

Abstract
Cholangiocarcinoma (CCA) is an aggressive biliary tract malignancy with a poor overall prognosis. There is a critical need to develop effective targeted therapies for the treatment of this lethal disease. In an effort to address this challenge, preclinical in vivo studies have become paramount in understanding CCA carcinogenesis, progression, and therapy. Various CCA animal models exist including carcinogen-based models in which animals develop CCA after exposure to a carcinogen, genetically engineered mouse models in which genetic changes are induced in mice leading to CCA, murine syngeneic orthotopic models, as well as xenograft tumors derived from xenotransplantation of CCA cells, organoids, and patient-derived tissue. Each type has distinct advantages as well as shortcomings. In the ideal animal model of CCA, the tumor arises from the biliary tract in an immunocompetent host with a species-matched tumor microenvironment. Such a model would also be time-efficient, recapitulate the genetic and histopathological features of human CCA, and predict therapeutic response in humans. Recently developed biliary tract transduction and orthotopic syngeneic transplant mouse models encompass several of these elements. Herein, we review the different animal models of CCA, their advantages and deficiencies, as well as features which mimic human CCA.
Keywords: Orthotopic, transgenic, transduction, xenotransplantation
INTRODUCTION
Cholangiocarcinoma (CCA) is the most common biliary malignancy and the second most common hepatic malignancy after hepatocellular carcinoma. CCAs are epithelial tumors likely originating from the biliary tree and are classified into three subtypes based on their anatomic location [1]. Intrahepatic cholangiocarcinoma (iCCA) originates above the second order bile ducts within the hepatic parenchyma [2]. Perihilar CCA (pCCA) arises between the second-order bile ducts and the cystic duct; pCCA is the most common type of CCA, accounting for approximately 50-70% of CCAs in different series [3, 4]. Distal CCA (dCCA), arises distal to the cystic duct [5]. Each anatomic subtype has a distinct epidemiology, molecular pathogenesis, and management. Overall, CCA is a relatively rare malignancy, representing 3% of all gastrointestinal cancers [6]. Although the incidence of CCA has increased over the past three decades, the prognosis remains poor with a 5-year overall survival of less than 10% [6, 7]. Potentially curative options such as surgical resection and liver transplantation (for pCCA) are limited to the subset of patients with early stage disease. For patients with advanced disease not amenable to surgical options, the standard of care is systemic chemotherapy with gemcitabine and cisplatin. However, the median overall survival with this regimen is <1 year [8]. Hence, there is a critical need for development of effective targeted molecular therapies for CCA. Such a precision medicine approach requires greater insight into the molecular pathogenesis of CCA.
In cancer research, in vitro studies utilizing cell culture are generally used to investigate the biochemical processes of cancer cells [9]. However, this approach is not sufficient to investigate the myriad of biologic process occurring in tumors including cell survival, inflammation, angiogenesis, and mutations. Although cell culture models have advantages including high reproducibility, homogeneity and highly controlled experimental conditions, cultured cells typically have uniform phenotypic and genetic characteristics; hence, it is challenging to study interactions between different cell types within a tumor or investigate the role of various biologic processes. Moreover, investigating novel therapeutic targets ultimately requires preclinical studies in animal models. Hence, the use of in vivo models is necessary in cancer research. The ideal animal model of CCA would originate from the biliary tract in an immunocompetent host with a species-matched microenvironment; have time-efficient tumor development; recapitulate the genetic, anatomic, and phenotypic features of human CCA (Table 1). Assessment of such a model would require anatomic, histopathologic, and genetic characterization as well as evaluation of liver injury or dysfunction (Table 2). Herein, we review the available animal models of cholangiocarcinoma, summarizing the strengths and weaknesses of the different models with a focus on carcinogen-based, xenograft, allograft, and genetic models. These animal models represent iCCA; we are still in need of pCCA and dCCA animal models of CCA.
Table 1.
Ideal Characteristics of an Animal Model Of CCA.
• Originates in an immunocompetent
host
|
• Genetic aberrations similar to a
subset of human CCA
|
• Phenotypic Expression of Markers
Widely Expressed in Human CCA
|
• Desmoplastic Syngeneic Stroma
|
• Mimics an Anatomic Subset of Human
CCA
|
• Metastatic Phenotype
|
Table 2.
Evaluation of Animal Models of CCA.
• Anatomic Localization within the
Liver
|
• Liver Injury/Dysfunction
|
• Histopathologic Characterization
|
• Genetic Characterization
|
• Animal Consequences
|
ALT, alanine aminotransferase; AST, aspartate aminotransferase; H&E, hematoxylin and eosin; IHC, immunohistochemistry; RNA-Seq, RNA sequencing
CARCINOGEN-BASED CCA ANIMAL MODELS
In chemical models, CCA is induced by administration of a carcinogen such as dimethylnitrosamine (DMN), thiocetamide (TAA) or furan. These carcinogenic compounds can be used to either induce a genotoxic effect with deoxyribonucleotide acid (DNA) structural changes or to enhance tumor formation via expansion of preneoplastic cells. Other carcinogens include infectious agents such as the liver flukes Opisthorchis viverrini (O. Viverrini) and Clonorchis sinensis, known risk factors for CCA in Southeast Asia [10].
Dimethylnitrosamine and diethylnitrosamine
DMN is a genotoxic compound metabolized by cytochrome P450 which is mainly active in the centrilobular hepatocytes [11]. DMN promotes alkylation of DNA structure and generation of reactive oxygen species known to induce protein, lipid and DNA damage [12]. Consequently, DMN induces not only CCA but also other gastrointestinal tumors, as well as skin, lung and hematopoietic tumors [13]. The DMN based CCA animal model was first described in 1978 by Thamavit et al. in Syrian Golden Hamsters [14]. In this model, the authors induced liver fluke infection via intragastric administration of O. Viverrini. Once parasitic eggs were detected in animal stools (typically 4 weeks after infection) 0.0025% DMN was added to the drinking water of animals for another ten weeks. Animals receiving either DMN alone or parasites alone did not develop tumors, whereas 100% of animals receiving both carcinogens developed tumors [14]. The authors hypothesized that O. viverrini infection promotes proliferation of altered biliary epithelial cells and the subsequent DMN administration exerts a carcinogenic effect on these altered cells. Long-term administration of DMN appears to be necessary as hamsters receiving a single oral dose of DMN plus O. viverrini infection developed CCA in only 20% and 44% of cases, respectively, in two different series [15, 16]. It has also been postulated that small intraportal “oval” cells, which appear in animals following liver injury, are activated by DMN administration and liver fluke infection subsequently stimulates proliferation of these dysplastic cells thereby promoting CCA [17]. DMN administration has also been combined with bile duct ligation in the Syrian hamster, resulting in CCA development in approximately 40% of animals [18].
Diethylnitrosamine (DEN), another carcinogen, may promote tumorigenesis via DNA methylation [19]. DEN administration results in formation of multifocal biliary cystic lesions in mice, and induces CCA in mice when combined with pentachlorophenol [20, 21]. In mice with liver-specific deletion of cylindromatosis (CYLD), a tumor suppressor gene which is mutated in familial cylindromatosis, DEN single intraperitoneal injection combined with phenobarbital in drinking water results in CCA development. These mice develop a ductular reaction which progresses to biliary fibrosis, and eventually 5 out of 8 mice developed nodules consistent with hepatocellular carcinoma (HCC) as well as CCA [22]. The formation of mixed CCA and HCC in these and other models is of interest in liver carcinogenesis but limits their applicability for studying iCCA.
Thioacetamide (TAA)
TAA is a chemical compound, which has been primarily used to induce liver fibrosis and cirrhosis in rodents [23]. Although the molecular mechanism of TAA carcinogenesis is not completely understood, it is likely that bio-activation of TAA induces reactive oxygen species which promote hepatotoxicity and interfere with ribosomal activity [24, 25]. TAA-induced CCA was first reported in 1984 by Praet et al. Male albino rats developed benign lesions such as cystadenomas after 8 months of TAA treatment, and 100% of animals developed CCA after 12 months of TAA treatment [26]. Subsequent studies have reported development of CCA microfoci with shorter duration (12-16 weeks) of TAA treatment (0.03% TAA administered in animal drinking water), and development of mass forming lesions occurring at longer treatment intervals (22-24 weeks) in essentially all animals [27–30]. Biliary dysplasia was observed at week 9 of TAA treatment, and microarray analysis of tumor versus non-tumor liver tissue demonstrated differential expression of genes involved in cell proliferation, apoptosis, and cell cycle [30]. Upregulation of stem cell factor and its receptor c-Kit, a proliferative and anti-apoptotic signaling system, has also been reported in animals with TAA-induced CCA [31]. Leptin, a hormone produced by adipose tissue, regulates caloric homeostasis and energy expenditure. Leptin is increased in obesity, which is a risk factor for CCA. Hence, TAA treated fa/fa Zucker rats which have faulty leptin receptors, had reduced tumor burden compared to TAA-treated lean Zucker rats [28]. TAA has also been utilized as an enhancer following DEN induction. However, the TAA and DEN combination tends to induce HCC, and CCA development was noted in only 10% of animals after 8 weeks of treatment [32]. The advantages of the TAA animal models include ease of carcinogen administration without the need for a surgical procedure and good reproducibility in different studies. However, this model has been limited primarily to rats.
Furan
Furan is a chemical compound used in manufacturing of other organic compounds such as herbicides, pharmaceutical compounds, and plastic [33]. Furan was described in 1991 for the first time as a cholangiocarcinogenic compound by the National Toxicology Program. Indeed, chronic furan administration at a high dose of 8 mg/Kg for 15 months induced CCA development in 98% of rats [34]. Moreover, six of the high-dose furan treated rats (n=13) developed distant metastasis in a variety of tissues including pancreas, abdominal blood vessels and adrenal capsule [34]. Higher doses of furan (15-60 mg/kg/per day) for 2-3 weeks result in rapid development of cholangiofibrosis, marked by the presence of bile ductular hyperplasia, intestinal metaplasia, and fibrosis [33]. Bile ductule hyperplasia appeared as early as 1 week following furan treatment initiation and intestinal metaplasia was noted after 2 weeks of treatment [35]. Strongly positive cytokeratin (CK)-19 staining is observed in both the hyperplastic bile ductular epithelial cells and the intestinal-like epithelial cells [33]. Furthermore, in rats with 3 weeks of treatment followed by 6 weeks without treatment, a marked persistent cholangiofibrosis was noted in the caudate liver lobe with the development of dysplastic glands which confirm the progressive nature of these lesions [33]. The investigators postulated that there are two different cell lineages in the histogenesis of cholangiocarcinoma in the furan treated rats; the major one being derived from intestinal like glands that appear during the early steps of cholangiofibrosis development and the less common one being derived from hyperplastic bile ductule-like structures which also develop during cholangiofibrosis [36]. The intestinal-like gland cells seem to have a greater proliferation than the hyperplastic bile ductule cells in the cholangiofibrotic tissue, which would explain the preferential selection for the intestinal-like gland cells [37]. This proliferative advantage could be linked with tyrosine kinase growth factor receptor overexpression in these cells, conferring a selective proliferative advantage over hyperplastic bile ductular epithelium [38]. It has been reported that intestinal metaplasia is associated with CCA development in humans with intestinal cell differentiation being observed in intra and extra-hepatic human CCA [39]. In summary, CCA generated in the furan rat model resembles human CCA and mimics the natural progression of iCCA from chronic bile duct lesions inducing cholangiofibrosis to subsequent CCA development. However, furan can induce other malignancies as well including malignant mesothelioma and mononuclear cell leukemia [40].
CHOLESTATIC MODELS OF CCA
Cholestasis, the retention of bile acids normally excreted into bile within the liver, elicits a toxic response leading to liver injury [41]. Cholestasis is a prominent component of several chronic biliary tract diseases such as primary sclerosing cholangitis, primary biliary cirrhosis, and biliary atresia [41]. Chronic cholestasis promotes CCA carcinogenesis by inducing genetic aberrations and pro-survival signaling pathways. Cholestasis alone or in combination with chemical carcinogens serves as the basis for several animal models of liver injury or CCA. One of the earlier cholestatic CCA models was reported by Thamavit et al. [18]. In this model, cholestasis was achieved via left and medial bile duct ligation (LMBDL). The combination of DMN and LMBDL resulted in CCA formation in approximately 40% of the mice after 40 weeks of treatment [18]. A more recent model utilized the combination of DEN and cholestasis [42]. Following two weekly DEN intraperitoneal injections, chronic cholestasis was induced by LMBDL. One week after LMBDL mice received DEN in corn oil via oral gavage. This combination resulted in CCA formation in 50% (5/10) of the animals by week 28, whereas none of the animals undergoing LMBDL alone or DEN administration alone developed CCA [42]. The advantages of this model include a higher tumor incidence and shorter duration to tumor development compared to prior similar models. However, it does require significant technical skill.
CCA XENOGRAFT AND ALLOGRAFT MODELS
Tumor graft models are commonly used in cancer studies. They permit investigation of novel therapeutic compounds, while remaining easy to use and cost effective. There are multiple types of graft models, the most common being the xenograft model, but many models implore the allograft as well [43].
Xenotransplantation models of CCA
The most common xenograft is a heterotopic graft, which entails xenotransplantation (via subcutaneous flank injection) of human cells or human tumor tissue into immunodeficient or nude mice. The first use of an ectopic CCA xenograft model was described in 1985 by Hudd et al., where cell lines derived from human metastatic CCA tissue were injected into the flank of a mouse to study the effect of a potential therapeutic compound. In this first ectopic CCA xenograft model, 26 of 30 mice developed tumors [44]. Subsequent studies have used cell lines derived from different CCA subtypes to study CCA pathogenesis and various therapies [45–65]. With their reproducibility, feasibility, modest cost, and time effectiveness, xenotransplantation models are useful to assess new drug efficacy and tolerability in vivo. The shortcomings of this model are that the host is immunocompromised and there is a species-mismatch between the tumor and the host microenvironment (Fig. 1).
Figure 1. Xenograft heterotopic models of CCA.
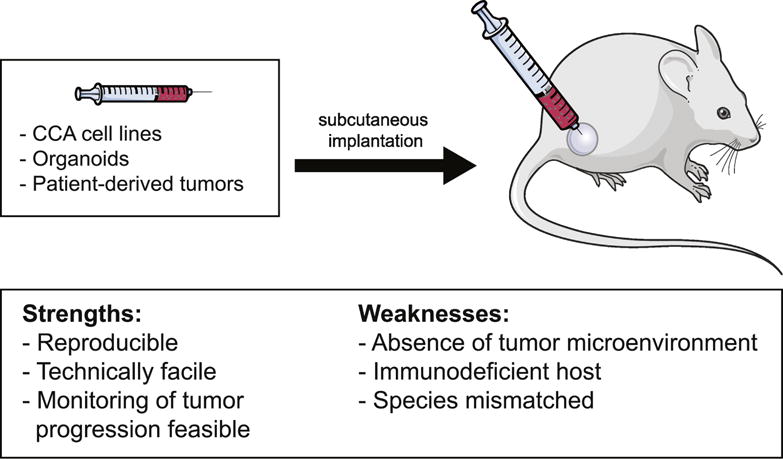
Schematic diagram depicting heterotopic implantation of CCA cell lines, organoids, or patient-derived tumors in mice, and the strengths and weaknesses of this model.
Xenograft models using CCA cells (Table 3)
Table 3.
Xenograft models of CCA using CCA cells.
| Xenotransplantation Model | Study Findings | Study |
|---|---|---|
| Injection of 3×106 Mz-ChA-1 cells +/− GABA intratumoral injection | GABA inhibits CCA growth in vivo | Fava et al., [47] |
| Injection of 3×106 Mz-IL-6 cells | IL-6 overexpression increases CCA xenograft tumor growth | Meng et al., [53] |
| Injection of 2×106 QBC939 cells +/− magnetic nanoparticle injections | Magnetic nanoparticles inhibit CCA tumor growth | Tang et al., [61] |
| Injection of 5×106 Sk-ChA-1 cells +/− intratumoral tamoxifen injection | Intratumoral tamoxifen injections decrease CCA growth | Pawar et al., [57] |
| Injection of 5×106 Mz-ChA-1 cells +/− green tea polyphenol EGCG i.p injection | EGCG decreases CCA tumor growth and increases tumor sensitivity to gemcitabine | Lang et al., [52] |
| Injection of 5×106 Mz-ChA-1 cells expressing Il-6 | Overexpression of Il-6 increases DNMT-1 expression in CCA | Braconi et al., [45] |
| Injection of 5×106 Mz-ChA-1 cells +/− resveratrol i.p injection | Resveratrol decreases Cyp1b1 expression in CCA tumors | Frampton et al., [48] |
| Injection of 5×106 Mz-ChA-1 cells +/− intratumoral anti-NYP antibody injection | NPY decreases CCA tumor growth | DeMorrow et al., [46] |
| Injection of 2×106 QBC939 cells +/− wortmannin i.p injection or +/− AdSi-beclin intratumor injection | Treatment with an autophagy inhibitor or knockdown of beclin-1 suppressed CCA xenograft tumor growth | Hou et al., [49] |
| Injection of M139 CypA-silenced CCA cell lines | CypA silencing decreases CCA xenograft tumor growth | Obchoei et al., [55] |
| Injection of HuCCT1 cell line overexpressing miR494 | miR494 decreases CCA xenograft tumor growth | Olaru et al., [56] |
| Injection of 1,5×106 CCLP1 cells +/− miR26-a overexpression | miR-26a overexpression increases in vivo CCA tumor growth | Zhang et al., [62] |
| Injection of 2×106 HuCCT-1 cells +/− WTAP-over or siWTAP | WTAP overexpression increases CCA tumor growth in an orthotopic xenograft model; WTAP knockdown decreases CCA tumor growth | Jo et al., [51] |
| Injection of 3×106 CCLP1 cells +/− miR92a or miR-17-92 | miR-19a overexpression induces CCA tumor growth in vivo | Zhu et al., [64] |
| Injection of 1×107 QBC939 cells +/− intratumoral rapamycin/salubrinal injection | Salubrinal with rapamycin has a synergistic antitumor effect in CCA | Zhao et al., [63] |
| Injection of 1×106 EGI1 cells +/− 1×107 Lent-SOX17 intratumoral injection | SOX17 overexpression decreases CCA xenograft tumor growth | Merino-Azpitarte et al., [54] |
| Injection of 5×106 HuCCT-1 cells +/−SC-43 gavage administration | SC-43 induces apoptosis in CCA via the SHP-1/STAT3 signaling pathway | Hu et al., [50] |
| Injection of 2×106 RBE cells +/− NRP-1 depletion | NRP- 1 depletion reduces tumor growth and metastasis | Zhu et al., [65] |
| Injection of 3×106 HuCCT-1 cells +/− telmisartan i.p injection | Telmisartan inhibits tumor growth in CCA | Samukawa et al., [58] |
| Injection of 1×107 RBE cells +/− shPTHLHx | Loss of PTHLH suppresses CCA tumorigenesis | Tang et al., [60] |
| Injection of 2×106 TFK-1 or HuCCT-1 cells +/− siAFAP1-AS1 | AFAP1-AS1 is oncogenic in CCA | Shi et al., [59] |
Several studies utilizing CCA xenograft models have investigated novel therapeutic agents for the treatment of CCA. These studies focused on compounds such as γ-aminobutyric acid (GABA) [47], combinations of salubrinal and rapamycin [63], SC-43 (a Sorafenib derivative) [50], telmisartan [58], and JQ1 (bromodomain inhibitor) [66]. Moreover, xenotransplantation of transfected cells can be used to investigate the upstream and downstream regulation of specific pathways [49, 51, 53, 54, 60] and microRNAs (miRs) [56, 62, 64, 65], with the hope of establishing a new anti-tumor treatment. In orthotopic CCA models, CCA cell lines are injected directly in the liver [51, 67]. This permits study of the biological behavior of CCAs within the liver’s physiological environment.
Patient-derived xenograft (PDX) model
There are limits to the effectiveness of xenograft models using continuously passaged cell lines. Such weaknesses include the potential loss of tumor heterogeneity due to selective pressures, loss of tumor representation due to culture specific mutations and gene silencing, and the lack of interaction with the tumor microenvironment and the immune system [66, 68–71]. To improve upon these models, the PDX model (Fig. 1) was developed [66]. Originally developed in the 1980’s to assist in adapting laboratory results to clinical oncology, PDX models are generated from primary human tumors being implanted directly into immunocompromised mice [66, 72–74]. Cavaloni et al. observed that fourth generation iCCA PDXs exhibit the same morphology, histology, and immunohistochemical reactivity that is seen in primary patient tumors. The authors went on to further characterize that fourth generation PDX and primary tumors have highly similar genetic and molecular profiles, highlighting similar stability of gene expression, tissue arrangement, ribonucleic acid (RNA) expression, and mutational similarities [64]. These similarities support that cell heterogeneity, architecture, genetic profile, and histology characteristics are maintained between PDX and primary tumors. They also allow for better understanding of pathway activations, cell interactions, and are excellent predictors of anti-tumor treatments [50, 54, 64, 75–77]. Nevertheless, CCA PDX models are expensive and time-consuming with a long engraftment period and a low rate of efficacy [78].
Xenograft models using CCA organoids
Recent studies have demonstrated that primary CCA can be grown in 3D in vitro culture to obtain organoids, referred to as “tumoroid” (Fig. 1). After long-term in vitro growth culturing, organoids show the same histological architecture and expression profile of the primary human CCA from which they were derived, and have a high engraftment rate with 100% of mice developing tumors [79]. Interestingly, organoid xenografts derived from patients with metastatic CCA generate the same metastatic profile in mice [75, 79]. An important advantage of organoid xenografts is the maintenance of mutational expression after long term culturing [79]. However, these models do not mimic the inflammatory and stromal microenvironment during in vitro or in vivo growth, which limits potential studies investigating cell-cell interactions in the tumor microenvironment [79].
Allograft models of CCA
Syngeneic orthotopic rat model of CCA
The syngeneic orthotopic rat model of CCA was developed by Sirica et al., and uses two rat cell lines, BDEneu and BDEsp. Although both cell lines were derived from the malignant cholangiocyte cell line BDE1, BDEneu exhibits an aggressive, highly malignant profile and CCA progression markers, while the BDEsp line is less aggressive. These cell lines were inoculated into the bile ducts of Fisher 344 rats, and after 21-26 days BDEsp inoculations resulted in small, non-metastatic iCCA, while BDEneu inoculations resulted in rapid tumor growth in all animals accompanied by bile duct obstruction and peritoneal metastasis [80]. This intrahepatic malignant cell implantation can be combined with common bile duct ligation to mimic the biliary obstruction and tumor progression observed in human CCAs [80]. In addition, BDEneu cells have biological and molecular features similar to human CCA, such as expression of tumor necrosis factor related apoptosis inducing ligand (TRAIL) and polo like kinase 2, as well as hedgehog pathway activation [51, 80, 81]. This model overcomes many of the immune and stromal limitations of other xenograft approaches. It is also an excellent model for investigating novel therapeutic agents, and has been used to characterize the effect of sorafenib treatment on CCA progression, where complete tumor regression was observed in 22% of animals [82]. Another study using this model demonstrated a reduction in CCA metastases with JP1584, a second mitochondria-derived activator of caspase mimetic, treatment [81]. Furthermore, a series of studies investigating the TRAIL and hedgehog pathways highlighted the role of the cancer-associated fibroblasts in CCA using this model [81, 83–86]. An organotypic culture model of CCA was established by using iCCA cells isolated from this orthotopic rat model with α-smooth muscle actin (α-SMA) positive cancer-associated fibroblasts [87]. This permits the in vitro study of interaction between key stromal cells and malignant CCA cells [87]. The advantages of the syngeneic orthotopic rat model include the presence of a tumor microenvironment and an immunocompetent host. This model can also be combined with bile duct ligation with resultant cholestasis, hence giving rise to CCA in a background of liver injury. However, the model does entail abdominal manipulation and consequent surgical risk to the animals (Fig. 2). Another limitation of this model is the absence of de novo CCA development as tumors arise following implantation of malignant cells. Moreover, as this is a rat model there are limited reagents available.
Figure 2. Syngeneic orthotopic models of CCA.
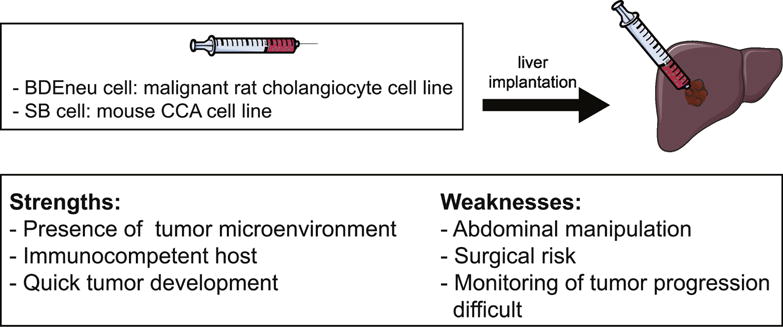
Schematic diagram depicting implantation of malignant rat or mouse CCA cells into mouse livers, and the strengths and weaknesses of this model.
Syngeneic orthotopic mouse model of CCA
A syngeneic orthotopic mouse model would overcome the disadvantage of limited reagents that the rat model poses. Such a model was recently described [88]; seven malignant mouse cell lines (SB1-7) were established from tumor nodules derived from a genetic transposon-based model of CCA (described below) [89]. All seven mouse cell lines can be implanted into the medial lobe of mouse livers with consequent orthotopic tumor formation [88]. The resulting tumors mimic histopathologic characteristics of human CCA including desmoplasia, malignant glands, as well as expression of CK-19 and SRY box 9 (SOX9) [88]. This is a unique mouse model as it allows genetic manipulation of cells prior to implantation. This model has the potential to be a valuable tool which can be utilized to enhance our understanding of CCA tumor-stroma interactions, pathogenesis, and therapeutics.
GENETIC MODELS OF CCA
Genetically engineered models (GEMs) of CCA include transgenic models and transduction models. Genetic models are an important tool in cancer investigation for elucidating complex biologic pathways, molecular processes, as well as for studying the role of oncogenes and tumor suppressor genes. Indeed, the possibility to generate animal models which have the potential to recapitulate specific genetic mutation as well as the biochemical, proteomic and phenotypic features observed in human CCA has enabled important progress in the understanding of CCA cancer. In these models, tumors develop spontaneously in situ in an immunocompetent organism with the appropriate tumor microenvironment. Before the development of Crispr/Cas9 system [90], the most commonly used systems to develop these models were Cre-Lox, tetracycline-dependent promoter regulation and FLP-mediated site-specific and spontaneous recombination methods [91–93]. Although genetic models can be quite informative in terms of elucidating mechanisms, they are not very time efficient and can be technically challenging and expensive. Moreover, these models may have uncontrolled pattern expression of the transgenes and their random integration can induce unexpected results [94].
Transgenic models of CCA
Mothers against decapentaplegic homolog 4 (Smad4)-Phosphatase and tensin homolog deleted on chromosome 10 (Pten) conditional knockout model
The Smad4-Pten model, with conditional disruption of both genes using Cre-loxP recombination, was developed in 2006 by Xu et al. [95]. Smad4 is one of the most frequently mutated tumor suppressor genes in human CCA. It is a protein activated in response to transforming growth factor-β (TGF-β) and is involved in the G1-S cell cycle arrest [96]. Smad4 gene expression is essential for the development of embryonic mice and Smad4 loss is embryonically lethal [97]. Therefore, to study the impact of Smad4 deletion, the investigators utilized a mouse strain carrying a conditional allele of Smad4 (Smad4Co/Co) and a tissue specific Cre expression system using albumin to induce liver-specific Smad4 deletion. Pten deletion induces constitutive activation of the PI3K/AKT pathway and extracellular signal regulated protein kinases 1/2 (ERK1/2) hyperphosphorylation, which has an essential role in cell proliferation and survival [98]. Pten confers a tumor suppressive effect via induction of G1-S cell cycle arrest [99]. To investigate the effect of liver-specific Smad4 and Pten deletion, the authors crossed Smad4Co/Co mice or mice carrying a Pten conditional allele (Ptenco) with a mouse strain expressing Cre-recombinase under the control of albumin regulatory elements (Alb-Cre). It should be noted that albumin-Cre is expressed in both hepatocytes and cholangiocytes during embryogenesis. Smad4 and Pten deletions were observed in both hepatocytes and cholangiocytes of the Smad4Co/Co/PtenCo/Co Alb-Cre phenotype. Consistently, Smad4 and Pten deletion resulted in bile duct hyperplasia at 2 months of age in these mice, and all animals had tumor development by 4-7 months of age. Histopathological analysis demonstrated sequential progression from bile ducts dysplasia to multifocal and invasive CCA. Molecular analyses of this model demonstrated increased levels of phosphorylated AKT and the downstream targets of the PTEN/PI3K/AKT pathways including mammalian target of rapamycin (mTOR) and glycogen synthase kinase-3β. Increased nuclear levels of cyclin D1 and ERK, which plays an important role in induction of cyclin D1 expression, were also observed. As AKT is activated in a majority of human CCAs, this model may mimic human disease. However, the authors did note that this might not be a pure CCA model as an earlier study had demonstrated that liver-specific disruption of Pten results in HCC in the majority of mice at 74-78 weeks of age [100]. Hence, if the Smad4Co/Co/PtenCo/Co Alb-Cre mice survived longer, they may have both CCA and HCC. In addition, unlike human CCA, the tumors in this model develop in the absence of a background of chronic liver injury and inflammation. Additionally, no metastases were noted in these mice.
Kras–Pten model
Mutations activating the proto-oncogene KRAS are some of the most frequent occurrences in human malignancies, including distal CCA; KRAS mutations are less frequent in iCCA. Activation of KRAS leads to upregulation of downstream pathways including the PI3k/AKT/mTOR pathway and the Raf/MEK/ERK pathways, and is implicated in a myriad of biological processes including cell proliferation, differentiation, and survival [101]. Accordingly, liver-specific Kras activation and Pten deletion promotes iCCA in mice [102]. Using the Cre-loxP system, Ikenoue et al. introduced an activating mutation of Kras (LSL-KrasG12D) plus deletion of Pten (Ptenflox) specifically in hepatocytes and hepatoblasts by crossing mice carrying a LSL-KrasG12D allele and/or a Ptenflox with Alb-Cre+ mice. The Alb-Cre+/LSL-KrasG12D/Ptenflox mice were referred to as AKPP mice. At 5 weeks of age, all of the AKPP mice developed abdominal distension secondary to hepatic enlargement and hemorrhagic ascites as well as jaundice and weight loss, mimicking symptoms frequently observed in human iCCA. The median survival of AKPP mice was 46 days and at autopsy these mice had multiple tumor nodules with a glandular morphology and an abundant desmoplastic stroma closely resembling well-differentiated iCCA. The mice had no evidence of metastatic disease. Immunohistochemical analyses demonstrated that AKPP mice develop iCCA exclusively whereas Alb-Cre+/LSL-KrasG12D/+/Ptenflox/+ mice which had deletion of only one Pten allele developed hepatocellular dysplasia in the majority of nodules with some nodules resembling iCCA. This is similar to several other genetically engineered mouse models which have features of HCC as well as CCA [95, 103, 104]. Mice without Pten deletion developed hepatocellular dysplasia but no features of CCA, suggesting that Pten may play a role in fate determination of hepatotumorigenesis. As activating Kras gene mutations and PI3K pathway alterations are frequently observed in human iCCA [105–107], this model recapitulates a subset of human iCCAs. Other advantages include relatively short duration to tumor development without the need for highly technical skills. However, this model does occur in the absence of any inflammation or chronic liver injury, frequent antecedent events in CCA. Additionally, no metastases were noted in these mice.
Kras-isocitrate dehydrogenase (IDH) model
Gain of function mutations of IDH1 and IDH2 occur in approximately 25% of iCCAs [108–110]. Mutant IDH blocks hepatocyte differentiation from progenitor cells via suppression of hepatocyte nuclear factor-4α, a transcriptional regulator of hepatocyte differentiation [111]. Intercrossing of mutant Idh2 (LSL-IDH2R172K), mice with activating Kras mutations, (LSL-KrasG12), and Alb-Cre+ mice resulted in palpable liver tumors in six out of six IDH2R172K/KrasG12D mice at 33-58 weeks of age [111]. Histopathologic analysis of these tumors confirmed iCCA with positive CK19 staining. Mice had multifocal liver masses as well as peritoneal metastases. In contrast, only one out of seven KRASG12D mice developed tumor nodules, and only HCC was detected in these nodules. As KRAS and IDH mutations occur frequently in human CCA, this genetically engineered model recapitulates a subset of human iCCAs. However, the time to tumor development was prolonged in these mice.
KrasG12D–p53L/L model
This model was established by intercrossing Alb-Cre mutants with KrasG12D mice with or without deletion of tumor protein 53 (TP53), a tumor suppressor gene frequently mutated in human cancer [104]. p53 is a nuclear DNA-binding phosphoprotein which can act as a transcriptional activator or repressor and is involved in the control of cellular response to DNA damage by cell cycle arrest or apoptosis [112]. Mice with the following genotypes were obtained, Alb-Cre/KrasG12D, Alb-Cre/KrasG12D/p53L/L, and Alb-Cre/KrasG12D/p53L/+. Mice with homozygous p53 deletion (Alb-Cre/KrasG12D/p53L/L) developed tumors at 9 weeks of age and had a mean survival of 19 weeks. Many of these mice had cystic fluid lesions, hemorrhagic ascites as well as evidence of tumor necrosis. Moreover, 75% of tumors displayed adjacent organ invasion or distant metastasis. Histopathological analysis revealed that overall 83% of the liver tumors had histological features of iCCAs (66% CCA exclusively, 17% mixed CCA-HCC phenotype, 17% HCC) [104]. Characterization of the molecular features demonstrated that the Alb-Cre/KrasG12D/p53L/L mice have activation of the MAPK/MEK and PI3K/AKT pathways, similar to a subset of human CCAs [107]. This model combines two of the most common genetic alterations observed in CCA. Moreover, malignant progression of precursor lesions in bile ducts noted in these animals is similar to human CCA. Notably, unlike several other genetic models, organ invasion or distal metastasis were present in this model. Nevertheless, this model does not reproduce the background of chronic liver injury nor the inflammatory and stromal microenvironment that is present in human CCA. Additionally, this is another model which is not exclusively a CCA model as 17% of the liver tumors in the Alb-Cre/KrasG12D/p53L/L were consistent with HCC.
p53−/−–carbon tetrachloride (CCL4) model
A frequent disadvantage of GEM CCA models is the absence of a background of chronic liver injury with fibrosis and inflammation during CCA development, as this microenvironment plays a major role in the aggressiveness of this malignancy in humans [113]. To address this issue, Farazi et al. treated mice harboring a p53 deletion with CCL4 [114]. CCL4 is a compound that used to induce liver fibrosis [115]. The investigators administrated CCL4 three times per week for four months to p53−/− mice. As expected, mice developed fibrosis with cholangiocyte proliferation. However, cholangiocyte apoptosis was observed only in the mice without a p53 deletion (p53+/+) and mice with deletion of only one p53 allele (p53+/−). Moreover, malignant cells were observed only in the p53−/− mice, appearing shortly after the end of CCL4 treatment. This model demonstrates that the loss of p53 expression combined with cholangiocyte hyperplasia induced by chronic injury is involved in the early phases of CCA carcinogenesis. CCA was detected in 54% of mice with homozygous p53 deletion and only 18% of mice with heterozygous p53 deletion, with a shorter tumor latency being observed in the p53−/− (29 weeks) compared with p53+/− mice (52 weeks). Moreover, 14% of the p53−/− mice developed metastasis. These findings support an important role for p53 deletion in CCA development. The toxic chronic liver injury associated with the genetic abnormality in this model reproduces the human CCA molecular profile with c-met activation, as well as COX-2 and cErbB2 overexpression. Moreover, the presence of chronic liver injury inducing fibrosis and inflammation is the strength of this model [113]. The limits of this model reside in the development of HCC as well as CCA and a relatively long duration of CCL4 treatment (4 months).
ErbB-2A model
ERBB2 is a receptor tyrosine kinase which induces RAS-ERK and PI3K-AKt pathway activation, hence playing a role in cell survival, proliferation and migration [116]. ERBB2 is frequently overexpressed in human CCA [116]. The expression of murine erbB2 was targeted to epithelial cells using the Bovine Keratin 5 (BK5) promoter in mice with constitutive expression of ErbB2 (ErbB-2A transgenic mice) [117]. Gallbladder adenocarcinoma was observed in 85% of these mice, appearing as early as 2-3 weeks of age. Mice greater than 4 months of age developed tumors in the adjacent biliary tract including the common bile duct and intrahepatic bile duct with an incidence of 87% and 30%, respectively. Analyses of the transgene expression showed that both the gallbladder adenocarcinoma and CCA expressed the transgene. The BK-ErbB-2A mice had increased COX-2 levels and MAPK pathway activation, similar to human CCA. The main disadvantage of this model is that it is primarily a model of gallbladder adenocarcinoma rather than CCA. Moreover, CCA tumor development required a longer duration as it was noted only in mice older than 4 months of age.
Notch1 model
Notch is a major protein regulator of mammalian hepatic cell fate. The Notch signaling pathway has been implicated in the development and proliferation of the biliary tree during the embryonic period [118]. Accordingly, aberrations in the pathway are associated with CCA carcinogenesis [119]. Zender et al. developed a transgenic mouse model with constitutive Notch expression in liver tissue. A transgenic mouse with tissue-specific overexpression of the intracellular domain of Notch 1 (NICD) (Rosa26Notch1C) was crossed with Alb-Cre mice for liver-specific expression [119]. Epithelial cells with features of hepatocyte and cholangiocyte differentiation were noted in livers of mice at 7 months of age. Changes in nuclear morphology were observed in primarily liver tissues from the Notch1C:AlbCre mice at 8 months of age. Xenotransplantation of these altered cells into flanks of immunodeficient mice resulted in formation of tumors with histopathologic features of human iCCAs including a desmoplastic stroma and CK7 and CK17 expression. These findings suggest that NICD expression in hepatic progenitor cells promotes differentiation of these cells towards a biliary lineage with prolonged NICD expression contributing to malignant transformation. However, given the high plasticity of these cells, the authors did not exclude that tumors have features of HCC or mixed CCA-HCC phenotype. The investigators also demonstrated that Notch induces cyclin E promoter activation leading to cyclin E expression and DNA instability, features found in human CCA [120–122].
Transduction models of CCA
NICD-AKT Model
In a subsequent study, Fan et al. further investigated the ability of the Notch pathway to convert hepatocytes into CCA cells. NOTCH signaling in the liver was activated via stable overexpression of NICD and plasmids for sleeping beauty transposase-mediated transgene integration to livers of mice were delivered by hydrodynamic tail vein injection with resultant tumor development in 100% of mice [123]. By 20 weeks after injection, cystic cholangiocellular tumors resembling human biliary cystadenomas were observed. As AKT overexpression is a frequent occurrence in human CCA, the investigators next injected the NICD plasmid with an AKT plasmid using hydrodynamic tail vein injection. At 3.5 week following injection, these mice developed cyst-like lesion on the liver surface. However, microscopic analyses revealed that malignant cells were detected as early as 1.5 weeks after injection. The lesions continued to grow and replaced most of the liver tissue by 5 weeks following injection. Additional signs of malignancy such as necrosis, strong mitotic activity and tissue invasion (features found in human iCCA) were also observed. These findings suggest that NOTCH and AKT signaling pathways collaborate to promote hepatocytes transformation to iCCA.
AKT/YAP model
A subsequent murine oncogene transduction model featured in situ tumor development from the biliary tract using biliary instillation of oncogenes. Mouse Akt and human yes-associated protein (YAP) were the oncogenes used to transduce the biliary epithelium. YAP is a transcriptional co-activator which has been implicated in liver carcinogenesis [124, 125]. Mouse constitutively active, myristoylated Akt and human YAP were instilled in the biliary tree of wild type C57BL/6 mice using a surgical approach (Fig. 3). During the surgery, the biliary duct draining the left lobe of the liver was ligated to allow retention of the transduction oncogenes within the lobe. For three consecutive days postoperatively, animals received intraperitoneal injections of interleukin-33 (IL-33). IL-33 is a potent biliary mitogen which promotes cholangiocyte proliferation [126] and has been implicated in liver fibrosis and inflammation [127]. Tumor development was noted beginning at week 6. Ten weeks following the surgery, tumors were noted in 72% of mice which received transduction of both oncogenes and systemic IL-33 administration. In contrast, tumors were present in only 20% of mice which were transduced with both oncogenes but did not receive systemic IL-33 indicating that IL-33 is essential for tumor development [89]. Morphologic and phenotypic analysis indicated that these tumors recapitulate human CCA with features such as hyperplasia of irregular glands, pancytokeratin and SOX9 expression, abundant α-SMA positive myofibroblasts and a common profile of gene expression between human CCA and this model. Mechanistic studies demonstrated that IL-33 enhances IL-6 expression in human cholangiocytes and facilitates tumor development by an IL-6 sensitive process. This is a robust model which is time efficient and CCA arises in situ from the biliary epithelium. The model recapitulates many features of the human disease including a desmoplastic stroma and alterations in signaling pathways known to be implicated in CCA genesis. However, the model does require considerable technical skill. Nonetheless, it should serve as an invaluable tool for the study of novel therapeutic agents for the treatment of this lethal cancer.
Figure 3. AKT/YAP BDL model.
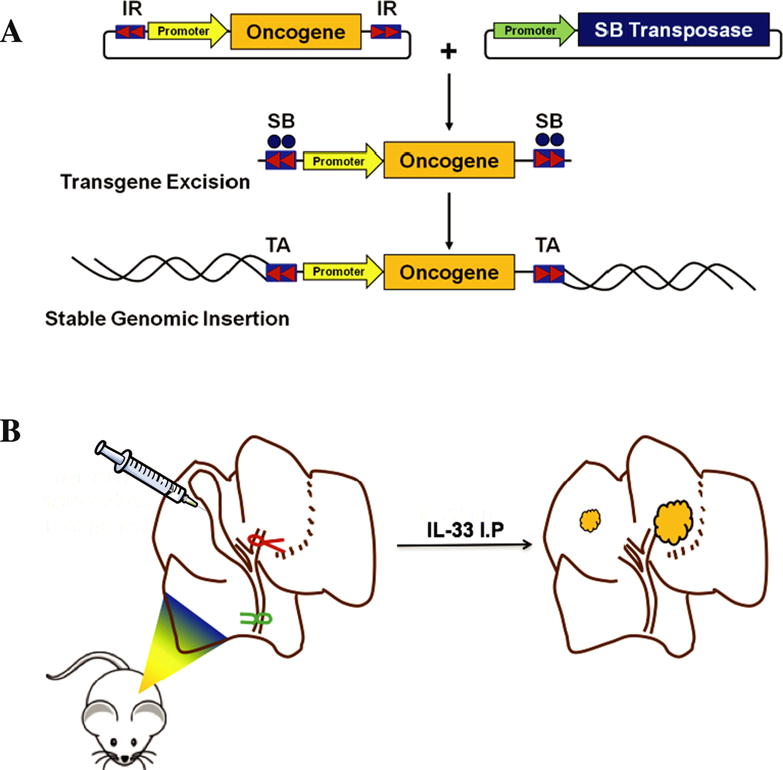
(A) Schematic diagram depicting a transposon-transposase transduction system. The transposon carrying the oncogene is cotransfected with the transposase. When transduced into the same cell, the transposase excises the target oncogene, allowing its incorporation into the host cells genome. (B) Schematic diagram depicting the surgical approach utilized in the AKT-YAP biliary transduction model [89].
CONCLUSION AND FUTURE DIRECTIONS
Animal models are essential tools in cancer research that permit the study of cancer biology and novel therapeutic agents. Historically, animal models of CCA were carcinogen based or xenograft models. Carcinogen based models are not specific for CCA development as various carcinogens can give rise to other tumor types. Xenograft models with xenotransplantation of CCA cell lines develop in a species mismatched immunodeficient host. A syngeneic orthotopic transplantation model in rats overcame these shortcomings. However, the primary limitation of this model was that there are fewer reagents for rats than there are for mice. There are several transgenic mouse models which can be quite informative in terms of elucidating mechanisms. However, these models are not very time efficient, can be technically challenging and expensive. In a recently developed transduction model in which oncogenes (AKT/YAP) are instilled directly into the biliary tree [89], tumors arise from the biliary tract in immunocompetent hosts with species-matched tumor microenvironment. This model is time efficient and closely mimics features of human CCA. Malignant cell lines derived from these tumors can in turn be implanted into mice resulting in a unique syngeneic orthotopic mouse model of CCA that overcomes the shortcomings of the orthotopic rat model. The vast majority of available CCA models are those of iCCA. PDX models can allow the study of pCCA/dCCA, however, there remains the issue of species mismatch as these are xenografts. As the majority of CCAs are pCCA/dCCA there is a need to develop specific animal models for these subtypes.
Animal Models of Cholangiocarcinoma Highlights.
Animal models of CCA are essential to understand the human disease
Ideal model would be in immunocompetent host, time-efficient, and mimic human CCA
Transgenic CCA models help elucidate mechanisms
Vast majority of available CCA models are those of iCCA
PDX models can allow study of pCCA/dCCA but are in immunocompromised host
Abbreviations
- α-SMA
α-smooth muscle actin
- AFAP1-AS1
actin filament associated protein 1 antisense RNA1
- BK5
bovine keratin 5
- CCA
cholangiocarcinoma
- CCL4
carbon tetrachloride
- CK
cytokeratin
- COX-2
cyclooxygenase-2
- CYLD
cylindromatosis
- Cyp1b1
cytochrome p450 1b1
- CypA
Cyclophilin A
- dCCA
distal cholangiocarcinoma
- DEN
diethylnitrosamine
- DMN
dimethylnitrosamine
- DNA
deoxyribonucleotide acid
- DNMT-1
DNA methyltransferase-1
- EGCG
epigallocatechin-gallate
- ERK1/2
extracellular signal regulated protein kinases 1/2
- GABA
γ-aminobutyric acid
- GEM
genetically engineered mouse
- HCC
hepatocellular carcinoma
- iCCA
intrahepatic cholangiocarcinoma
- IDH
isocitrate dehydrogenase
- IL
interleukin
- KRAS
kristen rat sarcoma virus
- LMBDL
left and median bile duct ligation
- MAPK
mitogen activated protein kinase
- miR
microRNA
- mTOR
mechanistic target of rapamycin
- NICD
Notch intracellular domain
- Notch1
notch homolog 1
- NPY
neuropeptide Y
- NRP-1
neuropilin-1
- O. viverrini
Opisthorchis viverrini
- pCCA
perihilar cholangiocarcinoma
- PDX
patient-derived xenograft
- PI3K
phosphoinositide 3 kinase
- Pten
phosphatase and tensin homolog
- PTHLH
parathyroid hormone-like hormone
- RNA
ribonucleic acid
- Smad4
mothers against decapentaplegic homolog 4
- SOX9
SRY box 9
- TAA
thioacetamide
- TGF-β
tumor growth factor-β
- TP53
tumor protein 53
- TRAIL
TNF related apoptosis inducing ligand
- WTAP
Wilms’ tumor1-associating protein
- YAP
yes associated protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors have nothing to disclose.
Conflict of Interest Form
We will complete off line and submit.
References
- 1.Welzel TM, McGlynn KA, Hsing AW, O’Brien TR, Pfeiffer RM. Impact of classification of hilar cholangiocarcinomas (Klatskin tumors) on the incidence of intra- and extrahepatic cholangiocarcinoma in the United States. J Natl Cancer Inst. 2006;98:873–875. doi: 10.1093/jnci/djj234. [DOI] [PubMed] [Google Scholar]
- 2.Blechacz B, Komuta M, Roskams T, Gores GJ. Clinical diagnosis and staging of cholangiocarcinoma. Nat Rev Gastroenterol Hepatol. 2011;8:512–522. doi: 10.1038/nrgastro.2011.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nakeeb A, Pitt HA, Sohn TA, Coleman J, Abrams RA, Piantadosi S, Hruban RH, Lillemoe KD, Yeo CJ, Cameron JL. Cholangiocarcinoma. A spectrum of intrahepatic, perihilar, and distal tumors. Ann Surg. 1996;224:463–473. doi: 10.1097/00000658-199610000-00005. discussion 473-465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DeOliveira ML, Cunningham SC, Cameron JL, Kamangar F, Winter JM, Lillemoe KD, Choti MA, Yeo CJ, Schulick RD. Cholangiocarcinoma: thirty-one-year experience with 564 patients at a single institution. Ann Surg. 2007;245:755–762. doi: 10.1097/01.sla.0000251366.62632.d3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rizvi S, Khan SA, Hallemeier CL, Kelley RK, Gores GJ. Cholangiocarcinoma - evolving concepts and therapeutic strategies. Nat Rev Clin Oncol. 2017 doi: 10.1038/nrclinonc.2017.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Everhart JE, Ruhl CE. Burden of digestive diseases in the United States Part III: Liver, biliary tract, and pancreas. Gastroenterology. 2009;136:1134–1144. doi: 10.1053/j.gastro.2009.02.038. [DOI] [PubMed] [Google Scholar]
- 7.Khan SA, Davidson BR, Goldin RD, Heaton N, Karani J, Pereira SP, Rosenberg WM, Tait P, Taylor-Robinson SD, Thillainayagam AV, Thomas HC, Wasan H, G. British Society of Guidelines for the diagnosis and treatment of cholangiocarcinoma: an update. Gut. 2012;61:1657–1669. doi: 10.1136/gutjnl-2011-301748. [DOI] [PubMed] [Google Scholar]
- 8.Weigt J, Malfertheiner P. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. Expert Rev Gastroenterol Hepatol. 2010;4:395–397. doi: 10.1586/egh.10.45. [DOI] [PubMed] [Google Scholar]
- 9.Cekanova M, Rathore K. Animal models and therapeutic molecular targets of cancer: utility and limitations. Drug Des Devel Ther. 2014;8:1911–1921. doi: 10.2147/DDDT.S49584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sripa B, Mairiang E, Thinkhamrop B, Laha T, Kaewkes S, Sithithaworn P, Tessana S, Loukas A, Brindley PJ, Bethony JM. Advanced periductal fibrosis from infection with the carcinogenic human liver fluke Opisthorchis viverrini correlates with elevated levels of interleukin-6. Hepatology. 2009;50:1273–1281. doi: 10.1002/hep.23134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koop DR. Oxidative and reductive metabolism by cytochrome P450 2E1. FASEB J. 1992;6:724–730. doi: 10.1096/fasebj.6.2.1537462. [DOI] [PubMed] [Google Scholar]
- 12.Yang CS, Smith T, Ishizaki H, Hong JY. Enzyme mechanisms in the metabolism of nitrosamines. IARC Sci Publ. 1991:265–274. [PubMed] [Google Scholar]
- 13.Henderson JM, Zhang HE, Polak N, Gorrell MD. Hepatocellular carcinoma: Mouse models and the potential roles of proteases. Cancer Lett. 2017;387:106–113. doi: 10.1016/j.canlet.2016.03.047. [DOI] [PubMed] [Google Scholar]
- 14.Thamavit W, Bhamarapravati N, Sahaphong S, Vajrasthira S, Angsubhakorn S. Effects of dimethylnitrosamine on induction of cholangiocarcinoma in Opisthorchis viverrini-infected Syrian golden hamsters. Cancer Res. 1978;38:4634–4639. [PubMed] [Google Scholar]
- 15.Flavell DJ, Lucas SB. Promotion of N-nitrosodimethylamine-initiated bile duct carcinogenesis in the hamster by the human liver fluke, Opisthorchis viverrini. Carcinogenesis. 1983;4:927–930. doi: 10.1093/carcin/4.7.927. [DOI] [PubMed] [Google Scholar]
- 16.Thamavit W, Pairojkul C, Tiwawech D, Shirai T, Ito N. Strong promoting effect of Opisthorchis viverrini infection on dimethylnitrosamine-initiated hamster liver. Cancer Lett. 1994;78:121–125. doi: 10.1016/0304-3835(94)90040-x. [DOI] [PubMed] [Google Scholar]
- 17.Lee JH, Rim HJ, Sell S. Heterogeneity of the “oval-cell” response in the hamster liver during cholangiocarcinogenesis following Clonorchis sinensis infection and dimethylnitrosamine treatment. J Hepatol. 1997;26:1313–1323. doi: 10.1016/s0168-8278(97)80467-9. [DOI] [PubMed] [Google Scholar]
- 18.Thamavit W, Pairojkul C, Tiwawech D, Itoh M, Shirai T, Ito N. Promotion of cholangiocarcinogenesis in the hamster liver by bile duct ligation after dimethylnitrosamine initiation. Carcinogenesis. 1993;14:2415–2417. doi: 10.1093/carcin/14.11.2415. [DOI] [PubMed] [Google Scholar]
- 19.Heindryckx F, Colle I, Van Vlierberghe H. Experimental mouse models for hepatocellular carcinoma research. Int J Exp Pathol. 2009;90:367–386. doi: 10.1111/j.1365-2613.2009.00656.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Umemura T, Kai S, Hasegawa R, Kanki K, Kitamura Y, Nishikawa A, Hirose M. Prevention of dual promoting effects of pentachlorophenol, an environmental pollutant, on diethylnitrosamine-induced hepato- and cholangiocarcinogenesis in mice by green tea infusion. Carcinogenesis. 2003;24:1105–1109. doi: 10.1093/carcin/bgg053. [DOI] [PubMed] [Google Scholar]
- 21.Umemura T, Kodama Y, Kanki K, Iatropoulos MJ, Nishikawa A, Hirose M, Williams GM. Pentachlorophenol (but not phenobarbital) promotes intrahepatic biliary cysts induced by diethylnitrosamine to cholangio cystic neoplasms in B6C3F1 mice possibly due to oxidative stress. Toxicol Pathol. 2003;31:10–13. doi: 10.1080/01926230390173806. [DOI] [PubMed] [Google Scholar]
- 22.Urbanik T, Boger RJ, Longerich T, Becker K, Ehrenberg KR, Hovelmeyer N, Hahn M, Schuchmann M, Jager D, Waisman A, Worns MA, Schulze-Bergkamen H. Liver specific deletion of CYLDexon7/8 induces severe biliary damage, fibrosis and increases hepatocarcinogenesis in mice. J Hepatol. 2012;57:995–1003. doi: 10.1016/j.jhep.2012.06.017. [DOI] [PubMed] [Google Scholar]
- 23.Yang MC, Chang CP, Lei HY. Induction of liver fibrosis in a murine hepatoma model by thioacetamide is associated with enhanced tumor growth and suppressed antitumor immunity. Lab Invest. 2010;90:1782–1793. doi: 10.1038/labinvest.2010.139. [DOI] [PubMed] [Google Scholar]
- 24.Barker EA, Smuckler EA. Altered microsome function during acute thioacetamide poisoning. Mol Pharmacol. 1972;8:318–326. [PubMed] [Google Scholar]
- 25.Hajovsky H, Hu G, Koen Y, Sarma D, Cui W, Moore DS, Staudinger JL, Hanzlik RP. Metabolism and toxicity of thioacetamide and thioacetamide S-oxide in rat hepatocytes. Chem Res Toxicol. 2012;25:1955–1963. doi: 10.1021/tx3002719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Praet MM, Roels HJ. Histogenesis of cholangiomas and cholangiocarcinomas in thioacetamide fed rats. Exp Pathol. 1984;26:3–14. doi: 10.1016/s0232-1513(84)80063-8. [DOI] [PubMed] [Google Scholar]
- 27.Al-Bader A, Mathew TC, Abul H, Al-Sayer H, Singal PK, Dashti HM. Cholangiocarcinoma and liver cirrhosis in relation to changes due to thioacetamide. Mol Cell Biochem. 2000;208:1–10. doi: 10.1023/a:1007082515548. [DOI] [PubMed] [Google Scholar]
- 28.Fava G, Alpini G, Rychlicki C, Saccomanno S, DeMorrow S, Trozzi L, Candelaresi C, Venter J, Di Sario A, Marzioni M, Bearzi I, Glaser S, Alvaro D, Marucci L, Francis H, Svegliati-Baroni G, Benedetti A. Leptin enhances cholangiocarcinoma cell growth. Cancer Res. 2008;68:6752–6761. doi: 10.1158/0008-5472.CAN-07-6682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jan YY, Yeh TS, Yeh JN, Yang HR, Chen MF. Expression of epidermal growth factor receptor, apomucins, matrix metalloproteinases, and p53 in rat and human cholangiocarcinoma: appraisal of an animal model of cholangiocarcinoma. Ann Surg. 2004;240:89–94. doi: 10.1097/01.sla.0000129492.95311.f2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yeh CN, Maitra A, Lee KF, Jan YY, Chen MF. Thioacetamide-induced intestinal-type cholangiocarcinoma in rat: an animal model recapitulating the multi-stage progression of human cholangiocarcinoma. Carcinogenesis. 2004;25:631–636. doi: 10.1093/carcin/bgh037. [DOI] [PubMed] [Google Scholar]
- 31.Mansuroglu T, Ramadori P, Dudas J, Malik I, Hammerich K, Fuzesi L, Ramadori G. Expression of stem cell factor and its receptor c-Kit during the development of intrahepatic cholangiocarcinoma. Lab Invest. 2009;89:562–574. doi: 10.1038/labinvest.2009.15. [DOI] [PubMed] [Google Scholar]
- 32.Romualdo GR, Grassi TF, Goto RL, Tablas MB, Bidinotto LT, Fernandes AAH, Cogliati B, Barbisan LF. An integrative analysis of chemically-induced cirrhosis-associated hepatocarcinogenesis: Histological, biochemical and molecular features. Toxicol Lett. 2017;281:84–94. doi: 10.1016/j.toxlet.2017.09.015. [DOI] [PubMed] [Google Scholar]
- 33.Elmore LW, Sirica AE. Phenotypic characterization of metaplastic intestinal glands and ductular hepatocytes in cholangiofibrotic lesions rapidly induced in the caudate liver lobe of rats treated with furan. Cancer Res. 1991;51:5752–5759. [PubMed] [Google Scholar]
- 34.Maronpot RR, Giles HD, Dykes DJ, Irwin RD. Furan-induced hepatic cholangiocarcinomas in Fischer 344 rats. Toxicol Pathol. 1991;19:561–570. doi: 10.1177/019262339101900401. [DOI] [PubMed] [Google Scholar]
- 35.Elmore LW, Sirica AE. Sequential appearance of intestinal mucosal cell types in the right and caudate liver lobes of furan-treated rats. Hepatology. 1992;16:1220–1226. [PubMed] [Google Scholar]
- 36.Elmore LW, Sirica AE. “Intestinal-type” of adenocarcinoma preferentially induced in right/caudate liver lobes of rats treated with furan. Cancer Res. 1993;53:254–259. [PubMed] [Google Scholar]
- 37.Sirica AE. Biliary proliferation and adaptation in furan-induced rat liver injury and carcinogenesis. Toxicol Pathol. 1996;24:90–99. doi: 10.1177/019262339602400113. [DOI] [PubMed] [Google Scholar]
- 38.Radaeva S, Ferreira-Gonzalez A, Sirica AE. Overexpression of C-NEU and C-MET during rat liver cholangiocarcinogenesis: A link between biliary intestinal metaplasia and mucin-producing cholangiocarcinoma. Hepatology. 1999;29:1453–1462. doi: 10.1002/hep.510290524. [DOI] [PubMed] [Google Scholar]
- 39.Kozuka S, Kurashina M, Tsubone M, Hachisuka K, Yasui A. Significance of intestinal metaplasia for the evolution of cancer in the biliary tract. Cancer. 1984;54:2277–2285. doi: 10.1002/1097-0142(19841115)54:10<2277::aid-cncr2820541037>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 40.Von Tungeln LS, Walker NJ, Olson GR, Mendoza MC, Felton RP, Thorn BT, Marques MM, Pogribny IP, Doerge DR, Beland FA. Low dose assessment of the carcinogenicity of furan in male F344/N Nctr rats in a 2-year gavage study. Food Chem Toxicol. 2017;99:170–181. doi: 10.1016/j.fct.2016.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kahraman A, Barreyro FJ, Bronk SF, Werneburg NW, Mott JL, Akazawa Y, Masuoka HC, Howe CL, Gores GJ. TRAIL mediates liver injury by the innate immune system in the bile duct-ligated mouse. Hepatology. 2008;47:1317–1330. doi: 10.1002/hep.22136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang H, Li TW, Peng J, Tang X, Ko KS, Xia M, Aller MA. A mouse model of cholestasis-associated cholangiocarcinoma and transcription factors involved in progression. Gastroenterology. 2011;141:378–388. 388 e371–374. doi: 10.1053/j.gastro.2011.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ruggeri BA, Camp F, Miknyoczki S. Animal models of disease: pre-clinical animal models of cancer and their applications and utility in drug discovery. Biochem Pharmacol. 2014;87:150–161. doi: 10.1016/j.bcp.2013.06.020. [DOI] [PubMed] [Google Scholar]
- 44.Hudd C, Euhus DM, LaRegina MC, Herbold DR, Palmer DC, Johnson FE. Effect of cholecystokinin on human cholangiocarcinoma xenografted into nude mice. Cancer Res. 1985;45:1372–1377. [PubMed] [Google Scholar]
- 45.Braconi C, Huang N, Patel T. MicroRNA-dependent regulation of DNA methyltransferase-1 and tumor suppressor gene expression by interleukin-6 in human malignant cholangiocytes. Hepatology. 2010;51:881–890. doi: 10.1002/hep.23381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.DeMorrow S, Onori P, Venter J, Invernizzi P, Frampton G, White M, Franchitto A, Kopriva S, Bernuzzi F, Francis H, Coufal M, Glaser S, Fava G, Meng F, Alvaro D, Carpino G, Gaudio E, Alpini G. Neuropeptide Y inhibits cholangiocarcinoma cell growth and invasion. Am J Physiol Cell Physiol. 2011;300:C1078–1089. doi: 10.1152/ajpcell.00358.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fava G, Marucci L, Glaser S, Francis H, De Morrow S, Benedetti A, Alvaro D, Venter J, Meininger C, Patel T, Taffetani S, Marzioni M, Summers R, Reichenbach R, Alpini G. gamma-Aminobutyric acid inhibits cholangiocarcinoma growth by cyclic AMP-dependent regulation of the protein kinase A/extracellular signal-regulated kinase 1/2 pathway. Cancer Res. 2005;65:11437–11446. doi: 10.1158/0008-5472.CAN-05-1470. [DOI] [PubMed] [Google Scholar]
- 48.Frampton GA, Lazcano EA, Li H, Mohamad A, DeMorrow S. Resveratrol enhances the sensitivity of cholangiocarcinoma to chemotherapeutic agents. Lab Invest. 2010;90:1325–1338. doi: 10.1038/labinvest.2010.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hou YJ, Dong LW, Tan YX, Yang GZ, Pan YF, Li Z, Tang L, Wang M, Wang Q, Wang HY. Inhibition of active autophagy induces apoptosis and increases chemosensitivity in cholangiocarcinoma. Lab Invest. 2011;91:1146–1157. doi: 10.1038/labinvest.2011.97. [DOI] [PubMed] [Google Scholar]
- 50.Hu MH, Chen LJ, Chen YL, Tsai MS, Shiau CW, Chao TI, Liu CY, Kao JH, Chen KF. Targeting SHP-1-STAT3 signaling: A promising therapeutic approach for the treatment of cholangiocarcinoma. Oncotarget. 2017;8:65077–65089. doi: 10.18632/oncotarget.17779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jo HJ, Shim HE, Han ME, Kim HJ, Kim KS, Baek S, Choi KU, Hur GY, Oh SO. WTAP regulates migration and invasion of cholangiocarcinoma cells. J Gastroenterol. 2013;48:1271–1282. doi: 10.1007/s00535-013-0748-7. [DOI] [PubMed] [Google Scholar]
- 52.Lang M, Henson R, Braconi C, Patel T. Epigallocatechin-gallate modulates chemotherapy-induced apoptosis in human cholangiocarcinoma cells. Liver Int. 2009;29:670–677. doi: 10.1111/j.1478-3231.2009.01984.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meng F, Yamagiwa Y, Ueno Y, Patel T. Over-expression of interleukin-6 enhances cell survival and transformed cell growth in human malignant cholangiocytes. J Hepatol. 2006;44:1055–1065. doi: 10.1016/j.jhep.2005.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Merino-Azpitarte M, Lozano E, Perugorria MJ, Esparza-Baquer A, Erice O, Santos-Laso A, O’Rourke CJ, Andersen JB, Jimenez-Aguero R, Lacasta A, D’Amato M, Briz O, Jalan-Sakrikar N, Huebert RC, Thelen KM, Gradilone SA, Aransay AM, Lavin JL, Fernandez-Barrena MG, Matheu A, Marzioni M, Gores GJ, Bujanda L, Marin JJG, Banales JM. SOX17 regulates cholangiocyte differentiation and acts as a tumor suppressor in cholangiocarcinoma. J Hepatol. 2017;67:72–83. doi: 10.1016/j.jhep.2017.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Obchoei S, Weakley SM, Wongkham S, Wongkham C, Sawanyawisuth K, Yao Q, Chen C. Cyclophilin A enhances cell proliferation and tumor growth of liver fluke-associated cholangiocarcinoma. Mol Cancer. 2011;10:102. doi: 10.1186/1476-4598-10-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Olaru AV, Ghiaur G, Yamanaka S, Luvsanjav D, An F, Popescu I, Alexandrescu S, Allen S, Pawlik TM, Torbenson M, Georgiades C, Roberts LR, Gores GJ, Ferguson-Smith A, Almeida MI, Calin GA, Mezey E, Selaru FM. MicroRNA down-regulated in human cholangiocarcinoma control cell cycle through multiple targets involved in the G1/S checkpoint. Hepatology. 2011;54:2089–2098. doi: 10.1002/hep.24591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pawar P, Ma L, Byon CH, Liu H, Ahn EY, Jhala N, Arnoletti JP, McDonald JM, Chen Y. Molecular mechanisms of tamoxifen therapy for cholangiocarcinoma: role of calmodulin. Clin Cancer Res. 2009;15:1288–1296. doi: 10.1158/1078-0432.CCR-08-1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Samukawa E, Fujihara S, Oura K, Iwama H, Yamana Y, Tadokoro T, Chiyo T, Kobayashi K, Morishita A, Nakahara M, Kobara H, Mori H, Okano K, Suzuki Y, Himoto T, Masaki T. Angiotensin receptor blocker telmisartan inhibits cell proliferation and tumor growth of cholangiocarcinoma through cell cycle arrest. Int J Oncol. 2017;51:1674–1684. doi: 10.3892/ijo.2017.4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shi X, Zhang H, Wang M, Xu X, Zhao Y, He R, Zhang M, Zhou M, Li X, Peng F, Shi C, Shen M, Wang X, Guo X, Qin R. LncRNA AFAP1-AS1 promotes growth and metastasis of cholangiocarcinoma cells. Oncotarget. 2017;8:58394–58404. doi: 10.18632/oncotarget.16880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tang J, Liao Y, He S, Shi J, Peng L, Xu X, Xie F, Diao N, Huang J, Xie Q, Lin C, Luo X, Liao K, Ma J, Li J, Zhou D, Li Z, Xu J, Zhong C, Wang G, Bai L. Autocrine parathyroid hormone-like hormone promotes intrahepatic cholangiocarcinoma cell proliferation via increased ERK/JNK-ATF2-cyclinD1 signaling. J Transl Med. 2017;15:238. doi: 10.1186/s12967-017-1342-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tang T, Zheng JW, Chen B, Li H, Li X, Xue KY, Ai X, Zou SQ. Effects of targeting magnetic drug nanoparticles on human cholangiocarcinoma xenografts in nude mice. Hepatobiliary Pancreat Dis Int. 2007;6:303–307. [PubMed] [Google Scholar]
- 62.Zhang J, Han C, Wu T. MicroRNA-26a promotes cholangiocarcinoma growth by activating beta-catenin. Gastroenterology. 2012;143:246–256 e248. doi: 10.1053/j.gastro.2012.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhao X, Zhang C, Zhou H, Xiao B, Cheng Y, Wang J, Yao F, Duan C, Chen R, Liu Y, Feng C, Li H, Li J, Dai R. Synergistic antitumor activity of the combination of salubrinal and rapamycin against human cholangiocarcinoma cells. Oncotarget. 2016;7:85492–85501. doi: 10.18632/oncotarget.13408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhu H, Han C, Lu D, Wu T. miR-17-92 cluster promotes cholangiocarcinoma growth: evidence for PTEN as downstream target and IL-6/Stat3 as upstream activator. Am J Pathol. 2014;184:2828–2839. doi: 10.1016/j.ajpath.2014.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhu H, Jiang X, Zhou X, Dong X, Xie K, Yang C, Jiang H, Sun X, Lu J. Neuropilin-1 regulated by miR-320 contributes to the growth and metastasis of cholangiocarcinoma cells. Liver Int. 2017 doi: 10.1111/liv.13495. [DOI] [PubMed] [Google Scholar]
- 66.Garcia PL, Miller AL, Gamblin TL, Council LN, Christein JD, Arnoletti JP, Heslin MJ, Reddy S, Richardson JH, Cui X, van Waardenburg R, Bradner JE, Yang ES, Yoon KJ. JQ1 Induces DNA Damage and Apoptosis, and Inhibits Tumor Growth in a Patient-Derived Xenograft Model of Cholangiocarcinoma. Mol Cancer Ther. 2018;17:107–118. doi: 10.1158/1535-7163.MCT-16-0922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mita Y, Ajiki T, Kamigaki T, Okazaki T, Hori H, Horiuchi H, Hirata K, Fujita T, Fujimori T, Kuroda Y. Antitumor effect of gemcitabine on orthotopically inoculated human gallbladder cancer cells in nude mice. Ann Surg Oncol. 2007;14:1374–1380. doi: 10.1245/s10434-006-9191-9. [DOI] [PubMed] [Google Scholar]
- 68.Harris LC, von Wronski MA, Venable CC, Remack JS, Howell SR, Brent TP. Changes in O6-methylguanine-DNA methyltransferase expression during immortalization of cloned human fibroblasts. Carcinogenesis. 1996;17:219–224. doi: 10.1093/carcin/17.2.219. [DOI] [PubMed] [Google Scholar]
- 69.Hidalgo M, Amant F, Biankin AV, Budinska E, Byrne AT, Caldas C, Clarke RB, de Jong S, Jonkers J, Maelandsmo GM, Roman-Roman S, Seoane J, Trusolino L, Villanueva A. Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer Discov. 2014;4:998–1013. doi: 10.1158/2159-8290.CD-14-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Morton CL, Houghton PJ. Establishment of human tumor xenografts in immunodeficient mice. Nat Protoc. 2007;2:247–250. doi: 10.1038/nprot.2007.25. [DOI] [PubMed] [Google Scholar]
- 71.Taylor AC, Shu L, Danks MK, Poquette CA, Shetty S, Thayer MJ, Houghton PJ, Harris LC. P53 mutation and MDM2 amplification frequency in pediatric rhabdomyosarcoma tumors and cell lines. Med Pediatr Oncol. 2000;35:96–103. doi: 10.1002/1096-911x(200008)35:2<96::aid-mpo2>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 72.Braakhuis BJ, Sneeuwloper G, Snow GB. The potential of the nude mouse xenograft model for the study of head and neck cancer. Arch Otorhinolaryngol. 1984;239:69–79. doi: 10.1007/BF00454264. [DOI] [PubMed] [Google Scholar]
- 73.Fiebig HH, Neumann HA, Henss H, Koch H, Kaiser D, Arnold H. Development of three human small cell lung cancer models in nude mice. Recent Results Cancer Res. 1985;97:77–86. doi: 10.1007/978-3-642-82372-5_8. [DOI] [PubMed] [Google Scholar]
- 74.Povlsen CO, Rygaard J. Heterotransplantation of human epidermoid carcinomas to the mouse mutant nude. Acta Pathol Microbiol Scand A. 1972;80:713–717. doi: 10.1111/j.1699-0463.1972.tb00340.x. [DOI] [PubMed] [Google Scholar]
- 75.Lampis A, Carotenuto P, Vlachogiannis G, Cascione L, Hedayat S, Burke R, Clarke P, Bosma E, Simbolo M, Scarpa A, Yu S, Cole R, Smyth E, Mateos JF, Begum R, Hezelova B, Eltahir Z, Wotherspoon A, Fotiadis N, Bali MA, Nepal C, Khan K, Stubbs M, Hahne JC, Gasparini P, Guzzardo V, Croce CM, Eccles S, Fassan M, Cunningham D, Andersen JB, Workman P, Valeri N, Braconi C. MIR21 drives resistance to Heat Shock Protein 90 inhibition in cholangiocarcinoma. Gastroenterology. 2017 doi: 10.1053/j.gastro.2017.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Song P, Du Y, Song W, Chen H, Xuan Z, Zhao L, Chen J, Chen J, Guo D, Jin C, Zhao Y, Tuo B, Zheng S. KCa3.1 as an Effective Target for Inhibition of Growth and Progression of Intrahepatic Cholangiocarcinoma. J Cancer. 2017;8:1568–1578. doi: 10.7150/jca.18697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang C, Lv H, Yang W, Li T, Fang T, Lv G, Han Q, Dong L, Jiang T, Jiang B, Yang G, Wang H. SVCT-2 determines the sensitivity to ascorbate-induced cell death in cholangiocarcinoma cell lines and patient derived xenografts. Cancer Lett. 2017;398:1–11. doi: 10.1016/j.canlet.2017.03.039. [DOI] [PubMed] [Google Scholar]
- 78.Cavalloni G, Peraldo-Neia C, Sassi F, Chiorino G, Sarotto I, Aglietta M, Leone F. Establishment of a patient-derived intrahepatic cholangiocarcinoma xenograft model with KRAS mutation. BMC Cancer. 2016;16:90. doi: 10.1186/s12885-016-2136-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Broutier L, Mastrogiovanni G, Verstegen MM, Francies HE, Gavarro LM, Bradshaw CR, Allen GE, Arnes-Benito R, Sidorova O, Gaspersz MP, Georgakopoulos N, Koo BK, Dietmann S, Davies SE, Praseedom RK, Lieshout R, JNM IJ, Wigmore SJ, Saeb-Parsy K, Garnett MJ, Laan LJ van der, Huch M. Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nat Med. 2017;23:1424–1435. doi: 10.1038/nm.4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sirica AE, Zhang Z, Lai GH, Asano T, Shen XN, Ward DJ, Mahatme A, Dewitt JL. A novel “patient-like” model of cholangiocarcinoma progression based on bile duct inoculation of tumorigenic rat cholangiocyte cell lines. Hepatology. 2008;47:1178–1190. doi: 10.1002/hep.22088. [DOI] [PubMed] [Google Scholar]
- 81.Fingas CD, Blechacz BR, Smoot RL, Guicciardi ME, Mott J, Bronk SF, Werneburg NW, Sirica AE, Gores GJ. A smac mimetic reduces TNF related apoptosis inducing ligand (TRAIL)-induced invasion and metastasis of cholangiocarcinoma cells. Hepatology. 2010;52:550–561. doi: 10.1002/hep.23729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Blechacz BR, Smoot RL, Bronk SF, Werneburg NW, Sirica AE, Gores GJ. Sorafenib inhibits signal transducer and activator of transcription-3 signaling in cholangiocarcinoma cells by activating the phosphatase shatterproof 2. Hepatology. 2009;50:1861–1870. doi: 10.1002/hep.23214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fingas CD, Bronk SF, Werneburg NW, Mott JL, Guicciardi ME, Cazanave SC, Mertens JC, Sirica AE, Gores GJ. Myofibroblast-derived PDGF-BB promotes Hedgehog survival signaling in cholangiocarcinoma cells. Hepatology. 2011;54:2076–2088. doi: 10.1002/hep.24588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fingas CD, Mertens JC, Razumilava N, Bronk SF, Sirica AE, Gores GJ. Targeting PDGFR-beta in Cholangiocarcinoma. Liver Int. 2012;32:400–409. doi: 10.1111/j.1478-3231.2011.02687.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fingas CD, Mertens JC, Razumilava N, Sydor S, Bronk SF, Christensen JD, Rizvi SH, Canbay A, Treckmann JW, Paul A, Sirica AE, Gores GJ. Polo-like kinase 2 is a mediator of hedgehog survival signaling in cholangiocarcinoma. Hepatology. 2013;58:1362–1374. doi: 10.1002/hep.26484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Razumilava N, Gradilone SA, Smoot RL, Mertens JC, Bronk SF, Sirica AE, Gores GJ. Non-canonical Hedgehog signaling contributes to chemotaxis in cholangiocarcinoma. J Hepatol. 2014;60:599–605. doi: 10.1016/j.jhep.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Campbell DJ, Dumur CI, Lamour NF, Dewitt JL, Sirica AE. Novel organotypic culture model of cholangiocarcinoma progression. Hepatol Res. 2012;42:1119–1130. doi: 10.1111/j.1872-034X.2012.01026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rizvi S, Fischbach SR, Bronk SF, Hirsova P, Krishnan A, Dhanasekaran R, Smadbeck JB, Smoot RL, Vasmatzis G, Gores GJ. YAP-associated chromosomal instability and cholangiocarcinoma in mice. Oncotarget. 2018;9:5892–5905. doi: 10.18632/oncotarget.23638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yamada D, Rizvi S, Razumilava N, Bronk SF, Davila JI, Champion MD, Borad MJ, Bezerra JA, Chen X, Gores GJ. IL-33 facilitates oncogene-induced cholangiocarcinoma in mice by an interleukin-6-sensitive mechanism. Hepatology. 2015;61:1627–1642. doi: 10.1002/hep.27687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chen S, Sun H, Miao K, Deng CX. CRISPR-Cas9: from Genome Editing to Cancer Research. Int J Biol Sci. 2016;12:1427–1436. doi: 10.7150/ijbs.17421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci U S A. 1992;89:5547–5551. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sauer B. Functional expression of the cre-lox site-specific recombination system in the yeast Saccharomyces cerevisiae. Mol Cell Biol. 1987;7:2087–2096. doi: 10.1128/mcb.7.6.2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Schlake T, Bode J. Use of mutated FLP recognition target (FRT) sites for the exchange of expression cassettes at defined chromosomal loci. Biochemistry. 1994;33:12746–12751. doi: 10.1021/bi00209a003. [DOI] [PubMed] [Google Scholar]
- 94.He L, Tian DA, Li PY, He XX. Mouse models of liver cancer: Progress and recommendations. Oncotarget. 2015;6:23306–23322. doi: 10.18632/oncotarget.4202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Xu X, Kobayashi S, Qiao W, Li C, Xiao C, Radaeva S, Stiles B, Wang RH, Ohara N, Yoshino T, LeRoith D, Torbenson MS, Gores GJ, Wu H, Gao B, Deng CX. Induction of intrahepatic cholangiocellular carcinoma by liver-specific disruption of Smad4 and Pten in mice. J Clin Invest. 2006;116:1843–1852. doi: 10.1172/JCI27282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kang YK, Kim WH, Jang JJ. Expression of G1-S modulators (p53, p16, p27, cyclin D1, Rb) and Smad4/Dpc4 in intrahepatic cholangiocarcinoma. Hum Pathol. 2002;33:877–883. doi: 10.1053/hupa.2002.127444. [DOI] [PubMed] [Google Scholar]
- 97.Yang X, Li C, Xu X, Deng C. The tumor suppressor SMAD4/DPC4 is essential for epiblast proliferation and mesoderm induction in mice. Proc Natl Acad Sci U S A. 1998;95:3667–3672. doi: 10.1073/pnas.95.7.3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Downward J. Mechanisms and consequences of activation of protein kinase B/Akt. Curr Opin Cell Biol. 1998;10:262–267. doi: 10.1016/s0955-0674(98)80149-x. [DOI] [PubMed] [Google Scholar]
- 99.Radu A, Neubauer V, Akagi T, Hanafusa H, Georgescu MM. PTEN induces cell cycle arrest by decreasing the level and nuclear localization of cyclin D1. Mol Cell Biol. 2003;23:6139–6149. doi: 10.1128/MCB.23.17.6139-6149.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Horie Y, Suzuki A, Kataoka E, Sasaki T, Hamada K, Sasaki J, Mizuno K, Hasegawa G, Kishimoto H, Iizuka M, Naito M, Enomoto K, Watanabe S, Mak TW, Nakano T. Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. Journal of Clinical Investigation. 2004;113:1774–1783. doi: 10.1172/JCI20513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dias Carvalho P, Guimaraes CF, Cardoso AP, Mendonca S, Costa AM, Oliveira MJ, Velho S. KRAS Oncogenic Signaling Extends beyond Cancer Cells to Orchestrate the Microenvironment. Cancer Res. 2018;78:7–14. doi: 10.1158/0008-5472.CAN-17-2084. [DOI] [PubMed] [Google Scholar]
- 102.Ikenoue T, Terakado Y, Nakagawa H, Hikiba Y, Fujii T, Matsubara D, Noguchi R, Zhu C, Yamamoto K, Kudo Y, Asaoka Y, Yamaguchi K, Ijichi H, Tateishi K, Fukushima N, Maeda S, Koike K, Furukawa Y. A novel mouse model of intrahepatic cholangiocarcinoma induced by liver-specific Kras activation and Pten deletion. Sci Rep. 2016;6:23899. doi: 10.1038/srep23899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lee KP, Lee JH, Kim TS, Kim TH, Park HD, Byun JS, Kim MC, Jeong WI, Calvisi DF, Kim JM, Lim DS. The Hippo-Salvador pathway restrains hepatic oval cell proliferation, liver size, and liver tumorigenesis. Proc Natl Acad Sci U S A. 2010;107:8248–8253. doi: 10.1073/pnas.0912203107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.O’Dell MR, Huang JL, Whitney-Miller CL, Deshpande V, Rothberg P, Grose V, Rossi RM, Zhu AX, Land H, Bardeesy N, Hezel AF. Kras(G12D) and p53 mutation cause primary intrahepatic cholangiocarcinoma. Cancer Res. 2012;72:1557–1567. doi: 10.1158/0008-5472.CAN-11-3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zou S, Li J, Zhou H, Frech C, Jiang X, Chu JS, Zhao X, Li Y, Li Q, Wang H, Hu J, Kong G, Wu M, Ding C, Chen N, Hu H. Mutational landscape of intrahepatic cholangiocarcinoma. Nat Commun. 2014;5:5696. doi: 10.1038/ncomms6696. [DOI] [PubMed] [Google Scholar]
- 106.Fujimoto A, Furuta M, Shiraishi Y, Gotoh K, Kawakami Y, Arihiro K, Nakamura T, Ueno M, Ariizumi S, Nguyen HH, Shigemizu D, Abe T, Boroevich KA, Nakano K, Sasaki A, Kitada R, Maejima K, Yamamoto Y, Tanaka H, Shibuya T, Shibata T, Ojima H, Shimada K, Hayami S, Shigekawa Y, Aikata H, Ohdan H, Marubashi S, Yamada T, Kubo M, Hirano S, Ishikawa O, Yamamoto M, Yamaue H, Chayama K, Miyano S, Tsunoda T, Nakagawa H. Whole-genome mutational landscape of liver cancers displaying biliary phenotype reveals hepatitis impact and molecular diversity. Nat Commun. 2015;6:6120. doi: 10.1038/ncomms7120. [DOI] [PubMed] [Google Scholar]
- 107.Rizvi S, Gores GJ. Emerging molecular therapeutic targets for cholangiocarcinoma. J Hepatol. 2017;67:632–644. doi: 10.1016/j.jhep.2017.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Borger DR, Tanabe KK, Fan KC, Lopez HU, Fantin VR, Straley KS, Schenkein DP, Hezel AF, Ancukiewicz M, Liebman HM, Kwak EL, Clark JW, Ryan DP, Deshpande V, Dias-Santagata D, Ellisen LW, Zhu AX, Iafrate AJ. Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist. 2012;17:72–79. doi: 10.1634/theoncologist.2011-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Voss JS, Holtegaard LM, Kerr SE, Fritcher EG, Roberts LR, Gores GJ, Zhang J, Highsmith WE, Halling KC, Kipp BR. Molecular profiling of cholangiocarcinoma shows potential for targeted therapy treatment decisions. Hum Pathol. 2013;44:1216–1222. doi: 10.1016/j.humpath.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 110.Wang P, Dong Q, Zhang C, Kuan PF, Liu Y, Jeck WR, Andersen JB, Jiang W, Savich GL, Tan TX, Auman JT, Hoskins JM, Misher AD, Moser CD, Yourstone SM, Kim JW, Cibulskis K, Getz G, Hunt HV, Thorgeirsson SS, Roberts LR, Ye D, Guan KL, Xiong Y, Qin LX, Chiang DY. Mutations in isocitrate dehydrogenase 1 and 2 occur frequently in intrahepatic cholangiocarcinomas and share hypermethylation targets with glioblastomas. Oncogene. 2013;32:3091–3100. doi: 10.1038/onc.2012.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Saha SK, Parachoniak CA, Ghanta KS, Fitamant J, Ross KN, Najem MS, Gurumurthy S, Akbay EA, Sia D, Cornella H, Miltiadous O, Walesky C, Deshpande V, Zhu AX, Hezel AF, Yen KE, Straley KS, Travins J, Popovici-Muller J, Gliser C, Ferrone CR, Apte U, Llovet JM, Wong KK, Ramaswamy S, Bardeesy N. Mutant IDH inhibits HNF-4alpha to block hepatocyte differentiation and promote biliary cancer. Nature. 2014;513:110–114. doi: 10.1038/nature13441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mollereau B, Ma D. The p53 control of apoptosis and proliferation: lessons from Drosophila. Apoptosis. 2014;19:1421–1429. doi: 10.1007/s10495-014-1035-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Rizvi S, Gores GJ. Pathogenesis, diagnosis, and management of cholangiocarcinoma. Gastroenterology. 2013;145:1215–1229. doi: 10.1053/j.gastro.2013.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Farazi PA, Zeisberg M, Glickman J, Zhang Y, Kalluri R, DePinho RA. Chronic bile duct injury associated with fibrotic matrix microenvironment provokes cholangiocarcinoma in p53-deficient mice. Cancer Res. 2006;66:6622–6627. doi: 10.1158/0008-5472.CAN-05-4609. [DOI] [PubMed] [Google Scholar]
- 115.Lemoinne S, Cadoret A, Rautou PE, Mourabit H El, Ratziu V, Corpechot C, Rey C, Bosselut N, Barbu V, Wendum D, Feldmann G, Boulanger C, Henegar C, Housset C, Thabut D. Portal myofibroblasts promote vascular remodeling underlying cirrhosis formation through the release of microparticles. Hepatology. 2015;61:1041–1055. doi: 10.1002/hep.27318. [DOI] [PubMed] [Google Scholar]
- 116.Yu D, Hung MC. Overexpression of ErbB2 in cancer and ErbB2-targeting strategies. Oncogene. 2000;19:6115–6121. doi: 10.1038/sj.onc.1203972. [DOI] [PubMed] [Google Scholar]
- 117.Kiguchi K, Carbajal S, Chan K, Beltran L, Ruffino L, Shen J, Matsumoto T, Yoshimi N, DiGiovanni J. Constitutive expression of ErbB-2 in gallbladder epithelium results in development of adenocarcinoma. Cancer Res. 2001;61:6971–6976. [PubMed] [Google Scholar]
- 118.Hofmann JJ, Zovein AC, Koh H, Radtke F, Weinmaster G, Iruela-Arispe ML. Jagged1 in the portal vein mesenchyme regulates intrahepatic bile duct development: insights into Alagille syndrome. Development. 2010;137:4061–4072. doi: 10.1242/dev.052118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zender S, Nickeleit I, Wuestefeld T, Sorensen I, Dauch D, Bozko P, El-Khatib M, Geffers R, Bektas H, Manns MP, Gossler A, Wilkens L, Plentz R, Zender L, Malek NP. A critical role for notch signaling in the formation of cholangiocellular carcinomas. Cancer Cell. 2013;23:784–795. doi: 10.1016/j.ccr.2013.04.019. [DOI] [PubMed] [Google Scholar]
- 120.Akhoondi S, Sun D, von der Lehr N, Apostolidou S, Klotz K, Maljukova A, Cepeda D, Fiegl H, Dafou D, Marth C, Mueller-Holzner E, Corcoran M, Dagnell M, Nejad SZ, Nayer BN, Zali MR, Hansson J, Egyhazi S, Petersson F, Sangfelt P, Nordgren H, Grander D, Reed SI, Widschwendter M, Sangfelt O, Spruck C. FBXW7/hCDC4 is a general tumor suppressor in human cancer. Cancer Res. 2007;67:9006–9012. doi: 10.1158/0008-5472.CAN-07-1320. [DOI] [PubMed] [Google Scholar]
- 121.Koepp DM, Schaefer LK, Ye X, Keyomarsi K, Chu C, Harper JW, Elledge SJ. Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science. 2001;294:173–177. doi: 10.1126/science.1065203. [DOI] [PubMed] [Google Scholar]
- 122.Welcker M, Clurman BE. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer. 2008;8:83–93. doi: 10.1038/nrc2290. [DOI] [PubMed] [Google Scholar]
- 123.Fan B, Malato Y, Calvisi DF, Naqvi S, Razumilava N, Ribback S, Gores GJ, Dombrowski F, Evert M, Chen X, Willenbring H. Cholangiocarcinomas can originate from hepatocytes in mice. J Clin Invest. 2012;122:2911–2915. doi: 10.1172/JCI63212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Pan DJ. The Hippo Signaling Pathway in Development and Cancer. Dev Cell. 2010;19:491–505. doi: 10.1016/j.devcel.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Tao JY, Calvisi DF, Ranganathan S, Cigliano A, Zhou LL, Singh S, Jiang LJ, Fan BA, Terracciano L, Armeanu-Ebinger S, Ribback S, Dombrowski F, Evert M, Chen X, Monga SPS. Activation of beta-Catenin and Yap1 in Human Hepatoblastoma and Induction of Hepatocarcinogenesis in Mice. Gastroenterology. 2014;147:690–701. doi: 10.1053/j.gastro.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Li J, Razumilava N, Gores GJ, Walters S, Mizuochi T, Mourya R, Bessho K, Wang YH, Glaser SS, Shivakumar P, Bezerra JA. Biliary repair and carcinogenesis are mediated by IL-33-dependent cholangiocyte proliferation. J Clin Invest. 2014;124:3241–3251. doi: 10.1172/JCI73742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Marvie P, Lisbonne M, L’Helgoualc’h A, Rauch M, Turlin B, Preisser L, Bourd-Boittin K, Theret N, Gascan H, Piquet-Pellorce C, Samson M. Interleukin-33 overexpression is associated with liver fibrosis in mice and humans. J Cell Mol Med. 2010;14:1726–1739. doi: 10.1111/j.1582-4934.2009.00801.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Farshidfar F, Zheng S, Gingras MC, Newton Y, Shih J, Robertson AG, Hinoue T, Hoadley KA, Gibb EA, Roszik J, Covington KR, Wu CC, Shinbrot E, Stransky N, Hegde A, Yang JD, Reznik E, Sadeghi S, Pedamallu CS, Ojesina AI, Hess JM, Auman JT, Rhie SK, Bowlby R, Borad MJ, N. Cancer Genome Atlas. Zhu AX, Stuart JM, Sander C, Akbani R, Cherniack AD, Deshpande V, Mounajjed T, Foo WC, Torbenson MS, Kleiner DE, Laird PW, Wheeler DA, McRee AJ, Bathe OF, Andersen JB, Bardeesy N, Roberts LR, Kwong LN. Integrative Genomic Analysis of Cholangiocarcinoma Identifies Distinct IDH-Mutant Molecular Profiles. Cell Rep. 2017;18:2780–2794. doi: 10.1016/j.celrep.2017.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]