efficient manner. Anal Chem. 2014;86:7421–7427. [DOI] [PubMed] [Google Scholar]
- [50].Maass S, Sievers S, Zuhlke D, et al. Efficient, global-scale quantification of absolute protein amounts by integration of targeted mass spectrometry and two-dimensional gel-based proteomics. Anal Chem. 2011;83:2677–2684. [DOI] [PubMed] [Google Scholar]
- [51].Bonn F, Pane-Farre J, Schluter R, et al. Global analysis of the impact of linezolid onto virulence factor production in S. aureus USA300. Int J Med Microbiol. 2016;306:131–140. [DOI] [PubMed] [Google Scholar]
- [52].Vizcaino JA, Csordas A, del-Toro N, et al. update of the PRIDE database and its related tools. Nucleic Acids Res. 2016;44(D1):D44–-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.