

Abstract
The erlin1/2 complex is a ∼2-MDa endoplasmic reticulum membrane–located ensemble of the ∼40-kDa type II membrane proteins erlin1 and erlin2. The best defined function of this complex is to mediate the ubiquitination of activated inositol 1,4,5-trisphosphate receptors (IP3Rs) and their subsequent degradation. However, it remains unclear how mutations of the erlin1/2 complex affect its cellular function and cause cellular dysfunction and diseases such as hereditary spastic paraplegia. Here, we used gene editing to ablate erlin1 or erlin2 expression to better define their individual roles in the cell and examined the functional effects of a spastic paraplegia–linked mutation to erlin2 (threonine to isoleucine at position 65; T65I). Our results revealed that erlin2 is the dominant player in mediating the interaction between the erlin1/2 complex and IP3Rs and that the T65I mutation dramatically inhibits this interaction and the ability of the erlin1/2 complex to promote IP3R ubiquitination and degradation. Remarkably, we also discovered that the erlin1/2 complex specifically binds to phosphatidylinositol 3-phosphate, that erlin2 binds this phospholipid much more strongly than does erlin1, that the binding is inhibited by T65I mutation of erlin2, and that multiple determinants within the erlin2 polypeptide comprise the phosphatidylinositol 3-phosphate–binding site. Overall, these results indicate that erlin2 is the primary mediator of the cellular roles of the erlin1/2 complex and that disease-linked mutations of erlin2 can affect both IP3R processing and lipid binding.
Keywords: inositol trisphosphate receptor (InsP3R), ubiquitylation (ubiquitination), phosphatidylinositol phosphatase, endoplasmic reticulum (ER), endoplasmic-reticulum-associated protein degradation (ERAD), erlin1, erlin2, mutation, phospholipid signaling, spastic paraplegia
Introduction
Inositol 1,4,5-trisphosphate (IP3)3 receptors (IP3Rs) are tetrameric, IP3- and Ca2+-gated Ca2+ channels located in the endoplasmic reticulum (ER) membrane of vertebrate cells (1–3). IP3Rs function in a plethora of cellular processes, including secretion, proliferation, apoptosis, and fertilization (3). There are three subtypes, IP3R1, IP3R2, and IP3R3, which are expressed in different proportions in different cell types and can assemble into homo- or heterotetramers (1, 4, 5). Each IP3R is ∼2700 amino acids in length, contains six transmembrane domains (1, 2), and is activated by signal transduction cascades beginning at cell surface receptors (e.g. G protein–coupled receptors) that generate IP3 (1–3). The coordinated binding of IP3 and Ca2+ facilitates channel opening and allows for flow of Ca2+ ions stored within the ER lumen to the cytosol (1–3, 6).
Upon stimulation of IP3/IP3R-dependent signaling, a fraction of IP3Rs are polyubiquitinated and thus targeted for proteasomal degradation (7) via the ubiquitin–proteasome pathway (UPP) (8). More specifically, degradation is mediated by the ER-associated degradation (ERAD) pathway, the facet of the UPP responsible for selective degradation of aberrant ER proteins (9, 10). This results in a rapid reduction in the steady-state level of IP3Rs (t½ ∼15–60 min), termed “IP3R down-regulation” (4, 7), and may also be the route by which IP3Rs are turned over under basal conditions (7, 11). Our studies on the mechanism of IP3R processing by the UPP have revealed that a complex formed from an assemblage of the ER membrane proteins erlin1 and erlin2 mediates IP3R polyubiquitination and degradation by binding to activated IP3Rs and recruiting the ubiquitin ligase RNF170 (7, 11–14).
Erlin1 and -2 are ∼40-kDa type II ER membrane proteins with an N-terminal transmembrane domain and the bulk of the polypeptide in the ER lumen (12, 13, 15). They oligomerize into a toroidal, ∼2-MDa complex to which RNF170 is constitutively bound (7, 13, 14). By far the best defined function of erlins to date is to mediate IP3R processing (7), but other possible roles have been proposed, including binding cholesterol (16), regulation of lipid metabolism (16, 17), and regulation of proteins that mediate SV40 exit from the ER (18). Interestingly, mutations to erlin1 and, more commonly, erlin2 have been linked to rare neurodegenerative diseases, e.g. hereditary spastic paraplegia (7, 19–27). As yet, however, it has not been possible to define how the mutations affect the cell biological function of erlin2 or the erlin1/2 complex and cause cell dysfunction (7).
Here, we used gene editing to delete either erlin1 or -2 to better define the role of each protein in terms of IP3R processing. We found that erlin2 is the dominant partner within the erlin1/2 complex and that a novel hereditary spastic paraplegia–linked mutation to erlin2 (threonine to isoleucine at position 65; T65I)4 dramatically inhibits the IP3R-processing function of the complex. Remarkably, we also show that the erlin1/2 complex binds specifically to phosphatidylinositol 3-phosphate (PI(3)P) in a manner inhibited by the T65I mutation to erlin2.
Results
Effects of erlin1 and erlin2 deletion on complex assembly
To define the roles of erlin1 and erlin2 in the erlin1/2 complex, we used TAL-effector nuclease- (TALEN) and CRISPR/Cas9-mediated gene editing technologies (11, 28–30) to ablate expression of either erlin2 or erlin1, respectively, in αT3 mouse pituitary cells, creating E2KO and E1KO cell lines (Fig. 1A). αT3 cells were used because we have previously observed therein robust gonadotropin-releasing hormone (GnRH)-induced, erlin1/2 complex–mediated IP3R1 ERAD (11–14, 31). The ablation of erlin1 or erlin2 was specific because no other pertinent proteins (e.g. the ERAD-related proteins ubiquitin, p97, Hrd1, gp78, and RNF170) (9–14) were affected (Fig. 1A). An exception was IP3R1, the level of which was consistently increased by ∼73 and ∼94% in the absence of either erlin1 or erlin2, respectively (Fig. 1A), suggesting that in addition to their role in processing activated IP3Rs (7, 11–14) the erlins also play a role in basal IP3R1 turnover. This is consistent with the ∼65% elevation of IP3R1 levels seen after RNF170 deletion (11).
Figure 1.

The effects of erlin1 and erlin2 deletion on assembly. A, lysates were made from control, E1KO, and E2KO αT3 cells and probed in immunoblots for ubiquitin, IP3R1, p97, Hrd1, gp78, erlin1, erlin2, and RNF170. B, nondenaturing PAGE of cell lysates probed with anti-erlin1, anti-erlin2, or anti-p97 as a loading control. C, anti-erlin1 or anti-erlin2 immunoprecipitates (IP) from control, E1KO, and E2KO αT3 cells probed for the proteins indicated. D, membrane preparations from control, E1KO, and E2KO αT3 cells (lanes 1–3) and immunoprecipitates from control αT3 cells, incubated either without (lanes 4–6) or with 100 nm GnRH for 5 min (lane 7), were probed in immunoblots for the proteins indicated, including erlin1 and erlin2 with anti-erlinpan (bottom panels). Note that several erlin-unrelated background bands were recognized by anti-erlinpan even in the membrane preparations (lanes 1–3) but that these were not present in immunopurified material (lanes 4–7).
To assess the assembly status of erlin1 and erlin2, lysates from control, E1KO, and E2KO cells were subjected to nondenaturing PAGE (Fig. 1B). This showed that both erlin1 alone (lane 3) and erlin2 alone (lane 5) were able to assemble into high-molecular-weight complexes of size similar to that of the native erlin1/2 complex (lanes 1 and 4). p97, which associates into an ∼600-kDa hexamer (32), served as a loading control. Furthermore, to assess the association status of RNF170, anti-erlin1 and anti-erlin2 immunoprecipitates were examined (Fig. 1C). Interestingly, although RNF170 interacted strongly with immunoprecipitated native erlin1/2 complex (lanes 1 and 2), it bound only weakly with immunoprecipitated erlin2 or erlin1 alone (lanes 4 and 5), suggesting that only the native complex provides the ideal module for RNF170 binding.
Finally, the relative expression of erlin1 and erlin2 and the composition of the erlin1/2 complex were assessed with a newly developed erlinpan antibody, raised against a completely conserved region of erlin1 and -2. This antibody was not entirely specific and strongly cross-reacted with several erlin-unrelated proteins in cell lysates.5 This difficulty was ameliorated by analyzing membrane preparations. The relative intensity of the erlinpan-immunoreactive bands at 43 (erlin2) and 41 kDa (erlin1) revealed that the ratio of erlin2 to erlin1 in αT3 cell membrane extracts was ∼2:1 (Fig. 1D, lane 1), and about the same ratio was found in erlin1/2 complex immunopurified with either anti-erlin1 or anti-erlin2 (Fig. 1D, lanes 4 and 5) or complex associated with activated IP3R1 (lane 7), indicating that all cellular erlin1 and -2 exist in homogeneous complexes with an ∼2:1 ratio. A similar ratio was obtained by mass spectral analysis of erlin1/2 complex immunopurified as in Fig. 1D, lane 5; the number of unique erlin2 and erlin1 peptides identified (an index of relative abundance) was 19 and 13, respectively.
Erlin1 and erlin2 play different roles within the erlin1/2 complex
To define the role(s) of erlin1 and erlin2 in IP3R ERAD, control, E1KO, and E2KO αT3 cells were treated with GnRH, and IP3R1 immunoprecipitates were analyzed. In control cells, GnRH induced rapid ubiquitination of IP3R1 and binding of erlin1, erlin2, and RNF170, reflecting association of the erlin1/2 complex–RNF170 module with activated IP3R1 (Fig. 2A, lanes 1–5). E1KO cells retained the capacity to ubiquitinate IP3R1 upon stimulation with erlin2 and RNF170 clearly coimmunoprecipitating with activated IP3R1 (Fig. 2A, lanes 6–10), indicating that erlin2 alone is capable of binding to activated IP3R1 and recruiting RNF170.6 However, in these E1KO cells, IP3R1 ubiquitination and the association of erlin2 and RNF170 were substantially inhibited relative to control cells (Fig. 2, A, lanes 1–10, and B; ∼67 ± 2% (n = 4) inhibition of peak ubiquitination), indicating that the complex formed by erlin2 and RNF170 functions relatively inefficiently. In contrast, analysis of immunoprecipitates from E2KO cells showed that these cells did not retain the capacity to substantially ubiquitinate activated IP3R1, and, importantly, neither erlin1 nor RNF170 coimmunoprecipitated (Fig. 2A, lanes 11–15), suggesting that erlin1 alone cannot bind to activated IP3R1; IP3R1 ubiquitination was inhibited by ∼94 ± 2% (n = 4) (Fig. 2B).
Figure 2.

The effects of erlin1 and erlin2 deletion on IP3R1 ERAD. A, cells were treated with 100 nm GnRH for 0–20 min, and anti-IP3R1 immunoprecipitates and input lysates were probed in immunoblots for the proteins indicated. B, quantitated IP3R1-associated ubiquitin immunoreactivity graphed as a percentage of control cell maximum (n = 4). C, cells were treated with 100 nm GnRH for 0, 30, or 60 min, and cell lysates were probed for the proteins indicated. D, quantitated IP3R1 immunoreactivity graphed as a percentage of t = 0 values (n = 4). E, [Ca2+]c in control, E1KO, and E2KO αT3 cells exposed to 100 nm GnRH (n ≥ 9). Error bars represent S.E.
Measurement of IP3R1 down-regulation, the decline in steady-state IP3R1 levels that is the culmination of IP3R1 ubiquitination and proteasomal degradation (7), revealed that the response was lost in E2KO cells, whereas it was considerably inhibited in E1KO cells (Fig. 2, C and D). This correlated with the extent of IP3R1 ubiquitination in E2KO and E1KO cells (Fig. 2, A and B). Finally, to check that the reductions in ubiquitination and down-regulation seen after erlin1 or erlin2 deletion are not due to impairment of the IP3/IP3R signaling pathway, we measured Ca2+ mobilization in response to GnRH (Fig. 2E). This revealed that changes in cytosolic free [Ca2+] ([Ca2+]c) were unaffected by erlin1 or erlin2 KO, indicating that the IP3/IP3R signaling pathway is not impaired. Taken together, the data in Figs. 1 and 2 indicate that although both erlin1 and erlin2 alone can assemble into high-molecular-weight complexes and bind RNF170, only erlin2 contains the motif for interaction with activated IP3R1, thereby allowing for IP3R1 ubiquitination.
The T65I mutation to erlin2 inhibits erlin1/2 complex binding to activated IP3Rs and IP3R ERAD
The hereditary spastic paraplegia–linked T65I mutation is in a region highly conserved between erlin2 and erlin1 (Fig. 3A). To explore how the T65I mutation affects erlin2 function, WT human erlin2HA (E2HAWT) and T65I erlin2HA (E2HAT65I) were stably expressed in E2KO αT3 cells (Fig. 3B). Two lines of each were examined (Fig. 3, B–I). E2HAT65I migrated slightly more rapidly that E2HAWT, was expressed at approximately the same levels as E2HAWT (indicating that the mutation does not affect stability), and did not alter the expression level of other relevant proteins (Fig. 3B); notably, however, only E2HAWT restored IP3R1 expression to control levels (compare lanes 3 and 5 with lane 1). Further characterizations showed that both E2HAWT and E2HAT65I were able to assemble into high-molecular-weight species similar to endogenous erlin1/2 complex (Fig. 3C) and, via coimmunoprecipitation, interact normally with endogenous erlin1 and RNF170 (Fig. 3D). Taken together, these data indicate that both E2HAWT and E2HAT65I bind to, and assemble with, their endogenous partners normally.
Figure 3.

Effects of the T65I mutation on erlin2 function. A, ClustalO alignment of the N-terminal regions of erlin1 and erlin2 from various species (Homo sapiens, Mus musculus, and Rattus norvegicus). Amino acid identity is indicated by asterisks; colons and periods indicate strongly and weakly conservative differences, respectively. B, lysates from control and E2KO αT3 cells and E2KO cell lines stably expressing E2HAWT or E2HAT65I (two of each; lanes 3–6) were probed in immunoblots for ubiquitin, IP3R1, p97, erlin1, erlin2, HA, and RNF170. C, nondenaturing PAGE of cell lysates probed with anti-erlin1, anti-erlin2, or anti-p97 as a loading control. D, anti-erlin2 or anti-HA immunoprecipitates (IP) from cell lines stably expressing E2HAWT or E2HAT65I probed for the proteins indicated. E and F, cells were treated with 100 nm GnRH for 0, 5, or 10 min; anti-IP3R1 immunoprecipitates were probed in immunoblots for the proteins indicated; and ubiquitin immunoreactivity was quantitated and graphed (n = 2). G and H, cells were treated with 100 nm GnRH for 0, 30, or 60 min; lysates were probed for the proteins indicated; and IP3R1 immunoreactivity was quantitated and graphed (n = 7). I, [Ca2+]c in cell lines stably expressing E2HAWT or E2HAT65I exposed to 100 nm GnRH (n = 11). Error bars represent S.E.
Analysis of GnRH-induced IP3R1 processing showed that introduction of E2HAWT into E2KO cells restored IP3R1 ubiquitination, whereas, remarkably, E2HAT65I did not (Fig. 3, E and F). Further analysis of the immunoprecipitates showed that erlin1 and RNF170 coimmunoprecipitated with activated IP3R1 when E2HAWT was present (Fig. 3E, lanes 1–3 and 7–9), indicating that E2HAWT allows for the formation of a functional erlin1/2 complex–RNF170 module. In contrast, neither E2HAT65I, erlin1, nor RNF170 associated with activated IP3R1 when E2HAT65I was present (lanes 4–6 and 10–12), indicating that E2HAT65I is incapable of forming a functional erlin1/2 complex–RNF170 module. Consistent with these results, E2HAWT, but not E2HAT65I, was able to mediate IP3R1 down-regulation in response to GnRH (Fig. 3, G and H). The lack of GnRH-induced IP3R1 ubiquitination and down-regulation in E2HAT65I-expressing cells was not due to an impairment of IP3R1 function or IP3-dependent signaling as GnRH-induced Ca2+ mobilization was essentially identical in cells expressing E2HAWT and E2HAT65I (Fig. 3I). Thus, the T65I mutation to erlin2 appears to greatly impair the ability of the erlin1/2 complex to associate with activated IP3Rs and mediate their ERAD.
The erlin1/2 complex specifically binds PI(3)P, and the T65I mutation disrupts this binding
The erlin1/2 complex has been suggested to exist in lipid raft–like, or detergent-resistant, membrane domains within the ER and thus has been suggested to have some lipid-binding capacity, such as binding cholesterol (15, 16). Thus, endogenous erlin1/2 complex was immunopurified from αT3 cells with anti-erlin1, and its binding to a panel of immobilized lipids (33, 34) was examined by probing for erlin2 (Fig. 4). Surprisingly, no specific binding to cholesterol was discovered, but rather binding to phosphatidylinositol 4-phosphate (PI(4)P) was detected (Fig. 4A). Further analysis showed that erlin1/2 complex interacted selectively with monophosphorylated phosphoinositides with the strongest binding to PI(3)P, ∼5-fold greater than that to PI(4)P and phosphatidylinositol 5-phosphate (PI(5)P) (Fig. 4, B and C). This binding appears to be direct as immunopurified erlin1/2 complex is largely free of contaminants (13) with the exception of RNF170 (14), and erlin1/2 complex immunopurified from αT3 cells RNF170KO cells (11) bound phosphoinositides identically to that from control cells (Fig. 4C).
Figure 4.

The erlin1/2 complex binds specifically to PI(3)P. A and B, erlin1/2 complex was immunopurified from control αT3 cells, and binding to lipid arrays was assessed by probing with anti-erlin2. TG, triglyceride; DAG, diacylglycerol; PA, phosphatidic acid; PS, phosphatidylserine; PE, phosphatidylethanolamine; PC, phosphatidylcholine; PI, phosphatidylinositol, PI(x)Py, various PI phosphates; LPA, lysophosphatidic acid; LPC, lysophosphocholine; S1P, sphingosine 1-phosphate. C, erlin1/2 complex was immunopurified from control and RNF170KO αT3 cells (11), and binding of equal amounts of complex to lipid arrays was assessed by probing with anti-erlin2. In the repeat experiment with similar results, RNF170 was depleted using siRNA (14). D, mouse E2FLAGHAWT and E1FLAGHAWT were expressed transiently in αT3 E1/E2KO cells and immunopurified with anti-HA, and binding of equal amounts to lipid arrays was assessed with anti-FLAG. The lowest panel shows an immunoblot of the material added to the lipid arrays probed with anti-FLAG, demonstrating that equal amounts of E1FLAGHAWT and E2FLAGHAWT (migrating at ∼ 47 and 48 kDa, respectively) were used. E, WT and chimeric erlin1 and erlin2 constructs either FLAGHA-tagged or HA-tagged. The switches were made between erlin2 and erlin1 at amino acid positions 31 and 33 and positions 299 and 301. Amino acid homology (identity) between mouse erlin2 and -1 in the N-terminal (NT), intervening, and C-terminal (CT) regions is 33, 86, and 25%, respectively. F, the constructs shown in E were expressed transiently in αT3 E1/E2KO cells and immunopurified with anti-HA, and binding of equal amounts of protein to PI(3)P (800 or 400 pmol) was assessed with anti-FLAG (lanes 1–4), anti-erlin2 (lanes 5 and 6), or anti-erlin1 (lanes 7 and 8). The lower panels show immunoblots of the material added to the PI(3)P arrays probed with the corresponding antibodies, demonstrating that matched amounts of the exogenous proteins were used. The epitopes for anti-erlin1 and anti-erlin2 are located in the C-terminal regions of erlin1 and -2 (13).
To begin to examine the determinants of PI(3)P binding, E1FLAGHAWT or E2FLAGHAWT (Fig. 4E) were expressed in αT3 cells lacking both erlin1 and erlin2 (E1/2KO cells), immunopurified using anti-HA, and probed with anti-FLAG (Fig. 4D). This revealed that E2FLAGHAWT bound much more strongly than E1FLAGHAWT, suggesting that, within the erlin1/2 complex, erlin2 is the primary mediator of binding to PI(3)P. To further explore the PI(3)P-binding determinants in erlin2, we created chimeras in which the N and C termini of erlin2 were replaced with the corresponding regions of erlin1 because the termini are the only regions that differ substantially between erlin1 and -2 (Figs. 4E and 3A) (13). Switching the C termini showed that the C-terminal region of erlin2 contributes to PI(3)P binding because E2FLAGHA bound more strongly than E2(E1CT)FLAGHA (Fig. 4F, lanes 1 and 2), and E1FLAGHA bound less strongly than E1(E2CT)FLAGHA (lanes 3 and 4). Switching the N termini showed that the N-terminal region of erlin2 also contributes to PI(3)P binding because E2HA bound much more strongly than E2(E1NT)HA (Fig. 4F, lanes 5 and 6), and E1HA bound less strongly than E1(E2NT)HA (lanes 7 and 8). Overall, these data indicate that the PI(3)P-binding site in erlin2 is composed of an ensemble of polypeptide regions.
Remarkably, examination of the lipid-binding properties of E2HAT65I showed that it had significantly reduced binding capacity for PI(3)P (Fig. 5). This was the case with material immunopurified from αT3 E2KO cells stably transfected with E2HAWT and E2HAT65I (Fig. 5A), which contain some erlin1 (Fig. 5A, lowest panel), or material immunopurified from transiently transfected αT3 E1/2KO cells, which contain only the exogenous erlin2 constructs (Fig. 5B). Thus, mutation of the Thr-65 region inhibits the ability of complexes containing erlin2 to bind to PI(3)P.
Figure 5.
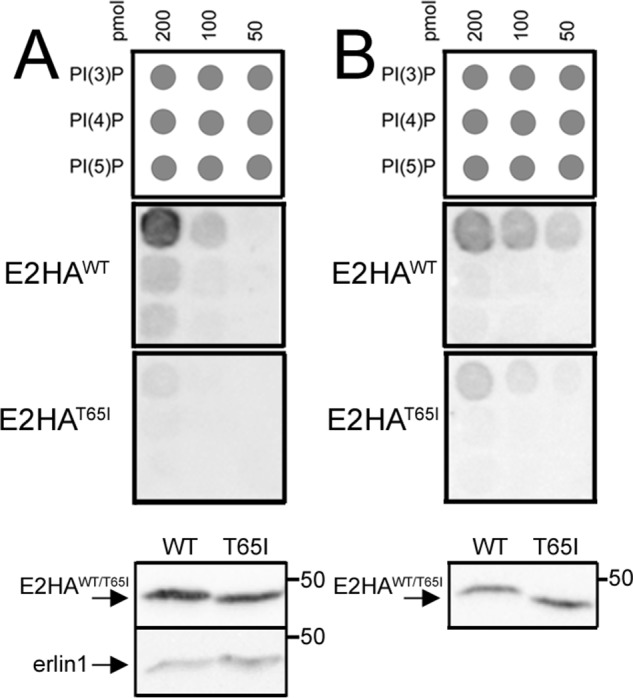
Effects of the T65I mutation on lipid binding. A, erlin complex was immunopurified with anti-HA from αT3 E2KO cell lines stably expressing E2HAWT or E2HAT65I, and binding to lipid arrays was assessed by probing with anti-erlin2. The lowest panels show immunoblots of material added to the lipid arrays probed with anti-erlin2 and anti-erlin1, demonstrating that approximately equal amounts of E1HAWT and E2HAT65I were used and that erlin1 coimmunoprecipitates with E2HAWT and E2HAT65I. B, E2HAWT and E2HAT65I were expressed transiently in αT3 E1/E2KO cells and immunopurified with anti-HA, and binding of equal amounts to lipid arrays was assessed with anti-erlin2. The lowest panel shows an immunoblot of material added to the lipid arrays probed with anti-erlin2, demonstrating that approximately equal amounts of E2HAWT and E2HAT65I were used.
Discussion
We have used gene editing to ablate erlin1 and erlin2 expression from αT3 cells to define the roles of these proteins in the functions of the erlin1/2 complex. We were able to show that although both erlin1 alone and erlin2 alone can form high-molecular-weight complexes and associate with RNF170, only erlin2 can bind efficiently to activated IP3Rs and mediate their ubiquitination and degradation. This indicates that the molecular determinants for the erlin1/2 complex interaction with activated IP3Rs lie within erlin2.
By far the best characterized cellular role of the erlin1/2 complex is to recruit RNF170 to activated IP3Rs and mediate IP3R ERAD (7). Thus, it was important to examine whether disease-linked erlin2 mutations affect this role because a defect in IP3R processing might help explain the pathology. Remarkably, the previously uncharacterized T65I mutation to erlin24 completely blocked the ability of the erlin1/2 complex to associate with activated IP3Rs and mediate their ERAD. This was not because the erlin1/2 complex–RNF170 module was grossly perturbed as T65I erlin2 was fully capable of forming high-molecular-weight complexes and associating with erlin1 and RNF170. That the T65I-containing module fails to associate with activated IP3Rs suggests that the region of erlin2 that binds to activated IP3Rs is in the vicinity of Thr-65 and that the T65I mutation perturbs the binding interface. Clearly, additional investigation of this region may reveal more about how the erlin1/2 complex interacts with its primary target. Unfortunately, however, it has been impossible, so far, to recapitulate the interaction between IP3Rs and erlins in vitro using purified proteins. This has impeded the rapid screening of mutants and indicates that the interaction requires conformational changes in IP3Rs (2) and membrane localization of IP3Rs and erlins only seen in living cells.
We have also made the surprising discovery that the erlin1/2 complex binds specifically to monophosphorylated phosphoinositides and PI(3)P in particular. These lipids play various roles within the cell; e.g. PI(4)P is fairly widely distributed and has a well-established role in controlling cargo exit from the Golgi, whereas PI(5)P distribution is much more restricted and may play a limited role in modulating nuclear functions (33, 34). Much attention has been paid to PI(3)P, which plays at least two major roles: by recruiting a range of adapter proteins to membranes it is critical both to the endocytic pathway and to autophagosome biogenesis at the ER (33–39). Given that the erlin1/2 complex seems to be localized exclusively to the ER (13, 15, 16) and that the erlin1/2 complex binds specifically to PI(3)P, it appears likely that the erlin1/2 complex–PI(3)P interaction has a role in the ER. A major puzzle that remains, however, is that PI(3)P is created and assumed to reside in the cytosol-facing leaflet of the ER membrane (33, 40), although available data indicate that erlin1 and 2 are oriented into the ER lumen by their N-terminal transmembrane domains with only a few residues protruding into the cytosol (13). Thus, it is not currently obvious how PI(3)P and the erlins can be arranged to interact. As with the interaction between the erlin1/2 complex and IP3Rs, the interaction of the erlin1/2 complex with PI(3)P is mediated primarily by erlin2. Analysis of erlin1/2 chimeras suggests that the PI(3)P-binding site in erlin2 is composed of various parts of the polypeptide and, thus, that structural analysis will be required to gain clear insight into how erlin2 and PI(3)P interact in living cells.
Interestingly, PI(3)P binding to erlin2 appears to be mediated, at least in part, by the Thr-65 region because the T65I mutation inhibits PI(3)P binding. Thus, some of the determinants of PI(3)P binding and activated IP3R binding are located in the same region of erlin2, raising the possibility that PI(3)P could be a cofactor for the recognition of activated IP3Rs by the erlin1/2 complex or even contribute to the mysterious events that allow for the retrotranslocation of activated, ubiquitinated IP3Rs from the ER membrane (7). Finally, it is noteworthy that erlin1 and erlin2 are members of the SPFH domain–containing protein family that includes stomatins, prohibitins, and flotillins (15). The association of these proteins with lipid raft–like membrane domains (15) has led to speculation that these proteins bind lipids, which has been borne out by studies showing that, for example, stomatin binds cholesterol (41), stomatin-like protein 2 binds cardiolipin (42), and prohibitin binds PI(3,4,5)P3 (43). Thus, an interaction of the erlin1/2 complex with PI(3)P fits with this familial trait.
In summary, we have shown that erlin2 is the dominant partner in the erlin1/2 complex and contains the determinants for binding to activated IP3Rs and PI(3)P. We have also identified two possible mechanisms by which the T65I mutation to erlin2 could cause hereditary spastic paraplegia. First, as T65I erlin2 is unable to interact with activated IP3Rs, IP3R ERAD is impaired. The expected chronic perturbation of IP3R levels and Ca2+ handling could contribute to neurodegeneration (44). This logic parallels the effects of the R199C mutation to RNF170 that perturbs Ca2+ signaling (11) and causes neurodegeneration and autosomal-dominant sensory ataxia (45). Second, as the T65I mutation reduces the PI(3)P-binding capacity of the erlin1/2 complex, PI(3)P metabolism and PI(3)P-dependent processes may be impaired in T65I-expressing cells with a deleterious effect on cell health.
Experimental procedures
Cells and antibodies
αT3 cells were cultured as described (31). Already available antibodies used were: mouse monoclonal anti-ubiquitin clone FK2 (BioMol International), anti-p97 (Research Diagnostics, Inc.), anti-HA clone HA11 (Covance), and anti-FLAG clone M2 (Sigma) and rabbit polyclonal anti-IP3R1, anti-erlin1, anti-erlin2, anti-RNF170, anti-HA, anti-gp78, and anti-Hrd1 (4, 12–14). An antibody that recognizes both erlin1 and erlin2 (anti-erlinpan) was raised against the conserved peptide HTLQEVYIELFDQIDENLK (residues 131–149 of erlin2) as described (4). Fura-2 AM, GnRH, G418, and other reagents were sourced as described (11–14).
Plasmids
Mouse erlin1 and erlin2 and human erlin1 cDNAs were subcloned from pCMV-Sport6 plasmids originally purchased from ATCC (12, 13). Human erlin2 cDNA was cloned from HeLa cells by RT-PCR as described (14). Human erlin2-HA (E2HAWT) was constructed by PCR amplification of human erlin2 cDNA such that an HA tag (GYPYDVPDYAG) was spliced to the C terminus. FLAGHA-tagged mouse erlin1 and erlin2 (E1FLAGHA and E2FLAGHA) and erlin1/2 chimeras (13) were created by PCR amplification such that a triple FLAGHA tag (DYKDHDGDYKDHDIDYKDDDDKGYPYDVPDYA) was spliced to the C termini. T65I human erlin2 (E2HAT65I) was created using mutagenic PCR. Primer sequences are available upon request.
Generation and analysis of erlin2 and erlin1 knockout and reconstituted αT3 cell lines
The TALEN (28) and CRISPR/Cas9 (29, 30) systems were used to target exons within the erlin2 and erlin1 genes, respectively. Erlin2 was targeted by inserting coding repeats into pcDNA3.1-Talen (+63) to generate erlin2-Talen-L (targeting CTGCTGACCTCCACCA) and erlin2-Talen-R (targeting TCTCATGCTCCCGTTC) constructs. αT3 cells were transfected using FuGENE HD transfection reagent (Promega) with each erlin2-Talen-R and -Talen-L and YFP-C1 (Clontech). For erlin1, oligonucleotides that contained the erlin1 target sequences (exon 1, CGATGCGTCACTGACCGGTGAGG, or exon 2, TCTTGTGGATGGAGGCGTACAGG) were annealed and then ligated into AflII-linearized gRNA vector (AddGene). αT3 cells were transfected with Erlin1-gRNA construct and vectors encoding hCas9 (AddGene) and EGFP (Clontech) (11). Two days after transfection, YFP- or EGFP-expressing cells were selected by FACS and plated at ∼1 cell/well in 96-well plates. Colonies were expanded and screened in immunoblots for erlin1 or erlin2 immunoreactivity, yielding E1KO or E2KO cell lines. To generate E1/2KO cells, erlin1 was targeted in E2KO cells as described. Stable reconstitution was obtained by transfecting αT3 E2KO cells with E2HAWT or E2HAT65I cDNAs using the NEON Transfection System (Invitrogen; 10 μg of total DNA in a 100-μl suspension of cells at 3 × 107/ml, 1 pulse, 20 ms, 1500 V) followed by selection in 1.3 mg/ml G418 for 72 h, plating at ∼1 cell/well in 96-well plates, colony expansion, screening in immunoblots with anti-erlin2 and anti-HA, and maintenance in ∼0.3 mg/ml G418. Two clones expressing each construct were characterized with essentially the same results.
Cell lysis, immunoprecipitation, and analysis by SDS-PAGE
To assess basic protein expression and IP3R down-regulation, αT3 cells were collected in HBSE buffer (155 mm NaCl, 10 mm HEPES, 1 mm EDTA, pH 7.4), incubated with Triton lysis buffer (150 mm NaCl, 50 mm Tris-HCl, 1 mm EDTA, 1% Triton X-100, 10 μm pepstatin, 0.2 mm phenylmethylsulfonyl fluoride, 0.2 μm soybean trypsin inhibitor, 1 mm DTT, pH 8.0), and centrifuged (16,000 × g for 10 min at 4 °C), and supernatant samples were subjected to SDS-PAGE and immunoblotting. Membrane fractions were prepared by harvesting cells in hypotonic homogenization buffer (10 mm Tris base, 1 mm EGTA, 10 μm pepstatin, 0.2 mm phenylmethylsulfonyl fluoride, 0.2 μm soybean trypsin inhibitor, 1 mm DTT, pH 7.4), sonication for 1 min, and centrifugation at 40,000 × g for 10 min at 4 °C. To analyze the interactions among the erlin1/2 complex, RNF170, and IP3R1, cells were disrupted using lysis buffer containing 1% CHAPS rather than Triton X-100. For analysis of IP3R1 ubiquitination, cells were incubated with CHAPS lysis buffer lacking DTT but supplemented with 5 mm N-ethylmaleimide for 30 min at 4 °C followed by addition of 5 mm DTT and centrifugation (16,000 × g for 10 min at 4 °C). Lysates were then incubated with anti-IP3R1 to immunoprecipitate IP3R1 as described (31), and complexes were heated to 37 °C for 30 min before being subjected to SDS-PAGE in 5% gels for ubiquitin conjugate analysis or in 10% gels for analysis of additional proteins. [Ca2+]c was measured as described (11) using cells loaded with 5 μm Fura2-AM for 1 h at 37 °C.
Nondenaturing PAGE
Cells were harvested with CHAPS lysis buffer and centrifuged (16,000 × g for 10 min at 4 °C), and lysates were mixed with 4× sample buffer and Coomassie Blue G-250 to 0.5% (w/v) (46). Proteins (10 μg) were electrophoresed in 3–12 or 4–16% gels, transferred to methanol-primed polyvinylidene difluoride membranes, and processed for immunoblotting (46).
Lipid binding
Immobilized lipid arrays used were either Membrane Lipid or PIP Strips (Echelon Biosciences Inc.) or were made by spotting PI(3)P, PI(4)P, and PI(5)P (all diC16, sodium salts; Echelon Biosciences Inc.) dissolved in chloroform/methanol/water (1:2:0.8, by volume) onto Hybond C or Protran Supported 0.2-μm nitrocellulose membrane (GE Healthcare) essentially as described (47). Arrays were incubated and washed with PBS plus 1% nonfat dried milk at 4 °C as follows: 1 h preincubation to block; 2 × 2-min washes; 6–20-h incubation with immunopurified protein; 2 × 2-min washes; 1-h incubation with anti-erlin2, anti-erlin1, or anti-FLAG; 2 × 2-min washes; 0.75-h incubation with secondary antibody; 3 × 2-min washes; 1 × 2-min wash with PBS; and exposure to enhanced chemiluminescence reagents. Erlin1/2 complex was purified from control and RNF170KO αT3 cells and cells stably transfected with E2HAWT and E2HAT65I using anti-erlin1 and erlin1 peptide for elution (13). HA-tagged and FLAGHA-tagged erlins were purified from NEON/transiently transfected E1/E2KO αT3 cells using anti-HA and HA peptide for elution (13).
Data presentation
All experiments were repeated at least once, and representative images of gels or lipid arrays are shown. Immunoreactivity was detected and quantitated using enhanced chemiluminescence reagents and a GeneGnome (SynGene Bio Imaging) or a Chemidoc (Bio-Rad). The migration positions of molecular mass markers (size in kDa) are shown to the right of immunoblot images. Quantitated data are expressed as mean ± S.E. of n independent experiments.
Author contributions
F. A. W., C. G. B., D. A. S., and R. J. H. W. conceptualization; F. A. W. and R. J. H. W. resources; F. A. W., C. G. B., D. A. S., and R. J. H. W. data curation; F. A. W., C. G. B., D. A. S., and R. J. H. W. formal analysis; F. A. W. and R. J. H. W. supervision; F. A. W. and R. J. H. W. funding acquisition; F. A. W., C. G. B., D. A. S., and R. J. H. W. validation; F. A. W., C. G. B., D. A. S., and R. J. H. W. investigation; F. A. W., C. G. B., D. A. S., and R. J. H. W. visualization; F. A. W., C. G. B., D. A. S., and R. J. H. W. methodology; F. A. W., C. G. B., and R. J. H. W. writing-original draft; F. A. W. and R. J. H. W. project administration; F. A. W., C. G. B., D. A. S., and R. J. H. W. writing-review and editing.
Acknowledgments
We thank Dr. Frédéric Darios (Institut du Cerveau et de la Moelle Epinière, INSERM U1127–CNRS UMR7225–UPMC UMRS1127, Hôpital de la Salpêtrière, 75013 Paris, France) for alerting us to the existence of the T65I mutation and for helpful editorial comments, Olivia Barrett and Dr. Tatyana Fedotova for assisting with lipid binding studies, Dr. Margaret Pearce for generating some of the chimeric constructs, and Jacqualyn Schulman and Laura Szczesniak for support and advice.
This work was supported by National Institutes of Health Grants DK107944 and GM121621. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
F. Darios, personal communication.
F. A. Wright and R. J. H. Wojcikiewicz, unpublished data.
It is intriguing that the amount of RNF170 that coimmunoprecipitates with activated IP3R1 via erlin2 in E1KO cells (Fig. 2A, lanes 6–10) is more than would be expected based on the low amount of RNF170 that coimmunoprecipitates when erlin2 is purified from E1KO cells (Fig. 1C, lane 4). This suggests that in stimulated E1KO cells (Fig. 2A, lanes 6–10) activated IP3R1 associates selectively with RNF170-bound erlin2 complexes.
- IP3
- inositol 1,4,5-trisphosphate
- IP3R
- inositol 1,4,5-trisphosphate receptor
- ER
- endoplasmic reticulum
- UPP
- ubiquitin–proteasome pathway
- ERAD
- ER-associated degradation
- PI(3)P
- phosphatidylinositol 3-phosphate
- GnRH
- gonadotropin-releasing hormone
- [Ca2+]c
- cytosolic Ca2+ concentration
- TALEN
- TAL-effector nuclease
- KO
- knockout
- PI(4)P
- phosphatidylinositol 4-phosphate
- PI(5)P
- phosphatidylinositol 5-phosphate
- PI(3,4,5)P3
- phosphatidylinositol 3,4,5-trisphosphate
- gRNA
- guide RNA
- YFP
- yellow fluorescent protein
- EGFP
- enhanced green fluorescent protein
- E1
- erlin1
- E2
- erlin2.
References
- 1. Foskett J. K., White C., Cheung K. H., and Mak D. O. (2007) Inositol trisphosphate receptor Ca2+ release channels. Physiol. Rev. 87, 593–658 10.1152/physrev.00035.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fan G., Baker M. L., Wang Z., Baker M. R., Sinyagovskiy P. A., Chiu W., Ludtke S. J., and Serysheva I. I. (2015) Gating machinery of InsP3R channels revealed by electron microscopy. Nature 527, 336–341 10.1038/nature15249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Berridge M. J. (2016) The inositol trisphosphate/calcium signaling pathway in health and disease. Physiol. Rev. 96, 1261–1296 10.1152/physrev.00006.2016 [DOI] [PubMed] [Google Scholar]
- 4. Wojcikiewicz R. J. (1995) Type I, II and III inositol 1,4,5 trisphosphate receptors are unequally susceptible to down-regulation and are expressed in markedly different proportions in different cell types. J. Biol. Chem. 270, 11678–11683 10.1074/jbc.270.19.11678 [DOI] [PubMed] [Google Scholar]
- 5. Monkawa T., Miyawaki A., Sugiyama T., Yoneshima H., Yamamoto-Hino M., Furuichi T., Saruta T., Hasegawa M., and Mikoshiba K. (1995) Heterotetrameric complex formation of inositol 1,4,5-trisphosphate receptor subunits. J. Biol. Chem. 270, 14700–14704 10.1074/jbc.270.24.14700 [DOI] [PubMed] [Google Scholar]
- 6. Alzayady K. J., Wang L., Chandrasekhar R., Wagner L. E. 2nd, Van Petegem F., and Yule D. I. (2016) Defining the stoichiometry of inositol 1,4,5-trisphosphate binding required to initiate Ca2+ release. Sci. Signal. 9, ra35 10.1126/scisignal.aad6281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wright F. A., and Wojcikiewicz R. J. (2016) Chapter 4—inositol 1,4,5-trisphosphate receptor ubiquitination. Prog. Mol. Biol. Transl. Sci. 141, 141–159 10.1016/bs.pmbts.2016.02.004 [DOI] [PubMed] [Google Scholar]
- 8. Komander D., and Rape M. (2012) The ubiquitin code. Annu. Rev. Biochem. 81, 203–229 10.1146/annurev-biochem-060310-170328 [DOI] [PubMed] [Google Scholar]
- 9. Ruggiano A., Foresti O., and Carvalho P. (2014) ER-associated degradation: protein quality control and beyond. J. Cell Biol. 204, 869–879 10.1083/jcb.201312042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Printsev I., Curiel D., and Carraway K. L. 3rd (2017) Membrane protein quality control at the endoplasmic reticulum. J. Membr. Biol. 250, 379–392 10.1007/s00232-016-9931-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wright F. A., Lu J. P., Sliter D. A., Dupré N., Rouleau G. A., and Wojcikiewicz R. J. (2015) A point mutation in the ubiquitin ligase RNF170 that causes autosomal dominant sensory ataxia destabilizes the protein and impairs inositol 1,4,5-trisphosphate receptor-mediated Ca2+ signaling. J. Biol. Chem. 290, 13948–13957 10.1074/jbc.M115.655043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pearce M. M., Wang Y., Kelley G. G., and Wojcikiewicz R. J. (2007) SPFH2 mediates the ERAD of IP3 receptors and other substrates in mammalian cells. J. Biol. Chem. 282, 20104–20115 10.1074/jbc.M701862200 [DOI] [PubMed] [Google Scholar]
- 13. Pearce M. M., Wormer D. B., Wilkens S., and Wojcikiewicz R. J. (2009) An endoplasmic reticulum (ER) membrane complex composed of SPFH1 and SPFH2 mediates the ER-associated degradation of inositol 1,4,5-trisphosphate receptors. J. Biol. Chem. 284, 10433–10445 10.1074/jbc.M809801200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lu J. P., Wang Y., Sliter D. A., Pearce M. M., and Wojcikiewicz R. J. (2011) RNF170, an endoplasmic reticulum membrane ubiquitin ligase, mediates inositol 1,4,5-trisphosphate receptor ubiquitination and degradation. J. Biol. Chem. 286, 24426–24433 10.1074/jbc.M111.251983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Browman D. T., Hoegg M. B., and Robbins S. M. (2007) The SPFH domain-containing proteins: more than lipid raft markers. Trends Cell Biol. 17, 394–402 10.1016/j.tcb.2007.06.005 [DOI] [PubMed] [Google Scholar]
- 16. Huber M. D., Vesely P. W., Datta K., and Gerace L. (2013) Erlins restrict SREBP activation in the ER and regulate cholesterol homeostasis. J. Cell Biol. 203, 427–436 10.1083/jcb.201305076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang G., Zhang X., Lee J.-S., Wang X., Yang Z.-Q., and Zhang K. (2012) Endoplasmic reticulum factor erlin2 regulates cytosolic lipid content in cancer cells. Biochem. J. 446, 415–425 10.1042/BJ20112050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Inoue T., and Tsai B. (2017) Regulated Erlin-dependent release of the B12 transmembrane J-protein promotes ER membrane penetration of a non-enveloped virus. PLoS Pathog. 13, e1006439 10.1371/journal.ppat.1006439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yıldırım Y., Orhan E. K., Iseri S. A., Serdaroglu-Oflazer P., Kara B., Solakoğlu S., and Tolun A. (2011) A frameshift mutation of erlin2 in recessive intellectual disability, motor dysfunction and multiple joint contractures. Hum. Mol. Genet. 20, 1886–1892 10.1093/hmg/ddr070 [DOI] [PubMed] [Google Scholar]
- 20. Alazami A. M., Adly N., Al Dhalaan H., and Alkuraya F. S. (2011) A nullimorphic erlin2 mutation defines a complicated hereditary spastic paraplegia locus (SPG18). Neurogenetics 12, 333–336 10.1007/s10048-011-0291-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Al-Saif A., Bohlega S., and Al-Mohanna F. (2012) Loss of erlin2 function leads to juvenile primary lateral sclerosis. Ann. Neurol. 72, 510–516 10.1002/ana.23641 [DOI] [PubMed] [Google Scholar]
- 22. Wakil S. M., Bohlega S., Hagos S., Baz B., Al Dossari H., Ramzan K., and Al-Hassnan Z. N. (2013) A novel splice site mutation in erlin2 causes hereditary spastic paraplegia in a Saudi family. Eur. J. Med. Genet. 56, 43–45 10.1016/j.ejmg.2012.10.003 [DOI] [PubMed] [Google Scholar]
- 23. Novarino G., Fenstermaker A. G., Zaki M. S., Hofree M., Silhavy J. L., Heiberg A. D., Abdellateef M., Rosti B., Scott E., Mansour L., Masri A., Kayserili H., Al-Aama J. Y., Abdel-Salam G. M. H., Karminejad A., et al. (2014) Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science 343, 506–511 10.1126/science.1247363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tian W. T., Shen J. Y., Liu X. L., Wang T., Luan X. H., Zhou H. Y., Chen S. D., Huang X. J., and Cao L. (2016) Novel mutations in endoplasmic reticulum lipid raft-associated protein 2 gene cause pure hereditary spastic paraplegia type 18. Chin. Med. J. 129, 2759–2761 10.4103/0366-6999.193444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fink J. K. (2013) Hereditary spastic paraplegia: clinico-pathologic features and emerging molecular mechanisms. Acta Neuropathol. 126, 307–328 10.1007/s00401-013-1115-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tunca C., Akçimen F., Coşkun C., Gündoğdu-Eken A., Kocoglu C., Çevik B., Bekircan-Kurt C. E., Tan E., and Başak A. N. (2018) ERLIN1 mutations cause teenage-onset slowly progressive ALS in a large Turkish pedigree. Eur. J. Hum. Genet. 26, 745–748 10.1038/s41431-018-0107-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rydning S. L., Dudesek A., Rimmele F., Funke C., Krüger S., Biskup S., Vigeland M. D., Hjorthaug H. S., Sejersted Y., Tallaksen C., Selmer K. K., and Kamm C. (2018) A novel heterozygous variant in ERLIN2 causes autosomal dominant pure hereditary spastic paraplegia. Eur. J. Neurol. 25, 943–e71 10.1111/ene.13625 [DOI] [PubMed] [Google Scholar]
- 28. Li T., Huang S., Zhao X., Wright D. A., Carpenter S., Spalding M. H., Weeks D. P., and Yang B. (2011) Modularly assembled designer TAL effector nucleases for targeted gene knockout and gene replacement in eukaryotes. Nucleic Acids Res. 39, 6315–6325 10.1093/nar/gkr188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mali P., Esvelt K. M., and Church G. M. (2013) Cas9 as a versatile tool for engineering biology. Nat. Methods 10, 957–963 10.1038/nmeth.2649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ran F. A., Hsu P. D., Wright J., Agarwala V., Scott D. A., and Zhang F. (2013) Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 10.1038/nprot.2013.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wojcikiewicz R. J., Xu Q., Webster J. M., Alzayady K., and Gao C. (2003) Ubiquitination and proteasomal degradation of endogenous and exogenous inositol 1,4,5-trisphosphate receptors in αT3-1 anterior pituitary cells. J. Biol. Chem. 278, 940–947 10.1074/jbc.M206607200 [DOI] [PubMed] [Google Scholar]
- 32. Dreveny I., Pye V. E., Beuron F., Briggs L. C., Isaacson R. L., Matthews S. J., McKeown C., Yuan X., Zhang X., and Freemont P. S. (2004) p97 and close encounters of every kind: a brief review. Biochem. Soc. Trans. 32, 715–720 10.1042/BST0320715 [DOI] [PubMed] [Google Scholar]
- 33. Kutateladze T. G. (2010) Translation of the phosphoinositide code by PI effectors. Nat. Chem. Biol. 6, 507–513 10.1038/nchembio.390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Choy C. H., Han B.-K., and Botelho R. J. (2017) Phosphoinositide diversity, distribution, and effector function: stepping out of the box. BioEssays 39, 1700121 10.1002/bies.201700121 [DOI] [PubMed] [Google Scholar]
- 35. Vanhaesebroeck B., Guillermet-Guibert J., Graupera M., and Bilanges B. (2010) The emerging mechanisms of isoform-specific PI3K signaling. Nat. Rev. Mol. Cell Biol. 11, 329–341 10.1038/nrm2882 [DOI] [PubMed] [Google Scholar]
- 36. Marat A. L., and Haucke V. (2016) Phosphatidylinositol 3-phosphates—at the interface between cell signaling and membrane traffic. EMBO J. 35, 561–579 10.15252/embj.201593564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Carlsson S. R., and Simonsen A. (2015) Membrane dynamics in autophagosome biogenesis. J. Cell Sci. 128, 193–205 10.1242/jcs.141036 [DOI] [PubMed] [Google Scholar]
- 38. Nascimbeni A. C., Codogno P., and Morel E. (2017) Local detection of PtdIns3P at autophagosome biogenesis membrane platforms. Autophagy 13, 1602–1612 10.1080/15548627.2017.1341465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Axe E. L., Walker S. A., Manifava M., Chandra P., Roderick H. L., Habermann A., Griffiths G., and Ktistakis N. T. (2008) Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 182, 685–701 10.1083/jcb.200803137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cheng J., Fujita A., Yamamoto H., Tatematsu T., Kakuta S., Obara K., Ohsumi Y., and Fujimoto T. (2014) Yeast and mammalian autophagosomes exhibit distinct phosphatidylinositol 3-phosphatre asymmetries. Nat. Commun. 5, 3207 10.1038/ncomms4207 [DOI] [PubMed] [Google Scholar]
- 41. Rungaldier S., Umlauf E., Mairhofer M., Salzer U., Thiele C., and Prohaska R. (2017) Structure-function analysis of human stomatin: a mutation study. PLoS One 12, e0178646 10.1371/journal.pone.0178646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Christie D. A., Lemke C. D., Elias I. M., Chau L. A., Kirchhof M. G., Li B., Ball E. H., Dunn S. D., Hatch G. M., and Madrenas J. (2011) Stomatin-like protein 2 binds cardiolipin and regulates mitochondrial biogenesis and function. Mol. Cell. Biol. 31, 3845–3856 10.1128/MCB.05393-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ande S. R., and Mishra S. (2009) Prohibitin interacts with phosphatidylinositol 3,4,5-trisphosphate (PIP3) and modulates insulin signaling. Biochem. Biophys. Res. Commun. 390, 1023–1028 10.1016/j.bbrc.2009.10.101 [DOI] [PubMed] [Google Scholar]
- 44. Egorova P. A., and Bezprozvanny I. B. (2018) Inositol 1,4,5-trisphosphate receptors and neurodegenerative disorders. FEBS J. 10.1111/febs.14366 10.1111/febs.14366 [DOI] [PubMed] [Google Scholar]
- 45. Valdmanis P. N., Dupré N., Lachance M., Stochmanski S. J., Belzil V. V., Dion P. A., Thiffault I., Brais B., Weston L., Saint-Amant L., Samuels M. E., and Rouleau G. A. (2011) A mutation in the RNF170 gene causes autosomal dominant sensory ataxia. Brain 134, 602–607 10.1093/brain/awq329 [DOI] [PubMed] [Google Scholar]
- 46. Sliter D. A., Kubota K., Kirkpatrick D. S., Alzayady K. J., Gygi S. P., and Wojcikiewicz R. J. (2008) Mass spectral analysis of type I inositol 1,4,5-trisphosphate receptor ubiquitination. J. Biol. Chem. 283, 35319–35328 10.1074/jbc.M807288200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dowler S., Currie R. A., Campbell D. G., Deak M., Kular G., Downes C. P., and Alessi D. R. (2000) Identification of pleckstrin-homology-domain-containing proteins with novel phosphoinositide-binding specificities. Biochem. J. 351, 19–31 10.1042/bj3510019 [DOI] [PMC free article] [PubMed] [Google Scholar]