

Abstract
Objective
To document the decline of upper and lower limb functions, mobility, and independence in daily living activities in adults with autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS) over a 2-year period.
Methods
An exploratory longitudinal design was used. Nineteen participants were assessed on 2 occasions 2 years apart. Assessments included the Standardized Finger Nose Test, Nine-Hole Peg Test, Lower Extremity Motor Coordination Test, Berg Balance Scale, 10-m walk test (10mWT), 6-minute walk test (6MWT), Scale for the Assessment and Rating of Ataxia (SARA), and Barthel Index.
Results
A significant decline was observed between baseline and follow-up for lower limb coordination, balance, walking abilities (10mWT and 6MWT), and overall disease severity (SARA). All differences were beyond measurement error documented in ARSACS. Results showed no significant decline for upper limb coordination and fine dexterity performance.
Conclusion
Although ARSACS is a slow, progressive disease, results showed that mobility, balance, and lower limb performance significantly decreased over the 2-year period and that selected outcome measures were able to capture this decline beyond measurement errors.
Autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS) is a progressive, hereditary disorder caused by mutations in the SACS gene1 located on chromosome 13q12. ARSACS was first described in the Saguenay–Lac-Saint-Jean and Charlevoix regions of Quebec (Canada). Worldwide, it is the most frequent recessive ataxia after Friedreich ataxia. ARSACS is characterized by the presence of symptoms in each of these 3 main components: pyramidal, cerebellar, and neuropathic. This characteristic leads to a large number of signs and symptoms with a high level of variability among individuals in terms of clinical presentation, severity, and progression. Manifestations include incoordination, impaired dexterity, gait ataxia, spasticity, and weakness, among others.2–6
The only published study addressing the evolution of ARSACS was conducted by Duquette et al.6 In this retrospective study, the team documented the evolution of gait/appendicular ataxia, spasticity, and neuropathy during childhood (before 18 years of age). Disease progression during adulthood has never been documented with a longitudinal design and standardized quantitative assessments. As pointed out by a recent US Food and Drug Administration report, studies documenting natural history are essential to improve our knowledge of prognosis and to better plan rehabilitation interventions and future clinical trials.
The aim of this study was to document the decline of upper and lower limb functions, mobility, and independence in daily living activities in adults with ARSACS over a 2-year period.
Methods
Study design
This was an exploratory longitudinal study.
Subjects
Participants were recruited in 2013 among patients followed up at the Neuromuscular Clinic of the Centre intégré universitaire de santé et de services sociaux du Saguenay–Lac-Saint-Jean (Québec, Canada). Inclusion criteria were age between 18 and 59 years, homozygote for the c.8844delT mutation in the SACS gene, and able to provide informed consent.
Standard protocol approvals, registrations, and patient consents
The study was approved by the Ethics Review Board of the Centre intégré universitaire de santé et de services sociaux Saguenay–Lac-Saint-Jean, and written informed consent was obtained from each participant.
Data collection
Participants were assessed 2 years apart by the same 2 physical therapists at both time points using standardized operational procedures. All assessments were conducted during 3 half-day sessions (within a maximum interval of 2 weeks) at both baseline and follow-up. Data on age, sex, mobility level, and walking aids used were collected. Disease stage of the participants was determined from their mobility level and the use of walking aid (according to the Scale for the Assessment and Rating of Ataxia [SARA] development study7) as follows: stage 1, no walking difficulty without any walking aid; stage 2, first walking difficulty, no walking aid; stage 3, walking with aid or support; and stage 4, using a wheelchair.
Outcome measures
Upper limb coordination was assessed with the Standardized Finger Nose Test (SFNT).4 Fine dexterity was measured with the Nine-Hole Peg Test (NHPT).8 Intrarater and interrater reliability of these tests was excellent (intraclass correlation coefficient [ICC] = 0.90–0.98), and their construct validity has been demonstrated in ARSACS.9 Lower limb coordination was measured with the Lower Extremity Motor Coordination Test (LEMOCOT).10,11 The intrarater and interrater reliability of the LEMOCOT and its construct validity are excellent in ARSACS (ICC = 0.92–0.97).11 Balance was assessed with the Berg Balance Scale,12 the construct validity of which was demonstrated in ARSACS.13 Walking ability was documented in terms of walking speed with the 10-m walk test (10mWT) at comfortable pace, and long-distance walking was assessed with the 6-minute walk test (6MWT).14 Both tests (10mWT and 6MWT) have excellent interrater reliability (ICC = 0.97–0.99), and construct validity was confirmed in the ARSACS population.13 Overall disease severity was measured with the SARA.7 Its interrater reliability is excellent (ICC = 0.97) in spinocerebellar ataxia7 and in recessive ataxia or nonprogressive cerebellar ataxia (ICC = 0.98).15 Independence in daily living activities was measured with the Barthel Index.16 Its validity and reliability in recessive ataxia are not known.
Statistical analysis
Data are expressed as mean ± SD for continuous variables and as frequency and percentage for categorical variables. When >1 trial was performed, the mean was used for analyses (SFNT, NHPT, LEMOCOT, 10mWT). Only results from the dominant hand/foot are presented here. Comparison between follow-up and baseline performance was made with the nonparametric Wilcoxon signed-rank test because the number of participants was <30. A value of p < 0.05 was considered significant. In addition, normality of the distribution of the difference was verified with the Kolmogorov-Smirnov statistic. Significant differences between follow-up and baseline were compared against the SEM of each outcome measure.7,9,11,13,17 Data were analyzed with IBM SPSS Statistics for Windows, version 24.0 (IBM Corp, Armonk, NY).
Data availability
Anonymized data will be shared by request from any qualified investigator.
Results
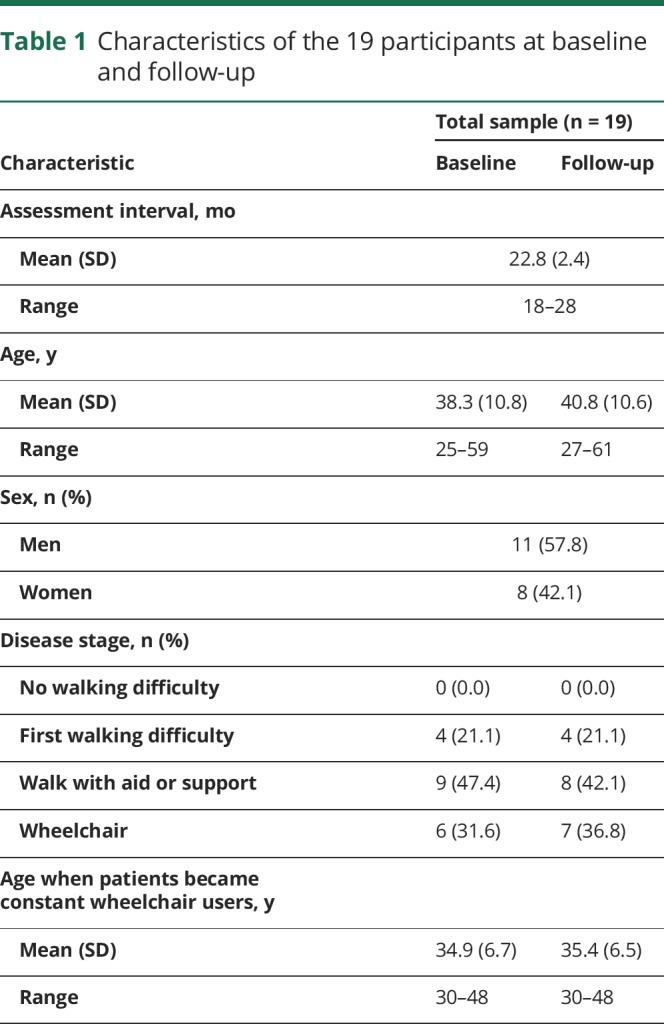
From the 28 participants at baseline, 19 participated in the follow-up. Reasons not to participate were lack of time (44.4%), lack of interest (44.4%), and health issues (11.1%). Individuals who refused to participate in the follow-up were not different from those who participated in terms of age (37.7 vs 38.8 years, p = 0.664), sex (56% vs 58% men, p = 0.612), and disease stage (p = 0.442). All participants' characteristics are presented in table 1. The mean age of the 19 participants at baseline was 38.3 years, and 58% were men. Eighteen participants were right-handed.
Table 1.
Characteristics of the 19 participants at baseline and follow-up
Comparisons of performance between follow-up and baseline are presented in table 2.
Table 2.
Performance of participants at baseline and follow-up

Lower limb coordination, balance, and walking ability significantly decreased during the 2-year period, and overall disease severity became worse; all these results are beyond measurement error. In addition, the distribution of differences was normal for all measures (p > 0.05) except for the NHPT (p = 0.001). According to walking ability results, an important percentage of participants lost their ability to perform walking tests at follow-up (10mWT 21.4% [3 of 14], 6MWT 28.6% [4 of 14]).
Discussion
A large population of people with ARSACS live in the Saguenay–Lac-Saint-Jean region, and an increasing number of people with recessive ataxias are now diagnosed as having ARSACS worldwide. Results of this first prospective natural history study will help clinicians to better inform their patients about disease progression. Despite the relatively small number of participants, we found a significant decline in lower limb coordination, balance, and walking ability, as well as an increase in overall disease severity, over 2 years. All results were beyond outcome measure measurement error. In addition, the normality of the distribution of the differences indicates that the presence of outliers did not significantly affect the results. These exploratory results will help plan future clinical trials in regard to the selection of outcome measures. Given the slow progression of the disease, the selection of outcome measures must be based on their sensitivity and accuracy to detect small changes. Otherwise, the tool can fail to detect a change that appears in the participant, or a significant difference can be found but reflects only the associated measurement error.
Regarding the absence of a significant decline of upper limb dexterity and coordination measured by the SFNT and NHPT, we hypothesize that participants were so impaired at baseline that they could not deteriorate further at follow-up (floor effect). Effectively, the mean number of targets touched in the SFNT by participants with ARSACS was less than half of that of healthy elderly people (women 11.5 and men 10.7 compared to 23.2 and 24.218), well illustrating the high level of impairment present in people with ARSACS. The same portrait is observed for the NHPT; participants in this study took an average of 58 seconds to complete the task compared to an average of 20.4 seconds for healthy elderly women and 22.4 seconds for healthy elderly men.19 However, given the small sample size, we cannot exclude a lack of power. Other outcome measures must be explored in this population to better track the decrease of upper limb functions.
This is the first study documenting prospectively the decline in functions in ARSACS. The small sample size limits the generalization of the results; individuals who accepted to participate in the study may be different in other characteristics from the ones who refused. However, the availability of metrologic properties for most outcome measures has permitted documentation of the SEM and has ensured that the changes were beyond measurement error. The next step will be to document the responsiveness of these outcome measures. In addition, the longitudinal documentation of ARSACS impairments must be continued with a larger cohort to take into account individual variability and with a longer period of time to have a better understanding of the disease progression.
Glossary
- ARSACS
autosomal recessive spastic ataxia of Charlevoix-Saguenay
- ICC
intraclass correlation coefficient
- LEMOCOT
Lower Extremity Motor Coordination Test
- NHPT
Nine-Hole Peg Test
- SARA
Scale for the Assessment and Rating of Ataxia
- SFNT
Standardized Finger Nose Test
- 6MWT
6-minute walk test
- 10mWT
10-m walk test
Author contributions
Cynthia Gagnon: study concept and design, study supervision, analysis and interpretation of data. Isabelle Lessard: acquisition, analysis and interpretation of data, writing of the manuscript. Caroline Lavoie: acquisition of data, review of the manuscript. Isabelle Côté: analysis of data, statistical analysis, writing of the manuscript. Raphaël St-Gelais: interpretation of data, writing of the manuscript. Jean Mathieu and Bernard Brais: study concept and design, critical revision of manuscript for intellectual content.
Study funding
Funded by the Canadian Institutes of Health Research in partnership with Fondation de l'Ataxie Charlevoix-Saguenay (Emerging Team Grant TR2-119189). C.G. holds career-grant funding from Fonds de recherche du Québec–santé (grants 22193 and 31011).
Disclosure
The authors report no disclosures relevant to the manuscript. Go to Neurology.org/N for full disclosures.
Publication history
Received by Neurology March 27, 2018. Accepted in final form July 4, 2018.
References
- 1.Engert JC, Berube P, Mercier J, et al. ARSACS, a spastic ataxia common in northeastern Quebec, is caused by mutations in a new gene encoding an 11.5-kb ORF. Nat Genet 2000;24:120–125. [DOI] [PubMed] [Google Scholar]
- 2.Bouchard JP, Barbeau A, Bouchard R, Bouchard RW. Autosomal recessive spastic ataxia of Charlevoix-Saguenay. Can J Neurol Sci 1978;5:61–69. [PubMed] [Google Scholar]
- 3.Gagnon C, Desrosiers J, Mathieu J. Autosomal recessive spastic ataxia of Charlevoix-Saguenay: upper extremity aptitudes, functional independence and social participation. Int J Rehabil Res 2004;27:253–256. [DOI] [PubMed] [Google Scholar]
- 4.Gagnon C, Mathieu J, Desrosiers J. Standardized finger-nose test validity for coordination assessment in an ataxic disorder. Can J Neurol Sci 2004;31:484–489. [DOI] [PubMed] [Google Scholar]
- 5.Gagnon C, Lavoie C, Lessard I, et al. The Virtual Peg Insertion Test as an assessment of upper limb coordination in ARSACS patients: a pilot study. J Neurol Sci 2014;347:341–344. [DOI] [PubMed] [Google Scholar]
- 6.Duquette A, Brais B, Bouchard JP, Mathieu J. Clinical presentation and early evolution of spastic ataxia of Charlevoix-Saguenay. Mov Disord 2013;28:2011–2014. [DOI] [PubMed] [Google Scholar]
- 7.Schmitz-Hubsch T, du Montcel ST, Baliko L, et al. Scale for the Assessment and Rating of Ataxia: development of a new clinical scale. Neurology 2006;66:1717–1720. [DOI] [PubMed] [Google Scholar]
- 8.Mathiowetz V, Weber K, Kashman N, Volland G. Adult norms for the Nine Hole Peg Test of finger dexterity. Occup Ther J Res 1985;5:24–38. [DOI] [PubMed] [Google Scholar]
- 9.Gagnon C, Lessard I, Brais B, et al. Validity and reliability of outcome measures assessing dexterity, coordination, and upper limbs strength in autosomal recessive spastic ataxia of Charlevoix-Saguenay. Arch Phys Med Rehabil Epub 2018 Feb 17. [DOI] [PubMed]
- 10.Desrosiers J, Rochette A, Corriveau H. Validation of a new lower-extremity motor coordination test. Arch Phys Med Rehabil 2005;86:993–998. [DOI] [PubMed] [Google Scholar]
- 11.Lessard I, Lavoie C, Côté I, Mathieu J, Brais B, Gagnon C. Validity and reliability of the LEMOCOT in the adult ARSACS population: a measure of lower limb coordination. J Neurol Sci 2017;377:193–196. [DOI] [PubMed] [Google Scholar]
- 12.Berg K, Wood-Dauphinee S, Williams JI, Gayton D. Measuring balance in elderly: preliminary development of an instrument. Physiother Can 1989;41:304–311. [Google Scholar]
- 13.Lessard I, Brais B, Côté I, et al. Assessing mobility in autosomal recessive spastic ataxia of Charlevoix-Saguenay population: validity and reliability of four outcome measures. J Neurol Sci 2018;390:4–9. [DOI] [PubMed] [Google Scholar]
- 14.Butland RJ, Pang J, Gross ER, Woodcock AA, Geddes DM. Two-, six-, and 12minute walking tests in respiratory disease. Br Med J 1982;284:1607–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weyer A, Abele M, Schmitz-Hubsch T, et al. Reliability and validity of the Scale for the Assessment and Rating of Ataxia: a study in 64 ataxia patients. Mov Disord 2007;22:1633–1637. [DOI] [PubMed] [Google Scholar]
- 16.Mahoney FI, Barthel DW. Functional evaluation: the Barthel Index. Md State Med J 1965;14:61–65. [PubMed] [Google Scholar]
- 17.Winser S, Smith CM, Hale LA, et al. Psychometric properties of a core set of measures of balance for people with cerebellar ataxia secondary to multiple sclerosis. Arch Phys Med Rehabil 2017;98:270–276. [DOI] [PubMed] [Google Scholar]
- 18.Desrosiers J, Hebert R, Bravo G, Dutil E. Upper-extremity motor co-ordination of healthy elderly people. Age Ageing 1995;24:108–112. [DOI] [PubMed] [Google Scholar]
- 19.McKay MJ, Baldwin JN, Ferreira P, Simic M, Vanicek N, Burns J. Reference values for developing responsive functional outcome measures across the lifespan. Neurology 2017;88:1512–1519. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymized data will be shared by request from any qualified investigator.