

Abstract
Clinically, traumatic brain injury (TBI) results in complex heterogeneous pathology that cannot be recapitulated in single pre-clinical animal model. Therefore, we focused on evaluating utility of nanoparticle (NP)-based therapeutics following three diffuse-TBI models: mildclosed-head injury (mCHI), repetitive-mCHI and midline-fluid percussion injury (FPI). We hypothesized that NP accumulation after diffuse TBI correlates directly with blood–brainbarrier permeability. Mice received PEGylated-NP cocktail (20–500 nm) (intravenously) after single- or repetitive-(1 impact/day, 5 consecutive days) CHI (immediately) and midline-FPI (1 h, 3 h and 6 h). NPs circulated for 1 h before perfusion/brain extraction. NP accumulation was analyzed using fluorescent microscopy in brain regions vulnerable to neuropathology. Minimal/no NP accumulation after mCHI/RmCHI was observed. In contrast, midlineFPI resulted in significant peak accumulation of up to 500 nm NP at 3 h post-injury compared to sham, 1 h, and 6 h groups in the cortex. Therefore, our study provides the groundwork for feasibility of NP-delivery based on NPinjection time and NPsize after mCHI/RmCHI and midline-FPI.
Keywords: Nanoparticles, Traumatic brain injury, Blood-brain barrier, Mild closed head injury, Fluid percussion injury, Optimal therapeutic window, Drug delivery, Pharmacodynamics
Traumatic brain injury (TBI) is a leading cause of disability worldwide with 1.7 million TBIs reported annually with an annual estimated economic cost of $76.5 billion in the United States alone.1,2 TBI occurs due to damage to the brain resulting from an external mechanical force, including rapid acceleration or deceleration, blast waves, crushing force, an impact or penetration by a projectile.3 Broadly, TBI can be classified into focal injury, associated with cerebral contusion and hematoma, and diffuse brain injury, associated with multifocal and widespread microscopic pathology.4 Over time, research has revealed TBI to be a complex disease process and not just a single pathophysiological event.5–7 Upon sustaining a TBI the mechanical forces from impact inflict heterogeneous tissue damage, referred to as the primary injury phase.7 This insult initiates a myriad of pathophysiological and biochemical secondary injury signaling cascades, including hypo- or hyper-perfusion, edema, blood–brainbarrier (BBB) dysfunction and inflammation that evolves from minutes to days post-trauma.5
The heterogeneous pathophysiology observed clinically following a TBI likely arises from the variations in the mode of impact such as the location, type and severity, as well as other factors including age, sex, and genetics.8 Pre-clinical animal models have been developed in the laboratory to effectively study and evaluate TBI pathology using a reproducible injury event while also controlling for above factors.9,10 However, a single injury model may not fully recapitulate all the facets of the secondary injury that are observed in human TBI.9 The most commonly used focal injury model is the controlled cortical impact (CCI), which produces a focal lesion, axonal injury, and necrosis.11 Pre-clinical models of diffuse brain injury include mild closed-head injury (mCHI), repetitive CHI (RmCHI) and midline fluid percussion injury (FPI). The RmCHI model produces early deficits in motor coordination and locomotor hyperactivity with increased astrocytic reactivity.12 Midline FPI produces a diffuse injury and results in neurological and physiological alterations.13–15 Clearly, RmCHI and FPI provide clinically-relevant, albeit different, injury phenotypes with distinct cellular alterations. Taken together, preclinical parameters such as injury phenotype is essential to successfully shepherd therapeutic approaches to clinical trials. Therefore, therapeutic strategies need to be evaluated in multiple TBI models while considering opportunities for different optimal therapeutic windows for each injury phenotype. However, a critical gap exists in understanding if/when BBB opening may occur after diffuse TBI, which can facilitate nanoparticle (NP) delivery.
The BBB dysfunction after injury may lead to extravasation of blood components into the brain parenchyma. Studies in different TBI animal models have demonstrated acute and delayed BBB disruption followed by restoration as evidenced by extravasation of endogenous serum immunoglobulins (IgG)16,17 and/or intravenously injected small molecule tracers, including Evans Blue18 and horseradish peroxidase (HRP).16,17 For example, Tanno et al established the transient BBB breakdown occurred after FPI (lateral) and the time course for re-establishment of the BBB varied based on the regions of the brain and proximity to the injury hub.16 Similarly, Schmidt et al.17 demonstrated that BBB disruption displayed regional differences following FPI (midline) with prominent HRP leakage in the cerebral cortex (proximal to injury hub) and corpus callosum.17 In contrast for RmCHI, a recent study reported no positive IgG immune-reactivity at 7 days after final impact and thus potentially no BBB breakdown.12 Yet, these results raise the question regarding the state of the BBB at acute time points after RmCHI. Collectively, these seminal studies support the notion that TBI compromises the BBB resulting in the extravasation of blood constituents into the normally impermeable brain parenchymal space.
BBB breakdown/permeability offers a unique opportunity to deliver drugs/nanoparticles that are normally excluded from the brain.19–22 Previously, we and others demonstrated the feasibility of delivering NPs to brain lesions after a focal brain injury via the disrupted BBB.23–25 The NP delivery to the brain through the transient BBB opening after focal TBI was dependent on the size of the particle, with smaller particles having prolonged permeability compared to larger particles.23,26 However, the utility of NPs to deliver diagnostic/therapeutic agents in other injury phenotypes such as diffuse brain injury is largely unknown. Therefore, in this study we assessed NP-size dependent accumulation in three different diffuse brain injury models: mild closed-head injury (mCHI), repetitive mild CHI (RmCHI) and midline fluid percussion injury (FPI). We hypothesized that NP accumulation in diffuse injury models would correlate directly with blood brain barrier permeability.
Methods
Materials
Carboxylated polystyrene NPs of different sizes were purchased from Life Technologies (Carlsbad, CA, USA). Specifically, the materials (with catalog numbers) used were 20 nm (F8783), 40 nm (F8793), 100 nm (F8797) and 500 nm (F8813) NPs with dark red (λex/λem = 660 nm/680 nm), red (λex/λem = 580 nm/605 nm), blue (λex/λem = 350 nm/440 nm) and yellow-green (λex/λem = 505 nm/515 nm) fluorescence, respectively. Methoxypolyethylene glycol amine 2000 (mPE-Gamine 2KDa) (06676), methoxypolyethylene glycol amine 750 (mPEGamine 750 Da) (07966), n-[3-dimethylaminopropyl]-n-ethyl, n-[3-dimethylaminopropyl]-n-ethyl [EDC] (E1769), MES hemisodium buffer (M8902), N-Hydroxysuccinimide (NHS) (56405), and Peroxidase type II from horseradish (P8250–50KU) were purchased from Sigma Aldrich (St. Louis, MO, USA). ImmPACT DAB peroxidase (HRP) substrate (SK-4105) was purchased from Vector laboratories (Burlingame, CA, USA). Slide-A-Lyzer Cassettes (20 K) (66003) and Kimwipes (06–666) were purchased from ThermoFisher scientific (Waltham, MA, USA). Vectashield antifade mounting medium (H-1000) from Vector Labs (Burlingame, Ca, USA) and Anti-mouse IgG antibody 488 (ab150105) from Abcam (Cambridge, UK) were purchased.
Nanoparticle PEG conjugation
Carboxylated NPs of different sizes, 20 nm, 40 nm, 100 nm and 500 nm were PEGylated using EDC/NHS chemistry. See Supplementary for details.
Animals
Male C57BL/6 mice (20–24 g) (Envigo, Inc., Indianapolis, IN) were used for all experiments (n = 3 per group). Animal care was approved by the Institutional Animal Care and Use Committees at the University of Arizona (Tucson, AZ). Each study has been approved by an institutional review committee and the procedures followed are in accordance with institutional guidelines and humane treatment of the animals. See Supplementary for details.
Closed head injury (CHI)
Mice were subjected to mild CHI or repetitive mCHI using the protocol previously described.12,27 Briefly, mice were lightly sedated via isoflurane inhalation and placed on Kimwipe secured to a Plexiglas stage. External anatomical landmarks (such as ear canals, eyes) were used to carefully position the animal to center under the vertical aluminum guide tube. The impact weights (9 diameter) with the desired mass (100 g or 50 g) was positioned at the top of aluminum tube and was allowed to fall freely down (865 mm) the aluminum guide tube on to the head of the mouse. A cushion sponge was located below the Kimwipe stage to receive the falling mouse. Animals received a single impact of either 100 g or 50 g (n = 6, pooled across the different weights, animals that displayed hematoma/skull fracture were excluded from the study). The RmCHI cohort (n = 3) received 50 g impact, 1 per day for 5 consecutive days.
Midline fluid percussion injury (FPI)
For injury induction 24 h post-surgery for craniotomy (Supplementary), mice were re-anesthetized with 5% isoflurane delivered for three minutes. The cap was removed from the injury-hub assembly and the dura was visually inspected through the hub to make sure it was intact with no debris. The hub was then filled with normal saline and attached to an extension tube connected to the male end of the fluid percussion device (Custom Design and Fabrication, Virginia Commonwealth University, Richmond, VA). An injury pressure of 1.1–1.2 atm was administered by releasing the pendulum onto the fluid-filled cylinder. Sham-injured mice underwent the same procedure except the pendulum was not released. Mice were monitored for the presence of a forearm fencing response and righting reflex times were recorded for the injured mice as indicators of injury severity.28,29 The righting reflex time is the total time from the initial impact until the mouse spontaneously rights itself from a supine position. The fencing response is a tonic posturing characterized by extension and flexion of the forearms that has been validated as an overt indicator of injury severity.28,29
Furthermore, naïve animals (no impact/injury) were injected with the cocktail of nanoparticles (20, 40, 100 and 500 nm) and were perfused one hour post-injury.
Nanoparticle (NP) and horseradish peroxidase (HRP) injection – mCHI/RmCHI
Retro-orbital injections of the venous sinus in the mouse were performed for intravenous delivery of the NPs and HRP, an alternative technique to tail-vein injection.30 Animals were anesthetized with isoflurane (3%) and the NP cocktail (75 μl) of 20 and 40 nm NPs for mCHI/RmCHI (at a dose of 50 mg/kg b. w.) was injected to the right eye, one hour before perfusion. HRP (83 mg/kg b.w. in 25 μl) was injected behind the left eye ten minutes before perfusion. Animals were euthanized at 1 h after mCHI/RmCHI. The NP circulation time of 1 h was held constant for each of the cohorts. Schematics show the experimental timeline for mCHI (Figure 1, A) and RmCHI (Figure 1, B).
Figure 1.
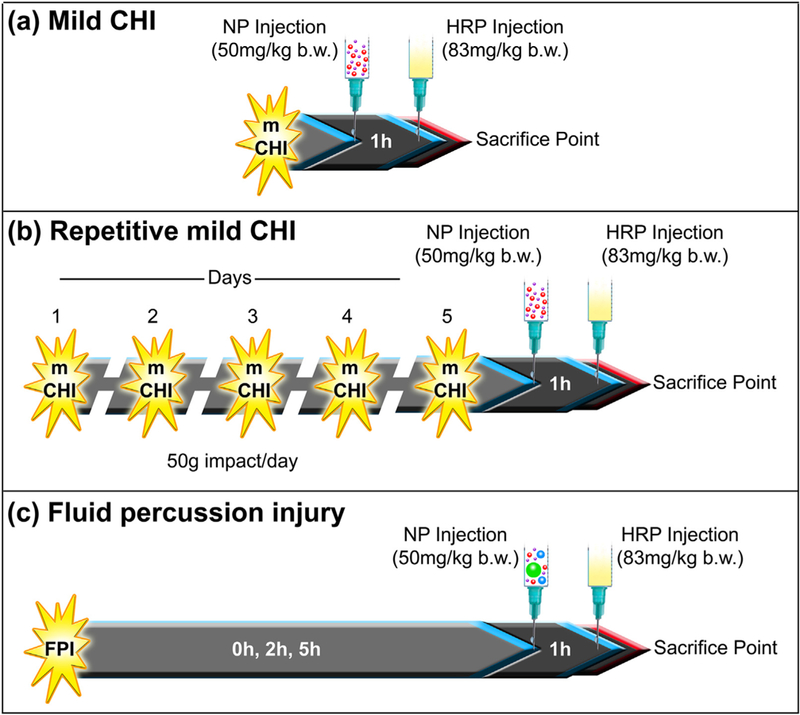
In vivo experimental study design: Cocktail of different size nanoparticles were intravenously injected (A) immediately after mild closed-head injury (mCHI), (B) immediately after the fifth impact of a repetitive mild RmCHI, and (C) immediately, 2 h and 5 h after midline fluid percussion injury (FPI). Animals were euthanized one hour after NP injection. The positive control permeability marker, horseradish peroxidase (HRP), was injected intravenously, 10 min before brain collection.
Nanoparticle (NP) and horseradish peroxidase (HRP) injection – midline FPI
Retro-orbital injections of the venous sinus in the mouse were performed for intravenous delivery of the NPs and HRP, an alternative technique to tail-vein injection.30 Animals were anesthetized with isoflurane (3%) and the NP cocktail (75 μl) of 20, 40, 100 and 500 nm NPs for the midline FPI study (at a dose of 50 mg/kg b.w.) was injected to the right eye, one hour before perfusion. HRP (83 mg/kg b.w. in 25 μl) was injected behind the left eye ten minutes before perfusion. Depending on the cohort group, animals were euthanized at 1 h, 3 h, and 6 h after midline FPI. The NP circulation time of 1 h was held constant for each of the cohorts. A schematic shows the experimental timeline for midline FPI (Figure 1, C).
Tissue collection
According to the injury groups, mCHI/RmCHI animals (1 h after final impact) animals were deeply anesthetized with isoflurane overdose. For the FPI study, animals (1 h, 3 h, and 6 h post-injury) were deeply anesthetized with lethal dose of sodium pentobarbital solution until a tail/toe pinch produced no reflex movement. Animals were transcardially perfused with cold phosphate-buffered saline (PBS), followed by 4% buffered paraformaldehyde solution. Brain tissue was collected and fixed overnight in 4% buffered paraformaldehyde followed by immersion in 30% sucrose solutions in 1X PBS for cryoprotection for 24 h. Samples were embedded in optimal cutting temperature (OCT) medium and frozen on dry ice. Samples were stored at −80 °C until sectioned coronally at a 25 μm thickness with a cryostat.
Analysis of HRP and NP accumulation after mCHI/RmCHI and FPI
Tissue sections were incubated in PBS buffer for 20mins at room temperature prior to use. For HRP analysis, freshly prepared DAB substrate solution (200 μl) was added to the tissue and incubated for ten mins at room temperature. Slides were then washed in PBS buffer three times (two mins each) and coverslipped after adding a drop of aqueous mounting media. Sections were imaged using Slide Scanner (PathScan Enabler IV, Meyer Instruments, TX, USA). See Supplementary for details.
For NP analysis, slides containing the frozen sections were incubated at room temperature for 20mins in 1X PBS to rehydrate the tissue and remove OCT compound. Slides were coverslipped after adding one drop of fluorescent mounting media (Vectashield). The whole brain sections were imaged with conventional epifluorescent/confocal microscopy at 10X/20X objective, respectively. See Supplementary for details.
Immunohistochemical analysis for mCHI/RmCHI
The sections were incubated with a solution made up of anti-mouse IgG secondary antibody 488 (1:200) with 2% goat serum and 0.1% triton X-100 for 2 h at room temperature in the dark. The sections were rinsed with PBS (4 times, 5 min each). A conventional epifluorescent microscope (Leica DMI6000 B, Leica Microsystems, Wetzlar, Germany) was used to image the stained sections. See supplementary for details.
Statistics
Statistical analyses were conducted in GraphPad Prism 5.0 (GraphPad Software, Inc., La Jolla CA). For the midline FPI study, the analysis of HRP/NP percent area in the regions of interest between the injured and sham groups at different time points was conducted using an ordinary two-way analysis of variance (ANOVA). Specifically, to compare the HRP/NP percent area between the injury and the sham group at each time point, Bonferroni’s post-hoc test was conducted. Tukey’s post-hoc test was conducted for pairwise comparison of HRP/NP percent area for each group (injury and sham) at different time points. For correlation analysis of HRP and NP, Pearson correlation test was conducted. The statistical values (P-value, F-value and degree of freedom) are included in the supplementary Table S2. The P-values are reported in the results section.
Results
Absence of HRP after mCHI/RmCHI
Our previous study using the CCI model of focal TBI, the highest level of HRP extravasation was observed within 1 h following impact.23 Therefore, we evaluated this time point in the mCHI model and observed minimal to no HRP accumulation (50 g and 100 g impact; Figure 2) suggesting an intact BBB acutely after mCHI (~50 mins after CHI). Furthermore, we did not observe any HRP staining within 1 h following the fifth impact in the RmCHI model (one per day for five consecutive days). Representative images of HRP staining after mCHI after single 50 g (Figure 2, A), and 100 g (Figure 2, B), and multiple impacts (50 g, 5X; Figure 2, C) were comparable to aïve brain tissue (Figure 2, D). Furthermore, to investigate if evidence of breach was not captured by the exogenous HRP tracer, we immunostained for endogenous IgG. We observed positive IgG staining at 1 h post-impact after single and multiple (5X) impacts (supplementary, Figure S2), suggesting that the BBB may have opened transiently prior to the HRP tracer injections.
Figure 2.
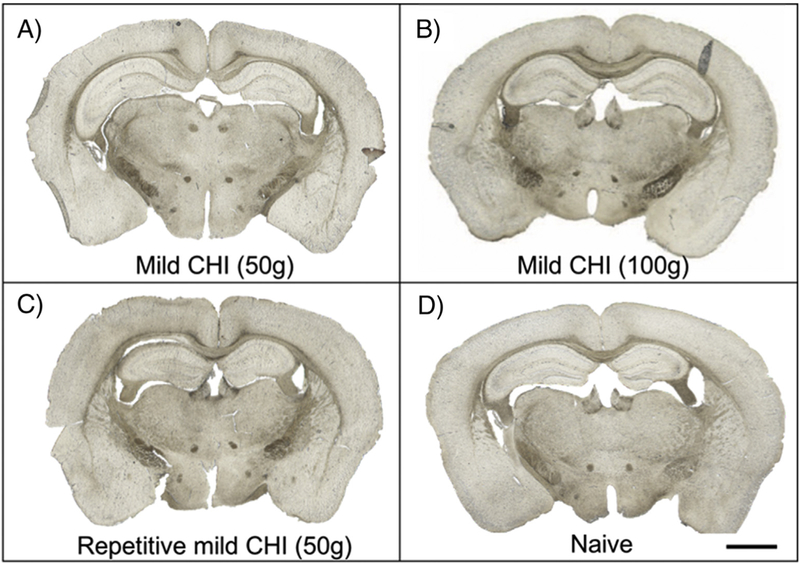
Representative images of HRP extravasation after mCHI and repetitive mCHI (A) single impact mCHI (50 g), (B) single impact mCHI (100 g), (C) multiple impacts (5X) RmCHI (50 g), (D) control (naïve). HRP circulated for 10mins prior to tissue collection at 1 h post-injury. Scale bar = 1500 μm.
Positive HRP extravasation after midline FPI
Representative images of HRP staining pattern of each FPI cohort at different time points are shown in Figure 3. The two-way ANOVA identified a significant difference between the two groups (injured and sham) for cortex (P = 0.0002) (See Table S2 for full statistical data). Also, a significant time dependent effect for both cortex (P = 0.0067) and corpus callosum (P = 0.0493) was observed. In the cortex, Bonferroni’s post hoc test comparing the injury and sham groups demonstrated a significant difference in the 3 h cohort (P = 0.0001). Comparing the cortex of the injured groups at different time points, using Tukey’s post-hoc test, revealed HRP staining at the 3 h time point was three fold greater than the 1 h (P = 0.0018) and 6 h time point (P = 0.007), Figure 3, II. In the corpus callosum, there was minimal to no HRP extravasation at 1 h and 6 h post-injury (Figure 3, III). However, a peak accumulation at 3 h post-injury was observed (about 15-fold increase) compared to the sham group (Bonferroni’s post hoc, P = 0.0021) in the corpus callosum. Moreover, Tukey’s post-hoc test demonstrated a peak accumulation at 3 h post-injury (about 15-fold increase) compared to the 1 h (P = 0.044) and 6 h (P = 0.0451); Figure 3, III. Therefore, HRP extravasation confirmed the BBB dysfunction at 3 h after FPI in the cortex and corpus callosum.
Figure 3.
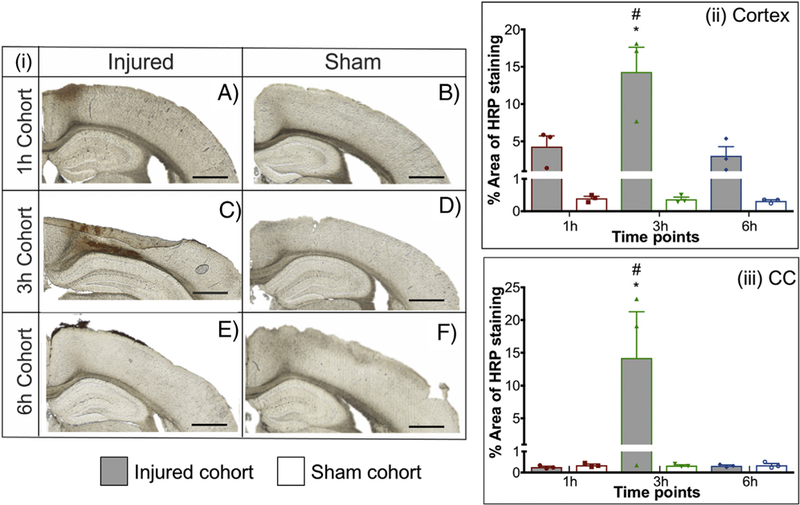
Representative images of HRP extravasation after FPI with injured and sham groups of 1 h (A-B), 3 h (C-D), 6 h (E-F). HRP was injected ten mins before sacrifice. Quantification of HRP extravasation after FPI. (ii) Cortex, (iii) Corpus callosum. * P < 0.05 compared to respective sham group, two-way ANOVA, Bonferroni’s post-hoc test. # P < 0.05 compared to 1 h and 6 h injured cohort, two-way ANOVA, Tukey’s post-hoc test. Two-segmented y-axis is used to display the sham groups. Error bars represent standard error of mean with n = 3. Scale bars = 200 μm.
Accumulation of NP after diffuse injury
To determine the extent of NP accumulation after acute TBI, we intravenously injected NP cocktail after mCHI/RmCHI (only 20 nm and 40 nm) and FPI (20, 40, 100 and 500 nm) and with a constant circulation time of 1 h.
Absence of NP accumulation after mCHI/RmCHI.
For the mCHI study, there was no fluorescent signal observed in any of the tissue sections after either single impact or multiple impact (Supplementary Figure S3).
Presence of NP accumulation after midline FPI.
We quantified the accumulation of each fluorescent NP via confocal microscopy for the midline FPI model (Figures 4–6). In the cortex, there was a significant effect between the injury and sham group for 20 nm (two-way ANOVA, P = 0.0002), 40 nm (P = 0.0006), 100 nm (P = 0.0071), and 500 nm (P = 0.0003). Moreover, the analysis demonstrated a significant time dependent effect for 20 nm (P = 0.0002), 40 nm (P = 0.0006), 100 nm (P = 0.0069), and 500 nm (P = 0.0013) (Figure 5, A-D). To examine the effect of each of these variables individually, post-hoc pair-wise analyses of critical comparisons are described below. It must be noted that the cocktail of NP injection contained equal fluorophore concentration yet varying total number of NP for each NP size, thereby preventing direct comparison across NPs with high fidelity. Therefore, our analysis focused on direct comparison within each NP size at different time points and not across NP sizes.
Figure 4.
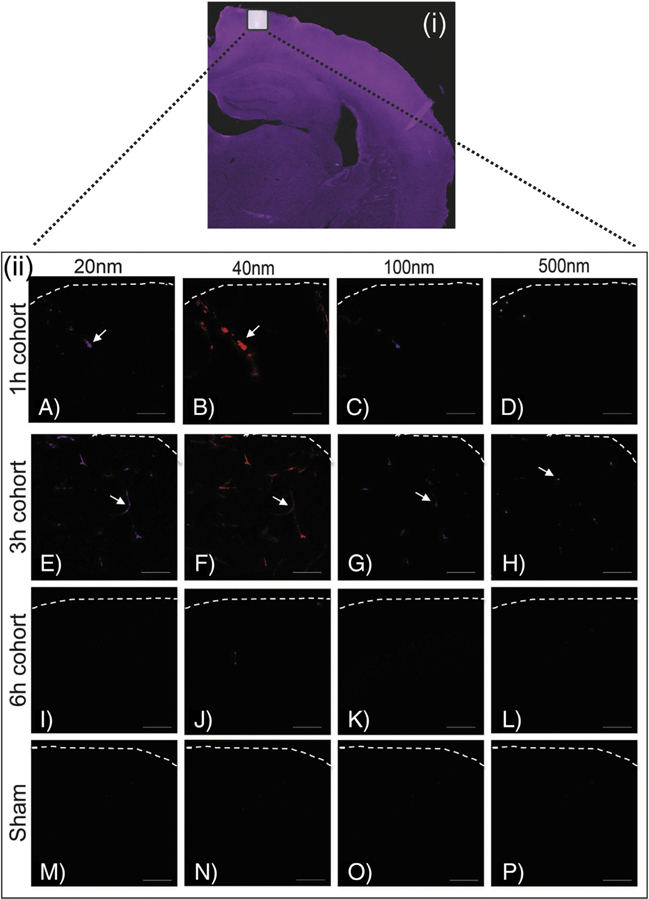
Accumulation of different size nanoparticles over time after FPI. (i) Representative image of whole brain scan and white box highlights the approximate location of the higher magnification cortical region. (ii) Representative images of NP accumulation at 1 h (A-D), 3 h (E-H), 6 h (I-L), and sham (control) (M-P). Columns display different size of NP accumulation over different time points post-midline-FPI. NPs were injected 1 h before sacrifice. Scale bar = 100 μm.
Figure 6.
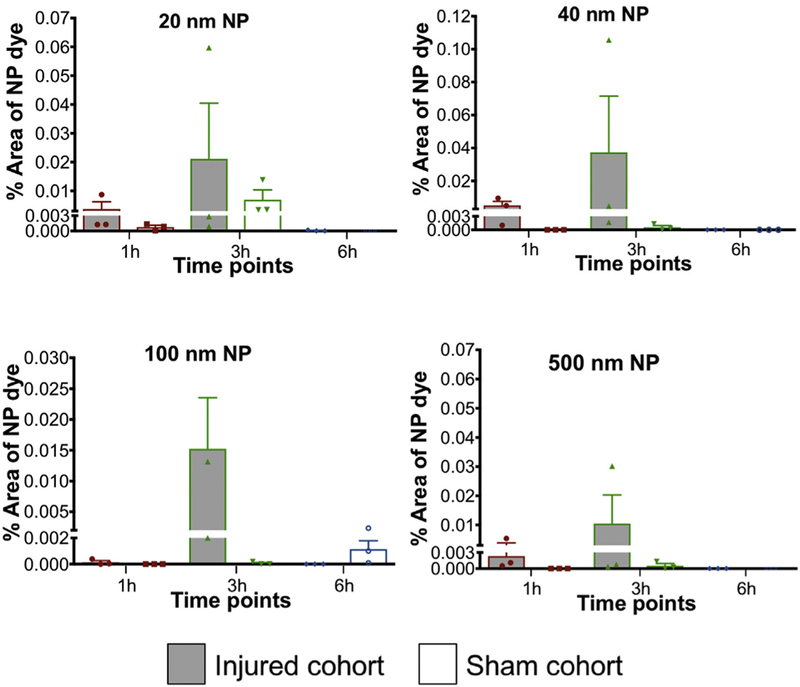
Nanoparticle accumulation after midline-FPI in corpus callosum. Accumulation of (a) 20 nm, (b) 40 nm, (c) 100 nm and (d) 500 nm nanoparticles at different time points (1 h, 3 h, and 6 h) after FPI. NPs were injected 1 h before sacrifice. Two-way ANOVA statistical test was conducted. Two-segmented y-axis is used to display the sham groups. Error bars represents standard error of mean with n = 3 animals per group.Correlation of HRP and NP accumulation after midline-FPI.
Figure 5.
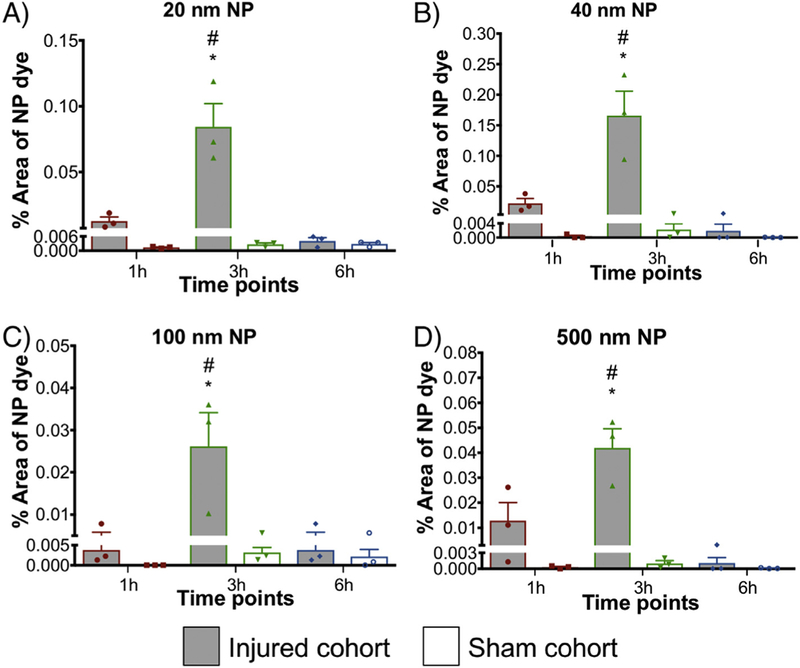
Nanoparticle accumulation after FPI in cortex. Accumulation of (A) 20 nm, (B) 40 nm, (C) 100 nm and (D) 500 nm nanoparticles at different time points (1 h, 3 h, and 6 h) after FPI. NPs were injected 1 h before sacrifice. * P < 0.05 compared to respective sham group, two-way ANOVA, Bonferroni’s post-hoc test. # P < 0.05 compared to 1 h and 6 h injured cohort, two-way ANOVA, Tukey’s post-hoc test. Two-segmented y-axis is used to display the sham groups. Error bars represent standard error of mean with n = 3 animals per group.
Analysis of the NP accumulation in midline FPI injured and sham brain tissue.
The first pairwise analysis focused on comparing NP accumulation in the injured and sham group across time points in the two ROIs (cortex and corpus callosum). Specifically, in the cortex, Bonferroni’s post-hoc test displayed significant difference in 20 nm (P < 0.0001), 40 nm (P < 0.0001), 100 nm (P < 0.0021) and 500 nm (P < 0.0001) NPs accumulation at 3 h post-injury as compared to their respective sham groups (Figure 5, (A–D)). No significant difference was observed at 1 h and 6 h post-injury compared to their respective sham groups for any NP. Furthermore, in the corpus callosum ROI there was no significant difference between the injured and the sham group for all NP sizes. Overall, all the NPs displayed significant accumulation at 3 h post-FPI compared to the sham group in the cortex.
Analysis of the NP accumulation in midline FPI injured group across different time points.
The second critical comparison focused on the NP accumulation within the injured group across different time points in the two ROIs: cortex and corpus callosum. In the cortex, for all the NP sizes, 20, 40, 100 and 500 nm, Tukey’s post-hoc test demonstrated a significant increase at 3 h post-injury for 20 nm (P < 0.0001), 40 nm (P ≤ 0.0001), 100 nm (P = 0.002) and 500 nm (P ≤ 0.001) compared to both h and 6 h cohorts (P < 0.05). The peak increase of accumulation in the cortex at 3 h post-injury was at least twice that at 1 h and 6 h for each NP size (Figure 5, A-D). In the corpus callosum, although there was no significant difference among the NP accumulation (20, 40, 100 and 500 nm) across time points, there was a trend with increased accumulation at 3 h post-FPI compared to that at 1 h and 6 h (Figure 6, A-D).
To further validate our hypothesis that NP accumulation directly depends on blood-brain barrier permeability, we evaluated the relationship between HRP and NP staining with Pearson correlation coefficient. For the cortex ROI, in Figure 7, each scatter plot represents corresponding data points of the percent area of HRP staining versus percent area of NPs across all the time periods. The results (Figure 7) show a significant correlation between the HRP staining and 20 nm (P = 0.0005), nm (P < 0.0001), 100 nm (P < 0.0001) and 500 nm (P = 0.0002). Moreover, the Pearson correlation coefficient for: 20 nm (r = 0.9147), 40 nm (r = 0.9574), 100 nm (r = 0.9608), and nm (r = 0.9388) indicates a robust correlation between HRP and the NPs. Particularly, HRP staining was maximally observed at 3 h post-injury and those animals/brain tissue displayed peak NP accumulation (20 nm, 40 nm, 100 nm and 500 nm). Furthermore, the same analysis was performed for the corpus callosum, but no correlation was identified due to limited NP accumulation.
Figure 7.
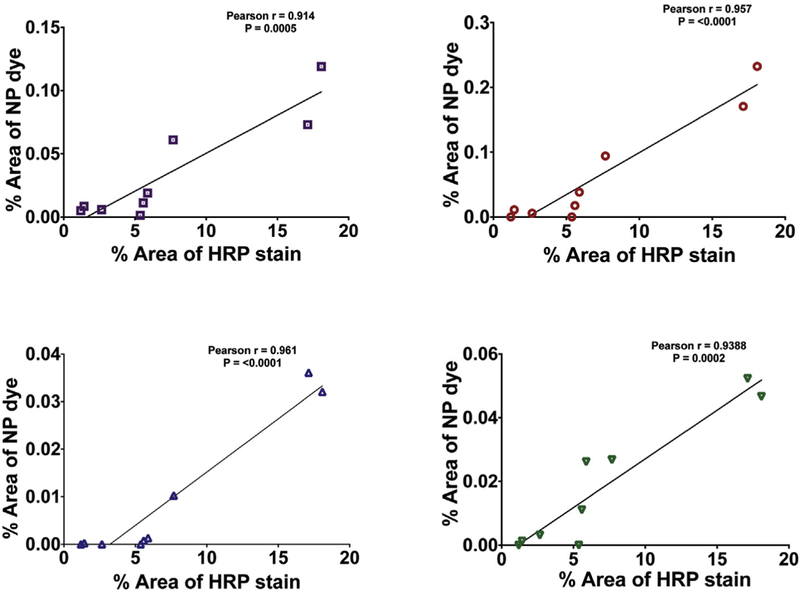
Correlation of HRP and NP accumulation after midline-FPI in cortex. The x-axis shows the percentage of the HRP stain (data from Figure 3) and the y-axis shows the percentage of NP accumulation (data from Figure 4–6) of (a) 20 nm (r = 0.9147), (b) 40 nm (r = 0.9574), (c) 100 nm (r = 0.9608), and (d) 500 nm (r = 0.9388). Pearson correlation coefficient (r) was close to 1, indicating a robust correlation between HRP and the NPs.
Discussion
Research on NP delivery after brain injury has gained traction over the last couple of years.22,31–37, 47,48, 23,25,35,38,39 TBI may lead to a transient breach in the BBB that can be capitalized to passively deliver NPs. Previously, using experimental focal injury (controlled cortical impact) our group has demonstrated the feasibility of NP delivery up to 500 nm in size with a peak NP accumulation at 1 h post-TBI.23 Yet, there is a critical gap in understanding the relationship between BBB opening and models of diffuse TBI for NP delivery. Here in this study, we used three models of diffuse brain injury (mild CHI, repetitive mild CHI and midline-fluid percussion injury (FPI)) to evaluate the effect of different size NP accumulation at acute time points after injury. We reported four key findings: 1) mCHI and RmCHI did not show any NP accumulation 1 h after final impact; 2) NPs up to 500 nm can be delivered to injured brain sustaining a diffuse TBI by midline-FPI model; 3) maximal accumulation occurred 3 h post-FPI for all NP sizes (20, 40, 100 and 500 nm); and 4) significant correlation was observed between HRP staining and NP accumulation in the cortex after midline FPI.
RmCHI is common in sports-related TBIs and war combat morbodities,40,41 with the potential for long-term consequences, including chronic traumatic encephalopathy.42 Evaluation and management of RmCHI remains a challenge due to the diffuse and microscopic pathology, where NP delivery for diagnostic and therapeutic approaches may provide a new strategy to improve detection of mCHI and clinical management. In this study we intravenously injected 20 and 40 nm NPs immediately following impact in mCHI and RmCHI with one-hour circulation time; HRP (BBB integrity marker) was injected ten minutes prior to sacrifice. We did not observe any HRP or NP accumulation at 1 h after single impact mCHI nor after 5 consecutive impacts in the rmCHI model. This observation of lack of the HRP/NP after mCHI has a few caveats and needs to be interpreted cautiously. First, it must be noted that we only probed a single time point after closed head injury and it could be possible that the disruption to the BBB was not captured at this time point. Second, we used large molecular weight tracers (~44 kDa) HRP as the marker of BBB integrity. A previous TBI study26 showed that BBB disruption and resolution is size dependent where small molecules (<10 kDa) showed prolonged permeability compared to larger tracers. Therefore, it could be possible that the BBB breach was not captured by larger molecular weight tracer. Lastly, in case of a minimal BBB disruption, the washout of NPs during pre-perfusion could be another possibility for lack of NP accumulation. This possibility is supported by a recent study using swine rapid rotation concussion model that demonstrated multifocal BBB disruption at 6–72 h post-injury.43 However, the role of differences in time post-injury, experimental impact models and species remains to be explored. As such, we acknowledge that probing BBB integrity via this approach captures a single snapshot in the dynamically complex response after impact. Therefore, future studies need to investigate changes in BBB integrity across additional time points post-injury, in different impact models/species and by using an array of varied size molecular weight tracers. Therefore, we further probed BBB integrity via immunohistochemistry for endogenous IgG within the parenchyma. IgG is not present in healthy brain tissue, but it may get trapped due to a transient BBB breakdown. Intriguingly, we observed IgG staining in the hippocampus in both single and multiple impact groups compared to staining in the naïve group (Supplementary, Figure S2). The presence of endogenous IgG and the absence of any exogenous HRP (injected 50mins post-impact) suggests one of two possibilities. One, a transient BBB breach occurs immediately after and/or during both single and multiple impacts, but sealed prior to the injection of HRP (i.e. within 50mins). Alternatively, the presence of IgG could possibly be due to active transport across the endothelial cells via deregulated endocytosis pathway such as transcytosis.44 Furthermore, the absence of any NP accumulation after immediate injection suggests that the BBB permeability was size dependent, where the 20 nm NPs may have been too large for extravasation as compared to the smaller sized IgG (estimated radius of 5 nm45).
In contrast to the mCHI study, the midline-FPI study showed both HRP and NP accumulation after injury. We observed extravasation of the HRP (~44 kDa with an estimated radius of ~3 nm45) in the cortex (proximal to injury hub) at 1 h, 3 h and 6 h post-injury. Significant HRP staining at 3 h post-injury was also noted in the corpus callosum. Previous rat FPI (midline and lateral) models reported similar observations of HRP extravasation at 1 h with marked reduction at 6 h post-injury in the cerebral cortex and corpus callosum.16,17 One key objective of this study was to establish the feasibility for NP delivery after diffuse brain injury to then further develop NP-based therapeutics and diagnostics. Demonstrating the time course of BBB disruption after diffuse brain injury was the first step in achieving this objective. In addition, although mCHI did not show any NP accumulation at 1 h post-injury, studies to assess NP accumulation at different time points is warranted.
In the recent years the interest for NP application for TBI has been increasing.23,25,35,38,39 For example, Bailey et al. used cerium oxide NPs (~10 nm) as an antioxidant agent in the lateral FPI model.39 Although this study demonstrated the feasibility for NP delivery following diffuse brain injury, little is known about the impact of NP size and optimal time window for effective delivery after TBI. We directly addressed this critical gap by evaluating a cocktail injection of different size PEGylated NPs at different time points after injury while maintaining a constant circulation time of 1 h. Furthermore, systemic NP delivery parameters such as size, charge and surface modification may influence the physiochemical stability and the rate of NP elimination from the circulation.46,47 Since the focus of this study was to evaluate the size and time dependent NP accumulation after diffuse brain injury, the NPs were PEGylated to reduce the negative charge on the carboxylated NPs for efficient systemic delivery and to minimize the influence of parameters outside of size. The zeta potentials of all different NP sizes were negatively charged within the range of −10 to −27 mV (See Supplementary Figure S4 and Table S1). As such, it must be noted that the zeta potential of the 40 nm size NP was −10 mV and significantly lower than that of the other NP sizes. The objective for using this set of NPs was to minimize variance introduced by the zeta potential to focus on NP size parameter, however, the 40 nm NP was −10 mV and less than negative that the other NPs. Furthermore, it must be noted that studies by Levchenko and group have shown that near neutral NPs have lower clearance and thus longer circulation time compared to negatively charged NPs.48 Therefore, future studies may evaluate the potential effect of NP zeta potential on NP accumulation following TBI. Moreover, additional parameters such as PEG density for each of the NP size may be explored as a delivery parameter to tune for optimal delivery. Maximal accumulation for all size NPs was observed at 3 h post-injury in the cortex (Figure 4). Our study is the first to report accumulation up to 500 nm PEGylated NPs following midline FPI. This finding supports our previous study using the focal brain injury model, controlled cortical impact (CCI), where we reported accumulation of ~500 nm NPs near the injured cortex out to 12 h post-injury.23 Taking our data one step further, we identified a direct correlation of the BBB integrity as measured through HRP staining with NP accumulation (Figure 6). Collectively, our results add to our current understanding regarding the size and time dependent NP delivery after diffuse TBI. Insight into such parameters is critical for determining the optimal delivery window for NPs following TBI.
An interesting finding from our study in the midline FPI cohort was the peak accumulation of the NPs at 3 h post-injury compared to 1 h and 6 h group. Acutely after FPI, regional cerebral blood flow reportedly decreases49 contributing to distressed endothelial cells and ultimately vasogenic and cytotoxic edema.6 This reduced cerebral blood flow may impede intravenously administered molecules to reach the injured area(s) of the brain.50 Furthermore, Lin and co-workers established a positive correlation between the BBB permeability and cerebral microvascular perfusion acutely after FPI in rat models.50 Immediately and 1 h post-injury, ischemic centers were prominent in the injured cortex indicating compromised capillary perfusion.50 However, by 4 h post-injury the blood vessels were significantly more perfused compared to immediate time point.50 Therefore, such altered blood vessels/perfusion phenomena that occurs post-injury further supports our observation of peak NP accumulation at 3 h.
Our main objective with this study was to investigate the feasibility of NP delivery and characterize the temporal and nanoparticle size dependent accumulation after experimental diffuse brain injury. As such, we did not directly probe the mechanism of NP accumulation in the brain after injury. The two major approaches for nanomaterials to enter the brain through the is via paracellular permeability and transcytosis.51 TBI pathology may lead to BBB dysfunction due to factors such as matrix metalloproteinases and lead to down regulation of tight junction expression and results in BBB dysfunction, thus facilitating paracellular diffusion.6,52 Another possible mechanism for the transport of particles across the BBB is via a transcellular route. Previous reports have suggested that transport across the BBB can occur without obvious changes associated with interendothelial junctions.53 These events include increase in caveolae vesicular formation and transcytosis in BBB endothelium in pathological conditions including cortical cold injury, contusion injury and ischemia.54–57 Additionally, transcellular events at the BBB can also involve extravasation of immune cells.58,59 Recent studies have suggested the existence of transcellular leukocyte migration and formation of invadosome-like protrusions into the endothelial cells.58,59 As such, future studies to probe the NP transport mechanism are warranted.
Another interesting finding from our study is the differential spatial NP accumulation within the brain regions (cortex and corpus callosum). The midline-FPI resulted in leaky vasculature in the cortex and the corpus callosum as visualized by HRP extravasation. Additionally, the NP accumulation was significant compared to the sham groups only in the cortex and not in the corpus callosum. Regional differences may be attributed to the variations in BBB structure in the cerebral cortex and white matter.60 Previous studies have reported not only a lower capillary density61 but also tighter barrier due to higher astrocytes and tight junction expressions in the corpus callosum compared to cortical regions.60 Thus, the heterogeneity in the structure could play a role in the absence of NP accumulation in the corpus callosum compared to the cortex.
In conclusion, we established that PEGylated polystyrene NPs (20 and 40 nm) failed to accumulate in the brain tissue after mCHI and RmCHI. In contrast, the FPI cohorts displayed NP accumulation of up to 500 nm in size. With a constant circulation time of 1 h, we observed a significant peak accumulation at 3 h post-FPI compared to 1 h and 6 h post-FPI. The NP accumulation was not only influenced by the NP injection time, but also on the spatial location in the brain tissue. There was significant NP accumulation only in the cortex and not in the BBB breached corpus callosum. Our current study provides the groundwork for the feasibility of NP delivery in terms of NP injection time and NP size after mCHI and FPI. Prospective application of our study ranges from contrast agents to therapeutic drug delivery after TBI and is grounded on an improved understanding of the BBB breakdown post-injury.
Supplementary Material
Acknowledgments
Authors would like to thank Dr. Jason Newbern for technical assistance-confocal microscopy; Amanda Witten for the artistic rendering-graphical abstract and Figure 1; and Kyle Offenbacher for drawing the ROIs-FPI analysis.
Authors would like to thank funding sources NIH NICHD (1DP2HD084067; SES), and Flinn Foundation (1976; VDK, SES, JL, PDA).
Footnotes
Authors declare no conflict of interest or disclosures.
References
- 1.Feigin VL, Barker-Collo S, Krishnamurthi R, Theadom A, Starkey N. Epidemiology of ischaemic stroke and traumatic brain injury. Best Pract Res Clin Anaesthesiol 2010;24(4):485–94. [DOI] [PubMed] [Google Scholar]
- 2.Langlois JA, Rutland-Brown W, Wald MM. The epidemiology and impact of traumatic brain injury: a brief overview. J Head Trauma Rehabil 2006;21(5):375–8. [DOI] [PubMed] [Google Scholar]
- 3.Maas AIR, Stocchetti N, Bullock R. Moderate and severe traumatic brain injury in adults. The Lancet Neurology 2008;7(8):728–41. [DOI] [PubMed] [Google Scholar]
- 4.Kelley BJ, Lifshitz J, Povlishock JT. Neuroinflammatory responses after experimental diffuse traumatic brain injury. Journal of Neuropathology & Experimental Neurology 2007;66(11):989–1001. [DOI] [PubMed] [Google Scholar]
- 5.Shlosberg D, Benifla M, Kaufer D, Friedman A. Blood-brain barrier breakdown as a therapeutic target in traumatic. Brain Inj 2010;6 (7):393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chodobski A, Zink BJ, Szmydynger-Chodobska J. Blood-brain barrier pathophysiology in traumatic brain injury. Transl Stroke Res 2011;2 (4):492–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Werner C, Engelhard K. Pathophysiology of traumatic brain injury. Br J Anaesth 2007;99(1):4–9. [DOI] [PubMed] [Google Scholar]
- 8.Margulies S, Hicks R. Combination therapies for traumatic brain injury: prospective considerations. Journal of Neurotrauma 2009;26(2):925–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xiong Y, Mahmood A, Chopp M. Animal models of traumatic brain injury. Nature Reviews Neuroscience 2013;14(2):128–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O’Connor WT, Smyth A, Gilchrist MD. Animal models of traumatic brain injury: a critical evaluation. Pharmacology and Therapeutics 2011;130(2):106–13. [DOI] [PubMed] [Google Scholar]
- 11.Smith DH, Soares HD, Pierce JS, Perlman KG, Saatman KE, Meaney DF, et al. A model of parasagittal controlled cortical impact in the mouse: cognitive and histopathologic effects. Journal of Neurotrauma 1995;12 (2):169–78. [DOI] [PubMed] [Google Scholar]
- 12.Kane MJ, Angoa-Pérez M, Briggs DI, Viano DC, Kreipke CW, Kuhn DM. A Mouse model of human repetitive mild traumatic brain injury. Journal of Neuroscience Methods, 203 (1). Elsevier BV; 2012:41–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dixon CE, Lighthall JW, Anderson TE. Physiologic, histopathologic, and cineradiographic characterization of a new fluid-percussion model of experimental brain injury in the rat. J Neurotrauma 1988;5(2):91–104. [DOI] [PubMed] [Google Scholar]
- 14.Rowe RK, Griffiths DR, Lifshitz J. Midline (central) fluid percussion model of traumatic brain injury. Methods Mol biol 2016;1462:211–30 [Chapter 13]. [DOI] [PubMed] [Google Scholar]
- 15.Rowe RK, Griffiths DR, Evilsizor MN, Thomas TC, Adelson PD, Lifshitz J, et al. Clinical relevance of midline fluid percussion brain injury: acute deficits, chronic morbidities and the utility of biomarkers. Brain injury 2016;30(11):1293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tanno H, Nockels RP, Pitts LH, Noble LJ. Breakdown of the blood-brain barrier after fluid percussive brain injury in the rat. Part 1: distribution and time course of protein extravasation. J Neurotrauma 1992;9(1):21–32. [DOI] [PubMed] [Google Scholar]
- 17.Schmidt RH, Grady MS. Regional patterns of blood-brain barrier breakdown following central and lateral fluid percussion injury in rodents. J Neurotrauma 1993;10(4):415–30. [DOI] [PubMed] [Google Scholar]
- 18.McIntosh TK, Noble L, Andrews B, Faden AI. Traumatic brain injury in the rat: characterization of a midline fluid-percussion model. Cent Nerv Syst Trauma 1987;4(2):119–34. [DOI] [PubMed] [Google Scholar]
- 19.de Boer AG, Gaillard PJ. Drug targeting to the brain. Annu Rev Pharmacol Toxicol 2007;47:323–55. [DOI] [PubMed] [Google Scholar]
- 20.Pardridge WM. Drug transport across the blood-brain barrier, 2nd ed. Journal of Cerebral Blood Flow & Metabolism 2012;32(11):1959–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alves JL. Blood-brain barrier and traumatic brain injury. J Neurosci Res 2013;92(2):141–7. [DOI] [PubMed] [Google Scholar]
- 22.Bharadwaj VN, Nguyen DT, Kodibagkar VD, Stabenfeldt SE. Nanoparticle-based therapeutics for brain injury. Adv Healthc Mater 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bharadwaj VN, Lifshitz J, Adelson PD, Kodibagkar VD, Stabenfeldt SE. Temporal assessment of nanoparticle accumulation after experimental brain injury: effect of particle size. Sci Rep 2016;629988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ping X, Jiang K, Lee S-Y, Cheng J-X, Jin X. PEG-PDLLA micelle treatment improves axonal function of the corpus callosum following traumatic brain injury. J Neurotrauma 2014;31(13):1172–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clond MA, Lee B-S, Yu JJ, Singer MB, Amano T, Lamb AW, et al. Reactive oxygen species-activated nanoprodrug of ibuprofen for targeting traumatic brain injury in mice. PLoS ONE 2013;8(4):e61819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Habgood MD, Bye N, Dziegielewska KM, Ek CJ, Lane MA, Potter A, et al. Changes in blood-brain barrier permeability to large and small molecules following traumatic brain injury in mice. Eur J Neurosci 2007; 21(1):231–8. [DOI] [PubMed] [Google Scholar]
- 27.Goddeyne C, Nichols J, Wu C, Anderson T. Repetitive mild traumatic brain injury induces ventriculomegaly and cortical thinning in juvenile rats. J Neurophysiol 2015;113(9):3268–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hosseini AH, Lifshitz J. Brain injury forces of moderate magnitude elicit the fencing response. Med Sci Sports Exerc 2009;41(9):1687–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carbonell WS, Maris DO, McCall T, Grady MS. Adaptation of the fluid percussion injury model to the mouse. Journal of Neurotrauma 1998;15 (3):217–29. [DOI] [PubMed] [Google Scholar]
- 30.Yardeni T, Eckhaus M, Morris HD, Huizing M, Hoogstraten-Miller S. Retro-orbital injections in mice. Lab Anim 2011;40(5):155–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yun X, Maximov VD, Yu J, Zhu H, Vertegel AA, Kindy MS. Nanoparticles for targeted delivery of antioxidant enzymes to the brain after cerebral ischemia and reperfusion injury. Journal of Cerebral Blood Flow and Metabolism 2013;33(4):583–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ruozi B, Belletti D, Sharma HS, Sharma A, Muresanu DF, Mössler H, et al. PLGA nanoparticles loaded Cerebrolysin: studies on their preparation and investigation of the effect of storage and serum stability with reference to traumatic brain injury. Mol Neurobiol 2015;52(2):899–912. [DOI] [PubMed] [Google Scholar]
- 33.Kwon EJ, Skalak M, Bu Lo R, Bhatia SN. Neuron-targeted nanoparticle for siRNA delivery to traumatic brain injuries. ACS Nano 2016;10 (8):7926–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khalin I, Alyautdin R, Wong TW, Gnanou J, Kocherga G, Kreuter J. Brain-derived neurotrophic factor delivered to the brain using poly (lactide-co-glycolide) nanoparticles improves neurological and cognitive outcome in mice with traumatic brain injury. Drug Deliv 2016;23(9):3520–8. [DOI] [PubMed] [Google Scholar]
- 35.Xu J, Ypma M, Chiarelli PA, Park J, Ellenbogen RG, Stayton PS, et al. Theranostic oxygen reactive polymers for treatment of traumatic brain injury. Adv Funct Mater 2016;26(23):4124–33. [Google Scholar]
- 36.Cruz LJ, Stammes MA, Que I, Beek ER, Knol-Blankevoort VT, Snoeks TJA, et al. Effect of PLGA NP size on efficiency to target traumatic brain injury. Journal of Controlled Release 2016;223:31–41. [DOI] [PubMed] [Google Scholar]
- 37.Cruz LJ, Que I, Aswendt M, Chan A, Hoehn M, Löwik C. Targeted nanoparticles for the non-invasive detection of traumatic brain injury by optical imaging and fluorine magnetic resonance imaging. Nano Res 2016;9(5):1276–89. [Google Scholar]
- 38.Bitner BR, Marcano DC, Berlin JM, Fabian RH, Cherian L, Culver JC, et al. Antioxidant carbon particles improve cerebrovascular dysfunction following traumatic brain injury. ACS Nano 2012;6(9):8007–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bailey ZS, Nilson E, Bates JA, Oyalowo A, Hockey KS, Sajja VSSS, et al. Cerium oxide nanoparticles improve outcome after in vitro and in vivo mild traumatic brain injury. J Neurotrauma 2016;33:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guskiewicz KM, Weaver NL, Padua DA, Garrett WE. Epidemiology of concussion in collegiate and high school football players. The American journal of sports medicine 2000;28(5):643–50. [DOI] [PubMed] [Google Scholar]
- 41.Okie S Traumatic brain injury in the war zone. The New England Journal of Medicine 2005;352(20):2043–7. [DOI] [PubMed] [Google Scholar]
- 42.McKee AC, Cantu RC, Nowinski CJ, Hedley-Whyte ET, Gavett BE, Budson AE, et al. Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol 2009;68(7):709–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Johnson VE, Weber MT, Xiao R, Cullen DK, Meaney DF, Stewart W, et al. Mechanical disruption of the blood–brain barrier following experimental concussion. Acta Neuropathologica 2018:1–16. [DOI] [PMC free article] [PubMed]
- 44.Villaseñor R, Ozmen L, Messaddeq N, Grüninger F, Loetscher H, Keller A, et al. Trafficking of endogenous immunoglobulins by endothelial cells at the blood-brain barrier. Science Reporter 2016:1–10. [DOI] [PMC free article] [PubMed]
- 45.Ban Y, Rizzolo LJ. A culture model of development reveals multiple properties of RPE tight junctions. Mol Vis 1997;3:18. [PubMed] [Google Scholar]
- 46.Li S-D, Huang L. Pharmacokinetics and biodistribution of nanoparticles. Mol Pharm 2008;5(4):496–504. [DOI] [PubMed] [Google Scholar]
- 47.Jo DH, Kim JH, Lee TG, Kim JH. Size, surface charge, and shape determine therapeutic effects of nanoparticles on brain and retinal diseases. Nanomedicine: nanotechnology, biology, and medicine 2015;11(7):1603–11. [DOI] [PubMed] [Google Scholar]
- 48.Levchenko TS, Rammohan R, Lukyanov AN, Whiteman KR, Torchilin VP. Liposome clearance in mice: the effect of a separate and combined presence of surface charge and polymer coating. Int J Pharm 2002;240 (1–2):95–102. [DOI] [PubMed] [Google Scholar]
- 49.Yamakami I, McIntosh TK. Effects of traumatic brain injury on regional cerebral blood flow in rats as measured with radiolabeled microspheres. J Cereb Blood Flow Metab 1989;9(1):117–24. [DOI] [PubMed] [Google Scholar]
- 50.Lin Y, Pan Y, Wang M, Huang X, Yin Y, Wang Y, et al. Blood-brain barrier permeability is positively correlated with cerebral microvascular perfusion in the early fluid percussion-injured brain of the rat. Lab Investig 2012;92(11):1623–34. [DOI] [PubMed] [Google Scholar]
- 51.Gao X, Gu X, Chen H. Chapter 3 - The distribution and elimination of nanomaterials in brain. In: Gao Jiang, editor. Neurotoxicity of Nanomaterials and Nanomedicine Academic Press; 2017. p. 59–74. [Google Scholar]
- 52.Main BS, Villapol S, Sloley SS, Barton DJ, Parsadanian M, Agbaegbu C, et al. Apolipoprotein E4 impairs spontaneous blood brain barrier repair following traumatic brain injury. Mol Neurodegener 2018:1–18. [DOI] [PMC free article] [PubMed]
- 53.De Bock M, Van Haver V, Vandenbroucke RE, Decrock E, Wang N, Leybaert L. Into rather unexplored terrain-transcellular transport across the blood-brain barrier. Glia 2016;64(7):1097–123. [DOI] [PubMed] [Google Scholar]
- 54.Nag SS, Takahashi JLJ, Kilty DWD. Role of vascular endothelial growth factor in blood-brain barrier breakdown and angiogenesis in brain trauma. J Neuropathol Exp Neurol 1997;56(8):912–21. [DOI] [PubMed] [Google Scholar]
- 55.Nag S, Kapadia A, Stewart DJ. Review: molecular pathogenesis of blood-brain barrier breakdown in acute brain injury. Neuropathology and Applied Neurobiology 2011;37(1):3–23. [DOI] [PubMed] [Google Scholar]
- 56.Li X, Han Y, Xu H, Sun Z, Zhou Z, Long X, et al. Aquaporin 4 expression and ultrastructure of the blood-brain barrier following cerebral contusion injury. Neural Regeneration Research 2013;8 (4):338–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Knowland D, Arac A, Sekiguchi KJ, Hsu M, Lutz SE, Perrino J, et al. Stepwise recruitment of transcellular and Paracellular pathways underlies blood-brain barrier breakdown in stroke. Neuron 2014;82 (3):603–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Carman CV. Mechanisms for transcellular diapedesis: probing and pathfinding by invadosome-like protrusions. Journal of Cell Science 2009;122(17):3025–35. [DOI] [PubMed] [Google Scholar]
- 59.Martinelli R, Zeiger AS, Whitfield M, Sciuto TE, Dvorak A, Van Vliet KJ, et al. Probing the biomechanical contribution of the endothelium to lymphocyte migration: diapedesis by the path of least resistance. Journal of Cell Science 2014;127(Pt 17):3720–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nyúl-Tóth Á, Suciu M, Molnár J, Fazakas C, Haskó J, Herman H, et al. Differences in the molecular structure of the blood-brain barrier in the cerebral cortex and white matter: an in silico, in vitro, and ex vivo study. Am J Physiol Heart Circ Physiol 2016;310(11):H1702–14. [DOI] [PubMed] [Google Scholar]
- 61.Wilhelm I, Nyúl-Tóth Á, Suciu M, Hermenean A, Krizbai IA. Heterogeneity of the blood-brain barrier. Tissue Barriers 2016;4(1): e1143544–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.