

Abstract
Cytoplasmic dynein binds its cargoes via the dynactin complex and cargo adapters, and the dynactin pointed-end protein p25 is required for dynein–dynactin binding to the early endosomal dynein adapter HookA (Hook in the fungus Aspergillus nidulans). However, it is unclear whether the HookA–dynein–dynactin interaction requires p27, another pointed-end protein forming heterodimers with p25 within vertebrate dynactin. Here, live-cell imaging and biochemical pulldown experiments revealed that although p27 is a component of the dynactin complex in A. nidulans, it is dispensable for dynein–dynactin to interact with ΔC-HookA (cytosolic HookA lacking its early endosome–binding C terminus) and is not critical for dynein-mediated early endosome transport. Using mutagenesis, imaging, and biochemical approaches, we found that several p25 regions are required for the ΔC-HookA–dynein–dynactin interaction, with the N terminus and loop1 being the most critical regions. Interestingly, p25 was also important for the microtubule (MT) plus-end accumulation of dynactin. This p25 function in dynactin localization also involved p25's N terminus and the loop1 critical for the ΔC-HookA–dynein–dynactin interaction. Given that dynactin's MT plus-end localization does not require HookA and that the kinesin-1–dependent plus-end accumulation of dynactin is unnecessary for the ΔC-HookA–dynein–dynactin interaction, our results indicate that p25 plays a dual role in cargo binding and dynactin regulation. As cargo adapters are implicated in dynein activation via binding to dynactin's pointed end to switch the conformation of p150, a major dynactin component, our results suggest p25 as a critical pointed-end protein involved in this process.
Keywords: dynein, kinesin, microtubule, molecular motor, cytoskeleton, Aspergillus, dynactin, endosome, intracellular transport, p25
Introduction
Cytoplasmic dynein is a multisubunit minus-end–directed microtubule (MT)2 motor that transports a variety of cargoes (1), and it is critical for the function of all eukaryotic cells, including neurons that are particularly sensitive to dynein defects (2). Dynein-powered transport requires the dynactin complex, which plays an important role in dynein–cargo interactions and dynein activation (3, 4). A dynein-interacting subunit of the dynactin complex is p150Glued (called p150 for simplicity), which contains an N-terminal MT-binding domain followed by coiled-coil domains CC1 and CC2 (5–9). The backbone of the dynactin complex is a mini-filament formed by multiple actin-related protein 1 (Arp1) subunits (10–12). One end of the Arp1 filament is capped by the barbed-end capping proteins (10, 13), and the other end binds to the pointed-end complex containing Arp11, p62, p25, and p27 (14). Although Arp11 and p62 are essential for Arp1 filament integrity and dynein function, the heterodimers p25 and p27 are unnecessary for Arp1 filament assembly and are only required for a subset of dynein functions (15–19). In filamentous fungi, p25 is critical for dynein-mediated transport of early endosomes and post-Golgi vesicles but not for dynein-mediated nuclear distribution (15, 17, 20). In Aspergillus nidulans, p25 is required for the physical interaction between dynein and early endosome (17), but the function of its binding partner p27 is unclear.
The dynein–early endosome interaction in filamentous fungi requires not only the dynactin complex but also the Hook complex (21–23). The C terminus of Hook and the two Hook-interacting proteins, FTS and FHIP, link Hook to early endosomes via the FHIP–Rab5 interaction (21–25). HookA (Hook in A. nidulans) interacts with dynein–dynactin, as revealed by biochemical pulldown assays (21), and this tripartite interaction requires p25 (21). Recently, mammalian Hook proteins and other dynein cargo adapters, including BICD2, have been shown to not only bind dynein–dynactin but also activate dynein's processive motility in vitro (26–29). Because dynein cargo adapters often contain similar domain arrangements, their mechanisms of dynein–dynactin activation may be similar although not identical (1, 26, 28, 30). In the EM structure of the BICD2–dynein–dynactin supercomplex, BICD2 directly contacts the Arp1 filament as well as its pointed end where p150's CC1A (the first part of CC1) domain also seems to contact when p150 is in its folded or “locked” conformation (3, 12). These observations led to an interesting hypothesis that cargo adapters may compete with p150's CC1A domain for binding to the pointed end, thereby causing p150 to assume its open or “unlocked” conformation, which in turn activates dynein (3, 29). However, the identity of pointed-end proteins involved in this process remains unknown. Although p25 is involved in the HookA–dynein–dynactin interaction, we have never tested the role of p27, the binding partner of p25, in this interaction. Because p25 and p27 are required for the stability of each other in mammalian cells (18), it has not been clear whether p27 mediates the function of p25.
In this work, we obtained evidence that p27 in A. nidulans is not critical for the HookA–dynein–dynactin interaction. Thus, within the p25–p27 heterodimer, only p25 is critical for the HookA–dynactin–dynein interaction. We identified several regions of p25 important for this interaction, including the two most critical regions, the N terminus and loop1. Importantly, we found that loss of p25 significantly decreases the MT plus-end accumulation of dynactin in live cells, and the two regions of p25 critical for HookA binding, namely the N terminus and the loop1 region, are also critical for dynactin's MT plus-end accumulation. In conjunction with published structural data (12), our results suggest that p25 is a key pointed-end component involved in both binding HookA and regulating dynactin.
Results
p27 is not critical for the HookA–dynein–dynactin interaction
Although p25 is known to be critical for early endosome transport (17), it has been unknown whether p27, the binding partner of p25 (4, 14, 18, 19), is involved in the same process. A previous sequence analysis indicates that both p25 and p27 adopt the left-handed parallel β-helix (LβH) fold (31). The structure of human p27 has more recently been solved at 2.2 Å resolution, and it indeed forms an LβH domain, followed by a disordered C-terminal segment (19). We identified the p27 protein homolog in A. nidulans as the product of An11815, based on its sequence homology with mammalian p27 and a predicted LβH domain (Fig. S1). To study the function of p27, we created a deletion mutant, Δp27 (Fig. S2). Loss of p27 does not apparently affect colony growth (Fig. 1A). To determine whether loss of p27 affects early endosome distribution, we introduced the Δp27 allele into the strain carrying the early endosome marker mCherry–RabA (32, 33). In filamentous fungi, cytoplasmic dynein and kinesin-3 drive bidirectional transport of early endosomes along microtubules (34, 35). Because microtubule plus-ends face the hyphal tip, dynein drives early endosomes away from the hyphal tip (34, 36–43). In this study, we compared the phenotypes of Δp27 with that of Δp25 under the same conditions. Although the Δp25 mutant defective in dynein-mediated early endosome transport exhibits a dramatic accumulation of early endosome in 100% of the hyphal tips, only about 38% of the Δp27 hyphal tips (n = 203) showed a very mild accumulation of early endosomes (Fig. 1B). Representative kymographs show that whereas early endosomes are mostly nonmobile and accumulated at the hyphal tip region in the Δp25 mutant, they move bi-directionally in the Δp27 mutant and WT cells (Fig. 1C). The overall frequency of dynein-mediated transport of early endosomes away from the hyphal tip is significantly decreased in the Δp25 mutant but not in the Δp27 mutant (Fig. 1D). Consistent with a very mild effect of p27 loss on dynein-mediated early endosome transport, the interaction between HookA and dynein–dynactin is not significantly affected by the loss of p27. Specifically, we introduced S-tagged ΔC-HookA (ΔC-HookA-S) (44) and GFP–dynein heavy chain (HC) (36, 45, 46) into the Δp25 and Δp27 mutants by genetic crosses and tested whether ΔC-HookA-S can be pulled down with GFP–dynein HC in the WT, Δp25, and Δp27 cell extracts (Note that the cytosolic ΔC-HookA is better than the full-length HookA for testing the direct HookA–dynein–dynactin interaction in pulldown assays because it does not bind early endosomes (21), thereby excluding the possibility of indirect interactions due to binding to the same early endosome.). The amount of ΔC-HookA-S pulled down from the Δp25 mutant extract was significantly diminished (Fig. 1, E and F), consistent with our previous conclusion that p25 is critical for the HookA–dynein–dynactin interaction (21). By contrast, loss of p27 did not affect significantly the amount of ΔC-HookA-S pulled down with GFP–dynein HC (Fig. 1, E and F).
Figure 1.

Analysis on the roles of A. nidulans p27 in early endosome movements and dynein–HookA interaction. A, colony phenotypes of a WT control strain, the Δp27 mutant, and the Δp25 mutant. B, microscopic images showing the distributions of mCherry–RabA-labeled early endosomes in WT and the Δp27 and Δp25 mutant strains. Yellow dotted lines show the hyphal shape. Bar, 5 μm. C, kymographs showing early endosome movements. For each kymograph, position of the hyphal tip is on the right side and indicated by a short arrow (hyphal tip is on the last kymograph). D, frequency of dynein-mediated retrograde early endosome movements in WT, the Δp27 mutant, and the Δp25 mutant. The frequency is defined as the number of mCherry–RabA-positive particles moving away from the hyphal tip in a single hypha within a 16-s period (n = 30 hyphae for each strain). The individual values are shown in a scatter plot together with the mean and S.D. values. **** indicates p < 0.0001 (Kruskal-Wallis test, one-way ANOVA, nonparametric test, unpaired, Prism 7). If p > 0.05, the difference is considered to be nonsignificant and not specifically indicated by any * (this applies to all figures in this paper). E, Western blots showing ΔC-HookA-S and dynactin p150 pulled down with GFP–dynein HC in the WT, Δp25, and Δp27 mutants. The bottom blot shows ΔC-HookA-S signals in total extract of each strain used for the pulldown experiment. F, quantitative analysis on the ratio of pulled-down dynactin p150 to GFP–dynein HC (dynactin p150/GFP–dynein HC, red, n = 3 individual pulldown experiments for each strain) and that of pulled-down ΔC-HookA-S to GFP–dynein HC (ΔC-HookA-S/GFP–dynein HC, blue, n = 4 individual pulldown experiments for each strain). Values of the mutants are relative to the values of the WT controls, which are set at 1. The individual values are shown in a scatter plot together with the mean and S.D. values. * indicates p < 0.05 (one-way ANOVA, unpaired).
To verify that A. nidulans p27 is indeed a dynactin component, we constructed a strain containing the p27–GFP fusion for GFP antibody-based pulldown experiments. In the same experiments, we also included a strain containing the p27–GFP fusion in the Δp25 background, a strain containing the p25–GFP fusion, and a strain containing the p25–GFP fusion in the Δp27 background. We found that both dynein heavy chain and dynactin p150 are pulled down with either p25–GFP or p27–GFP in WT extracts (Fig. 2A), and thus, both p25 and p27 are associated with dynactin. Interestingly, loss of p25 (Δp25) significantly lowered the level of p27–GFP in total extract (Fig. 2, A and B). More significantly, we could not even detect any dynein–dynactin pulled down with p27–GFP in the Δp25 mutant (Fig. 2A). In contrast, loss of p27 (Δp27) does not significantly affect the level of p25–GFP (Fig. 2, A and B) or the amounts of dynactin–dynein pulled down with p25–GFP (Fig. 2, A and C). Together, our results suggest that in A. nidulans p25 is required for p27 to bind dynactin, but not vice versa, and thus, p27 is more peripheral than p25 in the A. nidulans dynactin complex (Fig. 2D).
Figure 2.

Effect of Δp25 or Δp27 on the protein levels of p27 or p25 and the interaction of p25 or p27 with dynactin–dynein. A, Western blots showing dynein HC and dynactin p150 pulled down with p25–GFP in WT and Δp27 and with p27–GFP in WT and Δp25. Note that deletion of p27 (Δp27) does not apparently affect the level of p25–GFP or the association of p25 with dynactin p150 and dynein HC. However, deletion of p25 lowers the protein level of p27–GFP and abolishes its interaction with dynactin p150 and dynein HC. Ponceau S staining of the membrane is shown to indicate that similar amounts of total proteins were used as starting materials for different pulldown samples. B, quantitative analysis of the Western results on the protein level of p25 (red) or p27 (blue) in WT and the Δp27 or Δp25 mutant (n = 5 for each strain). Values of the mutants are relative to the values of the WT controls, which are set at 1. The individual values are shown in a scatter plot together with the mean and S.D. values. **** indicates p < 0.0001 (t test, unpaired). C, quantitative analysis on the ratio of pulled-down dynactin p150 to p25–GFP (p150/p25–GFP, red) and that of pulled-down dynein HC to p25–GFP (HC/p25–GFP, blue). Values of the mutants are relative to the values of the WT controls, which are set at 1 (n = 5 individual pulldown experiments for each strain). The individual values are shown in a scatter plot together with the mean and S.D. values. Because we could not detect any dynein HC and dynactin p150 pulled down with p27–GFP from the Δp25 extract (n = 5), we do not present a quantitative analysis of the data. D, cartoon showing the dynactin complex with p27 more peripheral than p25. The MT-binding domains of the p150 dimer are shown as the ear-like structures.
p25K4A/K7G/E9A/Y10G/E12A (N-GA), p25ΔL1 (ΔL1), p25ΔC (ΔC), and p25K36A/Y83A/H113A (KYH) mutants exhibit a defect in the HookA–dynein–dynactin interaction
To identify the p25 residues required for the HookA–dynein–dynactin interaction, we performed a structure–function study of A. nidulans p25. Based on homology modeling, A. nidulans p25 contains, besides the core LβH fold, two loop regions, loop1 (L1) and loop2 (L2), a C-terminal α-helix (C), and several clusters of conserved amino acids whose side chains face outside of the core structure, including the following: 1) Lys-36–Tyr-83–His-113; 2) Arg-82–Lys-146–Glu-147; and 3) Arg-21–Asp-42–Glu-118-Lys-135–Asp-153 (Fig. 3, A and B). We created the p25ΔL1 (called ΔL1 for simplicity) and p25ΔC (called ΔC for simplicity) strains containing deletion of the L1 region or the C-terminal α-helix, and also the p25R97A/R100A/V102A strain containing mutations in L2 (called RRV for simplicity) (Fig. 3, B and C). In addition, we also created the p25K36A/Y83A/H113A (called KYH for simplicity), p25R82A/K146A/E147A (called RKE for simplicity) and p25R21A/D42A/E118A/K135A/D153A (called RDEKD for simplicity) strains, which contain alanine replacements of the residues (Fig. 3, B and C). Finally, we also created the p25K4A/K7G/E9A/Y10G/E12A (called N-GA for simplicity) strain containing glycine and alanine substitutions of five N-terminal residues (Fig. 3, B and C). All the mutant p25 proteins were fused to GFP at the C terminus right before the stop codon, and the mutated DNA fragments replaced the WT p25 gene via homologous recombination (Fig. S3, showing the p25ΔL1-GFP allele as an example). They all form colonies that are healthier than the Δp25 mutant (Fig. 3C). Upon microscopic examination of the early endosome marker mCherry–RabA, we found that the RKE, RRV, and RDEKD mutations have no negative effect on early endosome distribution, and thus they were not further studied (Fig. 4A). However, mutants containing the ΔC, ΔL1, KYH, and N-GA mutations showed abnormal accumulation of early endosomes at the hyphal tip (Fig. 4, A and B). Among the four mutants, the ΔL1 and the N-GA mutants showed the most severe phenotype, although it was still less severe than that of the Δp25 mutant. Specifically, although almost 100% hyphal tips of the Δp25 mutant showed an abnormal accumulation of early endosomes, this was observed in about 75% (n >100) of the ΔL1 and N-GA mutant hyphal tips, 60% (n >200) of the ΔC mutant hyphal tips, and 40% (n >200) of the KYH mutant hyphal tips. These mutants exhibited a decrease in the frequency of dynein-mediated movements of early endosomes away from the hyphal tips, which was again most obvious in the ΔL1 and N-GA mutants (Fig. 4C).
Figure 3.

Analysis of A. nidulans p25 structure and colony phenotypes of various p25 mutants. A, sequence alignment of p25. The first four amino acids of the β-strands are highlighted in gray (each β-strand contains six amino acids with the last two form the turn). Three turns in between strands are colored as yellow, green, and cyan, respectively. The two loops (loop1 and loop2) and the C-terminal α-helix (α-helix) are underlined. The alignment was done using CLUSTALW. Residues that are identical (*), strongly similar (:) or weakly similar (.) are indicated below the sequences. B, model of A. nidulans p25 and positions of the amino acids mutated in this study. The names of the corresponding mutants are labeled in the same color below the names of the regions or the amino acid clusters (each of them is highlighted using the same color as that of the label). The β-strands of the LβH as well as the C-terminal α-helix (labeled on top) are highlighted in gray, and the position of loop1 is indicated with an arrow. C, colony phenotypes of a WT control strain containing p25–GFP (wild type) and various p25 mutant strains.
Figure 4.

Phenotypic analysis of the p25 mutants. A, microscopic images showing the distributions of mCherry–RabA-labeled early endosomes in a WT control strain containing p25–GFP (wild type) and various p25 mutants. Yellow dotted lines show the hyphal shape. Bar, 5 μm. B, kymographs showing early endosome movements in WT, ΔC, ΔL1, KYH, N-GA, and the Δp25 mutants. For each kymograph, position of the hyphal tip is on the right side and indicated by a short arrow (the word hyphal tip is on the last kymograph). C, frequency of retrograde early endosome movement in the ΔC, ΔL1, KYH, and N-GA strains compared with that in the WT control strain. The frequency is defined as the number of mCherry–RabA-positive particles moving away from the hyphal tip in a single hypha within a 16-s period (n = 54 hyphae for WT, n = 46 hyphae for ΔC, n = 50 hyphae for ΔL1, n = 48 hyphae for KYH, n = 30 hyphae for N-GA). The individual values are shown in a scatter plot together with the mean and S.D. values. **** indicates p < 0.0001; *** indicates p < 0.0005; ** indicates p < 0.005; and * indicates p < 0.05 (Kruskal-Wallis test, one-way ANOVA, nonparametric test, unpaired, Prism 7). D, Western blots showing that the ΔC, ΔL1, KYH, and N-GA mutations decrease the amounts of ΔC-HookA-S but not dynactin p150 pulled down with p25–GFP. E, quantitative analysis on the ratio of the pulled-down ΔC-HookA-S to p25–GFP (ΔC-HookA-S/p25–GFP, red) and that of the pulled-down dynactin p150 to p25–GFP (p150/p25–GFP, blue). Note that the individual ratio was calculated by dividing the intensity value of ΔC-HookA-S or p150 by the corresponding value of the WT or the mutant p25–GFP band in the same lane. The ratios were then normalized to the WT ratio, which was set as 1. The individual values are shown in a scatter plot together with the mean and S.D. values. For ΔC-HookA-S/p25–GFP, n = 6 for N-GA and n = 8 individual pulldown experiments for the other strains. For p150/p25–GFP, n = 5 for each strain. **** indicates p < 0.0001 (one-way ANOVA, unpaired).
To determine whether the ΔC, ΔL1, KYH, and the N-AG mutations affect HookA binding, we introduced the ΔC-HookA-S allele into these mutants by genetic crosses and tested whether it can be pulled down with p25–GFP containing one of these mutations (Fig. 4D). In the same assays, we also tested whether the amounts of dynactin p150 are normal in the pulled-down material. Although the WT p25–GFP pulled-down ΔC-HookA-S, the amounts of ΔC-HookA-S pulled down with ΔC-p25–GFP, ΔL1-p25–GFP, KYH-p25–GFP, and N-GA-p25–GFP were significantly decreased (Fig. 4, D and E). However, the amounts of p150 pulled down were not significantly different from normal (Fig. 4, D and E). These results indicate that the defects in binding HookA do not result from the inability of the mutant p25 proteins to bind dynactin. Rather, these mutations specifically affect the ability of p25 to mediate the HookA–dynein–dynactin interaction.
Δp25, p25K4A/K7G/E9A/Y10G/E12A (N-GA), and p25ΔL1 (ΔL1) mutants exhibit a defect in the MT plus-end accumulation of dynactin
The p150 protein, a key component of dynactin (3, 4), is always with the dynactin complex (18, 47), and its stability depends on Arp1 in filamentous fungi (16, 48). In mammalian cells and in A. nidulans, p150–GFP proteins form comet-like structures representing the accumulation of the dynactin complex at the MT plus-ends (49–52). This accumulation depends on the MT-binding domain of p150 as well as kinesin-1 (KinA) (50, 52). In the absence of p25, the comets were obviously less bright, although the organization of MTs labeled by CFP–TubA (53) appeared normal (Fig. 5, A–C, and Fig. S4A). Overall, loss of p25 caused the dynactin signals to appear less concentrated at the MT plus-ends but more obvious along MTs (Fig. 5A and Fig. S4B). This change of the dynactin localization pattern was also obvious in the p25 N-GA and ΔL1 mutants and less so in the ΔC or KLH mutant, although dynactin comet intensity was moderately decreased in the ΔC mutant as indicated by our quantitative analysis (Fig. 5, D–F).
Figure 5.

MT plus-end localization of dynactin is defective in the p25 mutants. A, microscopic images showing the localization pattern of p150–GFP in a WT control strain (wild type) and the Δp25 mutant. Two examples are shown for each. Yellow arrows indicate positions of hyphal tips. B, images of CFP–TubA-labeled MTs in WT and the Δp25 mutant. Arrows indicate positions of hyphal tips. C, quantitative analysis of the comet intensity of p150–GFP in WT and the Δp25 mutant. All the values are relative to the mean value of the WT, which is set at 1. The individual values are shown in a scatter plot together with the mean and S.D. values (n = 13 for WT, n = 14 for the Δp25 mutant). **** indicates p < 0.0001 (t test, unpaired). D, images showing the localization pattern of dynactin (p150–GFP and p25–GFP) in the WT control, ΔC, ΔL1, KYH, and N-GA mutants. Arrows indicate positions of hyphal tips. E, images showing dynactin and CFP–TubA-labeled MT in the ΔL1 mutant. In the merged image, MT is pseudo-colored red and dynactin is green. Yellow arrows indicate positions of hyphal tips. Bar, 5 μm. F, quantitative analysis of the comet intensity of GFP-labeled dynactin (p150–GFP and p25–GFP) in WT and various p25 mutants. All the values are relative to the mean value of the WT, which is set at 1. The individual values are shown in a scatter plot together with the mean and S.D. values (n = 25 for WT; n = 16 for ΔC; n = 17 for ΔL1; n = 17 for KYH; and n = 13 for N-GA). **** indicates p < 0.0001; *** indicates p < 0.0005; ** indicates p < 0.005 (one-way ANOVA, unpaired).
This defect in dynactin localization is not caused by the defect in HookA binding because HookA is unnecessary for the MT plus-end localization of dynactin (Fig. 6A). In fact, the p150–GFP comet intensity in the ΔhookA mutant was even a little higher than that in WT (Fig. 6B). Moreover, because p25 is important for the MT plus-end accumulation of dynactin, we were concerned about the possibility that the requirement of p25 for the HookA–dynein–dynactin interaction could be nonspecifically due to a defect in dynactin localization. To rule out this possibility, we tested whether ΔC-HookA is able to bind dynein–dynactin in the ΔkinA (kinesin-1) mutant where dynactin localizes along MTs instead of the plus-ends, a pattern similar to that in the Δp25 mutant (50, 52). We found that the physical interaction between ΔC-HookA and dynein–dynactin is clearly not defective in the ΔkinA mutant (Fig. 6, C and D). This result demonstrates that the requirement of p25 for the HookA–dynein–dynactin interaction is specific rather than being due to the requirement of p25 for the MT plus-end accumulation of dynactin.
Figure 6.

MT plus-end accumulation of dynactin and the binding of ΔC-HookA to dynein–dynactin are independent of each other. A, microscopic images showing p150–GFP signals in a WT control and the ΔhookA mutant. In both the WT control and the ΔhookA mutant, p150–GFP proteins form comet-like structures near the hyphal tip, representing the MT plus-end accumulation of dynactin. Yellow dotted lines show the hyphal shape. Bar, 5 μm. B, quantitative analysis of the comet intensity in WT and the Δp25 mutant. All the values are relative to the mean value of the WT, which is set at 1 (n = 23 for both strains). The individual values are shown in a scatter plot together with the mean and S.D. values. * indicates p < 0.05 (t test, unpaired). C, Western blots showing that ΔkinA does not apparently affect the interaction between ΔC-HookA–GFP and dynein–dynactin. The Δp25 strain was used as a negative control. D, quantitative analysis on the ratio of the pulled-down p150 to ΔC-HookA–GFP. The ratios of the ΔkinA mutant were normalized to the WT ratios, which were set as 1. The individual values are shown in a scatter plot together with the mean and S.D. values (n = 3 individual pulldown experiments for both strains).
We next determined whether loss of p25 causes any defect in dynactin–KinA (kinesin-1) interaction using pulldown assays. We found that the interaction is weakened in the Δp25 mutant as evidenced by a decrease in the amount of S-tagged KinA (KinA-S) pulled down with p150–GFP (Fig. 7, A and B). In contrast, the amount of S-tagged KinA (KinA-S) pulled down with p150–GFP appeared normal in the alcA–nudAHC strain grown on glucose that shuts off the expression of the dynein HC driven by the alcA promoter (Fig. 7, A and B). Thus, although dynein is not needed for the dynactin–KinA interaction, p25 affects dynactin–KinA interaction. In a separate experiment, we also tested whether dynactin is needed for the dynein–KinA interaction by using the alcA–nudKArp1 strain grown on glucose that shuts off the expression of Arp1. We found that the amount of KinA-S pulled down with GFP-labeled dynein HC is reduced in the absence of Arp1 (Fig. 7, C and D). Together, our results suggest that p25 enhances the dynactin–KinA interaction, and dynactin enhances the dynein–KinA interaction.
Figure 7.
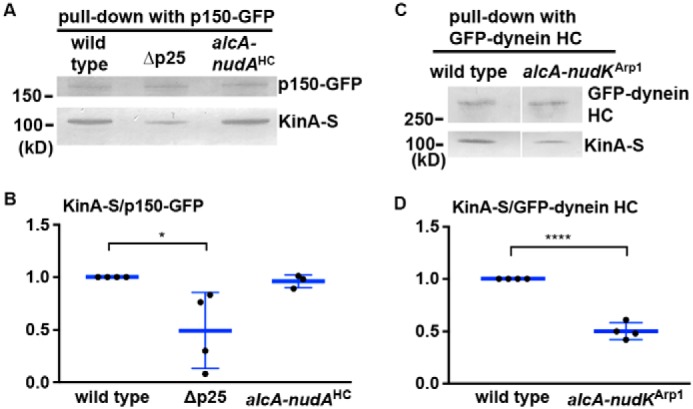
Loss of p25 affects the interaction between dynactin and KinA (kinesin-1), and loss of dynactin affects the interaction between dynein and KinA. A, Western blotting showing that the Δp25 mutation weakens the ability of p150–GFP to pull down KinA-S. In contrast, loss of dynein HC in the alcA–nudAHC strain grown on glucose (a repressive medium for the alcA promoter) does not significantly affect the amount of KinA-S pulled down with p150–GFP. B, quantitative analysis on the ratio of the pulled-down KinA-S to p150–GFP. The ratios of the Δp25 and the alcA–nudAHC mutants were normalized to the WT ratios, which were set as 1. The individual values are shown in a scatter plot together with the mean and S.D. values (n = 4 individual pulldown experiments for WT and Δp25 and n = 3 for alcA–nudAHC). * indicates p < 0.05 (one-way ANOVA, unpaired). C, Western blotting showing that the loss of Arp1 (the backbone of dynactin) in the alcA–nudKArp1 strain weakens the ability of GFP–dynein HC to pull down KinA-S. D, quantitative analysis on the ratio of the pulled-down KinA-S to GFP–dynein HC. The ratios of the alcA–nudKArp1 mutant were normalized to the WT ratios, which were set as 1. The individual values are shown in a scatter plot together with the mean and S.D. values (n = 4 individual pulldown experiments). **** indicates p < 0.0001 (t test, unpaired).
Discussion
In this study, we analyzed two proteins at the pointed end of the Arp1 filament, p25 and p27, in the A. nidulans dynactin complex, and we provided new insights into the function of p25 in cargo binding and dynactin regulation. Both p25 and p27 are conserved proteins that form structures containing an LβH domain (Fig. 3, A and B; Fig. S1) (19). However, although p25 is critical for dynein-mediated early endosome transport (17), p27 is largely dispensable for this process. We also confirmed that only p25 is crucial for the interaction between dynein–dynactin and ΔC-HookA (21). Although p27 appears to be more peripheral than p25 within the dynactin complex, it is not needed for this interaction. We have identified several regions of p25 important for the interaction with HookA, and among them, the N terminus and the loop1 region are the most important.
Our result that p27 is not critical for dynein-mediated early endosome movement in A. nidulans comes as a surprise in light of the results in mammalian cells that knocking down p27 with RNAi significantly lowers the level of p25 and affects early endosome distribution (18). The reason behind this difference in the results remains unclear, but we should point out that although the core structures of p25 proteins are conserved evolutionarily, regions that form interacting sites with other proteins may differ subtly. For example, part of the loop1 region of A. nidulans p25 rich in amino acids Gln and Ser is not present in mammalian p25 (Fig. 3A), although it is present in Ustilago maydis or Neurospora crassa p25 homolog. In addition, whereas the human p25–p27 interaction is likely mediated by the interaction between the p27 hydrophobic ridge on face B and the p25 hydrophobic ridge on face C of their respective triangular structures (19), the analogous hydrophobic ridges in A. nidulans p25 and p27 differ slightly from that in human p25 and p27. In human p25, the hydrophobic ridge contains six hydrophobic residues from position 2 to 7, with Glu at position 1. Unlike human p25, A. nidulans p25 misses part of the rung (Note that a rung contains 3 β strands and 18 amino acids.) that gives rise to the analogous position 1, and its position 2 has Gln instead of a hydrophobic residue (Fig. 8A). Moreover, the hydrophobic residue at position 6 in human or mouse p27 is replaced by Lys in A. nidulans p27 (Fig. S1), and the N terminus of A. nidulans p27 contains a stretch of 38 amino acids not present in its mammalian homologs.
Figure 8.
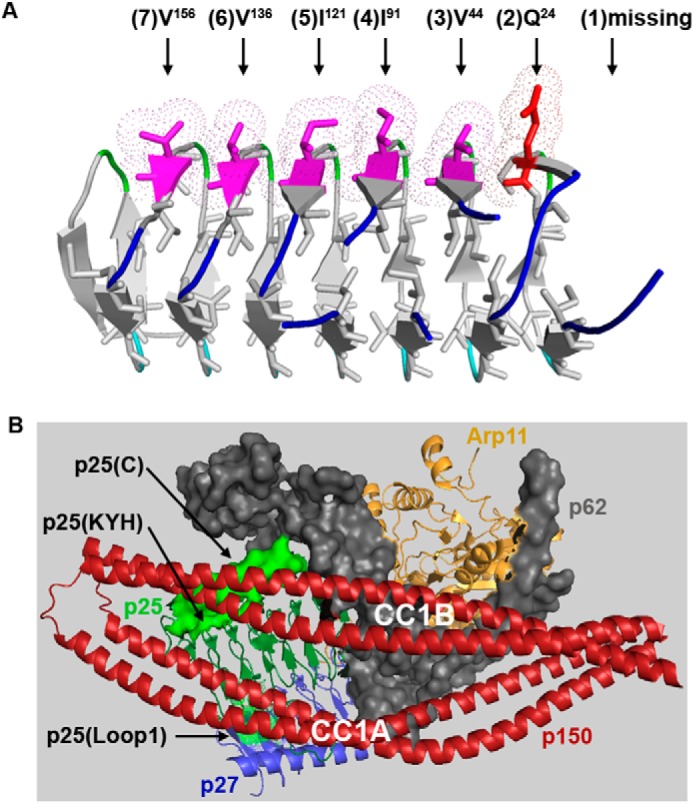
Structural analysis of p25 and the p25–p150 interaction. A, hydrophobic ridge pointing to the surface of the LβH core in the A. nidulans p25. In human p25, the hydrophobic ridge on face C of the triangular structure contains six hydrophobic residues from position 2 to 7 (indicated by black arrows), with Glu at position 1 (19). In comparison, p25 in A. nidulans misses part of the rung (three β strands and 18 amino acids) that gives rise to the analogous position 1, and its position 2 has Gln instead of a hydrophobic residue. B, p25 and p150 proteins are close to each other when p150 is in its folded conformation (12). The published structural data (12) were used for making this figure. The positions of the C terminus (p25(C)) and the KYH (p25(KYH)) cluster (indicated by black arrows) are close to p150's CC1B, whereas that of loop1 (p25(loop1)) (indicated by a black arrow) is close to p150's CC1A. The N terminus was not visible in the structure (12). Other pointed-end proteins such as Arp11, p62, and p27 are indicated by labels using colors corresponding to those in the structure figure.
Despite the subtle difference between the fungal p25/p27 dimer and mammalian p25/p27 dimer, it is most likely that the requirement of p25 in the HookA–dynein–dynactin interaction is evolutionarily conserved. The Hook protein was initially discovered in Drosophila, and three mammalian homologs (Hook1, Hook2, and Hook3) were subsequently identified (54, 55). The Hook–dynein interaction was first suggested by a yeast two-hybrid study showing the interaction between the N terminus of Zyg12, the Caenorhabditis elegans Hook homolog, and dynein light intermediate chain (56). The biochemical evidence for the interaction of ΔC-HookA with both dynein and dynactin was first obtained from pulldown assays using A. nidulans proteins (21). Such an interaction is conserved in mammalian cells (26, 28, 29). In addition, it was first revealed in A. nidulans by using different mutants that both the dynein and dynactin complexes are required for HookA to bind either dynein or dynactin (21). Such a tripartite complex is similar to the BICD2–dynein–dynactin complex (57), and indeed, recent structural studies have revealed that Hook3 and BICD2 interact with dynein–dynactin in a similar albeit nonidentical fashion (29, 30). Hook3 and BICD2 use their N-terminal domains to bind dynein light intermediate chain (29, 58, 59), and the coiled-coil domains of these cargo adapters locate along the Arp1 mini-filament (12, 30, 60). The N-terminal domains in both the A. nidulans HookA and mammalian Hook3 are necessary for the Hook–dynein–dynactin interaction, and an amino acid inside the Hook N termini involved in this interaction is conserved from A. nidulans to mammalian cells (21, 29). Currently, there is no experimental evidence from any higher eukaryotic system to suggest that the Hook–dynein–dynactin interaction requires p25. In structural studies, although BICD2 contacts the pointed end of the Arp1 mini-filament, such a contact was not observed in the case of mammalian Hook3, which could be because the C-terminal part of Hook3 was missing in the structure (30). However, consistent with our data demonstrating the importance of p25 in mediating the HookA–dynein–dynactin interaction, a recent modeling study also suggests a role of mammalian p25 in interacting with the three mammalian Hook proteins (Hook1, Hook2, and Hook3) (61). Based on these studies and our current data, we suggest that both fungal and mammalian p25 could contact Hook proteins directly, and/or it is critical for initiating and/or maintaining the Hook–dynein–dynactin interaction.
The p25 protein in A. nidulans is dispensable for fungal nuclear distribution that requires both dynactin and dynein, but it is required for dynein-mediated transport of not only early endosomes but also post-Golgi vesicles whose movement is HookA-independent (17, 20). Thus, p25 may be required for dynein–dynactin to bind other cargo adapters, an idea consistent with the result obtained from the modeling study suggesting a role of p25 in interacting with not only the Hook proteins but also the BICD proteins (BICD1, BICD2, and BICDR1) (61). However, the amino acids in p25 required for these interactions may not be conserved. In the modeling study, it was predicted that three basic amino acids of mammalian p25, Lys-74, Lys-75, and Lys-78, are involved in binding to the N-terminal part of BICD2 (61). These three amino acids are located in the loop2 region, and Lys-74 corresponds to Arg-97 in loop2 of A. nidulans p25 (Fig. 3A). However, we have not detected any obvious defect in dynein-mediated early endosome distribution exhibited by the p25R97A/R100A/V102A (RRV) mutant (Fig. 4A), suggesting that the two basic amino acids, Arg-97 and Arg-100, in loop2 of A. nidulans p25, are not important for HookA binding. Instead, the N terminus, loop1, the C-terminal α-helix, and the KYH cluster are important with the N terminus, with loop1 being the most critical for HookA binding.
In this study, we revealed that p25 has an important role in MT plus-end accumulation of dynactin, which is consistent with our previous finding that loss of p25 causes a noticeable reduction in the accumulation of dynein at the MT plus-end (17). The defect in dynactin's plus-end accumulation is partly explained by a decreased interaction between dynactin and KinA (kinesin-1) when p25 is absent. We suggest that p25 could either be directly involved in initiating or maintaining the KinA–dynactin interaction, or it is required for maintaining a proper structure of dynactin for its interaction with KinA. Whether p25 affects the integrity of the Arp1 mini-filament in mammalian cells has been in dispute. In one study, knocking down p25 with RNAi does not significantly disrupt the core dynactin complex as evidenced by its apparently normal sucrose gradient sedimentation pattern (18), and in addition, mammalian p25 is not required for the Arp1–dynamitin interaction (63). However, another study suggests that p25 RNAi causes a reduction of the level of Arp1 (64). Nevertheless, the data from A. nidulans using a deletion mutant of p25 strongly argues against a role of p25 in maintaining the integrity of the Arp1 mini-filament, because the amount of Arp1 pulled down with p150 is normal from lysate lacking p25 (17). More interestingly, the amount of dynein pulled down with p150 is increased upon loss of p25 (17), which also argues against the role of p25 in maintaining the length of the Arp1 mini-filament because cryo-EM structures indicate that the Arp1 mini-filament provides sites for dynein to bind (30, 60). In light of these results and the structural analysis suggesting that p150's CC1A and CC1B are close to p25 when p150 is in its folded state (Fig. 8B) (12), we suggest that loss of p25 most likely affects p150 conformation directly.
Importantly, we also found that the N terminus and loop1 of p25 are the most critical for the MT plus-end accumulation of dynactin. In comparison, the C-terminal α-helix or the KYH cluster has a mild or unobvious effect on the plus-end accumulation of dynactin. Thus, although HookA is unnecessary for dynactin's MT plus-end accumulation and the accumulation is unnecessary for dynein–dynactin to bind the cytosolic ΔC-HookA (Fig. 6), both of these events independently require the N terminus and loop1 of p25. According to the structural data (Fig. 8B) (12), the C-terminal α-helix and the KYH cluster are close to p150's CC1B, although loop1 is close to CC1A. The N terminus of p25 was not clearly recognized from the structure but is likely to be close to CC1A just like loop1. It is possible that the N-GA or the Δloop1 mutation weakens the p25–CC1A interaction to unfold p150 abnormally, whereas the ΔC or KYH mutation affects p150 conformation less significantly with the p25–CC1A interaction keeping p150 folded. In light of the idea that a cargo adapter competes against p150's CC1A for binding to the pointed end of the Arp1 mini-filament where it switches the folded p150 to its unfolded state (3, 29), our current data suggest that p25 could be the key protein in this process. It is likely that the N terminus and loop1 of p25 bind directly to HookA as well as to CC1A of p150. In comparison, the C terminus or the KYH cluster of p25 may represent an additional but less critical HookA-binding site or indirectly facilitate HookA binding by subtly modulating the p150–p25 interaction.
Experimental procedures
A. nidulans strains and media
A. nidulans strains used in this study are listed in Table 1. For biochemical experiments, YG (yeast extract plus glucose) + UU (or YUU) liquid medium was used. For live cell imaging experiments, liquid minimal medium containing 1% glycerol or 0.1% glucose plus supplements was used, and cells were cultured at 32 °C overnight and observed at room temperature. Colonies were grown on YAG (YG + agar) plates for 2 days at 37 °C.
Table 1.
Aspergillus nidulans strains used in this study
| Strain | Genotype | Source |
|---|---|---|
| JZ498 | ΔhookA::AfpyrG; argB2::[argB*-alcAp::mCherry-RabA]; ΔnkuA::argB; pyrG89; pyroA4; wA2 | 21 |
| JZ575 | Δp25::AfpyrG; ΔC1-HookA-GFP-AfpyrG, argB2::[argB*-alcAp:: mCherry-RabA]; possibly ΔnkuA::argB; possibly pyrG89; wA2 | 21 |
| JZ569 | ΔC-HookA-GFP-AfpyrG; argB2::[argB*-alcAp::mCherry-RabA]; ΔnkuA::argB; pyrG89; pyroA4; wA2 | 21 |
| RQ2 | GFP-nudAHC; argB2::[argB*-alcAp::mCherry-RabA]; ΔnkuA::argB; pyrG89; pyroA4; yA2 | 62 |
| TNO2A3 | ΔnkuA::argB; pyrG89; pyroA4 | 66 |
| XX223 | Δp25::AfpyrG; GFP-nudAHC; argB2::[argB*-alcAp::mCherry-RabA]; yA2 | 17 |
| XY13 | p150-GFP-AfpyrG; ΔnkuA::argB; pyrG89; pyroA4 | 52 |
| XY21 | p150-GFP-AfpyrG; Δp25::AfpyrG; pyroA4; pyrG89; possibly ΔnkuA::argB | X. Yao |
| XY42 | argB2::[argB*-alcAp::mCherry-RabA]; ΔnkuA::argB; pyrG89; pantoB100; yA2 | X. Yao |
| JZ475 | Δp27::AfpyrG; GFP-nudAHC; argB2::[argB*-alcAp::mCherry-RabA]; ΔnkuA::argB; pyrG89; pyroA4; yA2 | This work |
| JZ463 | Δp27::AfpyrG; ΔnkuA::argB; pyrG89; pyroA4 | This work |
| JZ470 | p27-GFP-AfpyrG; ΔnkuA::argB; pyrG89; pyroA4 | This work |
| JZ679 | p150-GFP-AfpyrG; ΔhookA::AfpyrG; ΔnkuA::argB; argB2::[argB*-alcAp::mCherry-RabA] | This work |
| JZ702 | ΔC-HookA-GFP-AfpyrG; ΔkinA-pyr4; possibly ΔnkuA::argB; pyrG89, yA2 | This work |
| RQ11 | p25ΔC-GFP-AfpyrG; argB2::[argB*-alcAp::mCherry-RabA]; ΔnkuA::argB; pyrG89; pantoB100; yA2 | This work |
| RQ14 | p25ΔL1-GFP-AfpyrG; argB2::[argB*-alcAp::mCherry-RabA]; ΔnkuA::argB; pyrG89; pantoB100; yA2 | This work |
| RQ59 | p150-GFP-AfpyrG; p25ΔL1-GFP-AfpyrG; argB2::[argB*-alcAp::mCherry-RabA]; ΔnkuA::argB; pyrG89; pantoB100 | This work |
| RQ61 | p150-GFP-AfpyrG; p25ΔC-GFP-AfpyrG; argB2::[argB*-alcAp::mCherry-RabA]; ΔnkuA::argB; pyrG89; pyroA4 | This work |
| RQ63 | p150-GFP-AfpyrG; p25-GFP-AfpyrG; possibly pyrG89; possibly ΔnkuA::argB | This work |
| RQ69 | p25-GFP-AfpyrG; argB2::[argB*-alcAp::mCherry-RabA]; ΔnkuA::argB; pantoB100; yA2; possibly pyrG89 | This work |
| RQ83 | p25K36A/Y83A/H113A-GFP-AfpyrG; argB2::[argB*-alcAp::mCherry-RabA]; ΔnkuA::argB; pyrG89; pantoB100; yA2 | This work |
| RQ84 | p25R82A/K146A/E147A-GFP-AfpyrG; argB2::[argB*-alcAp::mCherry-RabA]; ΔnkuA::argB; pyrG89; pantoB100; yA2 | This work |
| RQ87 | p25R97A/R100A/V102A-GFP-AfpyrG; argB2::[argB*-alcAp::mCherry-RabA]; ΔnkuA::argB; pyrG89; pantoB100; yA2 | This work |
| RQ88 | p25R21A/D42A/E118A/K135A/D153A-GFP-AfpyrG; argB2::[argB*-alcAp::mCherry-RabA]; ΔnkuA::argB; pyrG89; pantoB100; yA2 | This work |
| RQ96 | p25-GFP-AfpyrG; Δp27::AfpyrG; argB2::[argB*-alcAp::mCherry-RabA]; possibly ΔnkuA::argB; possibly pyrG89 | This work |
| RQ104 | p27-GFP-AfpyrG; argB2::[argB*-alcAp::mCherry-RabA]; pyroA4; possibly ΔnkuA::argB; possibly pyrG89 | This work |
| RQ128 | ΔC-HookA-S-AfpyrG; argB2::[argB*-alcAp::mCherry-RabA]; ΔnkuA::argB; pyrG89; pyroA4; wA2 | This work |
| RQ132 | ΔC-HookA-S-AfpyrG; p25-GFP-AfpyrG; argB2::[argB*-alcAp::mCherry-RabA]; possibly ΔnkuA::argB | This work |
| RQ133 | ΔC-HookA-S-AfpyrG; p25ΔC-GFP-AfpyrG; argB2::[argB*-alcAp::mCherry-RabA]; possibly ΔnkuA::argB | This work |
| RQ134 | ΔC-HookA-S-AfpyrG; p25ΔL1-GFP-AfpyrG; argB2::[argB*-alcAp::mCherry-RabA]; possibly ΔnkuA::argB | This work |
| RQ135 | ΔC-HookA-S-AfpyrG; p25K36A/Y83A/H113A-GFP-AfpyrG; argB2::[argB*-alcAp::mCherry-RabA]; possibly ΔnkuA::argB | This work |
| RQ137 | ΔC-HookA-S-AfpyrG; GFP-nudAHC; argB2::[argB*-alcAp::mCherry-RabA]; possibly ΔnkuA::argB; possibly pyrG89; pabaA1; wA2 | This work |
| RQ141 | Δp27::AfpyrG; ΔC-HookA-S-AfpyrG; GFP-nudAHC; argB2::[argB*-alcAp::mCherry-RabA]; possibly ΔnkuA::argB; possibly pyrG89 | This work |
| RQ142 | Δp25::AfpyrG; ΔC-HookA-S-AfpyrG; GFP-nudAHC; argB2::[argB*-alcAp::mCherry-RabA]; possibly ΔnkuA::argB; possibly pyrG89 | This work |
| RQ175 | p27-GFP-AfpyrG; Δp25::AfpyrG; argB2::[argB*-alcAp::mCherry-RabA]; yA2; possibly ΔnkuA::argB; possibly pyrG89 | This work |
| RQ177 | p25K4A/K7G/E9A/Y10G/E12A (N-AG)-GFP-AfpyrG; argB2::[argB*-alcAp::mCherry-RabA]; ΔnkuA::argB; pyrG89; pantoB100; yA2 | This work |
| RQ178 | ΔC-HookA-S-AfpyrG; p25K4A/K7G/E9A/Y10G/E12A (N-AG)-GFP-AfpyrG; argB2::[argB*-alcAp::mCherry-RabA]; ΔnkuA::argB | This work |
| RQ182 | p150-GFP-AfpyrG; p25K4A/K7G/E9A/Y10G/E12A (N-AG)-GFP-AfpyrG; argB2::[argB*-alcAp::mCherry-RabA]; ΔnkuA::argB; pyrG89 | This work |
| RQ183 | p150-GFP-AfpyrG; p25K36A/Y83A/H113A-GFP-AfpyrG; argB2::[argB*-alcAp::mCherry-RabA]; ΔnkuA::argB; pyrG89 | This work |
| RQ196 | kinA-S-AfpyrG; argB2::[argB*-alcAp::mCherry-RabA]; ΔnkuA::argB; pyrG89; pantoB100; yA2 | This work |
| RQ203 | p150-GFP-AfpyrG; kinA-S-AfpyrG; argB2::[argB*-alcAp::mCherry-RabA]; possibly ΔnkuA::argB; possibly pyrG89 | This work |
| RQ205 | p150-GFP-AfpyrG; kinA-S-AfpyrG; argB2::[argB*-alcAp::mCherry-RabA]; Δp25::AfpyrG; possibly ΔnkuA::argB; possibly pyrG89 | This work |
| RQ213 | alcA-nudAHC; p150-GFP-AfpyrG; kinA-S-AfpyrG; possibly ΔnkuA::argB; possibly pyrG89; wA2 | This work |
| RQ223 | GFP-nudAHC; kinA-S-AfpyrG; possibly ΔnkuA::argB; possibly pyrG89 | This work |
| RQ224 | GFP-nudAHC; kinA-S-AfpyrG; alcA-nudKArp1; possibly ΔnkuA::argB; possibly pyrG89 | This work |
| RQ261 | p150-GFP-AfpyrG; Δp25::AfpyrG; alcA-CFP-tubA-pyr4; pyrG89; possibly ΔnkuA::argB | This work |
| RQ262 | p150-GFP-AfpyrG; alcA-CFP-tubA-pyr4; pyrG89; possibly ΔnkuA::argB | This work |
| RQ264 | p150-GFP-AfpyrG; p25ΔL1-GFP-AfpyrG; alcA-CFP-tubA-pyr4; pyrG89; possibly ΔnkuA::argB | This work |
Live cell imaging and analyses
Fluorescence microscopy of live A. nidulans hyphae was as described (21). All images were captured at room temperature using an Olympus IX70 inverted fluorescence microscope linked to a Sensicam QE cooled CCD camera (PCO/Cooke Corp.). An UplanApo ×100 objective lens (oil) with a 1.35 numerical aperture was used. A filter wheel system with GFP/mCherry-ET Sputtered series with high transmission (Biovision Technologies) was used. Chroma 8600 filters for CFP were used for observing CFP signals as described previously (53). The IPLab software (Biovision Technologies) was used for image acquisition and analysis, including the measurement of comet intensity as described previously (17).
Construction of the Δp27 and p27–GFP strains
For constructing the Δp27 mutant, we obtained the deletion cassette for the An11815 gene provided by the Fungal Genetics Stock Center (FGSC) (65), which contains the A. fumigatus pyrG gene (AfpyrG) in the middle of the flanking sequences of both sides. The primers used for amplifying the flanking sequences were 5f (5′-GTAACGCCAGGGTTTTCCCAGTCACGACGGGAGTCGTTGGAGATCATAG-3′), 5r (5′-ATCCACTTAACGTTACTGAAATCATAGGGGCGGTTTAATGTCC-3′), 3f (5′-CTCCTTCAATATCATCTTCTGTCCTTTCTTTCGTCCCTACCAC-3′), and 3r (5′-GCGGATAACAATTTCACACAGGAAACAGCCCCTCTACTCCAGCATTTAC-3′). The cassette was transformed into the TNO2A3 strain carrying the ΔnkuA marker that greatly facilitates the selection of homologous recombination events (66). A strain containing the Δp27 allele was selected by PCR using two primers, 27N3 (5′-GAGTACTAAAACTGTGCG-3′) and 27C (5′-TTACCACCTGGCACCTA-3′), which are outside of the cassette. Site-specific integration of the fragment that replaces the WT p27 gene should generate a PCR product different in size from that obtained from a WT control, and this was what we found (Fig. S2). We further verified the Δp27 allele by sequencing both PCR products from WT and from the Δp27 mutant, which confirmed that the AfpyrG gene indeed replaced the An11815 gene encoding p27.
For constructing the p27–GFP fusion, we used the following six oligos to amplify genomic DNA and the GFP–AfpyrG fusion from the plasmid pFNO3 (deposited in the Fungal Genetics Stock Center by S. Osmani, Ohio State University, Columbus) (67): UP1C (5′-TCCCCTCTTCTTCCCAACAATCAGC-3′); UP1N (5′-CCCGCTGGAAGAGTTGCTGTTGAAA-3′); DW2N(5′-TGATATGGCCACTCTGGGGATTCTG-3′); DW2C (5′-TTAATGGGAGCTCGGATTATGAAG-3′); PYRG5 (5′-GCTGATTGTTGGGAAGAAGAGGGGAGGAGCTGGTGCAGGCGCTGGA-3′); and PYRG3 (5′-CAGAATCCCCAGAGTGGCCATATCACTGTCTGAGAGGAGGCACTGAT-3′). We then performed a fusion PCR (68), which generated the p27–GFP–AfpyrG fragment that we used to transform into the TNO2A3 strain carrying the ΔnkuA marker that greatly facilitates the selection of homologous recombination events (66).
Construction of the KinA-S strain
We constructed a DNA fragment containing kinA–S-AfpyrG (S-tag is inserted right before the stop codon of KinA) using the following six oligos: K5U (5′-AGTCTTTCAGAGACGCAGGG-3′); BW2 (5′-TCGTCTATCAAAAAACCAACTTGTG-3′); KF3 (5′-CACAAGTTGGTTTTTTGATAGACGAGGAGCTGGTGCAGGCG-3′); BW4 (5′-CCATCTAGATATCTGCAGGAAGGGGCTGTCTGAGAGGAGGCACTG-3′); BW5 (5′-CCCCTTCCTGCAGATATCTAGATGG-3′); and BW6 (5′-GCTGAAGTTGGTTGATTTGCGG-3′). KF3 and BW4 were used as primers to amplify the DNA containing S-AfpyrG using the ΔC-HookA–S-AfpyrG fragment (44) as a template, and the other oligos were used to amplify the genomic DNA of kinA flanking the S-AfpyrG. The final construct was made after a fusion PCR to fuse three fragments, and it was transformed into the XY42 strain carrying the ΔnkuA marker as well as alcA-mCherry–RabA (66, 69). Correct integration of the construct at the kinA locus was verified by PCR using primers K5UTR (5′-AACGACCTCACAGACTCAC-3′) and AFPyrG3 (5′-GTTGCCAGGTGAGGGTATTT-3′). The strains with the kinA-S fusion replacing the endogenous kinA gene have normal distribution of mCherry–RabA-labeled early endosomes, as opposed to the abnormal buildup of early endosomes at the hyphal tip in ΔkinA cells (69), suggesting that KinA-S is functional.
Homology modeling of A. nidulans p25
When we started this work in 2011, the p27 structure was not available (19). We first used A. nidulans p25 sequence to do a protein blast search against Protein Data Bank (PDB) protein database. The left-handed β-helix protein PDB 1v3w, a carbonic anhydrase from the archaeon Pyrococcus horikoshii, showed the highest sequence similarity with A. nidulans p25, and thus, this protein was selected as a template for p25 structure modeling using PHYRE2 (http://www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index)3 (70). This study led to the predication that A. nidulans p25 contains, besides the core structure of the left-handed β-helix folds, two loop regions and a C-terminal α-helix. We then identified several clusters of amino acids whose side chains face outside of the core structure.
Construction of strains carrying various p25 mutant alleles at the p25 locus
For constructing the p25 mutants containing the p25ΔL1–GFP allele, the following procedure was used. First, the F1-L fragment (∼1.2 kb) was produced by PCR using primers Q1 (5′-GGGATGCCGTTCAGATGATAGAG-3′) and Q7 (5′-TACTGATGGCGTCACCGCGGACGACGGC-3′) and using WT genomic DNA as template. Second, the F2-L fragment (∼4 kb) was produced by PCR using primers Q8 (5′-GTCGTCCGCGGTGACGCCATCAGTATCGGCCGATACACG-3′) and Q6 (5′-CGAATCTTCAACTCCTGGGTGCG-3′) and using genomic DNA template containing the p25–GFP–AfpyrG fusion gene (17). The two fragments were mixed and amplified by fusion PCR using primers Q2 (5′-TGGTAATAGACGGCAGTGGG-3′) and Q5 (5′-TCAACTCCTGGGTGCGAAAT-3′), which produced an ∼5-kb fragment. This fragment was transformed into the XY42 strain carrying the ΔnkuA marker that greatly facilitates the selection of homologous recombination events (66).
For constructing the p25 mutants containing the p25ΔC-GFP allele, the following procedure was used. First, the F1-C fragment (∼1.6 kb) was produced by PCR using primers Q1 (5′-GGGATGCCGTTCAGATGATAGAG-3′) and Q3 (5′-CTCCAGCGCCTGCACCAGCTCCCGCAATTGGCGTATCTCCC-3′) and using WT genomic DNA as template. Second, the F2-C fragment (∼3.6 kb) was produced by PCR using primers Q4 (5′-GGAGCTGGTGCAGGCGCTG-3′) and Q6 (5′-CGAATCTTCAACTCCTGGGTGCG-3′), and using genomic DNA template containing the p25–GFP–AfpyrG fusion gene (17). The two fragments were mixed and amplified by fusion PCR using primers Q2 (5′-TGGTAATAGACGGCAGTGGG-3′) and Q5 (5′-TCAACTCCTGGGTGCGAAAT-3′), which produced an ∼5-kb fragment. This fragment was transformed into the XY42 strain.
For constructing the p25 mutants containing the p25K36A/Y83A/H113A–GFP, p25R82A/K146A/E147A–GFP, and p25R21A/D42A/E118A/K135A/D153A–GFP strains, we first synthesized the DNA containing these point mutations using the DNA synthesis service from GenScript USA Inc. (Piscataway, NJ).
For making the p25K36A/Y83A/H113A mutations, we synthesizedAACACCCTCAAGGAAACCGGCAACAAAGTCTCTCGCCGGTCCCAGATCCATGGAACACACCACATCTCACTCGGGGGGGCCTCGATCATTATGGCCGACGCCGTCGTCCGCGGTGACCTTTTCCGCTCGTCATCCTCGCAATCGCAGTCGCAATCGCAGTCACAGTCACAATCTGGTAGCGGTGCCGGCAACAACAACATCGCCATCAGTATCGGCCGAGCCACGTTCATCTCACGGAGCGCAATCCTCCGCCCGCCCTCTCGTCTCTCGCGTGGTGTCCACACGTACACAACGCTGCATATCGGGTCTGCCGTTTTTGTAGGGGAGCGGAGTATCGTCGAGGCCGCGAAGGTGGAAGATAATGTGACGATTGGGAAGGATTGCGTGATTGGGTCGATGGCGATTCTGAAGGAAAGGTGTCAGGTACTAGACGGGTGCGTTGTGCCGGGGGGCATGGTGGTGCCGAGTCATTGTGTGGTTGGTGGGCAGCCGGCGCGGATTGTG (alanine substitution mutations are underlined).
For making the p25R21A/D42A/E118A/K135A/D153A mutations, we synthesized AACACCCTCAAGGAAACCGGCAACAAAGTCTCTGCCCGGTCCCAGATCCATGGAACACACCACATCTCACTCGGGGGGAAATCGATCATTATGGCCGCCGCCGTCGTCCGCGGTGACCTTTTCCGCTCGTCATCCTCGCAATCGCAGTCGCAATCGCAGTCACAGTCACAATCTGGTAGCGGTGCCGGCAACAACAACATCGCCATCAGTATCGGCCGATACACGTTCATCTCACGGAGCGCAATCCTCCGCCCGCCCTCTCGTCTCTCGCGTGGTGTCCACACGTACACAACGCTGCATATCGGGTCTCATGTTTTTGTAGGGGCCCGGAGTATCGTCGAGGCCGCGAAGGTGGAAGATAATGTGACGATTGGGGCCGATTGCGTGATTGGGTCGATGGCGATTCTGAAGGAAAGGTGTCAGGTACTAGCCGGGTGCGTTGTGCCGGGGGGCATGGTGGTGCCGAGTCATTGTGTGGTTGGTGGGCAGCCGGCGCGGATTGTG (alanine substitution mutations are underlined).
For making the p25R82A/K146A/E147A mutations, we synthesizedAACACCCTCAAGGAAACCGGCAACAAAGTCTCTCGCCGGTCCCAGATCCATGGAACACACCACATCTCACTCGGGGGGAAATCGATCATTATGGCCGACGCCGTCGTCCGCGGTGACCTTTTCCGCTCGTCATCCTCGCAATCGCAGTCGCAATCGCAGTCACAGTCACAATCTGGTAGCGGTGCCGGCAACAACAACATCGCCATCAGTATCGGCGCCTACACGTTCATCTCACGGAGCGCAATCCTCCGCCCGCCCTCTCGTCTCTCGCGTGGTGTCCACACGTACACAACGCTGCATATCGGGTCTCATGTTTTTGTAGGGGAGCGGAGTATCGTCGAGGCCGCGAAGGTGGAAGATAATGTGACGATTGGGAAGGATTGCGTGATTGGGTCGATGGCGATTCTGGCCGCCAGGTGTCAGGTACTAGACGGGTGCGTTGTGCCGGGGGGCATGGTGGTGCCGAGTCATTGTGTGGTTGGTGGGCAGCCGGCGCGGATTGTG (alanine substitution mutations are underlined).
For each of these mutants, the synthesized DNA was amplified to make fragment F2 (∼0.5 kb) using the primers p25NF2 (5′-CTCAAGGAAACCGGCAACAAAG-3′) and p25CR2 (5′-ACACAATGACTCGGCACCACCAT-3′). An upstream fragment (F1, ∼1.1 kb) was amplified from genomic DNA from the strain carrying p25–GFP using primers Q1 and p25NR2 (5′-CTTTGTTGCCGGTTTCCTTGAG-3′), and a downstream fragment (F3, ∼3.7 kb) was amplified using primers p25CF2 (5′-ATGGTGGTGCCGAGTCATTGTGT-3′) and Q6. F1, F2, and F3 were linked together by fusion PCR using the primers Q2 and Q5 to produce a product of ∼5 kb, which was transformed into XY42.
For constructing the strain carrying p25R97A/R100A/V102A–GFP, we used the primers Q1 and loop2R2 (5′-GGCGGAGGATTGCGCTCC-3′) pair and the loop2F (5′-GGAGCGCAATCCTCCGCCCGCCCTCTGCCCTCTCGGCCGGTGCCCACACGTACACAACGCTGCATATCG-3′) and Q6 pair to create ∼1.3- and a 3.9-kb fragments respectively, and we then used Q2 and Q5 for fusion PCR to create a product of ∼5 kb, which was transformed into XY42.
For constructing the strain carrying p25K4A/K7G/E9A/Y10G/E12A–GFP (or p25N-GA), we used the primers Q1 and p25NR (5′-ATTCCTGCGCCACCGGGTGCTGCAGGCGGCATTTTGATAGTTTG-3′) to make fragment 1 (855 bp), and primers p25NF (5′-CCGGTGGCGCAGGAATCGCAACGGTGCGGTCCTCT-3′) and Q6 to make a 4.4-kb fragment 2, and we then used Q2 and Q5 for fusion PCR to create a product of ∼5-kb, which was transformed into XY42.
For all these p25 mutants created by transformation, correct integration of the constructs into the p25 locus was verified by PCR using primers Q1 and GFP-5R (5′-CCAGTGAAAAGTTCTTCTCCTTTAC-3′) (Note that Q1 is at the p25 locus but not within the transformed fragment.) (Fig. S3 shows one example). All of the PCR products were sequenced to further verify the mutations.
Analyses of protein–protein interactions and Western blot analysis
The μMACS GFP-tagged protein isolation kit (Miltenyi Biotec) was used to determine whether GFP-tagged p25 pulls down S-tagged ΔC-HookA. This was done as described previously (21). Strains were grown overnight in liquid rich medium YG. Hyphal mass was harvested from overnight culture by filtering using Miracloth, and liquid was further removed by using paper towels. For each sample, 0.4 g of the hyphal mass was ground and mixed with 1.2 ml of cell lysis buffer containing 50 mm Tris-HCl, pH 8.0, and 10 μg/ml of a protease inhibitor mixture (Sigma). Cell extract was centrifuged at 8,000 × g for 20 min and then 16,000 × g for 10 min at 4 °C, and 1 ml of supernatant was used for the pulldown experiment. To pull down GFP-tagged p25 or p27 protein, 25 μl of anti-GFP MicroBeads was added into 1 ml of cell extract for each sample. To pull down GFP-tagged p150 or dynein HC, 40 μl of anti-GFP MicroBeads was added into 1 ml of cell extract for each sample. The microbeads/cell extracts mixture was rotated at 80 rpm for 30 min at 4 °C and then applied to the μColumn, followed by five times gentle wash with the lysis buffer. 50 μl of pre-heated (95 °C) SDS-PAGE sample buffer was used to elute the proteins, and 38 μl of the eluate was loaded onto each well of an SDS-polyacrylamide gel for Western analyses. Antibodies against GFP and S-tag used for Western analyses were from Clontech (polyclonal) and Cell Signaling Technology, respectively. The antibodies against dynein HC and dynactin p150 were described previously (16, 71). Western analyses were performed using the alkaline phosphatase system, and blots were developed using the alkaline phosphatase color development reagents from Bio-Rad. Quantitation of the protein band intensity was done using the IPLab software as described previously (52, 62).
Statistical analysis
Unpaired Student's t test was used for analyzing two data sets, and one-way ANOVA was used for analyzing multiple data sets (Prism 7 for Mac OS X, version 7.0c, 2017). For analyzing the frequency of dynein-mediated early-endosome movement, the Kruskal–Wallis test (one-way ANOVA, nonparametric test, unpaired) was performed following the recommendations of Prism 7 without assuming Gaussian distribution. All the p values are two-tailed.
Author contributions
R. Q. and X. X. conceptualization; R. Q. resources; R. Q., J. Z., and X. X. data curation; R. Q., J. Z., and X. X. formal analysis; R. Q. and X. X. supervision; R. Q., J. Z., and X. X. validation; R. Q., J. Z., and X. X. investigation; R. Q. and J. Z. methodology; R. Q., X. X., and J. Z. writing-review and editing; X. X. funding acquisition; X. X. and R. Q. writing-original draft.
Supplementary Material
Acknowledgments
We thank Xuanli Yao, Berl Oakley, Miguel Peñalva, Steve Osmani, and Bo Liu for strains and discussions, and the Fungal Genetic Stock Center for plasmids (deposited by Steve Osmani). We also thank Betsy Wang and Max Jin for technical assistance, and Sorana Raiciulescu for help with statistical analyses. National Institutes of Health Program Project Grant GM068087 supported the construction of the Δp27 cassette, which we purchased from the Fungal Genetic Stock Center.
This work was supported by National Institutes of Health Grants RO1 GM097580 and RO1 GM121850 (to X. X.) and a Uniformed Services University bridge grant (to X. X.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S4 and supporting Refs. 1–2.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- MT
- microtubule
- LβH
- left-handed parallel β-helix
- PDB
- Protein Data Bank
- oligo
- oligonucleotide
- ANOVA
- analysis of variance
- HC
- heavy chain
- CFP
- cyan fluorescent protein.
References
- 1. Reck-Peterson S. L., Redwine W. B., Vale R. D., and Carter A. P. (2018) The cytoplasmic dynein transport machinery and its many cargoes. Nat. Rev. Mol. Cell Biol. 19, 382–398 10.1038/s41580-018-0004-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Perlson E., Maday S., Fu M. M., Moughamian A. J., and Holzbaur E. L. (2010) Retrograde axonal transport: pathways to cell death? Trends Neurosci. 33, 335–344 10.1016/j.tins.2010.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cianfrocco M. A., DeSantis M. E., Leschziner A. E., and Reck-Peterson S. L. (2015) Mechanism and regulation of cytoplasmic dynein. Annu. Rev. Cell Dev. Biol. 31, 83–108 10.1146/annurev-cellbio-100814-125438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schroer T. A. (2004) Dynactin. Annu. Rev. Cell Dev. Biol. 20, 759–779 10.1146/annurev.cellbio.20.012103.094623 [DOI] [PubMed] [Google Scholar]
- 5. Holzbaur E. L., Hammarback J. A., Paschal B. M., Kravit N. G., Pfister K. K., and Vallee R. B. (1991) Homology of a 150K cytoplasmic dynein-associated polypeptide with the Drosophila gene glued. Nature 351, 579–583 10.1038/351579a0 [DOI] [PubMed] [Google Scholar]
- 6. Karki S., and Holzbaur E. L. (1995) Affinity chromatography demonstrates a direct binding between cytoplasmic dynein and the dynactin complex. J. Biol. Chem. 270, 28806–28811 10.1074/jbc.270.48.28806 [DOI] [PubMed] [Google Scholar]
- 7. Vaughan K. T., and Vallee R. B. (1995) Cytoplasmic dynein binds dynactin through a direct interaction between the intermediate chains and p150Glued. J. Cell Biol. 131, 1507–1516 10.1083/jcb.131.6.1507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Waterman-Storer C. M., Karki S., and Holzbaur E. L. (1995) The p150Glued component of the dynactin complex binds to both microtubules and the actin-related protein centractin (Arp-1). Proc. Natl. Acad. Sci. U.S.A. 92, 1634–1638 10.1073/pnas.92.5.1634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. King S. J., Brown C. L., Maier K. C., Quintyne N. J., and Schroer T. A. (2003) Analysis of the dynein–dynactin interaction in vitro and in vivo. Mol. Biol. Cell 14, 5089–5097 10.1091/mbc.e03-01-0025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schafer D. A., Gill S. R., Cooper J. A., Heuser J. E., and Schroer T. A. (1994) Ultrastructural analysis of the dynactin complex: an actin-related protein is a component of a filament that resembles F-actin. J. Cell Biol. 126, 403–412 10.1083/jcb.126.2.403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chowdhury S., Ketcham S. A., Schroer T. A., and Lander G. C. (2015) Structural organization of the dynein–dynactin complex bound to microtubules. Nat. Struct. Mol. Biol. 22, 345–347 10.1038/nsmb.2996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Urnavicius L., Zhang K., Diamant A. G., Motz C., Schlager M. A., Yu M., Patel N. A., Robinson C. V., and Carter A. P. (2015) The structure of the dynactin complex and its interaction with dynein. Science 347, 1441–1446 10.1126/science.aaa4080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cooper J. A., and Sept D. (2008) New insights into mechanism and regulation of actin capping protein. Int. Rev. Cell Mol. Biol. 267, 183–206 10.1016/S1937-6448(08)00604-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Eckley D. M., Gill S. R., Melkonian K. A., Bingham J. B., Goodson H. V., Heuser J. E., and Schroer T. A. (1999) Analysis of dynactin subcomplexes reveals a novel actin-related protein associated with the arp1 minifilament pointed end. J. Cell Biol. 147, 307–320 10.1083/jcb.147.2.307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee I. H., Kumar S., and Plamann M. (2001) Null mutants of the neurospora actin-related protein 1 pointed-end complex show distinct phenotypes. Mol. Biol. Cell 12, 2195–2206 10.1091/mbc.12.7.2195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang J., Wang L., Zhuang L., Huo L., Musa S., Li S., and Xiang X. (2008) Arp11 affects dynein–dynactin interaction and is essential for dynein function in Aspergillus nidulans. Traffic 9, 1073–1087 10.1111/j.1600-0854.2008.00748.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang J., Yao X., Fischer L., Abenza J. F., Peñalva M. A., and Xiang X. (2011) The p25 subunit of the dynactin complex is required for dynein–early endosome interaction. J. Cell Biol. 193, 1245–1255 10.1083/jcb.201011022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yeh T. Y., Quintyne N. J., Scipioni B. R., Eckley D. M., and Schroer T. A. (2012) Dynactin's pointed-end complex is a cargo-targeting module. Mol. Biol. Cell 23, 3827–3837 10.1091/mbc.e12-07-0496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yeh T. Y., Kowalska A. K., Scipioni B. R., Cheong F. K., Zheng M., Derewenda U., Derewenda Z. S., and Schroer T. A. (2013) Dynactin helps target Polo-like kinase 1 to kinetochores via its left-handed β-helical p27 subunit. EMBO J. 32, 1023–1035 10.1038/emboj.2013.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Peñalva M. A., Zhang J., Xiang X., and Pantazopoulou A. (2017) Transport of fungal RAB11 secretory vesicles involves myosin-5, dynein/dynactin/p25, and kinesin-1 and is independent of kinesin-3. Mol. Biol. Cell 28, 947–961 10.1091/mbc.e16-08-0566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang J., Qiu R., Arst H. N. Jr., Peñalva M. A., and Xiang X. (2014) HookA is a novel dynein–early endosome linker critical for cargo movement in vivo. J. Cell Biol. 204, 1009–1026 10.1083/jcb.201308009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yao X., Wang X., and Xiang X. (2014) FHIP and FTS proteins are critical for dynein-mediated transport of early endosomes in Aspergillus. Mol. Biol. Cell 25, 2181–2189 10.1091/mbc.e14-04-0873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bielska E., Schuster M., Roger Y., Berepiki A., Soanes D. M., Talbot N. J., and Steinberg G. (2014) Hook is an adapter that coordinates kinesin-3 and dynein cargo attachment on early endosomes. J. Cell Biol. 204, 989–1007 10.1083/jcb.201309022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xu L., Sowa M. E., Chen J., Li X., Gygi S. P., and Harper J. W. (2008) An FTS/Hook/p107(FHIP) complex interacts with and promotes endosomal clustering by the homotypic vacuolar protein sorting complex. Mol. Biol. Cell 19, 5059–5071 10.1091/mbc.e08-05-0473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Guo X., Farías G. G., Mattera R., and Bonifacino J. S. (2016) Rab5 and its effector FHF contribute to neuronal polarity through dynein-dependent retrieval of somatodendritic proteins from the axon. Proc. Natl. Acad. Sci. U.S.A. 113, E5318–E5327 10.1073/pnas.1601844113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McKenney R. J., Huynh W., Tanenbaum M. E., Bhabha G., and Vale R. D. (2014) Activation of cytoplasmic dynein motility by dynactin-cargo adapter complexes. Science 345, 337–341 10.1126/science.1254198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schlager M. A., Hoang H. T., Urnavicius L., Bullock S. L., and Carter A. P. (2014) In vitro reconstitution of a highly processive recombinant human dynein complex. EMBO J. 33, 1855–1868 10.15252/embj.201488792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Olenick M. A., Tokito M., Boczkowska M., Dominguez R., and Holzbaur E. L. (2016) Hook adaptors induce unidirectional processive motility by enhancing the dynein–dynactin interaction. J. Biol. Chem. 291, 18239–18251 10.1074/jbc.M116.738211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schroeder C. M., and Vale R. D. (2016) Assembly and activation of dynein–dynactin by the cargo adaptor protein Hook3. J. Cell Biol. 214, 309–318 10.1083/jcb.201604002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Urnavicius L., Lau C. K., Elshenawy M. M., Morales-Rios E., Motz C., Yildiz A., and Carter A. P. (2018) Cryo-EM shows how dynactin recruits two dyneins for faster movement. Nature 554, 202–206 10.1038/nature25462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Parisi G., Fornasari M. S., and Echave J. (2004) Dynactins p25 and p27 are predicted to adopt the LβH fold. FEBS Lett. 562, 1–4 10.1016/S0014-5793(04)00165-6 [DOI] [PubMed] [Google Scholar]
- 32. Abenza J. F., Galindo A., Pantazopoulou A., Gil C., de los Ríos V., and Peñalva M. A. (2010) Aspergillus RabB Rab5 integrates acquisition of degradative identity with the long distance movement of early endosomes. Mol. Biol. Cell 21, 2756–2769 10.1091/mbc.e10-02-0119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Abenza J. F., Pantazopoulou A., Rodríguez J. M., Galindo A., and Peñalva M. A. (2009) Long-distance movement of Aspergillus nidulans early endosomes on microtubule tracks. Traffic 10, 57–75 10.1111/j.1600-0854.2008.00848.x [DOI] [PubMed] [Google Scholar]
- 34. Lenz J. H., Schuchardt I., Straube A., and Steinberg G. (2006) A dynein loading zone for retrograde endosome motility at microtubule plus-ends. EMBO J. 25, 2275–2286 10.1038/sj.emboj.7601119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wedlich-Söldner R., Straube A., Friedrich M. W., and Steinberg G. (2002) A balance of KIF1A-like kinesin and dynein organizes early endosomes in the fungus Ustilago maydis. EMBO J. 21, 2946–2957 10.1093/emboj/cdf296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Han G., Liu B., Zhang J., Zuo W., Morris N. R., and Xiang X. (2001) The Aspergillus cytoplasmic dynein heavy chain and NUDF localize to microtubule ends and affect microtubule dynamics. Curr. Biol. 11, 719–724 10.1016/S0960-9822(01)00200-7 [DOI] [PubMed] [Google Scholar]
- 37. Konzack S., Rischitor P. E., Enke C., and Fischer R. (2005) The role of the kinesin motor KipA in microtubule organization and polarized growth of Aspergillus nidulans. Mol. Biol. Cell 16, 497–506 10.1091/mbc.e04-02-0083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Efimov V. P., Zhang J., and Xiang X. (2006) CLIP-170 homologue and NUDE play overlapping roles in NUDF localization in Aspergillus nidulans. Mol. Biol. Cell 17, 2021–2034 10.1091/mbc.e05-11-1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Egan M. J., Tan K., and Reck-Peterson S. L. (2012) Lis1 is an initiation factor for dynein-driven organelle transport. J. Cell Biol. 197, 971–982 10.1083/jcb.201112101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zeng C. J., Kim H. R., Vargas Arispuro I., Kim J. M., Huang A. C., and Liu B. (2014) Microtubule plus end-tracking proteins play critical roles in directional growth of hyphae by regulating the dynamics of cytoplasmic microtubules in Aspergillus nidulans. Mol. Microbiol. 94, 506–521 10.1111/mmi.12792 [DOI] [PubMed] [Google Scholar]
- 41. Xiang X., Qiu R., Yao X., Arst H. N. Jr., Peñalva M. A., and Zhang J. (2015) Cytoplasmic dynein and early endosome transport. Cell. Mol. Life Sci. 72, 3267–3280 10.1007/s00018-015-1926-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Steinberg G., Penalva M. A., Riquelme M., Wosten H. A., and Harris S. D. (2017) Cell biology of hyphal growth. Microbiol. Spectr. 5 10.1128/microbiolspec.FUNK-0034-2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Riquelme M., Aguirre J., Bartnicki-García S., Braus G. H., Feldbrügge M., Fleig U., Hansberg W., Herrera-Estrella A., Kämper J., Kück U., Mouriño-Pérez R. R., Takeshita N., and Fischer R. (2018) Fungal morphogenesis, from the polarized growth of hyphae to complex reproduction and infection structures. Microbiol. Mol. Biol. Rev. 82, e00068–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang J., Qiu R., and Xiang X. (2018) The actin capping protein in Aspergillus nidulans enhances dynein function without significantly affecting Arp1 filament assembly. Sci. Rep. 8, 11419 10.1038/s41598-018-29818-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhuang L., Zhang J., and Xiang X. (2007) Point mutations in the stem region and the fourth AAA domain of cytoplasmic dynein heavy chain partially suppress the phenotype of NUDF/LIS1 loss in Aspergillus nidulans. Genetics 175, 1185–1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Xiang X., Han G., Winkelmann D. A., Zuo W., and Morris N. R. (2000) Dynamics of cytoplasmic dynein in living cells and the effect of a mutation in the dynactin complex actin-related protein Arp1. Curr. Biol. 10, 603–606 10.1016/S0960-9822(00)00488-7 [DOI] [PubMed] [Google Scholar]
- 47. Zhang J., Twelvetrees A. E., Lazarus J. E., Blasier K. R., Yao X., Inamdar N. A., Holzbaur E. L., Pfister K. K., and Xiang X. (2013) Establishing a novel knock-in mouse line for studying neuronal cytoplasmic dynein under normal and pathologic conditions. Cytoskeleton 70, 215–227 10.1002/cm.21102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Minke P. F., Lee I. H., Tinsley J. H., Bruno K. S., and Plamann M. (1999) Neurospora crassa ro-10 and ro-11 genes encode novel proteins required for nuclear distribution. Mol. Microbiol. 32, 1065–1076 10.1046/j.1365-2958.1999.01421.x [DOI] [PubMed] [Google Scholar]
- 49. Vaughan P. S., Miura P., Henderson M., Byrne B., and Vaughan K. T. (2002) A role for regulated binding of p150(Glued) to microtubule plus ends in organelle transport. J. Cell Biol. 158, 305–319 10.1083/jcb.200201029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhang J., Li S., Fischer R., and Xiang X. (2003) Accumulation of cytoplasmic dynein and dynactin at microtubule plus ends in Aspergillus nidulans is kinesin dependent. Mol. Biol. Cell 14, 1479–1488 10.1091/mbc.e02-08-0516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wu X., Xiang X., and Hammer J. A. 3rd. (2006) Motor proteins at the microtubule plus-end. Trends Cell Biol. 16, 135–143 10.1016/j.tcb.2006.01.004 [DOI] [PubMed] [Google Scholar]
- 52. Yao X., Zhang J., Zhou H., Wang E., and Xiang X. (2012) In vivo roles of the basic domain of dynactin p150 in microtubule plus-end tracking and dynein function. Traffic 13, 375–387 10.1111/j.1600-0854.2011.01312.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Li S., Oakley C. E., Chen G., Han X., Oakley B. R., and Xiang X. (2005) Cytoplasmic dynein's mitotic spindle pole localization requires a functional anaphase-promoting complex, γ-tubulin, and NUDF/LIS1 in Aspergillus nidulans. Mol. Biol. Cell 16, 3591–3605 10.1091/mbc.e04-12-1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Krämer H., and Phistry M. (1996) Mutations in the Drosophila hook gene inhibit endocytosis of the boss transmembrane ligand into multivesicular bodies. J. Cell Biol. 133, 1205–1215 10.1083/jcb.133.6.1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Walenta J. H., Didier A. J., Liu X., and Krämer H. (2001) The Golgi-associated hook3 protein is a member of a novel family of microtubule-binding proteins. J. Cell Biol. 152, 923–934 10.1083/jcb.152.5.923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Malone C. J., Misner L., Le Bot N., Tsai M. C., Campbell J. M., Ahringer J., and White J. G. (2003) The C. elegans hook protein, ZYG-12, mediates the essential attachment between the centrosome and nucleus. Cell 115, 825–836 10.1016/S0092-8674(03)00985-1 [DOI] [PubMed] [Google Scholar]
- 57. Splinter D., Razafsky D. S., Schlager M. A., Serra-Marques A., Grigoriev I., Demmers J., Keijzer N., Jiang K., Poser I., Hyman A. A., Hoogenraad C. C., King S. J., and Akhmanova A. (2012) BICD2, dynactin, and LIS1 cooperate in regulating dynein recruitment to cellular structures. Mol. Biol. Cell 23, 4226–4241 10.1091/mbc.e12-03-0210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schroeder C. M., Ostrem J. M., Hertz N. T., and Vale R. D. (2014) A Ras-like domain in the light intermediate chain bridges the dynein motor to a cargo-binding region. Elife 3, e03351 10.7554/eLife.03351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lee I. G., Olenick M. A., Boczkowska M., Franzini-Armstrong C., Holzbaur E. L. F., and Dominguez R. (2018) A conserved interaction of the dynein light intermediate chain with dynein–dynactin effectors necessary for processivity. Nat. Commun. 9, 986 10.1038/s41467-018-03412-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Grotjahn D. A., Chowdhury S., Xu Y., McKenney R. J., Schroer T. A., and Lander G. C. (2018) Cryo-electron tomography reveals that dynactin recruits a team of dyneins for processive motility. Nat. Struct. Mol. Biol. 25, 203–207 10.1038/s41594-018-0027-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zheng W. (2017) Probing the energetics of dynactin filament assembly and the binding of cargo adaptor proteins using molecular dynamics simulation and electrostatics-based structural modeling. Biochemistry 56, 313–323 10.1021/acs.biochem.6b01002 [DOI] [PubMed] [Google Scholar]
- 62. Qiu R., Zhang J., and Xiang X. (2013) Identification of a novel site in the tail of Dynein heavy chain important for dynein function in vivo. J. Biol. Chem. 288, 2271–2280 10.1074/jbc.M112.412403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Cheong F. K., Feng L., Sarkeshik A., Yates J. R. 3rd, and Schroer T. A. (2014) Dynactin integrity depends upon direct binding of dynamitin to Arp1. Mol. Biol. Cell 25, 2171–2180 10.1091/mbc.e14-03-0842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Raaijmakers J. A., Tanenbaum M. E., and Medema R. H. (2013) Systematic dissection of dynein regulators in mitosis. J. Cell Biol. 201, 201–215 10.1083/jcb.201208098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. McCluskey K., Wiest A., and Plamann M. (2010) The Fungal Genetics Stock Center: a repository for 50 years of fungal genetics research. J. Biosci. 35, 119–126 10.1007/s12038-010-0014-6 [DOI] [PubMed] [Google Scholar]
- 66. Nayak T., Szewczyk E., Oakley C. E., Osmani A., Ukil L., Murray S. L., Hynes M. J., Osmani S. A., and Oakley B. R. (2006) A versatile and efficient gene-targeting system for Aspergillus nidulans. Genetics 172, 1557–1566 10.1534/genetics.105.052563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Yang L., Ukil L., Osmani A., Nahm F., Davies J., De Souza C. P., Dou X., Perez-Balaguer A., and Osmani S. A. (2004) Rapid production of gene replacement constructs and generation of a green fluorescent protein-tagged centromeric marker in Aspergillus nidulans. Eukaryot. Cell 3, 1359–1362 10.1128/EC.3.5.1359-1362.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Szewczyk E., Nayak T., Oakley C. E., Edgerton H., Xiong Y., Taheri-Talesh N., Osmani S. A., and Oakley B. (2006) Fusion PCR and gene targeting in Aspergillus nidulans. Nat. Protoc. 1, 3111–3120 [DOI] [PubMed] [Google Scholar]
- 69. Zhang J., Zhuang L., Lee Y., Abenza J. F., Peñalva M. A., and Xiang X. (2010) The microtubule plus-end localization of Aspergillus dynein is important for dynein–early-endosome interaction but not for dynein ATPase activation. J. Cell Sci. 123, 3596–3604 10.1242/jcs.075259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kelley L. A., and Sternberg M. J. (2009) Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 4, 363–371 10.1038/nprot.2009.2 [DOI] [PubMed] [Google Scholar]
- 71. Xiang X., Roghi C., and Morris N. R. (1995) Characterization and localization of the cytoplasmic dynein heavy chain in Aspergillus nidulans. Proc. Natl. Acad. Sci. U.S.A. 92, 9890–9894 10.1073/pnas.92.21.9890 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.