

Abstract
The yeast cytoplasmically-localized pGKL1/TP-DNAP1 plasmid/DNA polymerase pair forms an orthogonal DNA replication system whose mutation rate can be drastically increased without influencing genomic replication, thereby supporting in vivo continuous evolution. Here, we report that the pGKL2/TP-DNAP2 plasmid/DNA polymerase pair forms a second orthogonal replication system. We show that custom genes can be encoded and expressed from pGKL2, that error-prone TP-DNAP2s can be engineered, and that pGKL2 replication by TP-DNAP2 is both orthogonal to genomic replication in Saccharomyces cerevisiae and mutually orthogonal with pGKL1 replication by TP-DNAP1. This demonstration of two mutually orthogonal DNA replication systems with tunable error rates and properties should enable new applications in cell-based continuous evolution, genetic recording, and synthetic biology at large.
Keywords: orthogonal replication, polymerase engineering, in vivo mutagenesis, linear plasmids, DNA replication, protein-primed replication
Graphical Abstract
Introduction
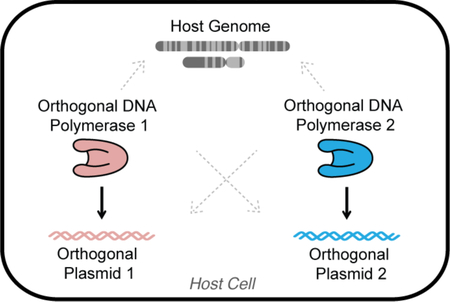
Expanding the capabilities of DNA polymerases (DNAPs) to change how they copy DNA in vitro has resulted in staple biotechnologies including PCR mutagenesis, DNA sequencing, nucleic acid diagnostics, and the evolution of synthetic genetic polymers.1–5 Engineering DNAPs with new properties in vivo could lead to similarly important advances, including cells that continuously evolve target genes, record lineage or exposure to custom stimuli through mutating barcodes, or use new genetic alphabets for biocontainment.6–10 However, the operation of highly engineered DNAPs in a living cell is challenging because they can easily harm genomic replication.11 We recently developed a special DNA replication system in Saccharomyces cerevisiae consisting of a DNAP/plasmid pair that is orthogonal to genomic replication and showed that engineered orthogonal DNAPs only replicate the orthogonal plasmid.6,7 Here, we demonstrate that an additional DNAP/plasmid pair is orthogonal to genomic replication in S. cerevisiae and that the two DNAP/plasmid pairs are mutually orthogonal to each other. This solidifies two platforms for independently expanding the properties of DNA replication in vivo.
Our two orthogonal replication systems are based on the cytoplasmically-localized pGKL1/pGKL2 (p1/p2) plasmids originating from Kluyveromyces lactis.11,12 Both p1 and p2 encode their own DNAPs, TP-DNAP1 and TP-DNAP2, respectively. We previously showed that engineered error-prone variants of TP-DNAP1 increase the mutation rate of p1 to ~10−5 substitutions per base (s.p.b.) without affecting the genomic mutation rate of ~10−10 s.p.b. in S. cerevisiae.6 We also found that p1 replication strictly requires TP-DNAP1.7 This allowed us to conclude that TP-DNAP1 and p1 constitute an orthogonal DNAP/plasmid pair such that engineered changes to TP-DNAP1 only act on p1 but not on the genome. However, it was not known whether orthogonality to genomic replication holds true for TP-DNAP2 and p2 nor whether the TP-DNAP1/p1 and TP-DNAP2/p2 pairs are mutually orthogonal. Here, we develop error-prone TP-DNAP2s, report the associated genetic techniques necessary to engineer the p2 plasmid and TP-DNAP2, and ultimately prove that the TP-DNAP2/p2 pair is both orthogonal to genomic replication and to p1 replication.
The existence of mutually orthogonal genetically tractable replication systems is significant for three main reasons. First, our finding of two mutually orthogonal DNA replication systems should lay the foundation for novel applications in synthetic biology. For example, in vivo accelerated evolution6 of different genes or sets of genes can now be carried out at two distinct custom mutation rates, which could be useful for evolving components in hierarchically organized signaling pathways. Another possibility includes using inducible error-prone orthogonal DNAPs to record multiple cellular events or external stimuli, where the number of mutational events in p1 and p2 would be independent readouts of the amount of exposure to two signals experienced by cells. In addition, the freedom to engineer two orthogonal DNAPs in vivo may enable propagation of different XNA’s in living cells, whereas current efforts are limited to either using novel base-pairs recognized by host DNAPs or engineering DNAPs to synthesize XNA with novel backbones in vitro.4,5,9 Second is a practical consideration. Our main motivation for developing an orthogonal replication (OrthoRep) system was to achieve continuous rapid evolution of target genes in vivo at extreme mutation rates that the genome cannot withstand.6 This was achieved by making TPDNAP1 highly error-prone so that it rapidly mutates the p1 plasmid but spares the genome. The mutual orthogonality demonstrated here ensures that the essential accessory genes encoded on p2 are also spared by error-prone TP-DNAP1s during directed evolution experiments of genes on p1 in OrthoRep. Third, our result provides in vivo evidence that DNA initiation of p1 and p2 use independent components. p1 and p2 both contain terminal proteins (TPs) linked to their 5’ termini, which act as origins of replication, akin to other protein-primed DNA replication systems like those found in bacteriophage Φ29 and adenovirus.14,15 The lack of homology between the TPs of p1 and p2 suggested that TP-DNAP1 and TP-DNAP2 may use distinct molecular interactions for plasmid initiation. In addition, preliminary in vitro biochemical data from our lab shows that TP-DNAP1 can initiate replication from p1’s inverted terminal repeat (ITR), hypothesized to act in concert with p1’s TP to form an original of replication, but cannot initiate replication from p2’s ITR (unpublished). Our observation of mutual orthogonality between p1 and p2 replication all but proves that highly specific TP-DNAP interactions with cognate TPs and ITRs govern plasmid initiation, encouraging future studies on the mechanisms of protein-primed DNA replication and suggesting a potential approach for engineering additional orthogonal replication systems that operate concurrently in the same cell.
Results and Discussion
Strategy for characterizing orthogonality
Our strategy for probing orthogonality of the TP-DNAP2/p2 DNAP/plasmid pair is based on engineering and using error-prone TPDNAP1 and TP-DNAP2 variants to measure whether they increase mutation rates of genes on p1, p2, and/or the host genome. If error-prone TP-DNAP1s only increase the mutation rate of p1 (but not of p2 and the host genome) and error-prone TP-DNAP2s only increase the mutation rate of p2 (but not of p1 and the host genome), then we may conclude that TPDNAP1/p1 and TP-DNAP2/p2 are mutually orthogonal DNA replication systems that are both orthogonal to genomic replication. This is indeed what we found.
Integration of heterologous genes onto p2 using CRISPR/Cas9
We first developed a reliable method for encoding and expressing user-defined genes on p2. To measure mutation rates of p2 replication, we needed to encode mKate2, URA3, and leu2* (mUL*) on p2. mKate2 would serve as a fluorescent reporter for copy number, URA3 as a selection marker, and leu2*, which contains a stop codon at a permissive site in LEU2 (Q180*), would serve as a reporter for substitution mutation rates in fluctuation tests that measure reversion to functional LEU2. We expected that mUL* could be integrated onto p2 via in vivo homologous recombination, following our procedures for manipulating p1.6,7 We constructed a DNA cassette encoding mUL* flanked by regions homologous to p2 (Figure S1) such that successful recombination would result in the replacement of the non-essential ORF1 found on wildtype (wt) p2.16 After transformation of this cassette into S. cerevisiae strain AR-Y292 containing wt p1 and p2 (Figure 1A, top), we isolated several clones exhibiting uracil prototrophy and detectable fluorescence from mKate2. Extraction of cytoplasmic plasmids from these clones confirmed presence of the recombinant p2-delORF1-mUL*, but only at low copy as confirmed by DNA gel electrophoresis and PCR with primers specific to p2-delORF1-mUL*. In contrast to similar cassette integrations into p1, passaging under selection for URA3 expression failed to cure the parental wt p2 plasmid and increase the copy number of p2-delORF1-mUL* to levels easily detectable by gel electrophoresis. Although p2-delORF1-mUL* encodes all the necessary genes for its own replication and was selected for through URA3, it is likely that the shorter size of wt p2 provided it with enough of a replicative advantage to be maintained.
Figure 1. Engineering recombinant p2.

(A) Top: AR-Y292 strain harboring p1 and p2, replicated by self-encoded TP-DNAP1 and TP-DNAP2, respectively. Middle: Intermediate strain derived from AR-Y292 by integration of mKate2, URA3, and leu2* over ORF1 of p2 and expression of Cas9 targeting ORF1 of p2, which is missing from the recombinant p2-delORF1-mUL*. Bottom: Complete curing of the parental p2 plasmid induced by Cas9 results in GA-Y021, which contains only p1 and recombinant p2-delORF1-mUL*. GA-Y021 is used as the base strain for subsequent measurements of p2 mutation rate. (B) Gel analysis (0.9% agarose) verifying integration of mKate2, URA3, leu2* and loss of parental p2 after Cas9 treatment. Lane 1: DNA extracted from AR-Y292, containing linear plasmids p1 and p2. Lane 2: DNA extracted from GA-Y021, confirming loss of p2 and its replacement by p2-delORF1-mUL* at high copy.
To cure the parental wt p2 plasmid, we attempted more active methods. We used a yeast CRISPR/Cas9 vector25 and three candidate sgRNAs to target ORF1 (Table S1), which is present in wt p2 but not in p2-delORF1-mUL* (Figure 1A, middle). One of the three sgRNA’s expressed in conjunction with a cytoplasmically-localized Cas9 achieved complete curing of p2 and a concomitant increase in the copy number of p2-delORF1-mUL*. This was evidenced by an increase in mKate2 fluorescent signal and a brighter p2-delORF1-mUL* DNA gel electrophoresis band (Figure 1B). Curing of parental p2 to undetectable levels was confirmed by lack of PCR amplification with primers specific to p2, yielding strain GA-Y021 (Figure 1A, bottom).
Using GA-Y021, we measured the mutation rate of p2 replication by wt TP-DNAP2 still encoded on p2-delORF1-mUL*, with a previously described fluctuation test where the number distribution of functional LEU2 mutants is used to calculate mutation rate by the MSS method.17–19 The mutation rate of p2-delORF1-mUL* replication was 5.96 × 10−10 s.p.b. (95% C.I.: 3.57 × 10−10–8.77 × 10−10) with a copy number of 128 per cell (Table 1, Entry 1; Table S3). This is similar to the wild type p1 mutation rate and copy number, which are 1.39 × 10−9 and 124, respectively.7
Table 1. TP-DNAP2 variants with elevated mutation rates.
Entry 1 is the native mutation rate of p2 replication measured in GA-Y021, where p2-delORF1-mUL* is replicated only by the self-encoded wt TP-DNAP2. Entries 2–18 are mutation rate measurements of p2 replication in presence of 17 error-prone TP-DNAP2 hits from library screening. All per-base substitution rates were measured using leu2* fluctuation analyses in GA-Y021, where p2-delORF1-mUL* is replicated in tandem by wt TP-DNAP2 and a TP-DNAP2 variant encoded in trans on a nuclear CEN6/ARS4 plasmid. Ranges in parentheses correspond to 95% confidence intervals, determined according to the MSS method17–19.
| Entry | TP-DNAP2 variant in trans | Per-base substitution rate of p2 (x10−10) | Fold increase |
|---|---|---|---|
| 1 | None | 5.96 (3.57–8.77) | − |
| 2 | S370Q | 83.0 (62.8–105) | 13.9 |
| 3 | Y424Q | 77.5 (56.9–100) | 13.0 |
| 4 | Y424E | 73.3 (55.2–93.3) | 12.3 |
| 5 | S370P | 50.1 (32.4–70.4) | 8.4 |
| 6 | Y424K | 44.4 (32.2–58.0) | 7.5 |
| 7 | S370R | 31.9 (22.4–42.5) | 5.3 |
| 8 | Y424G | 28.8 (20.0–38.7) | 4.8 |
| 9 | S370E | 27.9 (19.7–37.2) | 4.7 |
| 10 | S370K | 26.2 (18.0–35.4) | 4.4 |
| 11 | Y424R | 19.6 (13.0–27.1) | 3.3 |
| 12 | S370L | 14.8 (9.32–21.1) | 2.5 |
| 13 | L474D | 12.2 (7.33–17.9) | 2.0 |
| 14 | F882A | 11.7 (4.92–20.4) | 2.0 |
| 15 | F882V | 10.8 (6.30–16.2) | 1.8 |
| 16 | L474A | 8.37 (4.80–12.6) | 1.4 |
| 17 | F882R | 7.32 (3.91–11.5) | 1.2 |
| 18 | L474V | 5.76 (2.88–9.33) | 1.0 |
Replication of p2 by TP-DNAP2 in trans
To facilitate the straightforward testing of error-prone TP-DNAP2s, we showed that p2 replication could be fully sustained by TP-DNAP2 encoded in trans, on a standard yeast nuclear plasmid, rather than in cis, on p2. First, TP-DNAP2, which is ORF2 of p2, was deleted by homologous recombination of a synthetic cassette encoding URA3 (Figure S1). Since p2 is a multi-copy plasmid, the resulting strain (GA-Y069) harbored a mixture of the parental wt p2 and the recombinant p2 with ORF2 deleted (p2-delORF2-URA3), along with unaltered p1 (Figure 2A). In this strain, both the wt p2 and recombinant p2-delORF2-URA3 plasmids rely on TP-DNAP2 encoded on the parental wt p2 plasmid for replication. Thus, loss of wt p2 should disable replication of p2-delORF2-URA3. Indeed, when we cured the parental p2 plasmid by targeting ORF2 of wt p2 with Cas9 (Table S1), we found that all p2-delORF2-URA3 was also lost and that the strain could no longer grow in the absence of uracil (Figure 2B). Next, we repeated the same experiment but now in the presence of a codon-optimized TP-DNAP2 expressed in trans from a standard yeast CEN6/ARS4 nuclear plasmid (pGA55-reTP-DNAP2). After p2 was fully cured by Cas9, we found that p2 was not present, p2-delORF2-URA3 remained, and this strain could grow in the absence of uracil (Figure 2B). In addition, p1 was also maintained, indicating that the accessory genes encoded on p2-delORF2-URA3 necessary for replication of both p1 and p2 and transcription of TP-DNAP1 on p1 were still functional. Therefore, p2-derived plasmids can be replicated by TP-DNAP2 encoded on a standard nuclear plasmid, simplifying the characterization of p2 replication by error-prone TP-DNAP2 variants.
Figure 2. Functional complementation by recoded TP-DNAP2 in trans.

(A) Left: Linear plasmid system consisting of p1, p2, and recombinant p2-delORF2-URA3. Cas9 co-expressed with p2ORF2-sgRNA1 specifically targets double strand breaks to ORF2 of p2. Right: Complete curing of p2 by Cas9 results in loss of p2’s self-encoded TP-DNAP2. This deletion is functionally complemented by TP-DNAP2 encoded in trans, which can stably replicate p2. (B) Gel analysis (0.9% agarose) testing for stability of linear plasmid combinations in the absence (odd-numbered lanes) or the presence (even-numbered lanes) of Cas9 activity targeted to ORF2 of p2. Lanes 1 and 2: DNA extracted from AR-Y292, containing only wt p1 and p2, which are both lost in presence of Cas9. Lanes 3 and 4: DNA extracted from GA-Y069 containing p1, p2, and recombinant p2-delORF-URA3. Elimination of wt p2 by Cas9 curing eliminates the sole source of TPDNAP2, causing subsequent loss of p2-delORF1-URA3 and p1. Following loss of the linear plasmids, the strain in lane 4 was no longer prototrophic for uracil. Lanes 5 and 6: DNA extracted from a GA-Y069 derivative, which contains the linear plasmids p1, p2 and p2-delORF2-URA3 and encodes TP-DNAP2 on a nuclear CEN6/ARS4 plasmid in trans. Treatment with Cas9 results in a stable linear plasmid system lacking wt p2, instead consisting only of p1 and p2-delORF2-URA3 replicated entirely by TP-DNAP2 encoded in trans.
Discovery of error-prone TP-DNAP2 variants through library screening
To discover error-prone TP-DNAP2s, we screened a small library of TP-DNAP2s diversified at locations we hypothesized would be responsible for DNAP fidelity. An alignment between TPDNAP1 and TP-DNAP2 revealed that S370, Y424, L474, and F882 in TP-DNAP2 were homologous to residues in TP-DNAP1 that we previously found could be mutated to yield error-prone TP-DNAP1s (Figure S2).6,7 We therefore generated four distinct site-saturation mutagenesis libraries, each diversifying S370, Y424, L474, or F882 in TP-DNAP2 encoded on pGA55-reTPDNAP2. Due to unsuccessful attempts to generate a strain with a full deletion of p2-encoded TPDNAP2 and simultaneous integration of mKate, URA3, and leu2*, each library was transformed into GA-Y021 for convenient screening. However, since GA-Y021 still encodes wt TP-DNAP2 on p2-delORF1-mUL*, p2 mutation rates measured in this format are the result of in tandem replication of p2 by wt TP-DNAP2 and each TP-DNAP2 variant encoded in trans. We sampled each NNK library (theoretical diversity = 32 codon variants) at 6-fold coverage by picking 190 yeast colonies and passaging under selection for URA3 to stabilize the copy number of p2-delORF1-mUL* in the presence of the newly introduced TP-DNAP2 variants. To screen for TPDNAP2 variants with increased p2 mutation rate, each library member was subjected to a preliminary, small scale leu2* fluctuation test with six replicates. Seventeen candidate mutators with the highest expected number of mutants m calculated by the p0 method were then chosen for reconfirmation. Reconfirmation consisted of extracting the CEN6/ARS4 plasmids encoding TPDNAP2 variants, retransforming into a fresh GA-Y021 background and repeating leu2* fluctuation tests with 36 replicates to determine p2 mutation rate with higher precision (Table 1). Two error-prone TP-DNAP2 variants, S370Q and Y424Q, increased p2’s substitution mutation rate by ~14- and ~13-fold, respectively (Table 1, Entries 2 and 3). These variants were not active enough to fully complement a deletion of the native TP-DNAP2 and sustain p2-delORF2--URA3 replication on their own, making our measured mutation rates an underestimate of their true per-base substitution rate. We suspect that even slight decreases in p2 copy number when replicated by mutant polymerases may cause instability in the system due to disruption of expression levels of the nine essential genes encoded on p2. Despite this, these two error-prone TP-DNAP2 variants elevate p2 mutation rate to high enough levels for measuring orthogonality.
Orthogonality between p2 replication and genomic replication
To show that p2 replication is orthogonal to genomic replication, we measured the mutation rate of the host genome in the presence of error-prone TP-DNAP2 variants. Like p1 replication by TPDNAP1, p2 replication by TP-DNAP2 occurs in the cytoplasm via a protein-primed mechanism, making it likely that p2 replication is orthogonal to host genome replication. To test this, we transformed CEN6/ARS4 vectors lacking TP-DNAP2, or encoding codon optimized versions of TP-DNAP2WT, TP-DNAP2S370Q, or TP-DNAP2Y424Q into AR-Y436 (Figure S3). AR-Y436 contains wt p1 and p2, as well as an intact URA3 locus in the host genome. Genomic per-base substitution rates were determined via fluctuation tests based on the frequency of 5-FOA resistant clones arising from mutations in the genomic URA3 locus, as previously described,7,20 and are reported in Table 2. The substitution rich spectrum of URA3−disabling mutations makes this assay ideal for detecting whether the elevated substitution rate of TP-DNAP2S370Q or TP-DNAP2Y424Q contributes to genomic mutation. We observed no increase in the host genomic mutation rate when error-prone variants of TP-DNAP2S370Q and TP-DNAP2Y424Q were present.
Table 2. Genomic mutation rate is unaltered by error-prone TP-DNAP2 variants.
Entries 1–4 are per-base substitution rates of genomic replication in presence of TP-DNAP2 variants, measured using the genomic URA3 locus. Mutagenic TP-DNAP2 variants Y424Q and S370Q cause no statistically significant change in genomic mutation rate, relative to no TP-DNAP2 variant in trans (84% CI overlap method).21 Ranges in parentheses correspond to 95% confidence intervals, determined according to the MSS method.17–19
| Entry | TP-DNAP2 in trans | Per-base substitution rate at genomic URA3 locus (x10−10) |
|---|---|---|
| 1 | None | 1.6 (0.8–2.4) |
| 2 | WT | 2.2 (1.2–3.4) |
| 3 | Y424Q | 1.8 (1.0–2.9) |
| 4 | S370Q | 1.8 (1.0–2.8) |
Mutual orthogonality between p1 and p2 replication
Next, we wanted to test whether the replication mechanisms of p1 and p2 are mutually orthogonal in vivo by measuring p1 and p2 mutation rates in the presence of a panel of TP-DNAP1 and TPDNAP2 variants with varying mutation rates. Changes in p1 or p2 mutation rate induced by TPDNAP variants would therefore signal a degree of cross-replication between TP-DNAP1/p1 and TP-DNAP2/p2, if any. A panel of six polymerases was introduced on CEN6/ARS4 vectors into two separate base strains, AR-Y304 (p1 mutation rate reporter strain) and GA-Y021 (p2 mutation rate reporter strain), encoding mKate2, URA3, and leu2* on either p1 or p2, respectively (Figure 3A). Included in this panel were TP-DNAP1WT and two error-prone TP-DNAP1 variants found in previous screens: TP-DNAP1I777K, L900S and TP-DNAP1L477V, L640Y, I777K, W814N (Figure S4). Also included were TP-DNAP2WT and the error-prone TP-DNAP2S370Q and TP-DNAP2Y424Q variants found in this work. In AR-Y304 and GA-Y021, both p1 and p2 still encode their native wt TP-DNAP’s. Any contribution to replication by a third TP-DNAP encoded in trans is monitored by detecting changes in linear plasmid mutation rate. Importantly, these experiments afford each DNAP an opportunity to replicate its native plasmid and lose its attached TP, becoming “spent” and perhaps more likely to replicate its noncognate linear plasmid through DNAP exchange.13 The presence of TP-DNAP1WT or TP-DNAP2WT in trans had no effect on their noncognate linear plasmids’ mutation rate (Figures 3B and 3C, Table S2). Mutagenic variants TP-DNAP1I777K, L900S and TP-DNAP1L477V, L640Y, I777K, W814N increased p1’s mutation rate by 380- and 870-fold, respectively, but caused no statistically significant change in p2’s mutation rate (Figure 3B, 84% CI overlap method).21 Likewise, p1 mutation rate was unaltered in the presence of mutagenic TPDNAP2S370Q and TP-DNAP2Y424Q, which increased p2 mutation rate by 29- and 16-fold, respectively (Figure 3C). Thus, TP-DNAP1 replicates p1 with at least 870-fold specificity over p2, while TP-DNAP2 targets p2 with at least 29-fold specificity over p1. The level of mutual orthogonality measured here is limited by the error-rates of DNAPs used, especially that of TPDNAP2 variants. Future discovery of more mutagenic TP-DNAP1 or TP-DNAP2 variants may prove an even greater orthogonality between these two replication systems, which we expect to be the case.
Figure 3. Mutual orthogonality between p1 and p2 replication.

(A) Schematics detailing strains for measuring p1 mutation rate (left, red) and measuring p2 mutation rate (right, blue). Left: p1 mutation rate reporter strains derived from AR-Y304 encode mKate2, URA3 and leu2* on p1, enabling measurement of p1 copy number and mutation rate in presence of TP-DNAP variants in trans. Right: p2 mutation rate reporter strains derived from GA-Y021 encode mKate2, URA3 and leu2* on p2, enabling measurement of p2 copy number and mutation rate in presence of TP-DNAP variants in trans. (B) Mutagenic TP-DNAP1 variants increase p1 mutation rate (red) by 380- and 870-fold, without any increase in p2 mutation rate (blue). (C) Mutagenic TP-DNAP2 variants increase p2 mutation rate by 16- and 29-fold, without a similar increase in p1 mutation rate. Error bars correspond to 95% confidence intervals calculated according to the MSS method.17–19 See Table S2 for exact mutation rate values.
Conclusion
In summary, p1 replication by TP-DNAP1 and p2 replication by TP-DNAP2 are both orthogonal to genomic replication and to each other, resulting in two mutually orthogonal DNA replication systems in the same cell. We envision that this pair of orthogonal replication systems will enable the in vivo evolution of multiple genes at different elevated mutation rates, molecular recording of biological signals in two distinct DNA channels, and the establishment of additional mutually orthogonal DNAP/plasmid pairs by engineering new TPs.
Methods
DNA cloning
Plasmids cloned and used in this study are listed in Table S4. All oligonucleotide primers and synthesized gene fragments (gBlocks®) were purchased from IDT. Enzymes for PCR and cloning were obtained from NEB. All plasmids, including TP-DNAP2 libraries were cloned using Gibson assembly with overlap regions of 20–30 bp.22 Vectors harboring homologous recombination cassettes for p2 integrations were cloned as previously described for p1 integration cassettes.6,7
To clone pGA55, three gene fragments constituting recoded TP-DNAP2 were assembled with the vector backbone of pAR318, a yeast shuttle vector containing CEN6/ARS4 and HIS3 for propagation in yeast, and ColE1 and KanR for propagation in E. coli. The resulting vector was used for TP-DNAP2 complementation and generation of TP-DNAP2 libraries (see “TP-DNAP2 library generation” section).
Yeast strains
All yeast strains used in this work are listed in Table S5. S. cerevisiae strain AR-Y292 served as the parent for all strains used in this study and contains the wild type pGKL1 and pGKL2 (or p1 and p2) linear plasmids. GA-Y021 and GA-Y069 were created from AR-Y292 by p2 integration methods described below. AR-Y436 is a derivative of AR-Y292 encoding a functional copy of URA3 at the endogenous genomic locus, for 5-FOA-based fluctuation tests of genomic mutation rates in presence of mutagenic TP-DNAP2 variants.
Strains for testing mutual orthogonality were generated by transforming a panel of CEN6/ARS4 vectors encoding TP-DNAP1 or TP-DNAP2 variants into two base strains, AR-Y304 and GA-Y021. AR-Y304 (described previously7) contains recombinant p1 encoding mKate2, URA3 and leu2* without disturbing the native TP-DNAP1 ORF. Similarly, GA-Y021 encodes recombinant p2 that replaces ORF1 with mKate2, URA3 and leu2* without disturbing the native TP-DNAP2 ORF.
Yeast transformation
All yeast transformations were performed using the high-efficiency LiAc/SS carrier DNA/PEG method.23 For integrations into p2, 2–5 μg of plasmid containing the appropriate integration cassette was digested with ScaI, yielding a linearized cassette with blunt ends containing the genes of interest flanked by regions of homology to p2. The products of the digestion reaction were directly transformed into appropriate AR-Y292-derived strains harboring wild type p1 and p2, and plated on selective solid SC medium. Colonies appeared after 4–5 d of growth at 30 °C. CEN6/ARS4 plasmids were also transformed with the LiAc/SS carrier DNA/PEG method, but with only 500–3000 ng of DNA for individual vectors and with at least 10 μg of plasmid DNA for TP-DNAP2 library transformations, to maintain 6-fold coverage.
Linear plasmid DNA extraction
p1, p2, and all derived linear plasmids were extracted using a modified version of the yeast DNA extraction protocol detailed by Amberg et al..24 The modifications were as follows: (i) cells spun down from 40 mL of saturated culture were washed in 0.9% NaCl before treatment with Zymolyase (US Biological) to break up flocculated cells; (ii) 200 μg/mL proteinase K (Sigma) was supplemented during SDS treatment for degradation of TP; (iii) Rotation at ~10 r.p.m. was used during Zymolyase and proteinase K treatments. This large-scale extraction protocol was used for preparing DNA for absolute quantification by qPCR. For qualitative analysis by agarose gel electrophoresis, this extraction protocol was scaled down to extract DNA from only 1.5 mL of saturated yeast culture.
Curing of cytoplasmic plasmids using Cas9
To achieve active curing of the cytoplasmic p2 plasmid, the yeast Cas9 genomic modification system developed by Cate and coworkers was repurposed for cytoplasmic targeting.25 The SV-40 nuclear localization signal and 8× HIS tag were removed from the pCAS plasmid (Addgene plasmid # 60847) to localize Cas9 to the cytoplasm where p2 (and p1) plasmids propagate. Appropriate 20 nt spacers were cloned into this vector to target different sites in p2 (Table S1). These modified pCAS vectors were transformed into the strains harboring p2 plasmids to be cut, and plated on solid selective SC medium containing 1 g/L monosodium glutamate (MSG) as the nitrogen source and supplemented with G418 (400 μg/mL). Colonies that appeared after incubation at 30 °C for 2 d were inoculated into liquid selective SC medium with 1 g/L MSG and G418 (200 μg/mL) and passaged once at a 1:1000 dilution to cure the targeted p2 plasmid. The resulting cultures were then subjected to DNA extraction and analysis by gel electrophoresis to verify loss of the targeted p2 plasmid. To minimize potential toxicity due to Cas9 expression, final strains lacking the pCAS vector were isolated by passaging without G418 selection, and replica plating clones on solid medium with and without G418 to screen for loss of the pCAS vector.
TP-DNAP2 library generation
TP-DNAP1 and TP-DNAP2 peptide sequences were aligned using protein BLAST and four candidates residues for library generation were chosen by two criteria.26 First, candidate TPDNAP2 residues must match a residue in TP-DNAP1 known to affect fidelity, based on prior studies.6,7 Second, at least 25% of the 20 neighboring residues must align. This analysis yielded positions S370, Y424, L474 and F882.
To clone the expression vector for wild type TP-DNAP2 (pGA55), TP-DNAP2 was codon optimized for expression in S. cerevisiae with GenScript’s OptimumGene™ tool, and the recoded ORF was synthesized as three gene fragments, which were assembled downstream of the REV1 promoter in a CEN6/ARS4 vector containing selection markers HIS3 and KanR. The four TPDNAP2 NNK libraries were cloned from pGA55 via Gibson assembly. Gibson fragments containing the NNK codon at each library site were prepared with two sequential PCR’s, in order to limit bias in the NNK incorporation that may result from annealing between the degenerate codon and the plasmid template. The first PCR generated a linear fragment from pGA55 with a 5’ end that terminates immediately 3’ of the library codons, and a 3’ end in KanR. These linear amplicons were purified, diluted to 40 ng/μL, and re-amplified in a second PCR reaction with a forward primer containing Gibson overlap regions and NNK overhangs at the corresponding library site in TP-DNAP2, and the same reverse primer used in the initial PCR. These PCR products were then purified and treated with DpnI for 6 h at 37 °C to digest any pGA55 plasmid carry-through. The second Gibson fragment was PCR amplified from pGA55 to include the vector backbone starting 5’ in KanR and 3’ leading up to, but not including the library codon. For each library, 100 ng of corresponding PCR amplicons were combined in a 20 μL Gibson assembly reaction, and incubated at 50 °C for 1 h. The assemblies were purified and concentrated in 12 μL of ddH2O. 5 μL of the purified assembly products were then transformed into electrocompetent Top10 cells and recovered at 37 °C for 1 h. Each transformation was plated at 1×, 10× and 100× dilutions on solid LB medium supplemented with kanamycin (50 μg/mL). After overnight incubation at 37 °C, colony counts on the 10× an 100× plates were used to calculate the transformation efficiency. All transformations yielded more than 3200 transformants, corresponding to >100-fold coverage of each library in E. coli (each NNK library has a theoretical diversity of 32). The transformants from the 1× plates were then harvested by resuspension in 5 mL of sterile ddH2O. Library plasmid DNA was extracted from E. coli using the Zyppy™ Plasmid Miniprep Kit. Library quality was determined by verifying plasmid library sizes by agarose gel and Sanger sequencing of bulk library populations as well as 8 individual clones from each library.
p2 fluctuation tests using leu2*
For fluctuation tests used to measure p2 mutation rates, leu2* served as a marker for detecting mutational events. leu2* is a disabled version of LEU2, where Q180 is replaced with a TAA stop codon. Q180 is a permissive site where mutation to any other codon other than TAG and TGA results in functional reversion to LEU2. These mutational events can be detected by plating scores of parallel cultures on medium lacking leucine and counting the number distribution of functional LEU2 mutants.
For TP-DNAP2 library screening, each library member was subjected to small-scale leu2* fluctuation tests with six replicates. 190 library members from each yeast library transformation were arrayed and inoculated into liquid SC medium lacking uracil and histidine, and passaged three times at 1:10,000 dilutions. mKate2 fluorescence was measured at every passage on a microplate reader (TECAN Infinite® 200 PRO, settings: λex = 588, λem = 633) to track p2 copy number stabilization. To perform fluctuation tests, each library member was diluted 1:10,000 into liquid SC medium buffered to pH 5.8 and lacking uracil, histidine, tryptophan, and dilutions were split into six 100 μL replicates. Cultures were grown for 48 h at 30 °C to reach saturation. Saturated cultures were washed with 200 μL 0.9% NaCl to remove residual leucine and resuspended in 35 μL of 0.9% NaCl. 10 μL of this resuspension was spot plated onto solid SC medium buffered to pH 5.8 and lacking uracil, histidine, tryptophan and leucine. Spot plates were allowed to dry and incubated at 30 °C. Colonies were counted after 5 d. Colony counts were used to calculate the expected number of LEU2 functional mutants (m), using the p0 method.17
To precisely measure p1 and p2 per-base substitution mutation rates, several modifications were made to the small-scale protocol. First, 36 replicates were performed for reconfirmation of candidate TP-DNAP2 mutators, and 48 replicates for genomic and mutual orthogonality experiments. Titers were also determined for each strain after spot plating by pooling the residual volume from 4 replicates and plating dilutions on YPD. The expected number of LEU2 functional mutants (m) was determined by the Ma-Sandri-Sarkar maximum likelihood estimator (as calculated by the FALCOR tool and corrected for partial plating.17–19 The mean mKate2 fluorescence was determined from 50,000 event counts on Attune™ NxT Flow Cytometer and converted to p2 copy number by using a calibration curve (see below). To determine per-base substitution rates, the corrected m was normalized to the average cell titer, the p2 copy number, and the target size for functional leu2* reversion (2.33 bp). 95% confidence intervals were similarly scaled.
Genomic fluctuation tests using URA3
Fluctuation tests using genomically encoded URA3 were performed in the presence of TP-DNAP2 variants to determine the genomic per-base substitution rates, similarly to previously described protocols.20 AR-Y292 derived strains harboring the appropriate TP-DNAP2 variant encoded on a CEN6/ARS4 vector (with a HIS3 marker) were grown in liquid SC medium lacking uracil and histidine until saturation. Each strain was diluted 1:5,000 into SC medium lacking histidine and aliquoted into 48 replicates of 200 μL each. Cultures were grown for 48 hours at 30 °C to reach saturation. Saturated cultures were washed with 400 μL 0.9% NaCl and resuspended in 420 μL 0.9% NaCl. 400 μL from each replicate was spot plated on pre-dried solid SC medium lacking histidine and supplemented with 5-FOA (1 g/L). The residual 20 μL from six replicates were pooled, diluted, and plated on solid YPD medium to determine cell titers. Plates were allowed to dry before incubation at 30 °C. Colonies were counted on titer plates after 2 days, and on spot plates after 5 days of growth. The expected number of mutants (m) was calculated using the MSS maximum likelihood estimator method via the FALCOR tool, and corrected for partial plating, as described above. To determine per-base substitution rates, the corrected m was normalized to the average cell titer, the URA3 copy number (1 in haploid yeast), and the target size for 5-FOA resistance via substitutions in URA3 (104 bp). 95% confidence intervals were similarly scaled.
Calibration curve relating p2 copy number to mKate2 fluorescence
A standard curve relating p2 copy number to mKate2 fluorescence was prepared by combining quantitative PCR with flow cytometry. During the 1:10,000 back dilution step of the leu2* fluctuation tests for the mutual orthogonality experiment, six strains with mKate2 encoded on p2 were diluted into liquid SC medium buffered to pH 5.8 and lacking uracil, histidine and tryptophan to yield 50 mL of saturated culture. After 48 h of growth at 30 °C, a small portion of each culture was diluted 1:100 in 0.9% NaCl and analyzed on a flow cytometer (Attune™ NxT Flow Cytometer, settings: λex = 561, λem = 620; gain = 550) to determine the mean red fluorescence from 50,000 counts. Genomic DNA and linear plasmids were extracted from the remaining 40 mL of each culture using the large-scale DNA extraction protocol detailed in ‘Linear plasmid DNA extraction’ to ensure complete and unbiased extraction of linear plasmids relative to genomic DNA. All extracts were diluted 4000-fold for use in two distinct qPCR reactions, one to quantify p2-encoded leu2* and the other to quantify the genomic copy of LEU3 (see Table S3 for primers). Each 20 uL qPCR reaction consisted of 5 uL of template DNA, 2 uL forward primer (5 uM), 2 uL reverse primer (5 uM), 1 uL ddH2O, and 10 uL of Thermo Scientific™ Maxima SYBR Green/Fluorescein qPCR Master Mix (2×).
A standard curve for each primer set was prepared by performing qPCR on a dilution series of DNA extracted from F102–2 (25×, 125×, 625×, 3125×). Non-template controls with only ddH2O were included for each primer set to detect contamination. All qPCR’s were performed in triplicate on the Roche LightCycler® 480 System using the following protocol:
qPCR:
-
1)
95°C for 10 min
-
2)40×:
- 95 °C for 15 s
- 60 °C for 1 min
- measurement
-
3)
95 °C for 1 min
-
4)
55 °C for 1 min
Primer melting curve:
Ramp up to 95 °C at 0.11°C/s, with 5 measurements per °C.
Cycle threshold (Ct) values were determined by the LightCycler® 480 software (fit-points method, threshold = 1.75). Ct values from both standard curves were plotted against log (⌈DNA⌉). The slope and y-intercept were calculated using linear regression (Table S3). Each sample’s average Ct values were converted into copy number values by using the following equation: copy number = 10((sample Ct − yintercept)/slope). The calculated leu2* copy number was divided by the LEU3 copy number to normalize to genomes extracted and account for variance in DNA extraction efficiency across samples.
Supplementary Material
Figures S1-4: additional information for design of homologous recombination cassettes, TPDNAP alignment and DNA extractions for orthogonality experiments.
Tables S1-3: additional information for sgRNA design, exact mutation rate values for mutual orthogonality experiments and data collected for the p2 copy number vs. mKate2 calibration curve.
Tables S4-5: plasmids and yeast strains.
Acknowledgments:
We thank members of our group for helpful discussions and suggestions. This research was funded by the National Science Foundation (MCB1545158), the Defense Advanced Research Projects Agency (HR0011–15-2–0031), and the National Institutes of Health (1DP2GM119163–01).
Abbreviations:
- DNAP
DNA polymerase
- wt
wild type
- TP
terminal protein
Footnotes
Conflict of interest: A provisional patent has been filed on this work.
References:
- (1).Cadwell RC, and Joyce GF (1992) Randomization of genes by PCR mutagenesis. Genome Res. 2, 28–33. [DOI] [PubMed] [Google Scholar]
- (2).Metzker ML (2010) Sequencing technologies — the next generation. Nature Reviews Genetics 11, 31–46. [DOI] [PubMed] [Google Scholar]
- (3).Kranaster R, and Marx A (2010) Engineered DNA Polymerases in Biotechnology. ChemBioChem 11, 2077–2084. [DOI] [PubMed] [Google Scholar]
- (4).Taylor AI, Pinheiro VB, Smola MJ, Morgunov AS, Peak-Chew S, Cozens C, Weeks KM, Herdewijn P, and Holliger P (2015) Catalysts from synthetic genetic polymers. Nature 518, 427–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Pinheiro VB, Taylor AI, Cozens C, Abramov M, Renders M, Zhang S, Chaput JC, Wengel J, Peak-Chew S-Y, McLaughlin SH, Herdewijn P, and Holliger P (2012) Synthetic Genetic Polymers Capable of Heredity and Evolution. Science 336, 341–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Ravikumar A, Arzumanyan GA, Obadi MKA, Javanpour AA, and Liu CC (2018) Scalable continuous evolution of genes at mutation rates above genomic error thresholds. bioRxiv 313338 DOI: 10.1101/313338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Ravikumar A, Arrieta A, and Liu CC (2014) An orthogonal DNA replication system in yeast. Nature Chemical Biology 10, 175–177. [DOI] [PubMed] [Google Scholar]
- (8).Perli SD, Cui CH, and Lu TK (2016) Continuous genetic recording with self-targeting CRISPR-Cas in human cells. Science 353, DOI: 10.1126/science.aag0511. [DOI] [PubMed] [Google Scholar]
- (9).Malyshev DA, Dhami K, Lavergne T, Chen T, Dai N, Foster JM, Corrêa IR, and Romesberg FE (2014) A Semi-Synthetic Organism with an Expanded Genetic Alphabet. Nature 509, 385–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Ravikumar A, and Liu CC (2015) Biocontainment through Reengineered Genetic Codes. ChemBioChem 16, 1149–1151. [DOI] [PubMed] [Google Scholar]
- (11).Herr AJ, Ogawa M, Lawrence NA, Williams LN, Eggington JM, Singh M, Smith RA, and Preston BD (2011) Mutator Suppression and Escape from Replication Error–Induced Extinction in Yeast. PLOS Genetics 7, e1002282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Strak MJR, Boyd A, Mileham AJ, and Ramonos MA (1990) The plasmid-encoded killer system of Kluyveromyces lactis: A review. Yeast 6, 1–29. [DOI] [PubMed] [Google Scholar]
- (13).Klassen R, and Meinhardt F (2007) Linear Protein-Primed Replicating Plasmids in Eukaryotic Microbes, in Microbial Linear Plasmids, pp 187–226. Springer, Berlin, Heidelberg. [Google Scholar]
- (14).Rodríguez I, Lázaro JM, Salas M, and De Vega M (2004) phi29 DNA polymerase-terminal protein interaction. Involvement of residues specifically conserved among protein-primed DNA polymerases. J. Mol. Biol 337, 829–841. [DOI] [PubMed] [Google Scholar]
- (15).Mysiak ME, Holthuizen PE, and van der Vliet PC (2004) The adenovirus priming protein pTP contributes to the kinetics of initiation of DNA replication. Nucleic Acids Res 32, 3913–3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Schaffrath R, Stark MJR, Gunge N, and Meinhardt F (1992) Kluyveromyces lactis killer system: ORF1 of pGKL2 has no function in immunity expression and is dispensable for killer plasmid replication and maintenance. Curr Genet 21, 357–363. [DOI] [PubMed] [Google Scholar]
- (17).Foster PL (2006) Methods for Determining Spontaneous Mutation Rates, in Methods in Enzymology, pp 195–213. Academic Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Sarkar S, Ma WT, and Sandri GH (1992) On fluctuation analysis: a new, simple and efficient method for computing the expected number of mutants. Genetica 85, 173–179. [DOI] [PubMed] [Google Scholar]
- (19).Hall BM, Ma C-X, Liang P, and Singh KK (2009) Fluctuation AnaLysis CalculatOR: a web tool for the determination of mutation rate using Luria–Delbrück fluctuation analysis. Bioinformatics 25, 1564–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Lang GI, and Murray AW (2008) Estimating the Per-Base-Pair Mutation Rate in the Yeast Saccharomyces cerevisiae. Genetics 178, 67–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Zheng Q (2016) Comparing mutation rates under the Luria-Delbrück protocol. Genetica 144, 351–359. [DOI] [PubMed] [Google Scholar]
- (22).Gibson DG (2011) Enzymatic assembly of overlapping DNA fragments. Meth. Enzymol 498, 349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Gietz RD, and Schiestl RH (2007) High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc 2, 31–34. [DOI] [PubMed] [Google Scholar]
- (24).Amberg DC (2005) Methods in yeast genetics: a Cold Spring Harbor Laboratory course manual / 2005 ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y. [Google Scholar]
- (25).Ryan OW, Skerker JM, Maurer MJ, Li X, Tsai JC, Poddar S, Lee ME, DeLoache W, Dueber JE, Arkin AP, and Cate JHD (2014) Selection of chromosomal DNA libraries using a multiplex CRISPR system. Elife 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, and Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1-4: additional information for design of homologous recombination cassettes, TPDNAP alignment and DNA extractions for orthogonality experiments.
Tables S1-3: additional information for sgRNA design, exact mutation rate values for mutual orthogonality experiments and data collected for the p2 copy number vs. mKate2 calibration curve.
Tables S4-5: plasmids and yeast strains.